

Abstract
Astrocytes are critical regulators of neuronal function and an effective target for stroke therapy in animal models. Identifying individual targets with the potential for simultaneous activation of multiple downstream pathways that regulate astrocyte homeostasis may be a necessary element for successful clinical translation. Mitochondria and microRNAs each represent individual targets with multi-modal therapeutic potential. Mitochondria regulate metabolism and apoptosis, while microRNAs have the capacity to bind and inhibit numerous mRNAs. By combining strategies targeted at maintaining astrocyte function during and following cerebral ischemia, a synergistic therapeutic effect may be achieved.
Keywords: cerebral ischemia, miR, metabolism, chaperones, ATP, neuron
I. Introduction
Stroke remains a leading cause of death worldwide and the primary source of long-term neurological disability [1]. Despite hundreds of clinical trials investigating promising agents shown to improve neuronal survival in rodent models of stroke, the only effective clinical therapy remains minimizing the duration of ischemia via early thrombolysis [2]. One reason for the failure to translate successful findings in animal models to practical clinical therapies may reside in the complexity of signaling responses that occur within and between different cell types, reducing the likelihood that altering any single target will be effective. This suggests that identifying and testing single interventions that target multiple mechanisms to promote neuroprotection within the central nervous system (CNS) is a necessary next step in broadening the search for effective therapies to minimize injury and improve outcome subsequent to stroke.
As the most abundant cell in the human brain, astrocytes represent an attractive cellular candidate for stroke therapy. Individual astrocytes occupy discrete domains, with less than 5% overlap of processes with adjacent astrocytes [3]. In rodent hippocampal grey matter an individual astrocyte may contact up to 100,000 neurons [4, 5] and human astrocytes are even larger and more complex [6], emphasizing the significant role individual astrocytes play in neuronal regulation, synaptic transmission, and signal integration. Astrocytes also function as a coordinated syncytium by communicating with adjacent astrocytes via intercellular gap junctions located on extended processes [7], providing an additional astrocyte-dependent layer regulating the neuronal network. Because astrocytes have been shown to directly modulate neurotransmission, the association between astrocyte processes and neuronal synapses has earned the name “tripartite synapse” [8]. Astrocytes perform several aspects of neural housekeeping including: active uptake of extra-synaptic glutamate, regulating K+ homeostasis, maintaining the integrity of the blood-brain-barrier, clearing metabolic waste products, and buffering excess production of reactive oxidants (Fig. 1) [9-11]. During stroke, astrocytes have the potential to either protect neurons via these housekeeping mechanisms, or exacerbate injury by secreting glutamate, pro-inflammatory molecules and facilitating the formation of edema [12, 13]. The potential for astrocytes to either exert a multi-modal protective effect versus a damaging one further defines their critical role in stroke outcome; their multifaceted nature as potential targets for stroke therapy has been recently reviewed [14].
Figure 1.
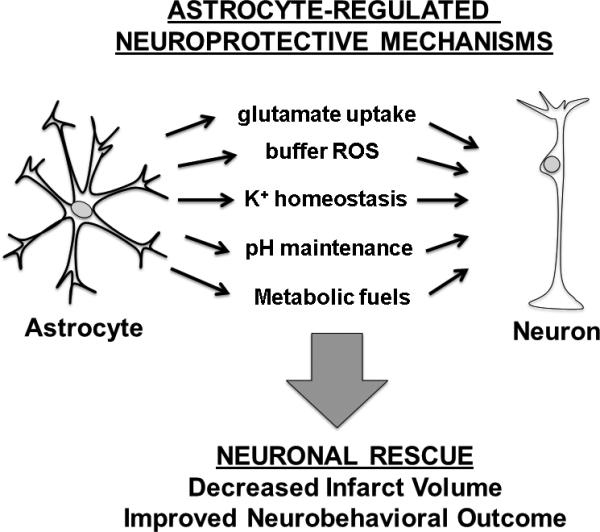
Astrocyte-targeted therapies can elicit multiple mechanisms of neuroprotection. ROS = reactive oxygen species.
Astrocyte-targeted strategies for stroke therapy
Cerebral ischemia occurs in a number of disease states, including embolic and hemorrhagic stroke, subdural and epidural hematoma, subarachnoid hemorrhage, traumatic brain injury, cerebral edema, vascular compression secondary to brain mass, cardiac arrest, or any physiologic condition resulting in a low cardiac output state. In the experimental setting, two widely utilized in vivo rodent models include transient middle cerebral artery occlusion (MCAO) to model ischemic stroke, and forebrain ischemia or four-vessel occlusion to model transient global cerebral ischemia, as seen clinically with cardiac arrest and resuscitation. MCAO is characterized by the rapid development of a core of irreversible necrotic cell death commonly in striatum and parts of the motor cortex, surrounded by a margin of hypoperfused tissue termed the penumbra (or ischemic boundary zone). Neurons in the penumbra may either survive by inducing pro-survival signaling pathways, or die later following reperfusion (delayed neuronal death) by initiating pro-apoptotic pathways [15]. This vulnerable region of brain therefore represents a potential target for therapeutic strategies that seek to improve clinical outcome by ultimately minimizing the total volume of infarct, and has been an area of intense scientific focus. However, concerns have been raised that in human ischemic stroke, reperfusion is more delayed and less complete than in the MCAO model.
