

Abstract
Objective
To identify the cause of a so-far unreported phenotype of infantile-onset multisystem neurologic, endocrine, and pancreatic disease (IMNEPD).
Methods
We characterized a consanguineous family of Yazidian-Turkish descent with IMNEPD. The two affected children suffer from intellectual disability, postnatal microcephaly, growth retardation, progressive ataxia, distal muscle weakness, peripheral demyelinating sensorimotor neuropathy, sensorineural deafness, exocrine pancreas insufficiency, hypothyroidism, and show signs of liver fibrosis. We performed whole-exome sequencing followed by bioinformatic analysis and Sanger sequencing on affected and unaffected family members. The effect of mutations in the candidate gene was studied in wild-type and mutant mice and in patient and control fibroblasts.
Results
In a consanguineous family with two individuals with IMNEPD, we identified a homozygous frameshift mutation in the previously not disease-associated peptidyl-tRNA hydrolase 2 (PTRH2) gene. PTRH2 encodes a primarily mitochondrial protein involved in integrin-mediated cell survival and apoptosis signaling. We show that PTRH2 is highly expressed in the developing brain and is a key determinant in maintaining cell survival during human tissue development. Moreover, we link PTRH2 to the mTOR pathway and thus the control of cell size. The pathology suggested by the human phenotype and neuroimaging studies is supported by analysis of mutant mice and patient fibroblasts.
Interpretation
We report a novel disease phenotype, show that the genetic cause is a homozygous mutation in the PTRH2 gene, and demonstrate functional effects in mouse and human tissues. Mutations in PTRH2 should be considered in patients with undiagnosed multisystem neurologic, endocrine, and pancreatic disease.
Introduction
Intellectual disability disorders affect 2–3% of the population and are often accompanied by comorbidities. Here, we report a novel intellectual disability phenotype of infantile-onset multisystem neurologic, endocrine, and pancreatic disease (IMNEPD) caused by homozygous mutations in the peptidyl-tRNA hydrolase 2 gene (PTRH2). Functional studies underline the pathogenicity of the identified mutation.
PTRH2 (synonym BIT1, Bcl-2 inhibitor of transcription 1) is an evolutionarily highly conserved protein that prevents the accumulation of dissociated peptidyl-tRNA, which could inhibit protein synthesis.1,2 PTRH2 moonlights in other functions that determine survival or death of a cell. It is part of an integrin signaling complex that mediates survival of cells attached to the extracellular matrix (ECM).3,4 Upon integrin-mediated cell attachment, PTRH2 promotes cell survival through interaction with focal adhesion kinase (FAK) and subsequent activation of the PI3K-AKT-NFkB pathway.4,5 PTRH2 expression also induces Bcl-2 transcription4 and blocks the intrinsic mitochondrial apoptotic pathway. On the other hand, PTRH2 is a key player in anoikis, a process defined as cell death caused by loss of cell attachment to the ECM.3,5 Upon loss of integrin-mediated cell attachment to the ECM, PTRH2 is phosphorylated, released from mitochondria into the cytosol, and complexes with the transcriptional regulator amino-terminal enhancer of split (AES) to promote apoptosis.3,6 PTRH2 inhibits extracellular signal-regulated kinase (ERK) phosphorylation,4,5 and it can be phosphorylated by protein kinase D1.6 Therefore, PTRH2 functions as a phosphoprotein that regulates NFkB and ERK signaling and thereby controls integrin-mediated apoptotic signals. The preliminary characterization of Ptrh2 mutant mice revealed homozygous Ptrh2 mutants develop a “runting” (dystrophy) syndrome postnatally and die within the first 2 weeks of life.5 The physiologic function of PTRH2, however, is unknown.
Subjects and Methods
Informed consent was obtained from the parents of the patients for the molecular genetic analysis, the publication of clinical data, photos, magnetic resonance images (MRI), and studies on immortalized lymphocytes and fibroblasts. The human study was approved by the local ethics committees of the Charité (approval no. EA1/212/08), and all animal experiments were carried out in accordance to the national ethic principles (registration no. T0344/12 and 07-023-7).
Genetic analyses
Homozygosity linkage intervals with LOD > 2 and length >1 Mb were identified using the Affymetrix High Wycombe, UK SNP array 6.0. For whole-exome sequencing, enriched genomic DNA was sequenced by single-end 101 bp using a Hiseq2000 sequencing machine with an output sequences amount of 12 Gb and >94% of the coding regions covered with >20-folds. The raw sequencing data can be retrieved from the Sequence Reads Archive (http://www.ncbi.nlm.nih.gov/sra; accession no. SRA385191). We used SOAP2.20 for reads alignment (http://soap.genomics.org.cn/) and the Medical Resequencing Analysis Pipeline (MERAP, http://sourceforge.net/projects/merap/) for variant calling, filtering, and prioritization. We detected single-nucleotide variants (SNVs), insertions and deletions (indels), and copy number variations (CNVs). We filtered and prioritized the variants based on the following criteria: (1) The variant was ruled out if its allele frequency was higher than 0.5% in the 1000-Genome database (http://www.1000genomes.org/), in the Exome Variant Server (http://evs.gs.washington.edu/EVS/), or in our in-house database with 521 exomes of Middle East origin; (2) Only variants predicted to be deleterious were retained, such as frameshift variants, nonsense variants, canonical splice sites variants, CNVs, and missense variants with positive predictions from a series of algorithms including phyloP (http://compgen.bscb.cornell.edu/phast/help-pages/phyloP.txt), GERP (http://mendel.stanford.edu/SidowLab/downloads/gerp/), SIFT (http://sift.jcvi.org/), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), and MutationTaster (http://www.mutationtaster.org/); (3) Variants needed to cohere with inheritance models and linkage intervals; (4) The disease-association of affected genes according to databases such as OMIM and HGMD was taken