

Abstract
AIM: To generate novel tumor models for preclinical validation of biomarkers that allow drug response prediction and individual therapeutic decisions.
METHODS: Cell line establishment was conducted by both direct in vitro culturing and in vivo xenografting followed by in vitro culturing procedure. A comprehensive characterization was subsequently performed. This included quality control, consisting of the confirmation of human and colorectal cancer (CRC) origin by DNA fingerprint and epithelial cell adhesion molecule (EpCAM) staining, as well as mycoplasma and human virus testing. Phenotypic analysis was done by light microscopy and multicolor flow cytometry. Histopathological examination (β-catenin and cytokeratin staining) was conducted in direct comparison to parental tumor tissues. Extensive molecular-pathological profiling included mutation analysis for CRC-associated driver mutations, assessment of chromosomal and microsatellite instability, and the grade of CpG island methylation. Additionally, an array-based comparative genomic hybridization analysis was performed. Drug responsiveness was assessed for a panel of classical and novel substances in clinical use for the treatment of solid cancers. Finally, tumorigenicity of the cell lines was tested by xenografting into immunocompromised nude mice.
RESULTS: Herein we describe the establishment of three ultra-low passage cell lines from two individual patients suffering from sporadic CRC. One cell line was derived directly from an early stage case (HROC18), whereas two cell lines could be established both direct from patient material and after xenografting from a late stage tumor (HROC32). All cell lines were free of contaminating mycoplasma and viruses. Molecular-pathological analysis allowed all cell lines to be classified as chromosomal instable (CIN+). They were aneuploid, with CpG island promoter methylation and microsatellite instability being absent. The following mutational profile was observed both in the cell lines and the parental tumor tissue: HROC18: APCmut, p53mut, K-raswt; HROC32: APCwt, p53mut, K-rasmut. All cell lines were characterized as epithelial (EpCAM+) cells, showing distinct morphology and migration speed, but comparable growth kinetics. The cell lines showed different patterns of response towards clinically approved and novel drugs, with HROC18 being more resistant than HROC32 cells. Finally, in vivo tumorigenicity was demonstrated.
CONCLUSION: We successfully established and characterized novel ultra-low passage patient-derived CRC models as useful instruments for analyzing biological characteristics associated with the CIN+ phenotype.
Keywords: Patient-derived tumor model, Colorectal cancer, Individualized medicine, Molecular characterization, Drug response, Translational research
Core tip: We ultra-low passage and well-characterized tumor models are considered the basis for modern preclinical research, but are still rare for colorectal carcinoma (CRC). Herein describe two novel ultra-low passage patient-derived CRC cell lines, HROC18 and HROC32, which were established both direct from patient material and after xenografting. We characterized these models according to phenotype, molecular, morphological, and growth characteristics, as well as by drug response profiles. These cell lines expand our comprehensive collection of tumor models, which in summary provide a useful instrument for basic and translational research. The models are available on request.
INTRODUCTION
The landmark paper by Vogelstein et al[1] ushered in a new era of understanding genetic alterations during colorectal tumor development and led to a new model of colorectal tumorigenesis. Since then, great leaps have been made in categorizing the main molecular classes of colorectal carcinoma (CRC) by pathogenetic mechanisms, namely those having chromosomal and microsatellite instability, as well as by CpG island methylator phenotype (CIMP)[2,3]. These mechanisms allow the definition of five molecular subtypes of CRC[4]. Initially, Fearon and Vogelstein proposed that inactivation of the adenomatous polyposis coli (APC) tumor suppressor gene occurs as a first step with the activation of KRAS mutations[5]. The subsequent malignant transformation is driven by additional mutations in the transforming growth factor PIK3CA and TP53 pathways[6-9]. As a consequence of these fundamental findings, further mutational analyses have shown that, in an individual cancer, only a limited number of pathways are dysregulated by “driver” mutations from a circumscribed number of about 80 candidate cancer genes (i.e., cancer genome landscapes)[10-12]. As tumors are highly heterogeneous and normal mutation rates are insufficient to account for the required number of mutations, Loeb et al[13] proposed a mutator phenotype of cancer cells that allows them to increase their rate of new mutations.