Another setting is that of global or transient forebrain ischemia, which mimics the global hypo-perfusion that occurs with cardiac arrest and resuscitation. When the ischemic time is short this results in delayed neuronal cell death occurring primarily in the hippocampus. In this model neuronal cell death occurs selectively in the hippocampal cornu ammonis 1 (CA1) subregion, while neurons in the nearby dentate gyrus (DG) are preserved, a phenomenon independent of regional perfusion differences.
Despite the clear role astrocytes play in maintaining neuronal homeostasis, few studies have investigated the effect of directly targeting astrocyte survival in the setting of cerebral ischemia. However, of the recent studies undertaken specifically targeting astrocytes, several strategies for improving outcome following stroke have been shown to be effective in rodent models. Induction of brain-derived neurotrophic factor (BDNF) in astrocytes reduced neuronal apoptosis and improved functional recovery [16]. Astrocytic pyruvate protected against glutamate excitotoxicity via a glutathione-dependent mechanism [17]. Ceftriaxone treatment, which induces astrocytic glutamate uptake via increased transporter (glutamate transporter 1/GLT-1, excitatory amino acid transporter 2/ EAAT2) expression, decreased neuronal injury [18]. We utilized a genetic approach to generate astrocyte-specific overexpression of superoxide dismutase 2 (SOD2), an endogenous mitochondrial antioxidant, using the astrocyte-specific glial fibrillary acidic protein (GFAP) promoter [19]. Overexpression of SOD2 in astrocytes was accompanied by preservation of GLT-1, and reduced evidence of oxidative stress in the CA1 region [19]. Similarly, selective overexpression of GLT-1 in astrocytes provided neuroprotection from moderate hypoxia ischemia [20]. Although astrocyte-targeted studies have been limited, successful strategies have encompassed several independent mechanisms. Approaches that harness a larger number of protective targets may prove to be more clinically effective.
III. Mitochondria: master regulators of cell survival
Therapeutic strategies seeking to optimize mitochondrial homeostasis may offer a single approach to influence multiple injury pathways. Mitochondria are central regulators of apoptosis, buffer increases in the concentration of free cytosolic calcium ([Ca2+]c), and maintain availability of ATP. Disruptions of mitochondrial homeostasis induce both apoptotic and necrotic cell death following cerebral ischemia. Depletion of energy reserves leads to a massive rise in free [Ca2+]c, both from the endoplasmic reticulum (ER) and from the extracellular space, which is transmitted to the matrix of mitochondria by voltage-dependent anion channels (VDACs) on the outer mitochondrial membrane and the mitochondrial Ca2+ uniporter (MCU) on the inner membrane (Fig. 2). Direct transfer of Ca2+ from ER to mitochondria can occur at sites of ER-mitochondrial apposition termed the mitochondrial-associated membrane (MAM) containing the ER-localized Ca2+-release channel inositol 1,4,5-trisphosphate receptor (IP3R), mitochondrial VDAC, and several other proteins (Fig. 2) [21]. When mitochondrial matrix Ca2+ exceeds buffering capacity, mitochondrial function becomes impaired and can result in opening of the mitochondrial permeability transition pore [22], releasing cytochrome c [23, 24] and other pro-apoptotic factors into the cytoplasm. MPTP opening can result in variable effects on free radical production, either increased or decreased, depending in part on the duration of ischemia [25]. Moreover, mitochondrial calcium handling and cellular buffering of reactive oxygen species appear to be intimately related [26], further emphasizing the central role of mitochondria in cell survival.
Figure 2.
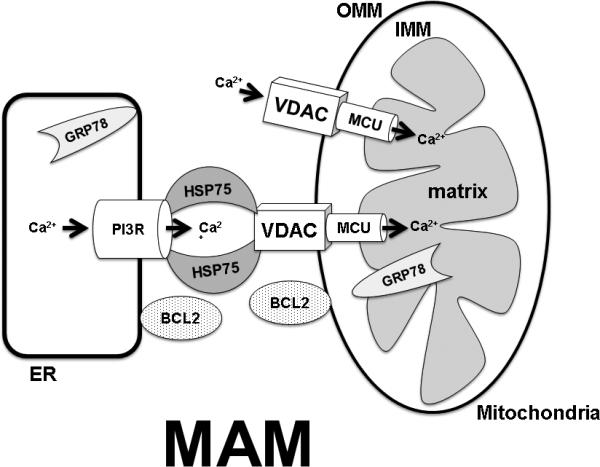
Mechanisms for mitigating cell death include targeting endogenous regulators of mitochondrial homeostasis and intracellular Ca2+ handling, HSP75, GRP78 and BCL2. When mitochondrial matrix Ca2+ exceeds buffering capacity mitochondrial function becomes impaired and results in opening of the mitochondrial permeability transition pore releasing cytochrome c and other pro-apoptotic factors into the cytoplasm. With energy depletion, increased free cytosolic Ca2+ is transmitted to the matrix of mitochondria by voltage-dependent anion channels (VDACs) on the outer mitochondrial membrane (OMM) and the mitochondrial Ca2+ uniporter (MCU) located on the inner mitochondrial membrane (IMM). Direct transfer of Ca2+ from the endoplasmic reticulum (ER) to mitochondria can also occur via the mitochondrial-associated membrane (MAM) complex, formed by the ER inositol 1,4,5-trisphosphate receptor (IP3R), mitochondrial VDACs, and other proteins. HSP75 modulates the activity of MAM while GRP78 serves to mitigate Ca2+ release from the ER to cytosol. BCL2 coordinates both mitochondrial function and induction of apoptosis.