into account; (5) reports on tolerance of homozygous loss-of-function mutations in respective genes were assessed; (6) Reports on physical interaction of candidate gene with any known causal genes were consulted. For a detailed description of the filtering procedure, please refer to Hu et al.7 The data analysis pipeline7,8 applied resulted in the identification of a homozygous PTRH2 gene mutation (chr17q23.1). The index pedigree has two generations and two patients. There are three homozygosity linkage intervals defined with a size >5 Mb and LOD >1.8: (1) chr16:12770901-20056282, LOD:2.056; (2) chr17:54833348-64807228, LOD:2.056; (3) chr7:54618943-77101532, LOD:2.056. We employed the aforementioned criteria for filtering and prioritization. Thus, there are 1, 1, and 0 rare homozygous variants in these three linkage regions, respectively. Among these two variants, PTRH2 harbors a frameshift deletion, while the other variant, namely, chr16:19533219A>G, causes a missense change (c.68T>C,p.L23P) in the gene GDE1 (NM_016641), which is predicted by SIFT, PolyPhen2, and MutationTaster as “tolerated,” “benign,” and “polymorphism,” respectively. We confirmed the absence of homozygous deleterious variants (missense, nonsense, frameshift, splice site change) in PTRH2 in the Exome Variant Server (http://evs.gs.washington.edu/EVS/), the 1000 Genome database (http://www.1000genomes.org/), the dbSNP138 (http://www.ncbi.nlm.nih.gov/SNP/), and our in-house 521 Middle East origin exome database. We also checked the haplotype data in the 1000 Genome database and found no compound heterozygote in PTRH2. Even heterozygous deleterious variants in PTRH2 have very low allele frequency (<0.1%), indicating strong purifying (negative) selection in population. Also, the haploinsufficiency index for PTRH2 (https://decipher.sanger.ac.uk/) is 18%, not likely to be haploinsufficiency. Patient 250690 in the Decipher database with a 2.4 Mb homozygous deletion covering 36 genes including PTRH2 was reported to display abnormal facial features and intellectual disability/developmental delay.
Sanger sequencing of the PTRH2 gene (NM_016077) was applied to confirm the mutation in the patients, establish the genotype in the other family members, and evaluate the presence of potential mutations in additional families.
Functional analyses of patient and control specimen
Epstein–Bar virus-transformed lymphocytes (LCLs) and fibroblast cultures were established from patients and healthy controls according to standard protocols.9 We analyzed cellular PTRH2 localization, cell viability, proliferation, and apoptosis as well as protein levels of PTRH2, pAKT/AKT, pMAPK/MAPK, pFAK/FAK, Bcl-2, and pS6.
Analysis of Ptrh2 mutant and wild-type mice
Ptrh2-mutant mice were generated by breeding Ptrh2flox/flox mice5 with MORE-Cre mice B6.129S4-Meoxtm1(cre)Sor/J (stock no. 003755, Jackson Laboratory, Bar Harbour, ME)10 to obtain heterozygote Ptrh2flox/− MeoxCre/− mice, which were subsequently backcrossed to wild-type C57BL/6 mice and further intercrossed to obtain the pure C57BL/6 genetic background Ptrh2-mutant mice Ptrh2flox/flox Meoxcre/−.4 Ptrh2flox/flox mice have one loxP sequence in the intron that lies between exons 1 and 2 and a second loxP sequence as well as neomycin-resistant gene introduced downstream of the second Ptrh2 exon. MeoxCre/Cre mice express the Cre recombinase from the endogenous Meox2 promoter, and expression of Cre recombinase is observed in epiblast-derived tissues as early as E5.10 Genotyping was performed using the primers G2F 5′-TGG GTC TTT GAA TCA ACT AG-3′, G1R 5′-ACA TGC CAC AAG CAA CTC CA-3′, and 30d 5′-TTT GAG ACC CTA TCA CTC CAC ACG-3′ with an expected 250 bp band in wild-type, a 200 bp band in homozygous Ptrh2 mutant mice, and both bands in heterozygous mice. C57BL/6 mice were obtained from the animal facility of the Charité – Universitätsmedizin Berlin, Germany. The breeding was performed during the day, the day of insemination was considered as embryonic day (E) 0, and the day of birth was designated as postnatal day (P) 0.
Viability and breeding of various genotypes (wild type [WT], heterozygous, homozygous) of Ptrh2 mice was monitored continuously, and their overall behavior was studied through home cage observation and basic neurological assessment. Gross organ malformation was analyzed through dissection of six mice from each genotype at P7. For further analysis of gross morphological organ abnormalities, Ptrh2 knockout mice and control littermates were sacrificed at P0 and/or P7, and their organs snap-frozen for RNA and protein extraction and/or immersed in 4% paraformaldehyde for histological analysis as reported previously.11 Coronal brain cryostat sections of 10 μm thickness were cut, collected on Superfrost plus slides and stained with cresyl violet. In addition, liver, pancreas, muscle, and diaphragm specimen were embedded in paraffin and analyzed following hematoxylin and eosin staining of 10 μm sections. mRNA and protein levels were analyzed through quantitative real-time PCR and Western blotting, respectively.
Elastase measurements
Elastase activity was measured in the stool of Ptrh2 KO and WT littermates at P7 using the Elastase Assay (Molecular Probes Grand Island, NY, USA) according to the manufacturer's instructions.
Quantitative real-time PCR
RNA extraction and cDNA synthesis were performed with established methods reported previously.12 To specifically amplify and detect Ptrh2, Hprt (Hypoxanthine-guanine phosphoribosyl-transferase, reference gene), and RpII (RNA polymerase II, reference gene) cDNA, we designed sets of primers and TaqMan probe using the GenScript real-time PCR (TaqMan) Primer Design online software (http://www.genscript.com). Primers and PCR conditions are available from the authors upon request. Ct values were calculated using the 7500 Fast System sodiumdodecyl sulfate (SDS) Software (Applied Biosystems Darmstadt, Germany) and further statistical calculations were performed on GraphPad Prism 5 Software (GraphPad Software Inc., La Jolla, CA). The 2−ΔΔCt method was applied for the quantification of the relative Ptrh2 mRNA expression.