This chromosomal instability phenotype (CIN+) is observed in 65%-70% of sporadic CRC and indicates an accelerated rate of gains or losses of whole or large portions of chromosomes, resulting in a karyotypic variability[14]. The consequence of CIN+ is an imbalance in the number of chromosomes (aneuploidy), subchromal genomic amplifications, and a high frequency of heterozygosity loss[15].
Transfer of these insights into clinical practice led to the development of therapies targeting epidermal growth factor (EGF)-receptor (EGFR) blockade (with the pre-requisite of KRAS mutational analysis)[16,17], and it is highly probable that further “individual” molecular testing of patients’ tumor tissue will become standard to guide and improve anticancer therapy regimens in the near future.
In this study, we describe the establishment and functional characterization of novel CIN+ cell lines with their corresponding xenograft model. A broad analysis of tumor biology, genetic alterations, and assessment of chemosensitivity towards an extended range of chemotherapeutic drugs is shown. Considering these aspects, the described cell lines provide an excellent tool for further development of individual therapy regimens.
MATERIALS AND METHODS
Tumor preparation and cell line establishment
Primary CRC resection specimens of HROC18 and HROC32 were received fresh from surgery, with informed written patient consent. Both patients underwent surgery upon initial diagnosis; hence, neither of them received any kind of prior therapy (i.e., surgery, chemotherapy, or radiotherapy). All procedures were approved by the Ethics Committee of the University of Rostock (reference number II HV 43/2004) in accordance with generally accepted guidelines for the use of human material. Tumor samples were cut into small pieces. Parts of the tumor were immediately frozen in freezing medium [fetal calf serum (FCS) containing 10% DMSO; Sigma Aldrich, Munich, Germany] at -80 °C for subsequent xenografting. Other pieces were frozen in liquid nitrogen for molecular analysis. The cell line establishment protocol was adapted from that of Maletzki et al[18]. Tumor cells were released from surrounding tissue by scraping and passage through a nylon mesh (100 μm). Single cell suspensions were seeded on collagen-coated 6-well plates in Quantum tumor medium [supplemented with 10% FCS, 200 mmol/L L-glutamine, antibiotics (penicillin G 10.000 IU/L; streptomycin 130 mg/L), and antimycotics (amphotericin B 6 mg/L)], and incubated at 37 °C in a humidified atmosphere of 5% CO2. All cell culture reagents were obtained from PAA (Cölbe, Germany), antibiotics and antifungal agents were from the pharmacy of the University of Rostock hospital. The medium was changed every 3-4 d. Initial passage into a 25 cm2 culture flask was performed when tumor cell growth was observed. If stromal cell growth was noted in the initial cultures, differential trypsinization was used to obtain a pure tumor cell population. Continually growing cell cultures were further passaged; frozen samples were prepared from low passages regularly.
In vivo tumorigenicity
In vivo tumorigenicity of HROC18 and HROC32 cells was tested following subcutaneous (sc) injection of 5 × 106 cells (with or without Matrigel®, BD Biosciences, Heidelberg, Germany) into the right and left hind flank of six-week-old female NMRI Foxn1nu mice. Mice were bred in the University of Rostock’s animal facilities and maintained in specified pathogen-free conditions. Their care and housing were in accordance with guidelines as put forth by the German Ethical Committee. Tumor growth was monitored for at least 10 wk. Outgrowing tumors were removed for histological examination.
In a parallel experiment, in vivo xenografting of a HROC32 patients’ tumor was performed. Subcutaneous tumor implantation was performed as described[19]. Established xenografts (≥ 1500 mm³) were removed and subjected to in vitro culture protocols as described above. In the case of HROC18, the quantity of tumor material was insufficient for in vivo xenografting; hence, cell line establishment was only done on patient tumor material.
Histology and immunohistochemistry of original tumors and xenografts
Histopathological examination of HE-stained primary tumors and the corresponding xenografts was performed according to standard protocols for clinicopathological CRC staging[20], with additional staging information being compiled from patients’ clinical charts. Supplementary immunostainings from paraffin-embedded primary tumors were done for β-catenin (Cell Signaling Technology, Boston, United States) and MNF116 (cytokeratin; Dako, Hamburg, Germany).