Two families of well-known cell protective proteins, the heat shock protein 70 family (HSP70) of chaperones and the apoptosis-regulating B-cell lymphoma 2 (BCL2) family, have been shown to be integral to maintaining mitochondrial homeostasis. The HSP70 family of chaperones is a functionally related group of proteins that assist in the folding or unfolding of proteins, sequestration of denatured proteins, and assembly of protein complexes. HSP72, the strongly stress-inducible cytosolic member of the HSP70 family, is known to protect from both necrotic and apoptotic cell death, and affects several different steps in the apoptosis cascade including reduction of mitochondria-dependent apoptotic signaling [27]. Astrocyte-specific overexpression of HSP72 helped preserve GLT-1 and reduced evidence of oxidative stress in the hippocampal CA1 region following transient forebrain ischemia [19]. GRP75 (HSP75/mortalin), a mitochondrial-localized member of the HSP70 family is an important molecular chaperone that in involved in mitochondrial Ca2+ homeostasis by participating in the MAM complex with VDAC and IP3R (Fig. 2) which regulates direct Ca2+-transfer between the ER and mitochondria [28]. Overexpression of GRP75 reduced damage in both in vitro and in vivo models of ischemic stroke [29, 30]. GRP78/BiP, another member of the HSP70 family, regulates the ER unfolded protein response. GRP78 is largely localized to the endoplasmic reticulum but has been shown to translocate to mitochondria [31], suggesting a possible role in MAM-dependent Ca2+ transport between ER and mitochondria (Fig. 2). Overexpressing GRP78 protected astrocytes against ischemic injury and preserved respiratory activity and mitochondrial membrane potential after ischemic stress [32]. The BCL2 protein family is a central regulator of cell survival by helping maintain mitochondrial membrane integrity and function, and coordinating apoptotic-signaling [33, 34]. Overexpressing pro-survival BCL2 family members protected against cerebral ischemia in vivo [35, 36] and in vitro [37]. Cytosolic BCL2 was shown to contribute to MAM formation by localizing to both the ER and mitochondrial outer membranes (Fig. 2) [38], and to affect ER and mitochondrial Ca2+ homeostasis after cerebral ischemia [39, 40].
In addition to coordinating apoptosis, mitochondria are fundamental to maintaining ATP levels by oxidative phosphorylation. Neurons have a relatively high rate of ATP consumption, requiring a constant source of reducing equivalents to re-phosphorylate ATP from ADP and AMP. ATP is required to establish and maintain resting electro-chemical gradients, repolarize membranes after depolarization and synaptic transmission, and for a host of intracellular signaling and biosynthetic functions. During cerebral ischemia substrate for oxidative phosphorylation (i.e. oxygen and glucose) are reduced, and energy deprivation results in impaired cellular function and eventually cell death (for review see [41]. Historically, glucose has been considered the primary source of energy for neurons [42]. However, neurons do not normally store glucose as glycogen, and must rely on a steady exogenous delivery of substrate [43, 44]. Astrocytes, which can store glycogen, appear to be critical in maintaining a source of metabolic fuel to neurons during conditions of oxygen/glucose deprivation [43, 44]. Evidence now suggests that lactate generated by astrocytes is transported into neurons via the monocarboxylate transporter-2 (MCT-2, [42], which can serve as a metabolic fuel to maintain basal neuronal activity, particularly when the blood supply of glucose is interrupted as occurs during stroke [45, 46]. Triggering astrocytic glycolysis is at least in part due to adenosine monophosphate-activated protein kinase (AMPK), an evolutionarily conserved enzyme that functions as an energy sensor by coupling changes in ATP supply to ATP production [47]. A recent study [48] demonstrated a critical role for AMPK in neuronal-astrocyte energy coupling. In vitro ischemia (oxygen-glucose deprivation, OGD) induced neuronal release of tissue plasminogen activator (tPA), a strong activator of AMPK in astrocytes. This activation resulted in augmented astrocytic production and release of lactate, which was transported into neurons via a MCT-2-dependent mechanism, increasing neuronal survival. However, in the non-stressed state, and particularly in the post-stroke/recovery phase when energy requirements are high, a return to oxidative phosphorylation with glucose as the substrate is preferred [41].
The astrocytic syncytium may influence neuronal survival by coordinating the spatial delivery of metabolic fuels and thereby maintain both mitochondrial and cellular integrity. Gap junctions are permeable to both glucose and lactate [49], and have the potential to facilitate delivery of substrates to metabolically active neurons in local areas of decreased perfusion. However, the role of astrocytic gap junctions in stroke remains controversial: indiscriminate passage of molecules <1000 kDa may be either beneficial or harmful, influencing stroke outcome [50]. For example, if astrocytic gap junctions remain open following ischemia [51], they can allow pro-apoptotic factors and other molecules to spread through the syncytium, expanding the size of the infarct [52].