Western blot
Protein extraction and Western blots were performed with established methods reported previously11 antibodies are listed in Table S1.
Immunocytology and -histology
LCLs briefly plated on poly-L-lysine and fibroblasts plated on fibronectin-coated coverslips were fixed in 4% paraformaldehyde. Cryostat sections were air-dried briefly, and paraffin sections were deparaffinized prior to rinsing. Coverslips and cryostat sections were further incubated in staining buffer (0.2% gelatin, 0.25% Triton X-100, 10% donkey or goat normal serum) for 30 min for permeabilization and blocking, followed by an overnight incubation with primary antibodies and a 2h incubation with the corresponding secondary antibodies (antibodies listed in Table S1). The specificity of the Ptrh2 immunoreaction was ensured in control sections treated with the corresponding blocking peptide as well as in sections incubated only in the secondary antibodies. Nuclei were labeled with 4′,6-diamidino-2-phenylindole (DAPI, 1:1000, Sigma-Aldrich, Munich, Germany). Fluorescently labeled cells or tissues were analyzed and imaged by a fluorescent Olympus BX51 microscope with the software Magnafire 2.1B (Olympus, Hamburg, Germany) and confocal microscopy images were taken by an lsm5exciter Zeiss confocal microscope with the software Zen (Zeiss, Jena, Germany). All images were processed using Adobe Photoshop.
Cell viability assay
Cells cultured in microtiter wells were pulsed with 25 μL of a 2.5 mg/mL MTT stock (Sigma-Aldrich) in PBS and incubated for 4 h. Subsequently, 100 μL of a solution containing 10% SDS and 0.01 N HCl was added, the plates were then incubated overnight, and absorption was read on a VICTOR 3 multilabel plate reader (PerkinElmer Life Sciences; reference/test wavelength 650/590 nm). Test reagents were added to medium alone to provide a blank control.
Staurosporine-mediated apoptosis
Staurosporine (2 μmol/L; Sigma) was added to adherent cells in serum-containing media. 24 h later, the cells were analyzed by either an MTT assay, immunoblotting for caspase-3 activation, or activated caspase-3 ELISA (BioVision Milpitas, CA, USA). For the latter, 1 × 105 patient fibroblasts were plated per well in a 96 well plate, allowed to attach and exposed to +/− staurosporine for 24 h. Subsequently, they were lysed, incubated with DEVD-AFC substrate for 1 h at 37°C, and fluorescence was assessed using a plate reader with 400/505 nm excitation/emission filters.
Mitochondrial respiratory chain enzyme activity measurements in human fibroblasts
Fibroblasts were cultured in M199 medium (Life Technologies, Breda, the Netherlands) supplemented with 10% fetal calf serum and 1% penicillin/streptomycin in a humidified atmosphere of 95% air and 5% CO2 at 37°C. Measurement of the mitochondrial respiratory chain enzyme activities in skin fibroblasts was performed according to established procedures.13
Results and Discussion
We report for the first time that patients with a mutation in the PTRH2 gene develop an infantile multisystem neurologic, endocrine, and pancreatic disease (IMNEPD) with intellectual disability, postnatal microcephaly, progressive cerebellar atrophy, hearing impairment, polyneuropathy, failure to thrive, and organ fibrosis with exocrine pancreas insufficiency. Two affected children of healthy, consanguineous parents of Yazidian-Turkish descent were born at term without complications and with normal weight, height, and head circumference (II.1; II.4, Fig.1A). Fetal movements were decreased in II.1 when compared to those of her healthy siblings. At birth, bilateral hip dislocation, muscular hypotonia, brachycephaly, facial dysmorphism with midface hypoplasia, hypertelorism, exotropia, and thin upper lip vermilion, proximally placed thumbs, deformities of the fingers and toes, and a shawl scrotum in the affected male were apparent (Fig.1B–D, Table1). Both patients later presented with postnatal microcephaly, growth retardation, failure to thrive, delayed motor milestones, progressive debilitating ataxia, distal muscle weakness (Achilles tendon contracture in II.1), peripheral demyelinating sensorimotor neuropathy, and sensorineural deafness (Table1, Tables S2, S3). Pyramidal signs could not be elicited. The affected female patient (II.1) became wheel-chair dependent due to muscle weakness and ataxia by 15 years of age. Patients had moderate intellectual disability with an intelligence quotient of 39 (II.1) and 48 (II.4) and a severe expressive speech delay. Cranial magnetic resonance imaging (cMRI) revealed microencephaly and progressive cerebellar atrophy as a correlate for the ataxia (Fig.1E). On electroencephalography (EEG) recordings, the patients presented with abnormal rhythmic alpha-beta-waves with high amplitudes even during sleep (Fig.1F), a finding reported in neurodegenerative diseases and/or cortical malformations.14
Figure 1.
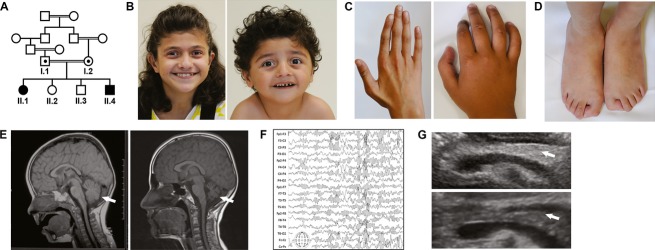
Phenotype of patients with infantile-onset multisystem neurologic, endocrine, and pancreatic disease (IMNEPD). (A) The index patients (II.1, II.4) are children of healthy, consanguineous Yazidian parents from Turkey. (B) Both patients displayed facial dysmorphism with midface hypoplasia, hypertelorism, exotropia, and thin upper lip (II.1: 13 years, II.4: 5 years) and deformities of (C) fingers and (D) toes (II.1). (E) Cranial MRI revealed progressive cerebellar atrophy (II.1: left at 2 and right at 12 years of age). (F) Abnormal rhythmic alpha-beta-waves with high amplitudes on EEG recording (II.1). (G) High echo intensity of the pancreas (arrow) of the affected female patient II.1 (top) compared to that of a healthy, age-matched control (bottom) in an abdominal ultrasound indicates organ fibrosis and/or increase in fatty tissue.