Mutational and methylation profile of tumor-associated target genes and determining the level of chromosomal instability
Molecular classification was done according to Ostwald et al[3] on an ABI 3500 genetic analyzer (Applied Biosystems, Darmstadt, Germany). Data analysis was carried out by taking advantage of SeqScape® Software v2.7 (Applied Biosystems). These data, as well as staging information compiled from the clinical charts, are summarized in Table 1. Mutations in tumor-associated APC, p53, KRAS, and BRAFV600E genes were analyzed as described. DNA-methylation in CIMP-sensitive promoters was traced by MethyLight with a modified marker panel originally published by Ogino et al[21]. The degree of chromosomal instability was assessed using SNP Array 6.0 from Affymetrix (Cleveland, OH) according to the manufacturer’s instructions.
Table 1.
Clinical and pathological characteristics of patients as well as cell line establishment protocol
| Tumor-ID | Age/gender | Tumor location | TNM-stage | UICC stage | Tumor type | Molecular type | β-catenin translocation | Corresponding xenograft | Direct cell line establishment | Cell line from xenograft | Paired B-LCL |
| HROC18 | 65/F | Caecum | G2T2N0 M0 | I | Primary adenocarcinoma | spStd | + | - | + | - | Yes |
| HROC32 | 83/F | Ascending colon | G2T4N2 M1 | IV | Primary adenocarcinoma | spStd | + | + | + | + | Yes |
F: Female; spStd: Sporadic standard; B-LCL: B lymphoid cell line; +: Positive; -: Negative.
DNA typing
Genomic DNA was isolated from cell lines at different passages, matched tumor and normal tissue, and corresponding B cells using the Wizard® Genomic DNA Purification Kit provided by Promega (Mannheim, Germany). Highly polymorphic short tandem repeat (STR) DNA marker (CSF1PO, TPOX, THO1, vWA, D16S539, D13S317, and D7S820) and the gender marker amelogenin were amplified in standard polymerase chain reaction (PCR) reactions and analyzed on an ABI Prism 3100 system. PCR primers were based on the original publication[22].
Generation of peripheral B cultures from patients
Peripheral blood lymphocytes were purified by Ficoll density-gradient centrifugation. B-lymphoid cell lines (B-LCLs) were generated by Epstein-Barr virus-transformation as described previously[23]. Outgrowing B-LCL cultures were harvested, expanded, characterized, and frozen.
In vitro growth kinetics, ploidy, and cell cycle analysis
Doubling time of HROC18 and HROC32 cells was determined from serial passages. Therefore, 5 × 105 viable cells were seeded into 25 cm2 flasks and the number of viable cells (defined by trypan blue exclusion) determined daily for seven days. Cultures were fed every 3 or 4 d. Ploidy and cell cycle analysis was performed by flow cytometry (FACSCalibur, BD Biosciences) using fixed cells (70% ethanol) after RNase A digestion (100 μg/mL; Sigma Aldrich) and propidium iodide (10 μg/mL; Sigma Aldrich) addition. Events (n = 10000) were measured for each sample, and cell cycle analysis was carried out by applying Modfit software (Verity Software House, Topsham, ME, United States). Matched B-LCLs were used as diploid controls.
Flow cytometric phenotyping of primary cell lines
Cell surface marker expression on CRC lines was traced by flow cytometry with and without interferon-gamma (IFN-γ) pre-treatment using a panel of FITC-, PE, or APC-conjugated Abs: CD11b, CD15, CD20, CD24, CD25, CD28, CD34, CD43, CD44, CD45, CD45ra, CD45ro, CD45rb, CD50, CD54, CD55, CD56, CD58, CD62L, CD66acde, CD69, CD71, CD80, CD86, CD102, CD154, CD178, CD326, MHC I (Immunotools, Friesoythe, Germany), MHC II (Miltenyi Biotec, Bergisch-Gladbach, Germany), and HLA-A2 (cell culture supernatant clone BB7.2). For HLA-A2, a secondary FITC-conjugated anti-mouse Ab was applied (Dako). Samples were analyzed using CellQuest software (BD Biosciences).
HLA typing
A two-digits encompassing resolution typing on patient-derived cell lines of HLA loci (HLA-A, -B and, -C, and HLA-DR and -DQ) was performed by Transfusion Medicine at the University of Rostock.