IV. The role of microRNAs in astrocyte-targeted strategies
microRNAs (miRs) are important post-transcriptional regulators that interact with multiple target messenger RNAs (mRNA) to coordinately regulate protein expression. A short (5-7 nucleotide-long) sequence in the mature miR determines the specificity of binding to mRNAs, so miRs can bind multiple mRNAs and mRNAs can be bound by multiple miRs, creating a new and complex layer of post-transcriptional control. As targets for stroke therapy, miR-based strategies provide the advantage of rapid onset of action, a critical element in developing effective clinical treatments for stroke. A successful phase 2 trial of the first miR-targeted drug, a locked nucleic acid targeting miR-122 to treat hepatitis C, has recently been completed [53], demonstrating that rapid translation of miR-based therapies from bench to clinic may be possible once candidate targets are identified. Studies investigating the role of miRs in cerebral ischemia are recent, and most have focused on changes in miR expression patterns with ischemia [54-56]. Cerebral ischemia induces several genes [57], which activate molecular cascades leading to both necrotic cell death in the anoxic core, and delayed neuronal cell death in the surrounding area [58, 59].
Subsequent studies have utilized profiling data to develop interventional strategies aimed at manipulating levels of miRs in the brain with the goal of mitigating injury and improving outcome following stroke. For example, miR-497 was increased in brain after MCAO, and knockdown of miR-497 was protective against MCAO-induced neuronal death [60]. To define the role of a miR, bioinformatically predicted molecular targets complementary to the binding sequence of the miR are testing for the ability of the miR to suppress expression when the 3’ untranslated region (3’UTR) of the putative target mRNA is placed downstream of a luciferase reporter construct. Using this approach, miR-497 was shown to directly target two anti-apoptotic genes, BCL-2 and BCL-w [60]. Our group utilized this approach to demonstrate that two brain-enriched miRs, miR-181a and miR-29a, are important mediators in the evolution of injury and determining outcome following stroke. Interestingly, a recent [61] microarray analysis of miR expression in the four principal cell types of the CNS (neurons, astrocytes, oligodendrocytes, and microglia) delineated a preferential cellular expression pattern of individual miRs; of note, miR-181a and miR-29a appear to be more highly expressed in astrocytes, corroborating both our own [62] and others’ [63] observations.
A recent review by our group [64] outlines in detail the effects of these astrocyte-localized miRs. Of interest, miR-181a and miR-29a each coordinate mitochondrial homeostasis via independent pathways. For example, miR-181a was shown to directly target GRP78 [65], anti-apoptotic members of the BCL2 family, BCL2 and myeloid cell leukemia 1 (MCL1) [66], and X-linked inhibitor of apoptosis (XIAP) [63], as well as additional targets involved in controlling mitochondrial function, redox state, and inflammatory pathways (for recent review see [67]. Overexpression of miR-181a in astrocytes subjected to glucose deprivation decreased mitochondrial membrane potential and increased reactive oxygen species (ROS) formation and cell death [66]. Concordant with the finding that miR-181a is expressed at greater levels in astrocytes, targeted reduction of miR-181a increased BCL2 and increased survival of primary astrocytes [66] while in primary neurons it failed to significantly change levels of BCL2 and did not improve survival after ischemia-like injury [68].
Regulation of mitochondrial homeostasis by miR-29 represents a more complex picture. We observed that miR-29a significantly increased in the resistant DG, but decreased in the vulnerable CA1 [62]. In the setting of in vitro ischemia, we demonstrated that miR-29a mimic protected and miR-29a inhibitor aggravated astrocyte injury and mitochondrial function by targeting the BCL2-family member p53 upregulated modulator of apoptosis (PUMA) [62]. However luciferase assays indicate that the miR-29 family targets both pro- and anti-apoptotic BCL2 family members [64]. While downregulation of miR-29 protected hearts against ischemia-reperfusion injury [69], upregulation of miR-29a protected neurons from apoptosis during both global [62] and focal cerebral ischemia [70]. Our results demonstrating targeting of several BCL2 family members strongly suggest that the reported pro-apoptotic and anti-apoptotic effects of the miR-29 family likely reflect inhibition of different targets in different cells and under different physiological or pathological settings. Indeed, verification of predicted miR targets by luciferase assay reflects only the potential to target in vivo; cell-type specific actions appear to play a substantial role in the physiological activity of individual miRs, and also require validation. However, cell-type specificity reveals an additional level of endogenous regulation that may be exploited for cell-type specific therapies aimed at reducing cerebral ischemic injury.
V. Future directions
The failure to translate successful findings in animal models to practical clinical therapies is likely multi-dimensional. Traditionally, stroke research has focused on exploiting a single mechanism, targeting a single cell type, the neuron. While methodologies overexpressing single genes, such as SOD2 and BCL2, have proven effective in minimizing injury in animal models, the clinical utility of such approaches has not been demonstrated. At this point in time, expanding the search for an effective clinical therapy to include multiple and alternative targets may be a new and helpful direction. Mitochondrial- and miR-based approaches each offer the unique potential for a single manipulation to evoke multiple, coordinated, and complementary cellular responses to improve astrocyte survival. Studies in our laboratory focusing on combining these modalities into a single intervention (i.e. altering levels of a single miR that targets mitochondria in astrocytes) have yielded promising results. However, as a result of their ability to target numerous genes systemic administration of miRs may also have unintended effects on other organ systems, which may limit the clinical applicability of a given miR. Another potential pitfall of RNA-based treatment paradigms is miR degradation by endogenous RNAases, thereby limiting their pharmacological efficacy. However, in addition to their rapid post-transcriptional effect, miRs can be readily chemically modified to enhance both stability and transfer across cell membranes. Moreover, miRs have been shown to exist endogenously in the circulation circulation [71] and may be a novel mode of astrocyte-neuronal communication via exosomes [72]. Exploring more clinically relevant approaches to alter endogenous miR production, such as intravenous or intraperitoneal delivery of chemically-stabilized miR mimics and inhibitors, and investigating post-treatment effects of manipulating these astrocyte-enriched miRs, are essential next steps for effective clinical translation.