Table 1.
Phenotype associated with homozygous PTRH2 mutation
| Pedigree ID (gender) | II.1 (f) | II.4 (m) | ||
|---|---|---|---|---|
| Age at last assessment (years) | 14.8 | 6.3 | ||
| Category | Feature | HPO1 | ||
| Growth | ||||
| Height | Postnatal growth retardation (years at onset) | 0008897 | + (11.4) | + (4) |
| Weight | Failure to thrive (years at onset) | 0001508 | + (11.4) | + (2.5) |
| Head and Neck | ||||
| Head | Postnatal microcephaly (OFC < P3; years at onset) | 0005484 | + (2.5) | + (0.3) |
| Brachycephaly | 0000248 | + | + | |
| Face | Abnormality of the midface | 0000309 | + | + |
| Myopathic facies | 0002058 | + | + | |
| Ears | Sensorineural hearing impairment | 0000407 | + | + |
| Eyes | Hypertelorism | 0000316 | + | + |
| Exotropia | 0000577 | + | + | |
| Mouth | Thin upper lip vermilion | 0000219 | + | + |
| Abdomen | ||||
| Liver | Hepatomegaly | 0002240 | − | + |
| Hepatic fibrosis or steatosis (on ultrasound) | 0001395/0001397 | + | + | |
| Pancreas | Exocrine pancreatic insufficiency | 0001738 | + | + |
| Pancreatic fibrosis (on ultrasound) | 0100732 | + | − | |
| Genitourinary | ||||
| External genitalia | Shawl scrotum | 0000049 | + | |
| Skeletal | ||||
| Pelvis | Congenital hip dislocation | 0001374 | + | − |
| Hands | Proximal placement of thumb | 0009623 | + | + |
| Long fingers | 0100807 | + | − | |
| Ulnar deviation of the 2nd and 3rd finger | 0009464, 0009463 | + | − | |
| Feet | Abnormality of the hallux | 0001844 | + | − |
| Talipes equinovalgus (incipient) | 0001772 | + | − | |
| Achilles tendon contracture | 0001771 | + | − | |
| Muscle, soft tissue | ||||
| Distal muscle weakness | 0002460 | + | + | |
| Skeletal muscle fibrosis (on ultrasound) | – | + | − | |
| Neurologic | ||||
| Central nervous system | Neonatal hypotonia | 0001319 | + | − |
| Motor delay | 0001270 | + | + | |
| Intellectual disability, moderate (IQ 39–45) | 0002342 | + (48) | + (39) | |
| Dysmetria | 0001310 | + | ||
| Ataxia | 0001251 | + | + | |
| Cerebellar hypoplasia (progressive) | 0001321 | + | + | |
| EEG abnormality: alpha-beta-waves even in sleep | 0002353 | + | + | |
| Peripheral nervous system | Demyelinating peripheral neuropathy (sensorimotor) | 0007108 | + | + |
| Endocrine features | ||||
| Hypothyroidism | 0000821 | + | + | |
| Prenatal manifestations | ||||
| Movement | Decreased fetal movement | 0001558 | + | − |
m, male; f, female; PTRH2, peptidyl-tRNA hydrolase 2; EEG, electroencephalogram; HPO, human phenotype ontology, OFC, occipito-frontal head circumference; P, percentile.
All symptoms are listed according to the nomenclature and the systematics of the Online Mendelian Inheritance in Man (OMIM) “Clinical Synopsis” and the Human Phenotype Ontology (HPO).22 The IQ was evaluated using the Kaufmann Assessment Battery for Children (K-ABC) in II.1 and the Bayley Scales of Infant Development (BSID-II) for II.4.
The mother had observed bulky steatorrhea since toddler age in her affected children. Further investigations revealed exocrine pancreatic insufficiency with subsequent deficiencies of lipophilic vitamins and abnormal clotting parameters and hypothyroidism (Table S4). The glycated hemoglobin (HbA1c) value was slightly elevated in patient II.1 at the age of 14 years, indicating elevated plasma glucose concentrations over prolonged periods of time and thus putatively beginning endocrine pancreas insufficiency. Oral supplementation of pancreas enzymes at a dose of 1000 units/kg per day and vitamins over a period of 1 year induced a recognizable weight gain and normalization of vitamin deficiencies and clotting parameters. Signs of liver and pancreas fibrosis and hepatomegaly were detected by ultrasound, while the morphology of the other organs was normal on abdominal MRI of patient II.1 (Fig.1G, data not shown). Slightly increased transaminase values, creatine kinase, and lactate dehydrogenase activities were observed in the plasma as indicators of a muscle and/or liver disease. Further assessment of the patients and their parents through electro- and echocardiography and lung function tests revealed normal cardiac and pulmonary function, respectively. The results of routine blood tests and extensive metabolic work-up also for a mitochondriopathy were normal (Table S4 and data not shown). Despite normal growth hormone levels and bone age, as revealed by conventional x-rays, both children had a short stature.
To identify the genetic basis of the disease, we performed high-throughput whole-exome sequencing followed by Sanger sequencing and bioinformatic analysis. We thereby identified a homozygous deletion of two base pairs in the PTRH2 gene in both affected children (c.269_270delCT, NM_016077; Fig.2A and B). This 2-base pair deletion in the coding sequence of exon 2 of PTRH2 causes a frameshift mutation, leading to a predicted protein truncated by 78 amino acids (p.Ala90fs, NP_057161) and is therefore predicted to be a null allele. The mutation lies within a highly conserved region of the protein (Fig.2C) and segregates with the disease phenotype, that is, is heterozygous in both parents and not observed in the healthy siblings. We confirmed the absence of the deletion in various whole-exome databases, as detailed in the methods. Here, even heterozygous PTRH2 deleterious variants have a very low allele frequency (<0.01%), indicating strong negative selection in population. This may explain why we did not detect PTRH2 gene mutations in 30 patients and families with exocrine pancreas insufficiency and intellectual disability as predominant symptoms. PTRH2 mRNA levels from fibroblasts of patients and controls did not differ significantly, while PTRH2 protein levels were strongly reduced in patient fibroblasts (Fig.2D and E).
