

Abstract
Objective
Research identified promising therapeutics in cell models of Amyotrophic Lateral Sclerosis (ALS), but there is limited progress translating effective treatments to animal models and patients, and ALS remains a disease with no effective treatment. One explanation stems from an acquired pharmacoresistance driven by the drug efflux transporters P-glycoprotein (P-gp) and breast cancer-resistant protein (BCRP), which we have shown are selectively upregulated at the blood-brain and spinal cord barrier (BBB/BSCB) in ALS mice and patients. Pharmacoresistance is well appreciated in other brain diseases, but overlooked in ALS despite many failures in clinical trials.
Methods
Here, we prove that a P-gp/BCRP-driven pharmacoresistance limits the bioavailability of ALS therapeutics using riluzole, the only FDA-approved drug for ALS and a substrate of P-gp and BCRP. ALS mice (SOD1-G93A) were treated with riluzole and elacridar, to block P-gp and BCRP, and monitored for survival as well as behavioral and physiological parameters.
Results
We show that riluzole, which normally is not effective when given at onset of symptoms, is now effective in the ALS mice when administered in combination with the P-gp/BCRP inhibitor elacridar. Chronic elacridar treatment increases riluzole Central nervous system (CNS) penetration, improves behavioral measures, including muscle function, slowing down disease progression, and significantly extending survival.
Interpretation
Our approach improves riluzole efficacy with treatment beginning at symptom onset. Riluzole will not provide a cure, but enhancing its efficacy postsymptoms by addressing pharmacoresistance demonstrates a proof-of-principle concept to consider when developing new ALS therapeutic strategies. We highlight a novel improved therapeutic approach for ALS and demonstrate that pharmacoresistance can no longer be ignored in ALS.
The limited progress in identifying successful therapies in Amyotrophic Lateral Sclerosis (ALS) has only resulted in one moderately effective pharmacological agent, riluzole.1,2 In research on the SOD1-G93A mouse model of ALS, riluzole showed a modest effect on survival when administrated prior to disease onset.3 Since then, follow-up studies have shown conflicting results,4–6 due to differences in trial design and lack of pharmacokinetic measurements. Riluzole brain disposition is limited in the ALS mouse model through interaction with the drug efflux transporters P-glycoprotein (P-gp) and breast cancer-resistant protein (BCRP) at the blood-brain/spinal cord barrier (BBB/BSCB).7,8 Accordingly, riluzole loses effectiveness as disease progresses in this model.9
Similarly, in patients with ALS, riluzole loses effectiveness in the later stages of disease.10 Studies in patients demonstrated that riluzole is particularly effective in the first 12 months of treatment, reducing mortality by 38% and that this initial efficacy is reduced to 19.4% at 21 months of treatment.11
The negligible therapeutic effect of riluzole in the ALS mice and its modest effect in patients could derive from acquired pharmacoresistance. In previous studies, we reported an ALS-specific and disease-driven increase in expression and function of two drug efflux transporters, P-gp and BCRP, in both ALS mouse spinal cord capillaries and in spinal cord tissue of ALS patients.12 The findings suggested that this increased expression potentially conferred an acquired pharmacoresistance, which could limit the bioavailability of ALS/CNS-targeted therapeutics.12,13
Here, we tested this hypothesis and determined whether improving riluzole CNS bioavailability through inhibition of P-gp and BCRP efflux transporter(s) improves and prolongs riluzole's therapeutic effect(s) in the ALS mice. Given that our ultimate goal is to translate this concept of inhibiting drug efflux to ALS patients' therapy, we further analyzed expression levels of P-gp in human ALS cases.
Materials and Methods
Animals
Mice were housed in accordance with Thomas Jefferson University IACUC and the NIH Guide for the Care and Use of Laboratory Animals. Protocol for this study received approval by \TJU Animal Care and Use Committee. Mutant SOD1-G93A mice modeling ALS [B6.Cg-Tg(SOD1-G93A)1Gur/J] were purchased from Jackson Laboratories (stock #004435). Male SOD1-G93A mice were bred with C57BL/6 females. Offspring were genotyped to determine presence of the human SOD1 transgene. As transgene copy number is known to fluctuate and decrease through subsequent breeding generations, quantitative PCR was performed to exclude mice with decreased SOD1 human transgene copy number.
P-glycoprotein knockout mice (P-gp−/−) on the FVB/N background were purchased from Taconic (stock #1487). Prior to crossing with SOD1-G93A mice, P-gp−/− mice were backcrossed to C57BL/6 mice (N = 5). After the fifth generation and a homogeneous C57BL/6 background, P-gp−/− mice were crossed with SOD1-G93A mice and genotyped to determine presence of the SOD1-G93A human transgene. As described above, mice were controlled for human transgene copy number via q-PCR.