Figure 3.
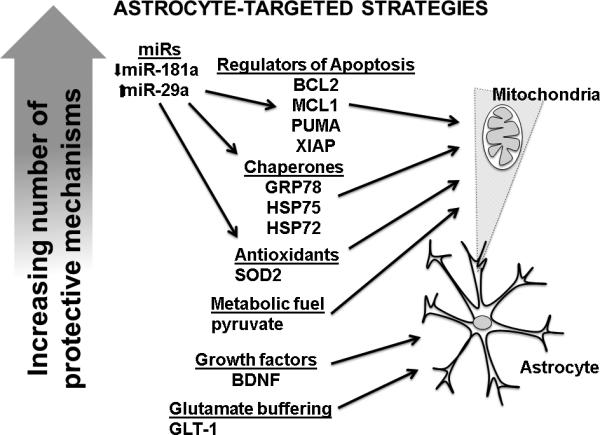
Effective astrocyte-targeted strategies for inducing neuroprotection vary in complexity. “Upstream” strategies, such as increasing or decreasing endogenous miRs to improve astrocytic mitochondrial homeostasis, target a greater number of mechanisms and may have a greater downstream effect on overall cellular homeostasis and neuroprotection. BCL2 = B cell lymphoma 2; BDNF = brain derived neurotrophic factor; GLT-1 = glutamate transporter 1; GRP78 = glucose regulated protein 78 (BiP); HSP72 = heat shock protein 72; HSP75 = heat shock protein 75 (GRP75 glucose regulated protein 75 also known as mortalin); MCL1 = Myeloid cell leukemia 1; PUMA = p53 upregulated modulator of apoptosis.ROS = reactive oxygen species; XIAP = X-linked inhibitor of apoptosis.
Acknowledgements
Supported by NIH T32-GM089626 to CMS, and NIH grants NS084396, NS053898, and NS080177 to RGG.
Footnotes
The authors have no conflicting financial interests.
References
- 1.Roger V, Go A, Lloyd-Jones D, Adams R, Berry J, Brown T, Carnethon M, Dai S, de Simone G, Ford E, Fox C, Fullerton H, Gillespie C, Greenlund K, Hailpern S, Heit J, Ho P, Howard V, Kissela B, Kittner S, Lackland D, Lichtman J, Lisabeth L, Makuc D, Marcus G, Marelli A, Matchar D, McDermott M, Meigs J, Moy C, Mozaffarian D, Mussolino M, Nichol G, Paynter N, Rosamond W, Sorlie P, Stafford R, Turan T, Turner M, Wong N, Wylie-Rosett J. Heart disease and stroke statistics--2011 update: a report from the American Heart Association. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blakeley J, Llinas R. Thrombolytic therapy for acute ischemic stroke. J Neurol Sci. 2007;261:55–62. doi: 10.1016/j.jns.2007.04.031. [DOI] [PubMed] [Google Scholar]
- 3.Ogata K, Kosaka T. Structural and quantitative analysis of astrocytes in the mouse hippocampus. Neuroscience. 2002;113:221–233. doi: 10.1016/s0306-4522(02)00041-6. [DOI] [PubMed] [Google Scholar]
- 4.Bushong E, Martone M, Jones Y, Ellisman M. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002;22:183–192. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halassa M, Fellin T, Takano H, Dong J, Haydon P. Synaptic islands defined by the territory of a single astrocyte. J Neurosci. 2007;27:6473–6477. doi: 10.1523/JNEUROSCI.1419-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bass N, Hess H, Pope A, Thalheimer C. Quantitative cytoarchitectonic distribution of neurons, glia, and DNa in rat cerebral cortex. J Comp Neurol. 1971;143:481–490. doi: 10.1002/cne.901430405. [DOI] [PubMed] [Google Scholar]
- 7.Rouach N, Glowinski J, Giaume C. Activity-Dependent Neuronal Control of Gap-Junctional Communication in Astrocytes. The Journal of Cell Biology. 2000;149:1513–1526. doi: 10.1083/jcb.149.7.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Araque A, Sanzgiri R, Parpura V, Haydon P. Astrocyte-induced modulation of synaptic transmission. Can J Physiol Pharmacol. 1999;77:699–706. [PubMed] [Google Scholar]
- 9.Giordano G, Kavanagh T, Costa L. Mouse cerebellar astrocytes protect cerebellar granule neurons against toxicity of the polybrominated diphenyl ether (PBDE) mixture DE-71. Neurotoxicology. 2009;30:326–329. doi: 10.1016/j.neuro.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noguchi Y, Shinozaki Y, Fujishita K, Shibata K, Imura Y, Morizawa Y, Gachet C, Koizumi S. Astrocytes protect neurons against methylmercury via ATP/P2Y(1) receptor-mediated pathways in astrocytes. PLoS One. 2013;8:e57898. doi: 10.1371/journal.pone.0057898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rathinam M, Watts L, Narasimhan M, Riar A, Mahimainathan L, Henderson G. Astrocyte mediated protection of fetal cerebral cortical neurons from rotenone and paraquat. Environ Toxicol Pharmacol. 2012;33:353–360. doi: 10.1016/j.etap.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- 13.Swanson R, Ying W, Kauppinen T. Astrocyte influences on ischemic neuronal death. Current molecular medicine. 2004;4:193–205. doi: 10.2174/1566524043479185. [DOI] [PubMed] [Google Scholar]
- 14.Zhao Y, Rempe D. Targeting astrocytes for stroke therapy. Neurotherapeutics. 2010;7:439–451. doi: 10.1016/j.nurt.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrer I, Planas A. Signaling of cell death and cell survival following focal cerebral ischemia: life and death struggle in the penumbra. J Neuropathol Exp Neurol. 2003;62:329–339. doi: 10.1093/jnen/62.4.329. [DOI] [PubMed] [Google Scholar]