Figure 2.

Homozygous PTRH2 gene mutation in index patients with IMNEPD. (A) By whole-exome sequencing, the homozygous mutation c.269_270delCT was identified in exon 2 of the PTRH2 gene (NM_016077), predicted to lead to a protein truncated by 78 amino acids (p.Ala90fs, NP_057161.1). (B) Electropherogram depicting homozygous deletion of two base pairs in the PTRH2 gene in the patient II.1, but not in a healthy control. (C) The mutation affects a highly conserved region of the protein. (D) Quantitative real-time PCR revealed normal mRNA levels in the patients when compared to controls, (E) while PTRH2 was strongly reduced on a protein immunoblot of fibroblast lysate from the affected subjects. IMNEPD, infantile-onset multisystem neurologic, endocrine, and pancreatic disease; PTRH2, peptidyl-tRNA hydrolase 2.
We further found a strong correlation between regions of high PTRH2 expression in the mouse (Fig.3) and sites of pathology suggested by the human phenotype and by that of Ptrh2-mutant mice (Ptrh2flox/flox MeoxCre/−).5 Ptrh2-mutant mice (KO) were indistinguishable from their healthy littermates at birth, but showed a progressive age-related relative decrease in length and weight, muscle weakness and wasting, and died by P8-10 (Fig.4A–C). By P7, the KO mice also presented with severe ataxia, their whole brain weight was slightly but significantly reduced, and cerebellar atrophy was prominent (Video S1, Fig.4D and E). As a cellular correlate, neuron soma size was reduced in the cerebral cortex (Fig.5A), while spontaneous apoptosis was not increased in KO brains at P7 (data not shown).
Figure 3.

Ptrh2 in the developing and adult murine brain. (A) Ptrh2 is abundantly expressed in the E14 brain, decreasing in the neocortex and cerebellum postnatally (qPCR, n = 5–8, one-way ANOVA, results compared to E14: ***P < 0.001; results compared to neocortex P0: ##P < 0.01, ###P < 0.001). (B) Throughout development, Ptrh2-positive cells were visualized at high density across all layers of the neocortex and also in basal ganglia and hippocampus (data not shown). Very low density of Ptrh2-positive cells was detected within the white matter and within proliferative zones such as the neocortical ventricular/subventricular zone or the rostral migratory stream (data not shown). (C) Ptrh2 is present predominantly in areas where postmitotic cells reside in the murine cerebellum from E14 to P56. (D) Ptrh2 is present in neurons (Map2, early neuron marker; NeuN, mature neuron marker), but not in M-phase proliferating, pH3-positive cells, or glial cells (Iba1, marker for microglia; Glial fibrillary acidic protein (GFAP), marker of astrocytes). (E) In the cerebellum, Ptrh2 is present predominantly in Calbindin-positive Purkinje cells, but not in GFAP-positive Bergman glia or the Calretinin-positive inner granular layer cells. Immunofluorescence images, scale bars 50 μm (B–E); confocal microscopy images, scale bars 10 μm (D). PTRH2, peptidyl-tRNA hydrolase 2; ANOVA, analysis of variance.
Figure 4.

Postnatal growth retardation, microencephaly, cerebellar atrophy, and ataxia in Ptrh2-mutant mice. (A) Ptrh2-mutant mice Ptrh2flox/flox MeoxCre/− (KO) are indistinguishable from their healthy littermates (WT) at birth, but show a runting syndrome by P7 with a significant decrease in (B) body weight (n = 8–13 animals/group, Student's t test) and (C) die by P8-10 (Kaplan–Meyer curve, n = 75). (D) While at P7 total brain weight was slightly reduced (n = 8–13 animals/group, Student's t test), (E) cerebellar atrophy was easily apparent in KO (n = 5–6 animals/group, Student's t test, scale bars 200 μm). See Video S1 for ataxia in KO at P7. *P < 0.05, ***P < 0.001.
Figure 5.
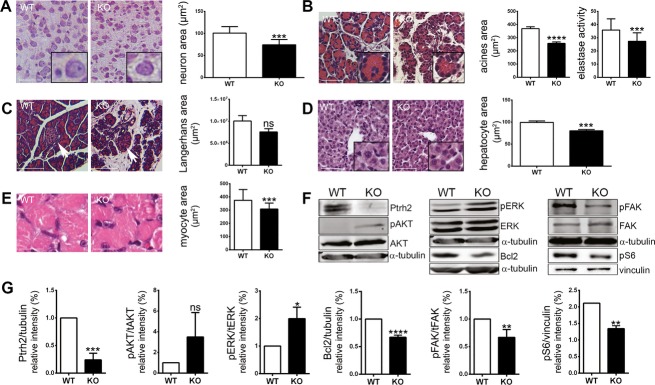
Effects of a loss of Ptrh2 in mice. (A) Neuron soma size area was reduced in the cortex (n = 21–47 cells/animal, three to four animals/group, Student's t test, scale bars 50 μm). (B) Exocrine pancreas insufficiency detected by reduced cross-section areas of the acini of the exocrine pancreas (n = 35–68 acini/animal, three animals/group, Student's t test, scale bars 50 μm) and reduced pancreas elastase levels in the stool of KO mice (n = 4 animals/group, run in triplicates, Student's t test). (C). Langerhans islet area did not differ significantly between KO and WT mice (n = 8–37 islands/animal, three animals/group, Student's t test, scale bars 100 μm). (i) Reduced soma size of hepatocytes (n = 7 cells/animal, 10 animals/group, Student's t test, scale bars 50 μm) and (D) reduced myocyte cross-section fiber area in KO compared to WT mice (n = 3, Student's t test, scale bars 20 μm). (E and F) Representative Western blots and quantification results for Ptrh2, pAKT/AKT, pERK/ERK, Bcl-2, pFAK/FAK, and pS6 in KO and WT brains (n = 3, Student's t test; see Table S1 for antibodies). ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Since further organs are involved in the human PTRH2-mutant phenotype, we analyzed murine pancreas, liver, and muscle at P7. The exocrine pancreas was insufficient in the KO mice as indicated by reduced pancreas elastase levels in the stool and by reduced cross-section areas of the pancreatic acini (Fig.5B). In contrast, Langerhans islets, associated with endocrine pancreas function, did not differ significantly in size between KO and WT mice (Fig.5C). Similar to the findings in neurons and pancreas acini, the soma size of hepatocytes and myocytes was also reduced significantly in KO mice (Fig.5D and E).