Drug treatment and survival analysis
Transgenic mutant SOD1-G93A male mice were used in a preclinical drug study format. Mice were enrolled into one of three treatment groups: control + placebo/Elacridar (n = 25), Riluzole + placebo (n = 25), or Riluzole + Elacridar (n = 25). Riluzole (Sigma-Aldrich, St. Louis, MO, USA) was administered via the chow (125 mg/kg of chow). Elacridar (Tocris, Bristol, United Kingdom) was administered via a time-controlled release pellet (50 mg/10 day release, Innovative Research of America, Sarasota, FL), which was implanted subcutaneously in the back of the neck. Control chow was prepared in the same manner as the Riluzole chow. To control for the effects of surgical pellet implantation, control, and riluzole-treated mice also received placebo pellets every 10 days. Treatment of Riluzole and Elacridar began at 100 days of age and continued for the lifespan of all mice. Baseline and study measures were recorded for weight, grip strength, food consumption, and survival. Determination of end stage was the inability for a mouse to right itself after 30 sec of being placed on its side.
Grip strength and weight assessment
Hindlimb grip strength was recorded 2–3 times per week for each mouse. The rate of grip strength deterioration for Riluzole + placebo and Riluzole + Elacridar groups was determined based on the average grip strength per day per mouse and fit to a linear curve. The weights for each mouse were recorded 2–3 times per week for each mouse and the rate of decline was quantified for Riluzole + placebo and Riluzole + Elacridar groups.
CMAP recordings
Mice were anesthetized with 1% isoflurane and 0.5% oxygen. Following dermatotomy, stainless steel-stimulating needle electrodes were inserted at the sciatic notch, near the sciatic nerve. A ground electrode was placed subcutaneously in the back and a reference electrode placed subcutaneously at the ankle. The sciatic nerve was stimulated (0.2 msec duration; 1.6 mV amplitude) and the response recorded via a needle electrode inserted into the plantar muscle in the medial half of the foot, following the line connecting the first and fifth tarsal/metatarsal joints. Data were collected using ADI Powerlab 8/30 stimulator and BioAMP amplifier (ADInstruments, Colorado Springs, CO) and analyzed using Scope 3.5.6 (ADInstruments). The compound muscle action potential (CMAP) (M-wave) amplitude was measured from baseline to peak. Data were averaged between left and right hindlimb traces.
In vivo LD800 imaging
LD800 (1 mg/kg) was injected i.p; 20 min later animals were anesthetized with a ketamine:xylazine mixture and sacrificed via cardiac perfusion with heparinized PBS. Spinal cord samples were collected and embedded in low-melting point agarose and sectioned in 750 μm thick sections using a tissue chopper (McIlwain Tissue Chopper, The Mickle Laboratory Engineering Company; Goose Green, United Kingdom). Spinal cord tissue sections were then imaged with an Odyssey infrared imager (LI-COR Biosciences, Lincoln, Nebraska).
Mass spectrometry
Age-matched, adult mice (150 days of age) were injected intraperitoneally with Riluzole-HCl (12 mg/kg). After 1 h, mice were sacrificed with carbon dioxide, blood was collected via cardiac puncture, and mice were flushed with saline (0.9%). Blood was placed in EDTA-coated eppendorf tubes and centrifuged to collect plasma. Brains and spinal cords were harvested and flash frozen. All samples were stored at −80°C until extraction. For plasma samples, nine volumes ice-cold, 100% ethanol was added to each sample, incubated overnight at 4°C and centrifuged for 25 min (130 g 4°C). Ethanol supernatant was removed and pellets were dried in a speed vacuum. Pellets were stored at −80°C. Pellets were resuspended in 0.1% formic acid, water bath sonicated for 20 min, and centrifuged at 15 rcf for 30 min. Supernatant was further diluted in 0.1% formic acid, and analyzed by LC-MS/MS on a Thermo LTQ-Orbitrap XL mass spectrometer at the Wistar Proteomics Facility. Riluzole-HCl had elution peaks at 17 min, peak areas were used at 5 ppm Mass Accuracy to find relative concentration amounts from standard curve.
Motor neuron counts
Mice were perfused with Dulbecco's phosphate-buffered saline (DPBS) and spinal cord lumbar segments were embedded in Optimal cutting temperature (OCT) freezing medium and stored at −20°C until sectioned. Forty micrometer fresh frozen tissue sections were dissected using a temperature controlled cryostat (MICROM HM 505 E, Thermo Scientific, Kalamazoo, MI). Tissue sections were coded blind and processed simultaneously. Sections were fixed with 2.5% paraformaldehyde for 10 min at room temperature. Every second section was stained about 3–4 min with 0.1% cresyl violet acetate and dipped in 95% Ethanol and 95% ethanol + glacial acetic acid to facilitate the staining. Finally, slides were mounted with permount and dried overnight at room temperature. Stained sections were visualized under a 60× oil immersion objective of a bright-field microscope (Olympus, Center Valley, PA). Four sections per animal were analyzed per group (n = 7). Large pyramidal motor neurons positively stained for cresyl violet with a prominent nucleus and size at least ≥20 μm2 were counted. Motor neurons were quantified using the optical fractionator workflow module of stereo investigator software (version 8) employing a grid size 75 × 75 μm and sampling grid size 100 × 100 μm. All analysis was carried out by an investigator blinded to the samples. Motor neuron counts per group were later quantified using Student's t-test and represented as average ± SEM.