- 16.Qu W, Wang Y, Wang J, Tang Y, Zhang Q, Tian D, Yu Z, Xie M, Wang W. Galectin-1 enhances astrocytic BDNF production and improves functional outcome in rats following ischemia. Neurochem Res. 2010;35:1716–1724. doi: 10.1007/s11064-010-0234-z. [DOI] [PubMed] [Google Scholar]
- 17.Miao Y, Qiu Y, Lin Y, Miao Z, Zhang J, Lu X. Protection by pyruvate against glutamate neurotoxicity is mediated by astrocytes through a glutathione-dependent mechanism. Mol Biol Rep. 2011;38:3235–3242. doi: 10.1007/s11033-010-9998-0. [DOI] [PubMed] [Google Scholar]
- 18.Ouyang Y, Voloboueva L, Xu L, Giffard R. Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J Neurosci. 2007;27:4253–4260. doi: 10.1523/JNEUROSCI.0211-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu L, Emery JF, Ouyang Y-B, Voloboueva LA, Giffard RG. Astrocyte targeted overexpression of Hsp72 or SOD2 reduces neuronal vulnerability to forebrain ischemia. Glia. 2010;58:1042–1049. doi: 10.1002/glia.20985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weller ML, Stone IM, Goss A, Rau T, Rova C, Poulsen DJ. Selective overexpression of excitatory amino acid transporter 2 (EAAT2) in astrocytes enhances neuroprotection from moderate but not severe hypoxia–ischemia. Neuroscience. 2008;155:1204–1211. doi: 10.1016/j.neuroscience.2008.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008;27:6407–6418. doi: 10.1038/onc.2008.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruiz A, Matute C, Alberdi E. Endoplasmic reticulum Ca2+ release through ryanodine and IP3 receptors contributes to neuronal excitotoxicity. Cell Calcium. 2009;46:273–281. doi: 10.1016/j.ceca.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 23.Green D, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333:1109–1112. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tait SWG, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 25.Penna C, Perrelli M-G, Pagliaro P. Mitochondrial pathways, permeability transition pore, and redox signaling in cardioprotection: therapeutic implications. Antioxidants & redox signalling. 2013;18:556–599. doi: 10.1089/ars.2011.4459. [DOI] [PubMed] [Google Scholar]
- 26.Webster KA. Mitochondrial membrane permeabilization and cell death during myocardial infarction: roles of calcium and reactive oxygen species. Future Cardiology. 2012;8:863–884. doi: 10.2217/fca.12.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giffard R, Han R, Emery J, Duan M, Pittet J. Regulation of apoptotic and inflammatory cell signaling in cerebral ischemia: the complex roles of heat shock protein 70. Anesthesiology. 2008;109:339–348. doi: 10.1097/ALN.0b013e31817f4ce0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. The Journal of Cell Biology. 2006;175:901–911. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Voloboueva LA, Duan M, Ouyang Y, Emery JF, Stoy C, Giffard RG. Overexpression of mitochondrial Hsp70/Hsp75 protects astrocytes against ischemic injury in vitro. J Cereb Blood Flow Metab. 2007;28:1009–1016. doi: 10.1038/sj.jcbfm.9600600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu L, Voloboueva LA, Ouyang Y, Emery JF, Giffard RG. Overexpression of mitochondrial Hsp70/Hsp75 in rat brain protects mitochondria, reduces oxidative stress, and protects from focal ischemia. J Cereb Blood Flow Metab. 2009;29:365–374. doi: 10.1038/jcbfm.2008.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun F-C, Wei S, Li C-W, Chang Y-S, Chao C-C, Lai Y-K. Localization of GRP78 to mitochondria under the unfolded protein response. Biochem J. 2006;396:31–39. doi: 10.1042/BJ20051916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ouyang Y, Xu L, Emery J, Lee A, Giffard R. Overexpressing GRP78 influences Ca2+ handling and function of mitochondria in astrocytes after ischemia-like stress. Mitochondrion. 2011;11:279–286. doi: 10.1016/j.mito.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adams J, Cory S. Bcl-2-regulated apoptosis: mechanism and therapeutic potential. Curr Opin Immunol. 2007;19:488–496. doi: 10.1016/j.coi.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parsons M, Green D. Mitochondria in cell death. Essays Biochem. 2010;47:99–114. doi: 10.1042/bse0470099. [DOI] [PubMed] [Google Scholar]
- 35.Kitagawa K, Matsumoto M, Tsujimoto Y, Ohtsuki T, Kuwabara K, Matsushita K, Yang G, Tanabe H, Martinou J, Hori M, Yanagihara T. Amelioration of hippocampal neuronal damage after global ischemia by neuronal overexpression of BCL-2 in transgenic mice. Stroke. 1998;29:2616–2621. doi: 10.1161/01.str.29.12.2616. [DOI] [PubMed] [Google Scholar]