Our further findings highlight a reduced PTRH2-mediated signaling in mutant mouse brains as molecular correlates for the observed phenotype (Fig.5F and G). We detected a reduction in both integrin-mediated signaling as determined by pFAK and Bcl-2 expression and an increase in pERK in Ptrh2-mutant mouse brains. Moreover, we identified a reduction in the mTOR activation marker pS6 levels in KO brains. Since mTOR is a key regulator of cell size,15 this and the reduced cell size in several KO organs link Ptrh2 to the mTOR/pS6 pathway.
To further analyze the effect of a loss of PTRH2 function in human tissues, we established lymphoblastoid cells and fibroblasts from our patients and healthy controls. The mitotic spindle apparatus morphology, cell cycle regulation, and respiratory chain enzymes and complex V activities did not differ between PTRH2 mutant and control fibroblasts (data not shown). While spontaneous apoptosis was not increased in the patient fibroblasts, apoptosis was increased significantly when these fibronectin-attached cells were treated with staurosporine, as detected by decreased cell viability and elevated activated caspase-3 levels (Fig.6A). PTRH2 overexpression in PTRH2-mutant patient fibroblasts was able to rescue this phenotype (Fig.6B). Integrin-mediated FAK activation was decreased in fibronectin-attached patient fibroblasts compared to controls (Fig6C and D). In line with our findings in the KO mice, pERK was increased in patient compared to control fibroblasts (Fig.6C and D). Moreover, we identified a reduction in pS6 levels in both patient fibroblasts as an indication of reduced mTOR pathway activation (Fig.6C and D).
Figure 6.
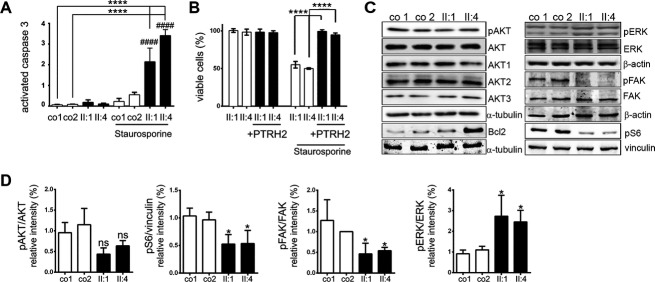
Effects of a loss of PTRH2 in human tissues. (A) While spontaneous apoptosis was not increased, staurosporine-induced apoptosis was significantly increased in PTRH2-mutant patient versus control fibroblasts (n = 4–6/group, activated caspase-3 levels, one-way ANOVA). (B) PTRH2 overexpression rescued the apoptotic effect of staurosporine in patient fibroblasts (cell viability assay, n = 3/group, one-way ANOVA). (C and D) Representative Western blots and quantification results for AKT1-3, pAKT/AKT, Bcl-2, pERK/ERK, pFAK/FAK, and pS6 (n = 3, one-way ANOVA; quantification of nonsignificant results for AKT1-3 and Bcl2 not shown; see Table S1 for antibodies). ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. PTRH2, peptidyl-tRNA hydrolase 2; ANOVA, analysis of variance.
Taken together, we demonstrate the physiological role of PTRH2 for the human and mouse organism. We identified mutations in PTRH2 as the cause of infantile multisystem neurologic, endocrine, and pancreatic disease (IMNEPD). This previously unidentified phenotype comprises postnatal microcephaly, progressive cerebellar atrophy, intellectual disability, failure to thrive, polyneuropathy, hearing impairment, and organ fibrosis with exocrine pancreas insufficiency. Future studies on further patients/pedigrees with similar phenotypes will be required to determine the full phenotype spectrum associated with the PTRH2 gene. We show that PTRH2 is essential for human cognitive function. The expression pattern of PTRH2 in the developing brain, the normal head circumference at birth, the development of secondary microcephaly and progressive cerebellar atrophy, and the abnormal EEG findings in patients all suggest that PTRH2 has an essential function in postmitotic cells, neurons, as opposed to neural progenitors. Progressive cerebellar atrophy in the patient and mutant mice is most striking as it suggests increased apoptosis in this region. This finding is in agreement with our observation that patient fibroblasts are more sensitive to staurosporine-mediated apoptosis compared to normal controls.
PTRH2 is a regulator of integrin-mediated cell survival and apoptosis and as such promotes survival of ECM-attached cells through activation of adhesion-initiated signal transduction pathways.4 We identified a reduced activation of the FAK-Pi3K-AKT-NFkB pathway in patients and mutant mice; this is in line with previous results in cancer cells.4 Mutations in other positive regulators of NFkB, which is involved in a variety of biological processes including neurogenesis and cell survival,16–19 have been reported in patients with intellectual disability.20,21 The role of NFkB signaling in the brain is not yet well understood, and PTRH2 provides an additional link as to how NFkB integrates signals from the ECM to intracellular processes. In addition, we report reduced cell sizes in various organs of Ptrh2-mutant mice and in line with this a reduced activation of the AKT/mTOR/pS6 pathway linked to the regulation of cell size.15 Our findings thereby identify PTRH2 as an essential factor in the regulation not only of cell survival, but also of cell growth (Fig.7).
Figure 7.