Periodic acid schiff staining
Saline perfused liver was postfixed with 4% paraformaldehyde for 24 h followed by 24 h incubation in 30% sucrose solution, embedded in Optimal cutting temperature (OCT) solution, and stored at −80°C until sectioned (10 μm thickness). Sections were hydrated with water, immersed in Periodic acid schiff (PAS) solutions (Sigma; St. Louis, MO 395) for 5 min at room temperature, and washed in water. Sections were immersed for 4 min in Schiff's reagent and washed in water for 5 min. Sections were counterstained with Hematoxylin solution, dehydrated in ethanol and HemoD solution, mounted, and imaged.
Immunofluorescence of patient tissue
OCT-embedded spinal cord and hippocampus tissue were cryostat sectioned at 10 μm. Slides were rinsed once in 1.5X TBS buffer and postfixed in 4% paraformaldehyde for 10 min at room temperature followed by treatment with antigen unmasking solution for 2 min at −20°C (33% acetic acid and 66% ethanol). After washing and blocking (2% BSA, 0.3% triton-X, 5% horse serum in 1.5X TBS), slides were incubated with primary antibodies (C219 from Covance 1:50; Bardford, CT and vWF from Dako 1:50; Carpinteria, CA). After washing, tissue was then incubated with fluorescent secondary antibodies, mounted with (4,6-diaminido-2-phenylindole) DAPI antifade solution, and imaged.
Results
Homogenates of lumbar spinal cords of two sporadic and two familial ALS patients displayed increased levels of P-gp compared to controls, including a control patient with Friedreich's Ataxia (Fig.1A). By immunohistochemistry, increases in P-gp expression are found in endothelial cells of the BSCB (Fig.1B and C). We also found a selective, tissue-specific increase of P-gp expression in areas affected by the disease, specifically spinal cord, while P-gp levels remained low in the unaffected hippocampus (Fig.1C). Microvascular leakage and reduced tight junction protein expression are known to occur in ALS and could contribute to motor neuron damage.14–19 Upregulation of P-gp could occur as a compensatory response to a leaking BSCB. However, activation of P-gp can still prevent effective drug delivery in the CNS, reducing drug bioavailability. To demonstrate this, we examined the in vivo accumulation of a specific P-gp substrate, LD800, in the spinal cord of the ALS mouse model where expression and function of P-gp increases as disease progresses4. Symptomatic mice have a marked decrease in LD800 accumulation compared to nontransgenic and P-gp−/− mice (Fig.2A and B).
Figure 1.

ALS patients have tissue-specific increases in P-gp protein expression. Protein expression of P-gp in ALS patients compared to controls and levels of P-gp in diseased versus nondiseased tissue areas were examined. (A) Densitometric analysis of P-gp protein expression in lumbar spinal cord shows an increase (1.2- to 1.5-fold depending on the patient) in sporadic (sALS) and familial (fALS) ALS compared to non-neuromuscular controls and a patient with Friedreich's Ataxia, a different neurological, spinal neuron disease. (B) Immunohistochemistry of lumbar spinal cord sections shows the colocalization of P-gp with the endothelial cell marker, von Willebrand Factor. P-gp expression is higher in endothelial cells of ALS patients in the ventral horn of lumbar spinal cord compared to controls (scale bar = 20 μm). (C) Levels of P-gp expression in the lumbar spinal cord and the hippocampus, an unaffected brain region, of the same ALS patient were compared. P-gp expression increases are tissue specific with higher levels of P-gp expression in the spinal cord of the ALS patient compared to the unaffected area (hippocampus) (scale bar = 20 μm). Thus, in ALS patients there is a tissue-specific increase in P-gp protein expression, which is selective to endothelial cells at the level of the BSCB.
Figure 2.