- 36.Zhao H, Yenari MA, Cheng D, Sapolsky RM, Steinberg GK. Bcl-2 overexpression protects against neuron loss within the ischemic margin following experimental stroke and inhibits cytochrome c translocation and caspase-3 activity. Journal of Neurochemistry. 2003;85:1026–1036. doi: 10.1046/j.1471-4159.2003.01756.x. [DOI] [PubMed] [Google Scholar]
- 37.Ouyang Y, Carriedo S, Giffard R. Effect of Bcl-x(L) overexpression on reactive oxygen species, intracellular calcium, and mitochondrial membrane potential following injury in astrocytes. Free Radic Biol Med. 2002;33:544–551. doi: 10.1016/s0891-5849(02)00912-7. [DOI] [PubMed] [Google Scholar]
- 38.Szegezdi E, MacDonald DC, Ní Chonghaile T, Gupta S, Samali A. Bcl-2 family on guard at the ER. American Journal of Physiology - Cell Physiology. 2009;296:C941–C953. doi: 10.1152/ajpcell.00612.2008. [DOI] [PubMed] [Google Scholar]
- 39.Ouyang Y, Giffard R. ER-Mitochondria Crosstalk during Cerebral Ischemia: Molecular Chaperones and ER-Mitochondrial Calcium Transfer. Int J Cell Biol. 2012;2012:493934. doi: 10.1155/2012/493934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ouyang Y, Stary C, Yang G, Giffard R. microRNAs: innovative targets for cerebral ischemia and stroke. Curr Drug Targets. 2013;14:90–101. doi: 10.2174/138945013804806424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hertz L. Bioenergetics of cerebral ischemia: a cellular perspective. Neuropharmacology. 2008;55:289–309. doi: 10.1016/j.neuropharm.2008.05.023. [DOI] [PubMed] [Google Scholar]
- 42.Simpson IA, Carruthers A, Vannucci SJ. Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J Cereb Blood Flow Metab. 2007;27:1766–1791. doi: 10.1038/sj.jcbfm.9600521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hertz L, Dienel G. Energy metabolism in the brain. Int Rev Neurobiol. 2002;51:1–102. doi: 10.1016/s0074-7742(02)51003-5. [DOI] [PubMed] [Google Scholar]
- 44.Wender R, Brown AM, Fern R, Swanson RA, Farrell K, Ransom BR. Astrocytic glycogen influences axon function and survival during glucose deprivation in central white matter. The Journal of neuroscience. 2000;20:6804–6810. doi: 10.1523/JNEUROSCI.20-18-06804.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fox P, Raichle M, Mintun M, Dence C. Nonoxidative glucose consumption during focal physiologic neural activity. Science. 1988;241:462–464. doi: 10.1126/science.3260686. [DOI] [PubMed] [Google Scholar]
- 46.Pellerin L, Magistretti P. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A. 1994;91:10625–10629. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Amato S, Man H. Bioenergy sensing in the brain: the role of AMP-activated protein kinase in neuronal metabolism, development and neurological diseases. Cell Cycle. 2011;10:3452–3460. doi: 10.4161/cc.10.20.17953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.An J, Haile W, Wu F, Torre E, Yepes M. Tissue-type plasminogen activator mediates neuroglial coupling in the central nervous system. Neuroscience. 2014;257:41–48. doi: 10.1016/j.neuroscience.2013.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rose CR, Ransom BR. Gap junctions equalize intracellular Na+ concentration in astrocytes. Glia. 1997;20:299–307. doi: 10.1002/(sici)1098-1136(199708)20:4<299::aid-glia3>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 50.Nakase T, Sohl G, Theis M, Willecke K, Naus C. Increased apoptosis and inflammation after focal brain ischemia in mice lacking connexin43 in astrocytes. Am J Pathol. 2004;164:2067–2075. doi: 10.1016/S0002-9440(10)63765-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martinez A, Saez J. Regulation of astrocyte gap junctions by hypoxiareoxygenation. Brain Res Brain Res Rev. 2000;32:250–258. doi: 10.1016/s0165-0173(99)00086-7. [DOI] [PubMed] [Google Scholar]
- 52.Lin J, Weigel H, Cotrina M, Liu S, Bueno E, Hansen A, Hansen T, Goldman S, Nedergaard M. Gap-junction-mediated propagation and amplification of cell injury. Nat Neurosci. 1998;1:494–500. doi: 10.1038/2210. [DOI] [PubMed] [Google Scholar]
- 53.Janssen H, Reesink H, Lawitz E, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer A, Patick A, Chen A, Zhou Y, Persson R, King B, Kauppinen S, Levin A, Hodges M. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368:1685–1694. doi: 10.1056/NEJMoa1209026. [DOI] [PubMed] [Google Scholar]
- 54.Dharap A, Bowen K, Place R, Li L, Vemuganti R. Transient focal ischemia induces extensive temporal changes in rat cerebral microRNAome. J Cereb Blood Flow Metab. 2009;29:675–687. doi: 10.1038/jcbfm.2008.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jeyaseelan K, Lim K, Armugam A. MicroRNA expression in the blood and brain of rats subjected to transient focal ischemia by middle cerebral artery occlusion. Stroke. 2008;39:959–966. doi: 10.1161/STROKEAHA.107.500736. [DOI] [PubMed] [Google Scholar]
