

Abstract
Macrophage migration inhibitory factor (MIF) is a homotrimeric proinflammatory cytokine implicated in chronic inflammatory diseases and malignancies, including cutaneous squamous cell carcinomas (SCC). To determine whether MIF inhibition could reduce UVB light–induced inflammation and squamous carcinogenesis, a small-molecule MIF inhibitor (CPSI-1306) was utilized that disrupts homotrimerization. To examine the effect of CPSI-1306 on acute UVB-induced skin changes, Skh-1 hairless mice were systemically treated with CPSI-1306 for 5 days before UVB exposure. In addition to decreasing skin thickness and myeloperoxidase (MPO) activity, CPSI-1306 pretreatment increased keratinocyte apoptosis and p53 expression, decreased proliferation and phosphohistone variant H2AX (γ-H2AX), and enhanced repair of cyclobutane pyrimidine dimers. To examine the effect of CPSI-1306 on squamous carcinogenesis, mice were exposed to UVB for 10 weeks, followed by CPSI-1306 treatment for 8 weeks. CPSI-1306 dramatically decreased the density of UVB-associated p53 foci in non–tumor-bearing skin while simultaneously decreasing the epidermal Ki67 proliferation index. In addition to slowing the rate of tumor development, CPSI-1306 decreased the average tumor burden per mouse. Although CPSI-1306–treated mice developed only papillomas, nearly a third of papillomas in vehicle-treated mice progressed to microinvasive SCC. Thus, MIF inhibition is a promising strategy for prevention of the deleterious cutaneous effects of acute and chronic UVB exposure.
Introduction
Chronic inflammation and carcinogenesis are two longstanding processes that are intertwined with and feed into each other (1). Although it has been widely recognized that long-standing inflammation (extrinsic to the tumor) can provide a favorable milieu for progression of transformed cells, recent studies have indicated that the tumor cells themselves can induce an inflammatory response (intrinsic; ref. 2). Various components of chronic inflammation foster the growth and progression of tumors by contributing to the generation of reactive oxygen species (ROS) and cytokines and ultimately promoting genomic instability, angiogenesis, cellular migration, invasion, metastasis, and evasion of tumor immunosurveillance.
Cytokines function as the principal mediators between tumor and the various inflammatory cells and may be produced by either or both. Macrophage migration inhibitory factor (MIF) is a pleotropic cytokine with predominantly proinflammatory properties that has been proposed to function as the primary link between inflammation and cancer (3, 4). MIF is a homotrimeric molecule that is expressed by a number of epithelial, mesenchymal, and inflammatory cells. MIF has been shown to play seminal roles in several chronic inflammatory diseases, including diabetes mellitus (5), multiple sclerosis (6), systemic lupus erythematosus, rheumatoid arthritis, and atherosclerosis (7). MIF expression levels are elevated within the tumor tissue and/or in the serum in a number of malignancies not limited to those arising from the lung, stomach, colon, ovary and liver, and squamous cell carcinoma (SCC) of esophageal and head and neck origin (8–13). This increased expression of MIF has been correlated with a negative prognosis in most of these malignancies.
MIF has recently been recognized to be an important player in a multitude of skin diseases of infectious, inflammatory, and neoplastic etiologies, including SCC (14, 15). UVB light functions as a complete carcinogen in skin by initiating genetic mutations through direct DNA damage and production of ROS as well as promoting the survival and growth of the transformed cells to produce tumors (16). Prolonged exposure to UVB light also causes chronic inflammation and immunosuppression, contributing to the persistence and proliferation of transformed cells. In skin, MIF is expressed in and secreted by epidermal keratinocytes, dermal fibroblasts, and infiltrating inflammatory cells (17). MIF is upregulated after UVB exposure, and through its proinflammatory and proangiogenic properties, MIF can abet the tumor-promoting effects of UVB (17–19). The other mechanisms by which MIF promotes tumorigenesis have not yet been elucidated.
In a BALB/c mouse model of UVB-induced SCC, lack of MIF decreased the acute inflammatory response and dermal edema. Compared with their wild-type (WT) counterparts, these mice also had diminished tumor number and burden, slower tumor progression, and lower VEGF levels (20). On the other hand, transgenic mice that overexpressed MIF were highly susceptible to UVB-induced carcinogenesis. Not only did they develop tumors earlier than their WT littermates, but they also developed more and larger tumors (21). In both of these mouse models, there was an inverse correlation between MIF expression and p53 levels. Although MIF is expressed at moderate levels in human skin (within keratinocytes and fibroblasts), actinic keratoses and SCCs express high levels of MIF. In addition, exposure to UVB radiation increased MIF expression in keratinocyte and SCC cell lines (19). These studies highlight the significance of MIF in the development and progression of cutaneous SCC. The following experiments were designed to examine whether inhibition of MIF via the administration of a CPSI-1306 can alleviate the inflammatory and carcinogenic effects of acute and chronic UVB exposure.
Materials and Methods
Mice
The experiments were performed using 6- to 8-week-old female Skh-1 hairless mice obtained from Charles River Laboratories. The mice were housed in cages in groups of 5 in the vivarium at The Ohio State University (Columbus, OH) according to the requirements established by the American Association for Accreditation of Laboratory Animal Care. All procedures were approved by the Ohio State University Institutional Animal Care and Use Committee before the initiation of the studies.
Drug administration
An orally bioavailable small-molecule inhibitor of MIF, CPSI-1306 (Cytokine PharmaSciences,), was used at a dose of 20 mg/kg/d per mouse. The mice were orally fed 100 μL per day of vehicle (15% DMSO, 0.5% methyl cellulose in water) or CPSI-1306 (in vehicle). The CPSI-1306 solution was prepared weekly based on the average mouse weight per cage.