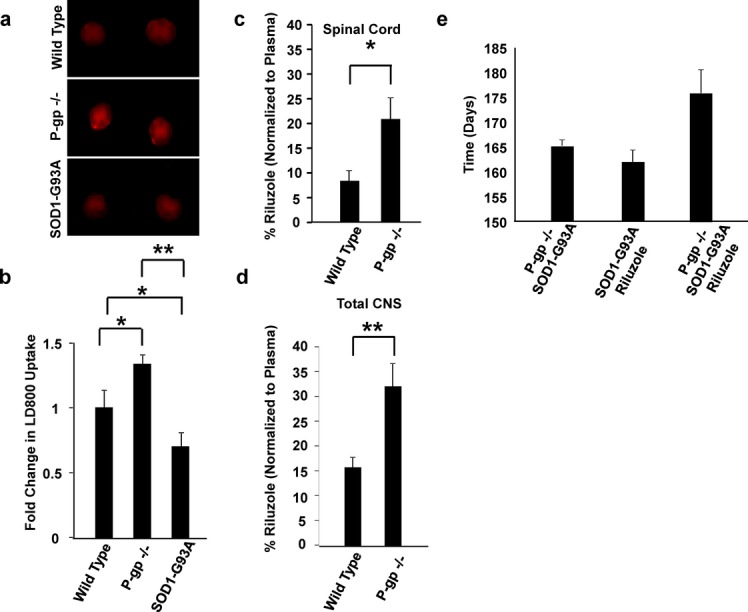
P-gp substrate disposition is altered in SOD1-G93A mice compared to wild-type (WT), and genetic inhibition of P-gp increases the effectiveness of riluzole in SOD1-G93A mice. A specific P-gp substrate, LD800, was injected intraperitoneally into WT, P-gp knockout (P-gp−/−), and symptomatic SOD1-G93A mice. Symptomatic SOD1-G93A mice have decreased accumulation of LD800 into the spinal cord compared to WT and P-gp−/− mice (P = 0.021 and P < 0.001, respectively; A and B). Riluzole is a P-gp substrate and has increased spinal cord (C) and total CNS (D) penetration in P-gp knockout mice. WT and P-gp−/− mice were acutely treated with intraperitoneal injections of riluzole, and the concentrations of riluzole in the plasma and spinal cord tissue were determined via mass spectrometry. (C) The percent of riluzole accumulation, normalized to riluzole plasma concentrations, in the spinal cords of P-gp−/− mice is significantly higher than the amount of riluzole in WT mice (20.9 ± 4.3% and 8.4 ± 2.2%, respectively; P = 0.028). (D) Accumulation of riluzole in the total CNS (brain and spinal cord) is also significantly higher in P-gp−/− compared to WT mice (31.9 ± 4.7% and 15.6 ± 2.0%, respectively; P = 0.010). (E) As compared to untreated P-gp−/−:: SOD1-G93A mice and SOD1-G93A riluzole-treated mice, P-gp−/−:: SOD1-G93A riluzole-treated mice have a trend toward increased survival (165.2 ± 2.89, 162.0 ± 2.51, and 176.0 ± 4.2, respectively; n = 5–6).
We then analyzed the extent to which P-gp alters riluzole disposition into the CNS and found that the percent of riluzole accumulation in the spinal cord and brain was higher in P-gp−/− mice compared to age-matched, nontransgenic mice (Fig.2C and D). Then, we crossed P-gp−/− and SOD1-G93A mice to obtain ALS mice lacking P-gp (P-gp−/−::SOD1-G93A+/−) and treated them with riluzole beginning at symptom onset (at ∼100 days of age). As very low numbers of P-gp−/−::SOD1-G93A mice are obtained from these crosses, we limited our analysis to 5–6 mice per group to allow initial evaluation of the impact of P-gp knock down on disease and riluzole penetration. Knocking out P-gp from the ALS mice improved significantly the therapeutic effect of riluzole (Fig.2E), further strengthening the assumption that as ALS progresses in mice and Pg-p and BCRP increase,12 riluzole penetration and efficacy decrease. To compensate for this progressive decrease in therapeutic levels of riluzole in the spinal cord of diseased mice, we pharmacologically inhibited the activity of Pg-p and BCRP. Inhibitors of drug efflux transporters are normally used to restore drug sensitivity in diseases in which the issue of pharmacoresistance is well characterized, such as leukemia, other forms of cancer, and epilepsy.20–22 Third-generation inhibitors,23 such as elacridar (GF120918), are highly specific24,25 and tolerated in patients.26–28
We treated the mice with elacridar, which provides dual inhibition of P-gp and BCRP.29 Chronic elacridar treatment, beginning at disease onset, is safe and does not alter disease progression in SOD1-G93A mice (Fig.3A). As expected, chronic treatment of elacridar did not affect overall expression of P-gp (Fig.3B) or BCRP (not shown); it did inhibit their function, though, resulting in a significant increase in riluzole spinal cord concentrations in diseased mice (Fig.3C). Despite higher levels of systemic riluzole, the riluzole + elacridar-treated mice did not show signs of liver pathology (Fig.3D), indicating that elacridar did not cause liver toxicity and that increasing systemic riluzole concentrations via elacridar were not hepatotoxic.
Figure 3.

Chronic elacridar treatment alone does not alter survival or P-gp expression levels, but does increase riluzole accumulation in SOD1-G93A mouse spinal cord. (A) Chronic treatment of elacridar, beginning at disease onset, is safe and does not alter disease progression in the SOD1-G93A ALS mice (Log-rank Mantel-Cox, χ2 = 0.046, P = 0.830). (B) P-gp expression levels in riluzole and riluzole + elacridar-treated mice are not altered. (C) Penetration of riluzole in the spinal cord of symptomatic 140-day-old mice co-treated with elacridar was measured by mass spectrometry compared to riluzole-only-treated mice. Significantly higher levels of riluzole were detected in riluzole + elacridar-treated mice compared to riluzole + placebo-treated aged-matched 140-day-old ALS mice (295 ± 70.9%; P = 0.016). (D) Periodic acid Schiff staining of liver sections from riluzole + placebo-treated mice and riluzole + elacridar-treated mice (scale bar = 50 μm). Chronic treatment with riluzole and elacridar does not cause overt toxicity to the liver of SOD1-G93A mice compared to riluzole treatment alone. (E) SOD1-G93A study mice have the same levels of the human transgene as quantified by qRT-PCR of DNA isolated from mouse tail. Mice displaying higher or lower copy numbers were excluded.