PTRH2 mediates cell survival and cell size. Simplified schematic drawing indicates model of putative role of PTRH2 in control of cell survival and cell size pathways. PTRH2, peptidyl-tRNA hydrolase 2; AES, amino-terminal enhancer of split; FAK, focal adhesion kinase; PI3K, phosphatidylinositide 3-kinase; AKT, serine/threonine-specific kinase or protein kinase B; ERK, extracellular signal-regulated kinase; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; Bcl2, B-cell lymphoma 2; mTOR, mechanistic target of rapamycin (serine/threonine kinase); S6K, ribosomal protein S6 kinase; 4EBP, eukaryotic initiation factor-4E-binding protein. Modified according to Jan et al.3 and Griffiths et al.4
Acknowledgments
The authors thank the family members who participated in this study. This work was supported by the German Research Foundation (SFB665), the Berlin Institute of Health (BIH), the Sonnenfeld Stiftung, the German Academic Exchange Service (DAAD), the NCRR P20-RR016453, the Robert C. Perry Fund (20061479), the Max-Planck Society, and the EU FP 7 project GENCODYS, (241995). We acknowledge the contribution of clinical data by W. Stäblein, U. Greef, A. Mühlig-Hofmann, S. Minh, and M. Classen, thank M. Zenker, M. Sukalo, and A. Steuerer for sending DNA samples of patients with IMNEPD-like phenotypes as well as A. Hübner, K. Handschug, J. Fassbender, S. Kosanke, L. Garcia-Halpin, W. Luck, and Joe W. Ramos for technical help and discussions.
Author Contributions
C. H. and A. M. K. recruited subjects, gathered patient history as well as clinical information and contributed clinical samples. H. H., L. M., T. F. W., and H. H. R. generated and analyzed whole-exome sequencing data and performed further mutation analyses in the cohort. L. I.-J., N. K., and O. N. examined DNA and protein expression in cell lines from affected individuals and controls; L. I.-J., M. S., B. E., S. S., and M. dlV. performed Western blots. L. I.-J. performed the immunocytology and -histology experiments and was aided by G. S.-D. M. L. M., M. J., L. I.–J., and M. dlV. performed experiments with Ptrh2 KO mice and patient cells. C. C. performed elastase assays. D. S., N. D., K. E., and L.vdH. performed cell cycle and mitochondrial enzyme studies. A. M. K., C. H., M. L. M., L. I.-J., and T. F. W. wrote the manuscript, which was read, corrected, and approved by all coauthors.
Conflict of Interest
Dr. Kaindl reports grants from German Research Foundation (DFG), grants from Berlin Institute of Health (BIH), during the conduct of the study. Dr. Hu reports grants from EU FP7 project GENCODYS, during the conduct of the study. The position of Dr. Musante is financed by EU FP 7 project GENCODYS, grant no 241995, during the conduct of the study.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Video S1. Ataxia and growth retardation in Ptrh2-mutant mouse at P7 compared to age-matched WT mouse.
Table S1. List of primary and secondary antibodies.
Table S2. Index patient development in first 2 years of life in comparison to the healthy siblings. Normal values according to Swaiman.23 n.d., not determined.
Table S3. Anthropometric data of index patients with IMNEPD. Centiles and SDS for head circumference, length/height, weight, and body mass index (BMI) are according to the Centers for Disease Control and Prevention (CDC) 2000 growth charts (http://www.cdc.gov/growthcharts/). Since here centiles and SDS are not available for the BMI of children under 2 years of age and for the head circumference of children above 3 years of age, these were extracted from Kromeyer-Hauschild et al. (http://www.mybmi.de/main.php)24 and Prader et al.25 BMI, body mass index; n.d., not determined; OFC, occipital-frontal head circumference; SDS, standard deviation score. *Under oral supplementation of pancreas enzymes at a dose of 1000 units/kg per day and vitamins.
Table S4. Laboratory blood values of index patients with IMNEPD. Values were obtained at ages 13 and 4 years of index patients II.1 and II.4, respectively. Normal values are age adapted. AST, aspartate aminotransferase; ALT, alanine aminotransferase; CK, creatine kinase; LDH, lactate dehydrogenase; AP, alkaline phosphatase; HbA1c, glycated hemoglobin; TSH, thyroid-stimulating hormone or thyrotropin; T3, triiodothyronine; fT4, free thyroxine; TPZ-INR, prothrombin ratio according to international normalized ratio; aPTT, activated partial thromboplastin time.