Acute UVB exposure study
Mice (n = 5 treated with vehicle or drug were used for each time point, 50 mice total) were treated with vehicle or CPSI-1306 for 5 consecutive days. Twenty-four hours after the last gavage, they were either euthanized (no UVB group) or dorsally exposed to 2,240 J/m2 (1 minimal erythemic dose; MED) of UVB light, after which they were euthanized at 30 minutes, 6, 24, or 48 hours. UVB light was emitted by Philips FS40 UV lamps (American Ultraviolet Company) and a UVX meter (UVP Inc.) was used to determine the UVB dose. The entire dorsal skin of the mouse was processed as follows: approximately 0.5 cm2 portion from the upper back was fixed in 10% neutral-buffered formalin for histologic examination and immunostains (2 hours at room temperature). The fixed tissues were processed and embedded in paraffin blocks. A 10-mm diameter punch was flash frozen for the myeloperoxidase (MPO) assay and all remaining skin was snap frozen.
Chronic UVB exposure study
A total of 36 mice (n = 5 non–UVB-exposed vehicle treated, n = 3 non–UVB-exposed CPSI-1306 treated, n = 13 UVB-exposed vehicle treated, and n = 15 UVB-exposed CPSI-1306 treated) were used in this study. Mice belonging to the UVB groups were exposed to 1 MED of UVB three times weekly on nonconsecutive days for 10 weeks. At the end of UVB exposure, vehicle or CPSI-1306 solution was fed to the respective groups for 5 consecutive days per week for 4 weeks. During the next 4 weeks, the frequency of feeding was reduced to three times weekly on nonconsecutive days. The mice were observed for the next 7 weeks and euthanized at the end of the 25-week study (Fig. 1). The individual tumor measuring at least 1 mm on each mouse was measured weekly to quantify tumor number, size, and total tumor area. The tumors were obtained and fixed separately in 10% neutral-buffered formalin (4 hours at room temperature) and the rest of the tissues were processed as above.
Figure 1.
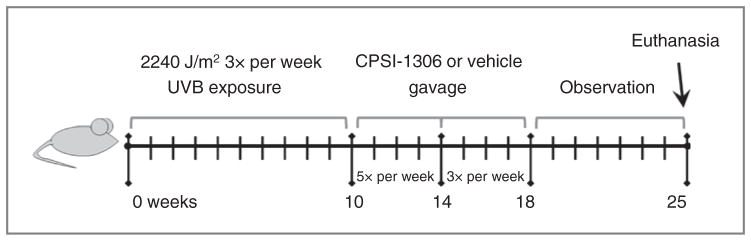
Schematic of chronic UVB exposure and CPSI-1306 treatment schedule. Female Skh-1 mice were exposed to UVB three times per week on nonconsecutive days for 10 weeks. UVB treatments were then halted and mice were gavaged with 20 mg/kg/d of CPSI-1306 or vehicle 5 days per week for 4 weeks, then 3 days per week for an additional 4 weeks then left untreated for the final 7 weeks, after which they were sacrificed.
Tumor grading
Hematoxylin and eosin-stained sections of the tumors were examined in a blinded manner by a board certified veterinary pathologist. As previously described, tumor grades were assigned as follows: benign papilloma (grades P1–3), malignant microinvasive SCC (grades MI1–3), or fully invasive SCC. Papillomas were exophytic tumors that showed no evidence of stromal invasion, whereas SCCs had a more endophytic appearance, with stromal invasion evidenced by loss of basement membrane continuity and development of an inflammatory stromal response. A grade 1 papilloma (P1) was composed primarily of epithelium without a pronounced papillary pattern; a grade 2 papilloma (P2) was a well-differentiated papillary mass; a grade 3 papilloma (P3) was similar to a grade 2 papilloma, except that a few finger-like projections of atypical cells at the base of the mass were present. Microinvasive SCCs (MI1-MI3) were subcategorized by depth of penetration into the dermis. Only tumors that invaded the panniculus carnosus were classified as fully invasive SCC. All grades of papilloma were considered benign (premalignant lesions), whereas all grades of microinvasive SCCs and SCCs were considered malignant (malignant tumors; ref. 22).
Myeloperoxidase activity assay
MPO is an enzyme produced by neutrophils and the levels reflect the degree of acute inflammation in a tissue. The activity was determined using a chemical assay as described previously (23).
IHC
The following primary antibodies and dilutions were used for IHC staining of the skin: p53- 1:500 dilution, 60 minutes at room temperature [clone CM5p; Novocastra (Leica Microsystems)]; Ki67- 1:200 dilution, 4°C overnight (clone TEC-3; Dako); cleaved caspase-3- 1:750 dilution, 4°C overnight (clone Asp175; Cell Signaling Technology), and phosphohistone H2A.X- 1:250 dilution, 4°C overnight (clone Ser139; Cell Signaling Technology). Five μm thick skin sections were deparaffinized and subjected sequentially to endogenous peroxidase blocking with 3% hydrogen peroxide, and antigen retrieval in Antigen Unmasking Solution (Vector Laboratories) for 15 minutes in a microwave. The slides were cooled for 20 minutes and blocked using the Avidin-Biotin Blocking Kit (Vector Laboratories – for cleaved caspase-3 and phosphohistone H2A. X only). The slides were then blocked with 1× casein (Vector Laboratories) for 30 minutes at room temperature, followed by incubation with the specific primary antibodies as mentioned above. After incubation with their respective biotinylated secondary antibodies, the stains were developed using the Vectastain ABC (Vector Laboratories) and diaminobenzidine (Vector Laboratories) kits, followed by hematoxylin counterstain. The slides were rinsed in 1× PBS containing 0.05% Tween-20 between the incubations.
IHC image analysis
The percentage of positive staining cells was calculated by manually counting positive and negative cells (ten ×60 magnification images for the acute study- p53, Ki67, cleaved caspase-3; five ×60 magnification images for chronic study, Ki67). For phosphohistone H2A.X, the percentage of positive epidermal area was determined using ImageJ from ten ×60 magnification images. The results are expressed as the average per group for the above. The number of p53 foci in the chronic study was determined by manually counting through the entire length of the dorsal skin section and expressed as average number per ×20 field.