These results looked promising, but ultimately, increased drug bioavailability is therapeutically meaningful only if it translates into improved efficacy. To find out, we tested riluzole as the candidate drug. As the only drug with a marginal, albeit variable, effect in the SOD1-G93A mice, it was an ideal candidate.
So we initiated riluzole and elacridar treatment in our mouse models. Most published preclinical studies in mice began riluzole treatment presymptomatically (between 50 and 60 days) and prior to disease onset.3,4,6,30,31 This is based on the assumption that high levels of mutant SOD1 expression in mice lead to an “enhanced” disease that needs to be managed aggressively (http://www.prize4life.org/page/prizes/Treatment_prize). But most of the compounds tested presymptomatically have only delayed disease onset rather than slowed disease progression with an overall extension of lifespan <10%.32,33 In fact, a delay in onset with pharmacological treatment is not likely to be predictive of how well a drug may perform clinically.
Our prior observations showed that P-gp and BCRP function increased beginning at symptom onset and peaked at symptomatic stage,12 so we initiated the riluzole and elacridar treatment at onset (P100). In addition, we wanted to mimic, to the extent possible, the therapeutic regimen of the patients who are treated with riluzole after they are diagnosed with the disease.
Our analysis of the SOD1-G93A mice consisted of three groups of mice (n = 25 males/group): (1) control, placebo-treated; (2) riluzole + placebo-treated; and (3) riluzole + elacridar-treated. Copy number of the SOD1-G93A human transgene was controlled (Fig.3E). We found that cotreatment using riluzole + elacridar beginning at symptoms onset significantly extended survival compared to control/placebo and riluzole/placebo groups (Fig.4A). Riluzole alone was not beneficial and did not extend survival compared to control mice. However, when given in combination with elacridar, it significantly slowed disease progression, extending mouse survival by 13% from onset of symptoms (Fig.4A). This specific effect on disease progression is superior to many pharmacological interventions in ALS mice,32 which primarily affected disease onset but not duration.
Figure 4.

Cotreatment with riluzole and elacridar increases survival, NMJ function, behavior, and motor neuron counts compared to riluzole treatment alone. (A) Cotreatment of riluzole + elacridar (165 ± 2.1 days) significantly extends survival compared to control + placebo (156.7 ± 2.8 days) and riluzole + placebo (159.7 ± 1.9 days) groups (Log-rank Mantel-Cox, χ2 = 7.292, P = 0.026; Logrank test for trend, χ2 = 6.615, P = 0.010; Gehan-Breslow-Wilcoxon, χ2 = 8.226, P = 0.016). (B and C) Compound muscle action potentials (CMAPs) have higher peak amplitudes in 140 day riluzole + elacridar-treated mice compared to age-matched riluzole + placebo-treated mice (7.48 ± 0.67 mV and 5.00 ± 0.67 mV, respectively; P = 0.017). (D) Riluzole + elacridar treatment sustains higher CMAP values throughout treatment compared to riluzole + placebo treatment. (E) In the riluzole + elacridar-treated mice the slope of hind limb strength decline is significantly decreased compared to the riluzole + placebo group (P < 0.001). (F and G) Quantification (neurons larger than 400 μm) and stained lumbar spinal cord tissue sections of motor neurons in the ventral horn of SOD1-G93A mice.
We also note the negative impact that consecutive surgeries of pellet implantation had on the mice. The life-span of placebo pellet-implanted SOD1-G93A mice decreased by ∼6 days compared to nonsurgery ALS mice (156.7 ± 2.8 days and 162.9 ± 0.69 days, respectively; −3.8%; P < 0.05), likely due to the negative impact of multiple pellet implantation surgeries. The full potential of the beneficial effect of riluzole + elacridar might have been negatively skewed by the pellet implantation regimen.
Importantly, treatment of riluzole and elacridar significantly improved vital parameters (Fig.4). We treated two additional cohorts of mice with riluzole + placebo (n = 10) or riluzole + elacridar (n = 11), from 100 to 140 days, and performed electrophysiological recordings from the plantar muscle following sciatic nerve stimulation to measure functional innervation by motor neurons of the lumbar spinal cord (Fig.4B and C). CMAPs had higher peak amplitudes in 140 day riluzole/elacridar mice compared to age-matched riluzole/placebo mice (Fig.4B and C). Throughout the 40-day treatment, the riluzole/elacridar mice maintained significantly higher CMAP values compared to the riluzole/placebo mice (Fig.4C). Accordingly, in the same mice, hindlimb grip strength was improved (Fig.4E). There was a trend, although not significant, for an increased number of remaining motor neurons in the lumbar spinal cord of P140 mice cotreated with riluzole and elacridar.
That we did not see a significant increase in motor neuron number, but did see significant physiological improvements, corresponds with previous studies that reported a dissociation between motor neuron death and disease progression in mutant SOD1 mice.34,35 Our study confirms that stopping motor neuron death may not be sufficient to slow disease, but that maintaining motor neuron function may be sufficient.
Discussion
The impact of P-gp and drug efflux is routinely considered in drug development,36 but tests are normally performed in healthy animals or healthy individuals to determine whether a drug can cross the BBB. The data presented here and in our previous studies,12 argue that proper pharmacodynamic and pharmacokinetic studies need to be analyzed in patients and disease-relevant models rather than in normal subjects and that P-gp and BCRP function, in particular, needs to be considered in the context of ALS over disease progression when these two drug efflux transporters begin to effectively pump out therapeutics and when their function increases incrementally with the disease.