References
- 1.Menninger JR. Peptidyl transfer RNA dissociates during protein synthesis from ribosomes of Escherichia coli. J Biol Chem. 1976;251:3392–3398. [PubMed] [Google Scholar]
- 2.De Pereda JM, Waas WF, Jan Y, et al. Crystal structure of a human peptidyl-tRNA hydrolase reveals a new fold and suggests basis for a bifunctional activity. J Biol Chem. 2004;279:8111–8115. doi: 10.1074/jbc.M311449200. [DOI] [PubMed] [Google Scholar]
- 3.Jan Y, Matter M, Pai JT, et al. A mitochondrial protein, Bit1, mediates apoptosis regulated by integrins and Groucho/TLE corepressors. Cell. 2004;116:751–762. doi: 10.1016/s0092-8674(04)00204-1. [DOI] [PubMed] [Google Scholar]
- 4.Griffiths GS, Grundl M, Leychenko A, et al. Bit-1 mediates integrin-dependent cell survival through activation of the NFkappaB pathway. J Biol Chem. 2011;286:14713–14723. doi: 10.1074/jbc.M111.228387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kairouz-Wahbe R, Biliran H, Luo X, et al. Anoikis effector Bit1 negatively regulates Erk activity. Proc Natl Acad Sci USA. 2008;105:1528–1532. doi: 10.1073/pnas.0711357105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biliran H, Jan Y, Chen R, et al. Protein kinase D is a positive regulator of Bit1 apoptotic function. J Biol Chem. 2008;283:28029–28037. doi: 10.1074/jbc.M803139200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu H, Wienker TF, Musante L, et al. Integrated sequence analysis pipeline provides one-stop solution for identifying disease-causing mutations. Hum Mutat. 2014 doi: 10.1002/humu.22695. doi: 10.1002/humu.22695. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 8.Najmabadi H, Hu H, Garshasbi M, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
- 9.Neitzel H. A routine method for the establishment of permanent growing lymphoblastoid cell lines. Hum Genet. 1986;73:320–326. doi: 10.1007/BF00279094. [DOI] [PubMed] [Google Scholar]
- 10.Tallquist MD, Soriano P. Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis. 2000;26:113–115. doi: 10.1002/(sici)1526-968x(200002)26:2<113::aid-gene3>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 11.Issa L, Kraemer N, Rickert CH, et al. CDK5RAP2 expression during murine and human brain development correlates with pathology in primary autosomal recessive microcephaly. Cereb Cortex. 2013;23:2245–2260. doi: 10.1093/cercor/bhs212. [DOI] [PubMed] [Google Scholar]
- 12.Kraemer N, Neubert G, Issa L, et al. Reference genes in the developing murine brain and in differentiating embryonic stem cells. Neurol Res. 2012;34:664–668. doi: 10.1179/1743132812Y.0000000060. [DOI] [PubMed] [Google Scholar]
- 13.Janssen AJ, Trijbels FJ, Sengers RC, et al. Measurement of the energy-generating capacity of human muscle mitochondria: diagnostic procedure and application to human pathology. Clin Chem. 2006;52:860–871. doi: 10.1373/clinchem.2005.062414. [DOI] [PubMed] [Google Scholar]
- 14.Bode H, Bubl R. EEG changes in type I and type II lissencephaly. Klin Padiatr. 1994;206:12–17. doi: 10.1055/s-2008-1046573. [DOI] [PubMed] [Google Scholar]
- 15.Kim DH, Sarbassov DD, Ali SM, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 16.Denis-Donini S, Dellarole A, Crociara P, et al. Impaired adult neurogenesis associated with short-term memory defects in NF-kappaB p50-deficient mice. J Neurosci. 2008;28:3911–3919. doi: 10.1523/JNEUROSCI.0148-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurland JF, Kodym R, Story MD, et al. NF-kappaB1 (p50) homodimers contribute to transcription of the bcl-2 oncogene. J Biol Chem. 2001;276:45380–45386. doi: 10.1074/jbc.M108294200. [DOI] [PubMed] [Google Scholar]
- 18.Rolls A, Shechter R, London A, et al. Toll-like receptors modulate adult hippocampal neurogenesis. Nat Cell Biol. 2007;9:1081–1088. doi: 10.1038/ncb1629. [DOI] [PubMed] [Google Scholar]
- 19.Widera D, Mikenberg I, Elvers M, et al. Tumor necrosis factor alpha triggers proliferation of adult neural stem cells via IKK/NF-kappaB signaling. BMC Neurosci. 2006;7:64. doi: 10.1186/1471-2202-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mochida GH, Mahajnah M, Hill AD, et al. A truncating mutation of TRAPPC9 is associated with autosomal-recessive intellectual disability and postnatal microcephaly. Am J Hum Genet. 2009;85:897–902. doi: 10.1016/j.ajhg.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsuda A, Suzuki Y, Honda G, et al. Large-scale identification and characterization of human genes that activate NF-kappaB and MAPK signaling pathways. Oncogene. 2003;22:3307–3318. doi: 10.1038/sj.onc.1206406. [DOI] [PubMed] [Google Scholar]
- 22.Robinson PN, Kohler S, Bauer S, et al. The human phenotype ontology: a tool for annotating and analyzing human hereditary disease. Am J Hum Genet. 2008;83:610–615. doi: 10.1016/j.ajhg.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swaiman KF. Neurologic examination after the newborn period until 2 years of age. In: Swaiman KF, Ashwal S, Ferriero DM, editors. Pediatric neurology: principles & practice. 4 ed. Philadelphia: Mosby; 2006. pp. 37–46. [Google Scholar]
- 24.Kromeyer-Hauschild K, Wabitsch M, Kunze D, et al. Perzentile für den Body-mass-Index für das Kindes- und Jugendalter unter Heranziehung verschiedener deutscher Stichproben. Monatsschr Kinderheilkd. 2001;149:807–818. [Google Scholar]
- 25.Prader A, Largo RH, Molinari L, Issler C. Physical growth of Swiss children from birth to 20 years of age. First Zurich longitudinal study of growth and development. Helv Paediatr Acta Suppl. 1989;52:1–125. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Ataxia and growth retardation in Ptrh2-mutant mouse at P7 compared to age-matched WT mouse.
Table S1. List of primary and secondary antibodies.
Table S2. Index patient development in first 2 years of life in comparison to the healthy siblings. Normal values according to Swaiman.23 n.d., not determined.
Table S3. Anthropometric data of index patients with IMNEPD. Centiles and SDS for head circumference, length/height, weight, and body mass index (BMI) are according to the Centers for Disease Control and Prevention (CDC) 2000 growth charts (http://www.cdc.gov/growthcharts/). Since here centiles and SDS are not available for the BMI of children under 2 years of age and for the head circumference of children above 3 years of age, these were extracted from Kromeyer-Hauschild et al. (http://www.mybmi.de/main.php)24 and Prader et al.25 BMI, body mass index; n.d., not determined; OFC, occipital-frontal head circumference; SDS, standard deviation score. *Under oral supplementation of pancreas enzymes at a dose of 1000 units/kg per day and vitamins.
Table S4. Laboratory blood values of index patients with IMNEPD. Values were obtained at ages 13 and 4 years of index patients II.1 and II.4, respectively. Normal values are age adapted. AST, aspartate aminotransferase; ALT, alanine aminotransferase; CK, creatine kinase; LDH, lactate dehydrogenase; AP, alkaline phosphatase; HbA1c, glycated hemoglobin; TSH, thyroid-stimulating hormone or thyrotropin; T3, triiodothyronine; fT4, free thyroxine; TPZ-INR, prothrombin ratio according to international normalized ratio; aPTT, activated partial thromboplastin time.