CPD dot blot
Epidermal DNA was extracted using the PureLink Genomic Extraction Kit (Invitrogen) according to manufacturer directions. DNA (100 ng) was chemically denatured with NaOH, heat denatured at 95°C for 10 minutes, then neutralized with ammonium acetate. DNA was mixed with SSC and blotted onto nitrocellulose membranes via gentle vacuum pressure in triplicate. Membranes were dried for 1 hour at 80°C then washed three times in Tris-Buffered Saline Tween-20 (TBST) at room temperature for 5 minutes each. Membranes were blocked in TBST containing 5% BSA for 1 hour at room temperature and incubated overnight at 4°C with a 1:500 dilution of antithymine dimer antibody (Kamiya Biomedical). Membranes were washed as above then incubated with secondary antibody at a 1:10,000 dilution for 45 minutes at room temperature. Finally, membranes were washed, incubated with Lumiglo (Invitrogen), photographed, washed, incubated with SYBR Gold (Invitrogen), and photographed. ImageJ analysis was used to generate pixel intensities for cyclobutane pyrimidine dimers (CPD) and SYBR gold and these values were expressed in a ratio to determine relative intensities.
Statistical analysis
For the acute exposure experiments, linear mixed effects models were applied to the data to allow for correlations among observations from the same mouse. For all models, the interaction between UV exposure time and outcome (cleaved caspase-3, Ki67, phosphohistone H2A.X, p53, CPD formation, skin thickness, and MPO) was assessed. In the presence of significant interaction, differences in outcome between CPSI-1306–treated mice and vehicle mice were estimated at each UV exposure time, with 95% confidence intervals (CI); otherwise, an overall group difference with 95% CI was estimated. For outcomes with nonzero values at baseline, interaction contrasts were also used to assess the relative effect of CPSI-1306 in the presence of UVB exposure versus no UVB exposure. Note that cleaved caspase-3, CPD formation, skin thickness, and MPO were first natural-log transformed to stabilize variances; for cleaved caspase-3, a small number (0.01) was added before log transformation due to zero values.
Categorized tumor burden and the proportion of mice with at least one tumor at week 18 were compared between CPSI-1306 and vehicle groups using Fisher exact test. Similar to the acute experiments, linear mixed effects models were used to assess differences in Ki67 and p53 between the chronic exposure CPSI-1306 and vehicle groups. For Ki67, an interaction contrast was used to assess the relative effect of CPSI-1306 in the presence of chronic exposure versus no UV exposure. For p53, group comparisons were made at various cells per field (1–2 cells, 3–5 cells, 6–10 cells, 11–20 cells, >20 cells).
All analyses were performed using SAS/STAT software, version 9.2 (SAS Institute Inc.).
Results and Discussion
Acute UVB exposure study
To determine whether inhibition of MIF could protect against the deleterious epidermal effects of UVB exposure, we utilized a small-molecule MIF inhibitor, CPSI-1306 (6). This molecule prevents the association of MIF monomers and prevents the generation of the biologically functional MIF homotrimer. We gavaged Skh-1 hairless mice with CPSI-1306 for 5 consecutive days and then exposed them to 1 MED of UVB. Exposure to UVB causes significant epidermal keratinocyte apoptosis in the form of sunburn cells, which is accompanied by an increase in p53 expression (24). In severe cases, the keratinocyte apoptosis may be extensive, resulting in full thickness epidermal necrosis, such as in blistering sunburns. Toward the end of apoptosis or soon after, the lost keratinocytes are replaced by enhanced epidermal proliferation. Disruption of these processes such as by decreased apoptotic clearance and/or increased epidermal proliferation can lead to accumulation of DNA damage. UVB exposure also leads to the release of proinflammatory cytokines, leading to a late inflammatory response, which may in turn induce additional cell damage through ROS production. We euthanized the mice at specific time points from 30 minutes to 48 hours post-UVB exposure and analyzed the levels of key epidermal markers of UVB-induced damage.
Inhibition of MIF decreased acute UVB-induced epidermal proliferation
To examine the net effect of acute UVB exposure on epidermal proliferation, we compared the levels of cleaved caspase-3, an apoptotic protein and Ki67, a marker of proliferation between vehicle-treated and CPSI-1306–treated mice. No significant differences were observed in the levels of either marker between the non–UVB-exposed vehicle and non–UVB-exposed CPSI-1306–treated mice (Fig. 2A and B). UVB-associated keratinocyte apoptosis peaked at 24 hours post UVB exposure, as shown by the increased level of cleaved caspase-3. CPSI-1306–induced keratinocyte apoptosis could be appreciated as early as 30 minutes after a single UVB exposure (Fig. 2A). At 6, 24, and 48 hours following UVB exposure, the CPSI-1306–treated mice showed significantly increased expression of cleaved caspase-3 compared with the vehicle-treated mice (P < 0.0001). Epidermal proliferation as determined by Ki67 staining was significantly decreased by CPSI-1306 at 24 (P = 0.0206) and 48 hours (P = 0.0010) compared with vehicle-treated mice at each time point (Fig. 2B). Though not statistically significant, suppression of Ki67 by CPSI-1306 compared with vehicle-treated mice within each time point could also be observed at 30 minutes and 6 hours after UVB exposure (Fig. 2B). Thus, inhibition of MIF led to enhanced keratinocyte apoptosis and decreased epidermal proliferation following UVB exposure.
Figure 2.

Inhibition of MIF increases keratinocyte apoptosis, while decreasing proliferation. Female Skh-1 mice were pretreated with vehicle or CPSI-1306 for 5 days, exposed to 1 MED of UVB, and sacrificed at 30 minutes or 6, 24, or 48 hours after UVB exposure. Dorsal skin was examined for expression of the proapoptotic protein cleaved caspase-3 or the proliferation marker Ki67 by IHC. The percentage of positively staining epidermal cells was determined (CPSI-1306 vs. vehicle). A, left: CPSI-1306 significantly enhanced epidermal cleaved caspase—mediated apoptosis at 6, 24, and 48 hours compared with vehicle (*, P < 0.0001). Right, representative cleaved caspase-3 IHC images (×60) at 24 hours post UVB exposure when the maximum difference was observed between CPSI-1306 and vehicle-treated mice. Arrows indicate cleaved caspase-3–positive epidermal cells. B, left: CPSI-1306 significantly decreased epidermal proliferation as examined by Ki67 IHC at both 24 (*, P = 0.0206) and 48 hours (*, P = 0.0010), relative to vehicle. Right: representative Ki67 IHC images (×60 magnification) at 48 hours post UVB exposure when the maximum difference was observed between CPSI-1306 and vehicle-treated mice. Bars represent SD; *, P < 0.05.