Clearly, an “improved” riluzole effectively maintained functional motor neurons, as shown by the CMAPs and behavioral results, and ultimately slowed disease. Thus, our study provides a proof-of-principle concept that pharmacologically inhibiting P-gp and BCRP transporter activity can improve ALS pharmacotherapy.
While we acknowledge that riluzole is not very efficacious, it served as a proof-of-principle compound to test the impact of drug efflux transporter-mediated pharmacoresistance in ALS. Riluzole is the only moderately effective drug available for ALS treatment and it is also a substrate for P-gp and BCRP. Additionally, riluzole is the only drug whose marginal and inconsistent effects in mice translated into a consistent effect in patients. Thus, it is, so far, the only candidate drug to use in the ALS mice to demonstrate that it is possible to enhance drug bioavailability specifically for ALS therapeutics by targeting drug efflux transporter-mediated pharmacoresistance. Our data clearly demonstrate that by blocking P-gp and BCRP, it is possible to enhance riluzole CNS penetration in mice, ultimately restoring its efficacy even when administration begins at onset. As our data indicate that riluzole penetration is inversely correlated to riluzole's responsiveness in mice: they also strongly suggest that the decrease in riluzole efficacy observed in patients as disease progresses11 may derive from the parallel disease-driven increase in pharmacoresistance. As seen in the clinic, there is variability in riluzole efficacy across patients, which may be attributable to individual variations in expression levels of drug efflux transporters. In future studies, there is an opportunity to evaluate functional levels of P-gp and BCRP in ALS patients and eventually establish a tailored dosing strategy for those patients with high pharmacoresistance potentials (i.e., high expression and activity levels of P-gp and BCRP). Therefore, revisiting riluzole therapy by inhibiting pharmacoresistance could improve quality of life of ALS patients until a more efficacious therapeutic strategy is identified. However, targeting P-gp and BCRP has broader implications for ALS therapeutics that go beyond riluzole itself. P-gp and, to a lesser extent, BCRP have broad substrate specificity. Their function has been shown to limit bioavailability of multiple drugs.36 Accordingly, inhibition of these transporters with elacridar26,27 or Tariquidar 37 has proven effective in increasing bioavailability and efficacy of different drugs in cancer and epileptic patients, without causing overt toxicity. Thus, our data indicate that the same mechanism of acquired pharmacoresistance might apply to other ALS therapeutics and that blocking P-gp and BCRP with elacridar or similar transporters' inhibitors might also improve delivery, and ultimately, efficacy, of other candidate drugs. We have identified P-gp and BCRP as the only two efflux transporters specifically upregulated in ALS. Thus, future candidate drugs should be tested to determine whether they are P-gp/BCRP substrates, and P-gp/BCRP inhibition should be considered when designing future pre- and clinical trials with drugs whose bioavailability is limited by these two transporters.
Our data also indicate that when designing new or redesigning previously failed ALS trials, rather than focusing on increasing the dose of the drug to maximize its effect, we should target P-gp and BCRP as an effective way to control and improve bioavailability as disease progresses. Again, using riluzole as an example, one may argue that to enhance riluzole CNS bioavailability and concentrations, thus maintaining its therapeutic effects, it would be possible to increase its dosage without blocking P-gp and/or BCRP. However, our data show that in the long run, increasing riluzole dose would not be as effective as it may be expected since disease pathogenic mechanisms would continue to incrementally upregulate P-gp and BCRP expression levels and function, effectively pumping riluzole out of the CNS regardless of the original dose. In fact, studies have attempted to increase riluzole dosage with no real improvement of efficacy (reviewed in 33) and it has already been demonstrated that riluzole clearance is independent of dose.37 Two additional factors suggest that increasing riluzole concentrations would not be effective. First, these transporters depend on ATP to extrude their substrates. This characteristic renders them less sensitive to saturation under conditions of ionic unbalances often occurring in disease or in situations of increased substrate concentrations. Regardless of the substrate concentrations (e.g., riluzole), ATP-dependent Pg-p and BCRP would still be able to function at no saturation. Second, higher systemic doses of riluzole might increase the chances of hepatotoxicity as the ALS-driven increase in P-gp and BCRP seems to be tissue specific, and using elacridar may be the way to just increase riluzole CNS concentrations without overt toxicity. Overall, our studies suggest that to maintain efficacious levels of therapeutics throughout disease progression adjusting doses of elacridar (or other more potent and selective P-gp/BCRP inhibitors yet to be developed) may be the most valuable solution.
In the short term, with no better pharmacological tools available, we have the opportunity to improve the effects of riluzole in patients as well as begin a reevaluation of other drugs that failed in preclinical and clinical trials. In the long-term, we will have the opportunity to evaluate on a drug-to-drug and patient-to-patient basis the contribution of P-gp and BCRP altering drug bioavailability and therapeutic efficacy of new ALS therapeutics.