- 56.Liu D, Tian Y, Ander B, Xu H, Stamova B, Zhan X, Turner R, Jickling G, Sharp F. Brain and blood microRNA expression profiling of ischemic stroke, intracerebral hemorrhage, and kainate seizures. J Cereb Blood Flow Metab. 2010;30:92–101. doi: 10.1038/jcbfm.2009.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kernagis D, Laskowitz D. Evolving role of biomarkers in acute cerebrovascular disease. Ann Neurol. 2012;71:289–303. doi: 10.1002/ana.22553. [DOI] [PubMed] [Google Scholar]
- 58.Chopp M, Li Y. Apoptosis in focal cerebral ischemia. Acta Neurochir Suppl. 1996;66:21–26. doi: 10.1007/978-3-7091-9465-2_4. [DOI] [PubMed] [Google Scholar]
- 59.Mattson M, Culmsee C, Yu Z. Apoptotic and antiapoptotic mechanisms in stroke. Cell Tissue Res. 2000;301:173–187. doi: 10.1007/s004419900154. [DOI] [PubMed] [Google Scholar]
- 60.Yin K-J, Deng Z, Huang H, Hamblin M, Xie C, Zhang J, Chen YE. miR-497 regulates neuronal death in mouse brain after transient focal cerebral ischemia. Neurobiology of Disease. 2010;38:17–26. doi: 10.1016/j.nbd.2009.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jovicic A, Roshan R, Moisoi N, Pradervand S, Moser R, Pillai B, Luthi-Carter R. Comprehensive expression analyses of neural cell-type-specific miRNAs identify new determinants of the specification and maintenance of neuronal phenotypes. J Neurosci. 2013;33:5127–5137. doi: 10.1523/JNEUROSCI.0600-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ouyang Y, Xu L, Lu Y, Sun X, Yue S, Xiong X, Giffard R. Astrocyte-enriched miR-29a targets PUMA and reduces neuronal vulnerability to forebrain ischemia. Glia. 2013;61:1784–1794. doi: 10.1002/glia.22556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hutchison E, Kawamoto E, Taub D, Lal A, Abdelmohsen K, Zhang Y, Wood Wr, Lehrmann E, Camandola S, Becker K, Gorospe M, Mattson M. Evidence for miR-181 involvement in neuroinflammatory responses of astrocytes. Glia. 2013;61:1018–1028. doi: 10.1002/glia.22483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ouyang Y, Xu L, Yue S, Liu S, Giffard R. Neuroprotection by astrocytes in brain ischemia: Importance of microRNAs. Neurosci Lett. 2014;565C:53–58. doi: 10.1016/j.neulet.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ouyang Y, Lu Y, Yue S, Xu L, Xiong X, White R, Sun X, Giffard R. miR-181 regulates GRP78 and influences outcome from cerebral ischemia in vitro and in vivo. Neurobiol Dis. 2012;45:555–563. doi: 10.1016/j.nbd.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ouyang Y, Lu Y, Yue S, Giffard R. miR-181 targets multiple Bcl-2 family members and influences apoptosis and mitochondrial function in astrocytes. Mitochondrion. 2012;12:213–219. doi: 10.1016/j.mito.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ouyang Y, Stary C, White R, Giffard R. The Use of microRNAs to Modulate Redox and Immune Response to Stroke. Antioxid Redox Signal:ePub ahead of print. 2014 doi: 10.1089/ars.2013.5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moon J, Xu L, Giffard R. Inhibition of microRNA-181 reduces forebrain ischemia-induced neuronal loss. J Cereb Blood Flow Metab. 2013;33:1976–1982. doi: 10.1038/jcbfm.2013.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ye Y, Perez-Polo JR, Qian J, Birnbaum Y. The role of microRNA in modulating myocardial ischemia-reperfusion injury. Physiological Genomics. 2011;43:534–542. doi: 10.1152/physiolgenomics.00130.2010. [DOI] [PubMed] [Google Scholar]
- 70.Khanna S, Rink C, Ghoorkhanian R, Gnyawali S, Heigel M, Wijesinghe D, Chalfant C, Chan Y, Banerjee J, Huang Y, Roy S, Sen C. Loss of miR-29b following acute ischemic stroke contributes to neural cell death and infarct size. J Cereb Blood Flow Metab. 2013;33:1197–1206. doi: 10.1038/jcbfm.2013.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sepramaniam S, Tan J-R, Tan K-S, DeSilva D, Tavintharan S, Woon F-P, Wang C-W, Yong F-L, Karolina D-S, Kaur P, Liu F-J, Lim K-Y, Armugam A, Jeyaseelan K. Circulating MicroRNAs as Biomarkers of Acute Stroke. International Journal of Molecular Sciences. 2014;15:1418–1432. doi: 10.3390/ijms15011418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morel L, Regan M, Higashimori H, Ng S, Esau C, Vidensky S, Rothstein J, Yang Y. Neuronal exosomal miRNA-dependent translational regulation of astroglial glutamate transporter GLT1. J Biol Chem. 2013;288:7105–7116. doi: 10.1074/jbc.M112.410944. [DOI] [PMC free article] [PubMed] [Google Scholar]