CPSI-1306 reduced acute UVB-induced keratinocyte DNA damage
UVB exposure induces DNA damage directly through generation of CPD and pyrimidine (6–4) pyrimidone photo adducts (25) as well as indirectly via production of ROS resulting in 8-oxo-2′-deoxyguanosine adduct formation (16). To determine the effect of MIF inhibition on UVB-induced keratinocyte DNA damage, we examined the repair kinetics of CPD by dot blot analysis. UVB-induced CPD DNA adducts were more abundant at every time point examined in vehicle versus CPSI-1306–treated epidermis (Fig. 3A, P = 0.0001). Phosphohistone H2A.X has been shown to be a surrogate marker for dsDNA breaks (DSB) induced by UVB (26). Following UVB exposure, the levels of phosphohistone H2A.X peaked at 6 hours in both vehicle-and CPSI-1306–treated epidermis, following which the levels returned to nearly baseline levels. At every time point, CPSI-1306 treatment reduced the expression of phosphohistone H2A.X in epidermal keratinocytes (Fig. 3B, P < 0.0001). Even the baseline phosphohistone H2A.X levels were decreased in the non–UVB-exposed epidermis of CPSI-1306 compared with vehicle-treated mice.
Figure 3.

CPSI-1306 treatment decreases UVB-induced DNA damage. Epidermal DNA isolated from the dorsal skin of mice treated with vehicle or CPSI-1306 for 5 days before a single UVB exposure was examined by dot blot analysis to determine levels of CPD. A, left, at all time points examined, CPSI-1306 significantly decreased UVB-induced CPD levels compared with vehicle-treated skin (P = 0.0001). Right, dot blot for CPD; each dot represents DNA from one mouse and the various treatment groups are labeled and outlined for comparison between CPSI-1306 and vehicle-treated mice. Dorsal skin was also examined immunohistochemically to determine changes in levels of phosphohistone H2A.X and p53. B, left, CPSI-1306 treatment decreased epidermal levels of phosphohistone H2A.X compared with vehicle-treated mice at baseline and at all time points following UVB exposure (P < 0.0001). Right, representative phosphohistone H2A.X IHC images (×60 magnification) at 6 hours post UVB exposure when the maximum difference was observed between CPSI-1306 and vehicle-treated mice. C, CPSI-1306 significantly increased epidermal p53 levels at both 30 minutes (P = 0.0009) and 6 hours (P = 0.0001) following UVB exposure relative to vehicle. Right, representative p53 IHC images (×60 magnification) at 6-hour post UVB exposure when the maximum difference is observed between CPSI-1306 and vehicle-treated mice. Bars represent SD; *, P < 0.05.
MIF has been shown to antagonize the expression as well as function of wild-type p53 (27, 28). An increased level of p53 after UVB exposure ensures appropriate repair of DNA damage and elimination of irreversibly damaged cells (29). This enhanced production of p53 occurs principally through stabilization of the p53 protein, which prevents its proteasome-mediated degradation. To examine the effect of CPSI-1306 on p53, we examined its expression by IHC. As expected, induction of p53 is an early event following UVB exposure, detectable within 30 minutes (Fig. 3C), and reaching a maximum between 6 hours and 24 hours. Inhibition of MIF activity enhanced p53 levels in epidermal keratinocytes at 30 minutes (P = 0.0009) and 6 hours (P = 0.0001) after UVB exposure (Fig. 3C). After this, there was no significant difference in the expression of p53 between the vehicle- and CPSI-1306–treated mice (Fig. 3C). Thus, our findings are consistent with the idea that inhibiting MIF resulted in a decrease in the accumulation of DNA damage via a p53 mechanism.
CPSI-1306 reduced UVB-induced acute inflammation
The acute inflammatory response induced by UVB exposure contributes to secondary epidermal changes and accumulation of DNA damage. This inflammation is characterized by production of proinflammatory and chemotactic factors, including MIF, from damaged keratinocytes and fibroblasts, leading to vasodilation resulting in dermal edema and influx of neutrophils, followed by other inflammatory cells such as macrophages. To determine whether inhibition of MIF could reduce this inflammation in murine skin, we compared the average MPO levels, a surrogate for the number of neutrophils that infiltrate into the skin following UVB exposure (Table 1). The MPO levels increased progressively up to 48 hours after UVB irradiation, at which time point the CPSI-1306–treated mice showed significantly lower MPO levels (P = 0.0293). We then compared the average bi-fold thickness of back skin, a physical manifestation of UVB-induced cutaneous dermal edema (Table 1). The skin thickness increased progressively, reaching a maximum at 48 hours in both groups. Pretreatment with CPSI-1306 reduced the average skin thickness at 48 hours, at an almost statistically significant level (P = 0.0656). Thus, there is some evidence that inhibition of MIF may reduce the UVB-associated acute inflammatory response. Interestingly, the skins of the vehicle-treated mice were significantly thinner than the CPSI-1306–treated mice at 30 minutes following UVB exposure (P = 0.0003).
Table 1.