Acknowledgments
We thank N. Maragakis and T. Heiman-Patterson for contributing ALS patient tissue; L. Kenyon for human control tissue and T. Hala for technical assistance of tissue collection. This work was funded by DOD W81XWH-11-1-0767 to P. P.; Landenberger Foundation to P. P.; National Institutes of Health- RO1-NS074886 to D. T.; National Institutes of Health- F31-NS080539 to M. R. J.; MDA Developmental Award to D. J.; Farber Family Foundation to P. P. and D. T.
Conflict of Interest
Dr. Jablonski reports grants from National Institutes of Health, during the conduct of the study; In addition, has a patent Novel Methods of Treating a Neurodegenerative Disease in a Mammal pending. Dr. Jacob reports grants from Muscular Dystrophy Association, during the conduct of the study. Dr. Trotti reports grants from National Institutes of Health, during the conduct of the study; In addition, has a patent Novel Methods of Treating a Neurodegenerative Disease in a Mammal pending. Dr. Pasinelli reports grants from Department of Defense, Landenberger Foundation, during the conduct of the study; In addition, has a patent Novel Methods of Treating a Neurodegenerative Disease in a Mammal pending.
References
- 1.Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14:248–264. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- 2.Sreedharan J, Brown RH. Amyotrophic lateral sclerosis: problems and prospects. Ann Neurol. 2013;74:309–316. doi: 10.1002/ana.24012. [DOI] [PubMed] [Google Scholar]
- 3.Gurney ME, et al. Benefit of vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann Neurol. 1996;39:147–157. doi: 10.1002/ana.410390203. [DOI] [PubMed] [Google Scholar]
- 4.Del Signore SJ, et al. Combined riluzole and sodium phenylbutyrate therapy in transgenic amyotrophic lateral sclerosis mice. Amyotroph Lateral Scler. 2009;10:85–94. doi: 10.1080/17482960802226148. [DOI] [PubMed] [Google Scholar]
- 5.Scott S, et al. Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Scler. 2008;9:4–15. doi: 10.1080/17482960701856300. [DOI] [PubMed] [Google Scholar]
- 6.Waibel S, Reuter A, Malessa S, et al. Rasagiline alone and in combination with riluzole prolongs survival in an ALS mouse model. J Neurol. 2004;251:1080–1084. doi: 10.1007/s00415-004-0481-5. [DOI] [PubMed] [Google Scholar]
- 7.Milane A, et al. Minocycline and riluzole brain disposition: interactions with P-glycoprotein at the blood-brain barrier. J Neurochem. 2007;103:164–173. doi: 10.1111/j.1471-4159.2007.04772.x. [DOI] [PubMed] [Google Scholar]
- 8.Milane A, et al. Interactions between riluzole and ABCG2/BCRP transporter. Neurosci Lett. 2009;452:12–16. doi: 10.1016/j.neulet.2008.12.061. [DOI] [PubMed] [Google Scholar]
- 9.Dunlop J, Beal McIlvain H, She Y, Howland DS. Impaired spinal cord glutamate transport capacity and reduced sensitivity to riluzole in a transgenic superoxide dismutase mutant rat model of amyotrophic lateral sclerosis. J Neurosci. 2003;23:1688–1696. doi: 10.1523/JNEUROSCI.23-05-01688.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zoccolella S, et al. Riluzole and amyotrophic lateral sclerosis survival: a population-based study in southern Italy. Eur J Neurol. 2007;14:262–268. doi: 10.1111/j.1468-1331.2006.01575.x. [DOI] [PubMed] [Google Scholar]
- 11.Bensimon G, Lacomblez L, Meininger V Group AS. A controlled trial of riluzole in amyotrophic lateral sclerosis. N Engl J Med. 1994;330:585–591. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- 12.Jablonski MR, et al. Selective increase of two ABC drug efflux transporters at the blood-spinal cord barrier suggests induced pharmacoresistance in ALS. Neurobiol Dis. 2012;47:194–200. doi: 10.1016/j.nbd.2012.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller DS. Regulation of P-glycoprotein and other ABC drug transporters at the blood-brain barrier. Trends Pharmacol Sci. 2010;31:246–254. doi: 10.1016/j.tips.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winkler EA, et al. Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 2013;125:111–120. doi: 10.1007/s00401-012-1039-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garbuzova-Davis S, et al. Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res. 2012;1469:114–128. doi: 10.1016/j.brainres.2012.05.056. [DOI] [PubMed] [Google Scholar]
- 16.Garbuzova-Davis S, et al. Reduction of circulating endothelial cells in peripheral blood of ALS patients. PLoS One. 2010;5:e10614. doi: 10.1371/journal.pone.0010614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nicaise C, et al. Impaired blood-brain and blood-spinal cord barriers in mutant SOD1-linked ALS rat. Brain Res. 2009;1301:152–162. doi: 10.1016/j.brainres.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 18.Zhong Z, et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci. 2008;11:420–422. doi: 10.1038/nn2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garbuzova-Davis S, et al. Ultrastructure of blood-brain barrier and blood-spinal cord barrier in SOD1 mice modeling ALS. Brain Res. 2007;1157:126–137. doi: 10.1016/j.brainres.2007.04.044. [DOI] [PubMed] [Google Scholar]