CPSI-1306 decreases UVB-induced acute inflammation
| UV exposure | Group | MPO activity: mean (SD) | Bi-fold skin thickness (mm): mean (SD) |
|---|---|---|---|
| No UV | Vehicle | 0.0041 (0.0015) | 0.64 (0.05) |
| CPSI-1306 | 0.0041 (0.0005) | 0.70 (0) | |
| 30 min | Vehicle | 0.0033 (0.0011) | 0.58 (0.04) |
| CPSI-1306 | 0.0035 (0.0009) | 0.78a (0.08) | |
| 6 h | Vehicle | 0.0042 (0.0013) | 0.68 (0.04) |
| CPSI-1306 | 0.0048 (0.0009) | 0.72 (0.11) | |
| 24 h | Vehicle | 0.0145 (0.0056) | 0.96 (0.15) |
| CPSI-1306 | 0.0128 (0.0053) | 0.92 (0.08) | |
| 48 h | Vehicle | 0.0205 (0.0037) | 1.34 (0.11) |
| CPSI-1306 | 0.0134b (0.0059) | 1.18c (0.23) |
P = 0.0003.
P = 0.0293.
P = 0.0656.
Chronic UVB exposure study
To determine whether inhibition of MIF can protect against the deleterious effects of long-term UVB exposure, we exposed Skh-1 mice to 1 MED of UVB three times a week for 10 weeks. The mice were then treated with CPSI-1306 for 5 consecutive days per week for 4 weeks. Because of a limited supply of CPSI-1306, the frequency of CPSI-1306 feeding was reduced to three nonconsecutive days per week during the subsequent 4 weeks. After 7 additional weeks of observation without CPSI-1306 treatment, the mice were euthanized at the end of 25 weeks (Fig. 1). Upon prolonged exposure to UVB, there was increased tumor burden in vehicle- versus CPSI-1306–treated mice (Fig. 4C) corresponding to increased accumulation of DNA mutations as indicated by mutant p53 foci (Fig. 5B) and enhanced epidermal proliferation (Fig. 5A).
Figure 4.

Inhibition of MIF decreases UVB-induced squamous carcinogenesis. Skh-1 mice were exposed to UVB three times per week on nonconsecutive days for 10 weeks. UVB treatments were halted, then mice were gavaged 5 days per week with 20 mg/kg/d CPSI-1306 for 4 weeks then 3 days per week for an additional 4 weeks, and finally monitored for an additional 7 weeks without further treatment. All tumors were counted and measured weekly after cessation of UVB exposure, excised at the end of the experiment, and histologically graded by a board certified veterinary pathologist to determine the grade and extent of invasion. A, inhibition of MIF decreased tumor multiplicity compared with vehicle; B, CPSI-1306 treatment delayed tumor onset relative to vehicle; C, treatment with CPSI-1306 decreased tumor burden compared with vehicle; D, CPSI-1306 treatment inhibited tumor progression compared with vehicle; P1-P3, benign papilloma 1–3; MI1-MI3, malignant microinvasive squamous cell carcinoma.
Figure 5.

CPSI-1306 treatment reduces proliferation and p53 foci formation in nontumor epidermis. Non–tumor-bearing dorsal skin harvested from chronically UVB-irradiated mice treated with either vehicle or CPSI-1306 was examined immunohistochemically to determine the percentage of Ki67-positive epidermal keratinocytes as well as the number of p53 foci. A, left, treatment with CPSI-1306 significantly inhibited epidermal proliferation in non–tumor-bearing skin compared with vehicle, relative to no UV exposure (P = 0.0168); right; representative Ki67 IHC images (×60 magnification) of chronic UVB-exposed CPSI-1306 and vehicle-treated mice. B, left, MIF inhibition after chronic UVB exposure significantly decreased the density of epidermal p53 foci (P = 0.0014); right, representative p53 IHC images (×20 magnification) of chronic UVB-exposed CPSI-1306 and vehicle-treated mice. Bars represent SD. *, P < 0.05.
Inhibition of MIF after chronic UVB exposure protected against SCC development
Following chronic UVB radiation, the mice were monitored continuously for skin tumor development. Though the difference in the number of tumors between the groups was less pronounced at the end of 25 weeks, at all time points examined, CPSI-1306–treated mice had fewer tumors compared with vehicle controls (Fig. 4A). At the completion of CPSI-1306 treatment (18 weeks), 62% of vehicle-treated mice were tumor free compared with 93% of CPSI-1306–treated mice (Fig. 4B, P = 0.0691). At the end of 25 weeks, 62% of vehicle-treated mice were tumor free compared with 60% of CPSI-1306–treated mice (Fig. 4B, P = 0.9337). Although the average tumor burden in the CPSI-1306–treated mice was visibly lower throughout the entire study compared with vehicle-treated mice (Fig. 4C), due to the lack of tumor development in some mice and the inherent variability of this tumor model, this differential tumor burden response did not achieve statistical significance. However, when classified on the basis of size (≤10 mm2, 11–20 mm2, ≥20 mm2), the categorized tumor burden in CPSI-1306–treated mice at the end of CPSI-1306 treatment (18 weeks) was significantly lower than that of the vehicle-treated mice (Supplementary Table S2, P = 0.0131). After 7 weeks of no CPSI-1306 (25 weeks), categorized tumor burden in CPSI-1306 compared with vehicle-treated mice was no longer significantly different (Supplementary Table S2, P = 0.5573). These data suggest that the efficacy of CPSI-1306 may be dependent upon continuous treatment. Of the tumors that developed in the vehicle-treated mice, 28% were microinvasive SCCs (14% MI1 and 14% MI2), whereas none of the tumors of the CPSI-1306–treated mice developed into microinvasive or frank SCCs (Fig. 4D). Thus, inhibition of MIF prevented UVB-induced cutaneous squamous carcinogenesis. In our model, the beneficial effects of CPSI-1306 were more apparent at 18 weeks than at 25 weeks. These data suggest that continued inhibition of MIF through administration of CPSI-1306 could have further prevented UVB-induced squamous carcinogenesis.
CPSI-1306 treatment after chronic UVB exposure diminished epidermal proliferation
To examine whether CPSI-1306 could affect chronic UVB-induced epidermal proliferation, the average Ki67 proliferation index of the non–tumor-bearing skin was determined. CPSI-1306 decreased the percentage of Ki67-positive epidermal keratinocytes in both non–UVB-exposed and UVB-exposed skin. This decrease was statistically significant in the skin of UVB-exposed mice relative to no UV exposure (Fig. 5A, P = 0.0168). Thus, inhibition of MIF decreased epidermal proliferation associated with chronic UVB exposure.
CPSI-1306 inhibition following chronic UVB exposure reduced the number of p53 foci
Chronic UVB exposure causes mutations of the p53 gene and the resultant abnormal p53 protein is resistant to proteasome-mediated degradation and can be easily detected (30, 31). Cells with the nonfunctional p53 protein are frequently impervious to normal DNA damage checkpoints and can persist for long periods of time. They may expand clonally to form UVB-induced “p53 foci” that can be detected by IHC. These clones may accumulate additional DNA mutations and progress to develop squamous papillomas and SCCs. In fact, the risk for SCC development correlates with the density of p53 foci (32, 33).
To examine whether CPSI-1306 could affect the epidermal accumulation of mutant p53, we examined the expression of p53 protein in non–tumor-bearing skin by IHC. The average number of epidermal p53 foci was significantly reduced in the CPSI-1306–treated mice compared with the vehicle-treated mice (Fig. 5B, P = 0.0014). The p53 foci were further classified on the basis of their size, that is, number of cells per foci as follows: 3–5, 6–10, 11–20, and > 20 cells. The majority of the foci contained less than ten cells. Although there was a statistically significant decrease only in the number of the smaller p53 foci, that is, those with 3–5 and 6–10 cells (P < 0.0001, P = 0.0578, respectively, data not shown), there were fewer foci of all sizes in the skin of CPSI-1306–treated mice compared with vehicle-treated mice (Fig. 5B, P = 00014). Thus, inhibition of MIF led to a lower density of p53 foci.
Our data indicate that inhibition of MIF before acute UVB exposure reduced epidermal proliferation while enhancing apoptotic removal of cells. This was accompanied by increased and early expression of p53 protein and an overall decrease in DNA damage and DNA DSBs. In addition to these beneficial effects on epidermal keratinocytes, CPSI-1306 also lessened the dermal edema and acute inflammation associated with UVB exposure. Thus, MIF inhibition protected against both the direct and indirect effects of acute UVB exposure-induced skin changes. Treatment of chronically UVB-exposed skin with CPSI-1306 decreased the density of p53 foci and significantly reduced epidermal proliferation. Inhibition of MIF also reduced tumor burden and malignancy rate. These effects were present even after cessation of CPSI-1306. Taken together, our data indicate that inhibition of MIF was protective against both the acute and chronic effects of UVB exposure.
Functional antagonism between MIF and p53
MIF was identified as one of the negative regulators of p53 activity and its inhibitory effect on p53 is manifold, including suppression of p53 transcriptional activity, reduction in p53-dependent apoptosis, and evasion of cell-cycle arrest (27). The exact mechanism by which this repression is mediated is still being elucidated and could be context dependent. MIF contributes to the progression of endotoxic shock by repressing p53 function and upregulating Cox-2 activity (34). On the other hand, NM23-H1, a suppressor of metastasis, physically interacts with MIF and relieves suppression of p53 (35). Jab1, a modulator of intracellular signaling, interacts with several cell-cycle regulators, including AP1, MIF, and p53, and as such may function as a mediator of functional interactions between MIF and p53 (36, 37). In addition to these indirect associations, MIF has been shown to interact directly with p53 through cysteine residues independent of Jab1 (38), and this interaction is determined by the redox state of the amino acid residues involved. The interaction of MIF and p53 promotes the association of p53 with Mdm2 and thus leading to the ubiquitination of p53 and its proteasome-mediated degradation. Thus, MIF and p53 play contradictory roles in cell-cycle regulation, inflammation, and tumorigenesis.
Proposed mechanism of action of CPSI-1306 in our model
After acute UVB exposure, there is an induction of MIF expression and trimerization in the skin (19) which promotes p53 degradation, thus antagonizing the function of p53 (39), resulting in increased inflammation and accumulation of DNA damage through increased epidermal proliferation and decreased keratinocyte apoptosis. CPSI-1306 prevents the formation of the MIF homotrimer and renders MIF biologically nonfunctional and incapable of blocking p53 protein expression and activity (Fig. 6A). In chronically UVB-exposed skin, there is accumulation of DNA damage indicated by increased formation of p53 foci. Inhibition of MIF activity alleviates its inhibitory effect on p53. Increased p53 activity slows cell-cycle progression, allowing time for the repair of DNA damage and, thus, decreasing p53 foci density. These processes together diminish the carcinogenic transformation and progression of transformed cells (Fig. 6B). Thus, our studies demonstrate that the inhibition of MIF protects against the deleterious effects of acute and chronic UVB exposure principally by relieving its inhibitor effects on p53.
Figure 6.

A proposed model for the role of MIF in UVB-mediated inflammation and skin carcinogenesis. A, acute UVB exposure induces MIF expression and trimerization in the skin, which inhibits p53 function by enhanced degradation of p53, increased inflammation and epidermal proliferation, and decreased apoptosis. CPSI-1306 treatment reverses the above-mentioned effects of MIF, decreasing the accumulation of DNA damage. B, chronic UVB exposure results in increased dermal MIF expression and trimerization, leading to increased epidermal proliferation and decreased apoptosis. As a consequence of genomic DNA damage and loss of p53 function, there is increased tumor formation and progression in the vehicle-treated mice. Treatment with CPSI-1306 prevents the trimerization of MIF, thus reducing the accumulation of DNA damage and eventually the squamous carcinogenesis.
Implications.
Macrophage migration inhibitory factor is a viable target for the prevention of UVB-induced cutaneous SSCs.
Acknowledgments
The authors thank Erin Burns, Katie Samijlenko, Evan Sommer, Kristen Duckro, and Jessica Cooperstone for their help with sample preparation.
Grant Support
This work was supported by grant R03CA164399.
Footnotes
Disclosure of Potential Conflicts of Interest
T. Sielecki has ownership interest (including patents) in Cytokine PharmaSciences. No potential conflicts of interest were disclosed by the other authors.