- 20.Brandt C, Bethmann K, Gastens AM, Löscher W. The multidrug transporter hypothesis of drug resistance in epilepsy: proof-of-principle in a rat model of temporal lobe epilepsy. Neurobiol Dis. 2006;24:202–211. doi: 10.1016/j.nbd.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 21.Bankstahl JP, Hoffmann K, Bethmann K, Loscher W. Glutamate is critically involved in seizure-induced overexpression of P-glycoprotein in the brain. Neuropharmacology. 2008;54:1006–1016. doi: 10.1016/j.neuropharm.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 22.Agarwal S, Hartz AMS, Elmquist WF, Bauer B. Breast cancer resistance protein and P-glycoprotein in brain cancer: two gatekeepers team up. Current Pharm Des. 2011;17:2793–2802. doi: 10.2174/138161211797440186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Potschka H. Role of CNS efflux drug transporters in antiepileptic drug delivery: overcoming CNS efflux drug transport. Adv Drug Deliv Rev. 2012;64:943–952. doi: 10.1016/j.addr.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 24.Kuntner C, et al. Dose-response assessment of tariquidar and elacridar and regional quantification of P-glycoprotein inhibition at the rat blood-brain barrier using (R)-[(11)C]verapamil PET. Eur J Nucl Med Mol Imaging. 2010;37:942–953. doi: 10.1007/s00259-009-1332-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shaik N, Giri N, Pan G, Elmquist WF. P-glycoprotein-mediated active efflux of the anti-HIV1 nucleoside abacavir limits cellular accumulation and brain distribution. Drug Metab Dispos. 2007;35:2076–2085. doi: 10.1124/dmd.107.017723. [DOI] [PubMed] [Google Scholar]
- 26.Kruijtzer CMF. Increased oral bioavailability of topotecan in combination with the breast cancer resistance protein and P-glycoprotein inhibitor GF120918. J Clin Oncol. 2002;20:2943–2950. doi: 10.1200/JCO.2002.12.116. [DOI] [PubMed] [Google Scholar]
- 27.Kuppens IELM, et al. A phase I, randomized, open-label, parallel-cohort, dose-finding study of elacridar (GF120918) and oral topotecan in cancer patients. Clin Cancer Res. 2007;13:3276–3285. doi: 10.1158/1078-0432.CCR-06-2414. [DOI] [PubMed] [Google Scholar]
- 28.Stokvis E, Rosing H, Causon RC, et al. Quantitative analysis of the P-glycoprotein inhibitor Elacridar (GF120918) in human and dog plasma using liquid chromatography with tandem mass spectrometric detection. J Mass Spectrom. 2004;39:1122–1130. doi: 10.1002/jms.687. [DOI] [PubMed] [Google Scholar]
- 29.Kawamura K, et al. Evaluation of limiting brain penetration related to P-glycoprotein and breast cancer resistance protein using [(11)C]GF120918 by PET in mice. Mol Imaging Biol. 2011;13:152–160. doi: 10.1007/s11307-010-0313-1. [DOI] [PubMed] [Google Scholar]
- 30.Gurney ME, Fleck TJ, Himes CS, Hall ED. Riluzole preserves motor function in a transgenic model of familial amyotrophic lateral sclerosis. Neurology. 1998;50:62–66. doi: 10.1212/wnl.50.1.62. [DOI] [PubMed] [Google Scholar]
- 31.Kriz J, Gowing G, Julien J-P. Efficient three-drug cocktail for disease induced by mutant superoxide dismutase. Ann Neurol. 2003;53:429–436. doi: 10.1002/ana.10500. [DOI] [PubMed] [Google Scholar]
- 32.Benatar M. Lost in translation: treatment trials in the SOD1 mouse and in human ALS. Neurobiol Dis. 2007;26:1–13. doi: 10.1016/j.nbd.2006.12.015. [DOI] [PubMed] [Google Scholar]
- 33.Traynor BJ, et al. Neuroprotective agents for clinical trials in ALS: a systematic assessment. Neurology. 2006;67:20–27. doi: 10.1212/01.wnl.0000223353.34006.54. [DOI] [PubMed] [Google Scholar]
- 34.Gould TW, et al. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci. 2006;26:8774–8786. doi: 10.1523/JNEUROSCI.2315-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parone P, et al. Enhancing mitochondrial calcium buffering capacity reduces aggregation of misfolded SOD1 and motor neuron cell death without extending survival in mouse models of inherited amyotrophic lateral sclerosis. J Neurosci. 2013;33:4657–4671. doi: 10.1523/JNEUROSCI.1119-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu X, Chen C, Smith BJ. Progress in brain penetration evaluation in drug discovery and development. J Pharmacol Exp Ther. 2008;325:349–356. doi: 10.1124/jpet.107.130294. [DOI] [PubMed] [Google Scholar]
- 37.Bruno R, et al. Population pharmacokinetics of riluzole in patients with amyotrophic lateral sclerosis. Clin Pharmacol Ther. 1997;62:518–526. doi: 10.1016/S0009-9236(97)90047-3. [DOI] [PubMed] [Google Scholar]