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
Authors’ Contributions
Conception and design: P. Nagarajan, K.L. Tober, T. Sielecki, A.R. Satoskar, T.M. Oberyszyn
Development of methodology: K.L. Tober
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): P. Nagarajan, K.L. Tober, J.A. Riggenbach, D.F. Kusewitt
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): P. Nagarajan, K.L. Tober, D.F. Kusewitt, A.M. Lehman
Writing, review, and or revision of the manuscript: P. Nagarajan, K.L. Tober, A.M. Lehman, T.M. Oberyszyn
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): K.L. Tober, J.A. Riggenbach, J. Pruitt, A.R. Satoskar
Study supervision: K.L. Tober, A.R. Satoskar, T.M. Oberyszyn
Other (pathologic diagnoses of skin tumors): D.F. Kusewitt
Other (synthesis and development of cpsi-1306): T. Sielecki
Other (designed, synthesized, and transferred antagonist to senior author): J. Pruitt
References
- 1.Candido J, Hagemann T. Cancer-related inflammation. J Clin Immunol. 2013;33 (Suppl 1):S79–84. doi: 10.1007/s10875-012-9847-0. [DOI] [PubMed] [Google Scholar]
- 2.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 3.Bucala R, Donnelly SC. Macrophage migration inhibitory factor: a probable link between inflammation and cancer. Immunity. 2007;26:281–5. doi: 10.1016/j.immuni.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 4.Conroy H, Mawhinney L, Donnelly SC. Inflammation and cancer: macrophage migration inhibitory factor (MIF)-the potential missing link. QJM. 2010;103:831–6. doi: 10.1093/qjmed/hcq148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanchez-Zamora Y, Terrazas LI, Vilches-Flores A, Leal E, Juarez I, Whitacre C, et al. Macrophage migration inhibitory factor is a therapeutic target in treatment of non-insulin-dependent diabetes mellitus. FASEB J. 2010;24:2583–90. doi: 10.1096/fj.09-147066. [DOI] [PubMed] [Google Scholar]
- 6.Kithcart AP, Cox GM, Sielecki T, Short A, Pruitt J, Papenfuss T, et al. A small-molecule inhibitor of macrophage migration inhibitory factor for the treatment of inflammatory disease. FASEB J. 2010;24:4459–66. doi: 10.1096/fj.10-162347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ayoub S, Hickey MJ, Morand EF. Mechanisms of disease: macrophage migration inhibitory factor in SLE, RA and atherosclerosis. Nat Clin Pract Rheumatol. 2008;4:98–105. doi: 10.1038/ncprheum0701. [DOI] [PubMed] [Google Scholar]
- 8.He XX, Chen K, Yang J, Li XY, Gan HY, Liu CY, et al. Macrophage migration inhibitory factor promotes colorectal cancer. Mol Med. 2009;15:1–10. doi: 10.2119/molmed.2008.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McClelland M, Zhao L, Carskadon S, Arenberg D. Expression of CD74, the receptor for macrophage migration inhibitory factor, in non-small cell lung cancer. Am J Pathol. 2009;174:638–46. doi: 10.2353/ajpath.2009.080463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lippitz BE. Cytokine patterns in patients with cancer: a systematic review. Lancet Oncol. 2013;14:e218–28. doi: 10.1016/S1470-2045(12)70582-X. [DOI] [PubMed] [Google Scholar]
- 11.Kindt N, Laurent G, Nonclercq D, Journe F, Ghanem G, Duvillier H, et al. Pharmacological inhibition of macrophage migration inhibitory factor interferes with the proliferation and invasiveness of squamous carcinoma cells. Int J Oncol. 2013;43:185–93. doi: 10.3892/ijo.2013.1944. [DOI] [PubMed] [Google Scholar]
- 12.Li H, Zang J, Wang P, Dai L, Zhang J, Wang K. Gastric cancer susceptibility in gastric cancer relatives: attributable risks of macrophage migration inhibitory factor promoter polymorphism and Helicobacter pylori. Cytokine. 2012;60:346–51. doi: 10.1016/j.cyto.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 13.Krockenberger M, Kranke P, Hausler S, Engel JB, Horn E, Nurnberger K, et al. Macrophage migration-inhibitory factor levels in serum of patients with ovarian cancer correlates with poor prognosis. Anticancer Res. 2012;32:5233–8. [PubMed] [Google Scholar]
- 14.Pazyar N, Feily A, Yaghoobi R. Macrophage migration inhibitory factor as an incriminating agent in dermatological disorders. Indian J Dermatol. 2013;58:157. doi: 10.4103/0019-5154.108068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilliver SC, Emmerson E, Bernhagen J, Hardman MJ. MIF: a key player in cutaneous biology and wound healing. Exp Dermatol. 2011;20:1–6. doi: 10.1111/j.1600-0625.2010.01194.x. [DOI] [PubMed] [Google Scholar]
- 16.Halliday GM. Inflammation, gene mutation and photoimmunosuppression in response to UVR-induced oxidative damage contributes to photocarcinogenesis. Mutat Res. 2005;571:107–20. doi: 10.1016/j.mrfmmm.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 17.Shimizu T. The role of macrophage migration inhibitory factor (MIF) in ultraviolet radiation-induced carcinogenesis. Cancers. 2010;2:1555–64. doi: 10.3390/cancers2031555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shimizu T. Role of macrophage migration inhibitory factor (MIF) in the skin. J Dermatol Sci. 2005;37:65–73. doi: 10.1016/j.jdermsci.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 19.Heise R, Vetter-Kauczok CS, Skazik C, Czaja K, Marquardt Y, Lue H, et al. Expression and function of macrophage migration inhibitory factor in the pathogenesis of UV-induced cutaneous nonmelanoma skin cancer. Photochem Photobiol. 2012;88:1157–64. doi: 10.1111/j.1751-1097.2012.01108.x. [DOI] [PubMed] [Google Scholar]
- 20.Martin J, Duncan FJ, Keiser T, Shin S, Kusewitt DF, Oberyszyn T, et al. Macrophage migration inhibitory factor (MIF) plays a critical role in pathogenesis of ultraviolet-B (UVB) -induced nonmelanoma skin cancer (NMSC) FASEB J. 2009;23:720–30. doi: 10.1096/fj.08-119628. [DOI] [PubMed] [Google Scholar]
- 21.Honda A, Abe R, Yoshihisa Y, Makino T, Matsunaga K, Nishihira J, et al. Deficient deletion of apoptotic cells by macrophage migration inhibitory factor (MIF) overexpression accelerates photocarcinogenesis. Carcinogenesis. 2009;30:1597–605. doi: 10.1093/carcin/bgp160. [DOI] [PubMed] [Google Scholar]
- 22.Thomas-Ahner JM, Wulff BC, Tober KL, Kusewitt DF, Riggenbach JA, Oberyszyn TM. Gender differences in UVB-induced skin carcinogenesis, inflammation, and DNA damage. Cancer Res. 2007;67:3468–74. doi: 10.1158/0008-5472.CAN-06-3798. [DOI] [PubMed] [Google Scholar]
- 23.Wilgus TA, Koki AT, Zweifel BS, Kusewitt DF, Rubal PA, Oberyszyn TM. Inhibition of cutaneous ultraviolet light B-mediated inflammation and tumor formation with topical celecoxib treatment. Mol Carcinog. 2003;38:49–58. doi: 10.1002/mc.10141. [DOI] [PubMed] [Google Scholar]
- 24.Lee JK, Kim JH, Nam KT, Lee SH. Molecular events associated with apoptosis and proliferation induced by ultraviolet-B radiation in the skin of hairless mice. J Dermatol Sci. 2003;32:171–9. doi: 10.1016/s0923-1811(03)00094-x. [DOI] [PubMed] [Google Scholar]
- 25.Cadet J, Mouret S, Ravanat JL, Douki T. Photoinduced damage to cellular DNA: direct and photosensitized reactions. Photochem Photobiol. 2012;88:1048–65. doi: 10.1111/j.1751-1097.2012.01200.x. [DOI] [PubMed] [Google Scholar]
- 26.Barnes L, Dumas M, Juan M, Noblesse E, Tesniere A, Schnebert S, et al. GammaH2AX, an accurate marker that analyzes UV genotoxic effects on human keratinocytes and on human skin. Photochem Photobiol. 2010;86:933–41. doi: 10.1111/j.1751-1097.2010.00744.x. [DOI] [PubMed] [Google Scholar]
- 27.Hudson JD, Shoaibi MA, Maestro R, Carnero A, Hannon GJ, Beach DH. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med. 1999;190:1375–82. doi: 10.1084/jem.190.10.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salminen A, Kaarniranta K. Control of p53 and NF-kappaB signaling by WIP1 and MIF: role in cellular senescence and organismal aging. Cell Signal. 2011;23:747–52. doi: 10.1016/j.cellsig.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 29.Lu YP, Lou YR, Yen P, Mitchell D, Huang MT, Conney AH. Time course for early adaptive responses to ultraviolet B light in the epidermis of SKH-1 mice. Cancer Res. 1999;59:4591–602. [PubMed] [Google Scholar]
- 30.Benjamin CL, Ananthaswamy HN. p53 and the pathogenesis of skin cancer. Toxicol Appl Pharmacol. 2007;224:241–8. doi: 10.1016/j.taap.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Gruijl FR, Rebel H. Early events in UV carcinogenesis–DNA damage, target cells and mutant p53 foci. Photochem Photobiol. 2008;84:382–7. doi: 10.1111/j.1751-1097.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- 32.Rebel H, Mosnier LO, Berg RJ, Westermande Vries A, van Steeg H, van Kranen HJ, et al. Early p53-positive foci as indicators of tumor risk in ultraviolet-exposed hairless mice: kinetics of induction, effects of DNA repair deficiency, and p53 heterozygosity. Cancer Res. 2001;61:977–83. [PubMed] [Google Scholar]
- 33.Melnikova VO, Pacifico A, Chimenti S, Peris K, Ananthaswamy HN. Fate of UVB-induced p53 mutations in SKH-hr1 mouse skin after discontinuation of irradiation: relationship to skin cancer development. Oncogene. 2005;24:7055–63. doi: 10.1038/sj.onc.1208863. [DOI] [PubMed] [Google Scholar]
- 34.Mitchell RA, Liao H, Chesney J, Fingerle-Rowson G, Baugh J, David J, et al. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc Natl Acad Sci U S A. 2002;99:345–50. doi: 10.1073/pnas.012511599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jung H, Seong HA, Ha H. Direct interaction between NM23-H1 and macrophage migration inhibitory factor (MIF) is critical for alleviation of MIF-mediated suppression of p53 activity. J Biol Chem. 2008;283:32669–79. doi: 10.1074/jbc.M806225200. [DOI] [PubMed] [Google Scholar]
- 36.Oh W, Lee EW, Sung YH, Yang MR, Ghim J, Lee HW, et al. Jab1 induces the cytoplasmic localization and degradation of p53 in coordination with Hdm2. J Biol Chem. 2006;281:17457–65. doi: 10.1074/jbc.M601857200. [DOI] [PubMed] [Google Scholar]
- 37.Shackleford TJ, Claret FX. JAB1/CSN5: a new player in cell cycle control and cancer. Cell Div. 2010;5:26. doi: 10.1186/1747-1028-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jung H, Seong HA, Ha H. Critical role of cysteine residue 81 of macrophage migration inhibitory factor (MIF) in MIF-induced inhibition of p53 activity. J Biol Chem. 2008;283:20383–96. doi: 10.1074/jbc.M800050200. [DOI] [PubMed] [Google Scholar]
- 39.Fingerle-Rowson G, Petrenko O. MIF coordinates the cell cycle with DNA damage checkpoints. Lessons from knockout mouse models. Cell Div. 2007;2:22. doi: 10.1186/1747-1028-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]