Cytokine secretion pattern
Spontaneous cytokine release by tumor cells was analyzed from duplicates (3 × 104 cells/well) in 24-well plates. Cell-free supernatants were harvested at different time-points and cytokine production (IL8, and IL10, Immunotools) was quantified by enzyme-linked immunosorbent assay according to the manufacturer’s instructions. Plates were coated with capture antibodies and incubated overnight (4 °C). Following five washing steps (1 × PBS) and blocking (2% BSA, 1 h, room temperature), standards and samples were transferred in duplicate wells and incubated (1 h, room temperature). Plates were washed again, incubated with biotinylated antibody (1:200, 1 h), and subjected to streptavidin-HRP conjugates (1:500; 30 min). Reaction visualization was done by adding TMB (3,5,3’,5’-tetramethylbenzidine) solution (100 μL/well) and incubation in the dark until color change. Reactions were stopped (100 μL of 2 mol/L H2SO4 solution) and plates immediately measured at a wavelength of 450 nm on a plate reader (Tecan Infinite® M200, Crailsheim, Germany). Measurement of carcinoembryonic antigen (CEA) and carbohydrate antigen 19-9 was kindly accomplished by the Institute of Clinical Chemistry and Laboratory Medicine (University of Rostock Medical Center).
Mycoplasma and human viral infection
Mycoplasma contamination was tested by the 16S-rRNA-gene-based PCR amplification method from whole cell lysates. For determining potential polyomavirus infection (JC/BK and SV40), gDNA was isolated using the Wizard genomic DNA purification kit (Promega). All procedures were performed as described by Maletzki et al[18].
Wound healing assay
Tumor cells were seeded in duplicates (6 × 105 cells/well, 6-well plates) and grown to 100% confluency. Monolayers were scratched with a 10 μL pipette tip to induce a wound. Wounded edges were imaged using a Zeiss inverted microscope. Images were taken daily for a period of 9 d under a × 10 objective lens. Migration property was calculated by measuring scratch diameter under a microscope within each well.
In vitro chemosensitivity analysis
Cells were seeded into 96-well microtiter plates at 5 × 103 or 1 × 104 cells/well. When cells reached 30%-40% confluency, cultures were exposed to increasing drug concentrations of 5-fluorouracil (5-FU), cisplatin, irinotecan, gemcitabine, taxol, trastuzumab, rapamycin, erlotinib (courtesy of the University of Rostock’s hospital pharmacy), or nilotinib (kindly provided by Novartis AG). Substances were selected based on the following criteria: (1) clinical approval for first-, second-, or third-line therapy of CRC patients (5-FU, irinotecan)[24]; (2) experimental and/or (pre-) clinical evidence of efficacy against CRC tumors (gemcitabine, taxol, cisplatin, and trastuzumab)[18,25]; and (3) promising novel targeted drugs, especially in case of metastatic disease (rapamycin, erlotinib, and nilotinib)[26,27].
After three days of exposure, media were removed and replaced by fresh medium supplemented with therapeutics. Following another three days, medium was removed, plates carefully washed, and they were then stained with crystal violet (0.2%, 10 min). Finally, drug effects from triplicate wells were determined at the level of 50% inhibition (IC50) in comparison to control, measured at 570 nm (reference wavelength: 620 nm).
Statistical analysis
Statistical analysis was done upon treatment with chemotherapeutics and targeted inhibitors. IC50 values were calculated using Sigma Plot 12.5 software (Systat Software Inc., San Jose, CA) applying the four parameter logistic function standard curve analysis for dose response. Values are given as absolute numbers.
RESULTS
Clinicopathologic characteristics of the primaries and cell line establishment
Parental tumors for HROC18 and HROC32 were primary colorectal carcinomas of the right colon. Histologically, both tumors were moderately well-differentiated tubular adenocarcinomas; one presenting at an early stage with infiltration into the muscular bowel wall, but without regional or distant metastasis (parental to HROC18), and the other at late stage with penetration of the peritoneum, regional lymph node metastases, and synchronous distant metastases to the liver and brain. Molecularly, both tumors were classified as sporadic standard (spStd) type and with nuclear beta-catenin translocation (Figure 1 and Table 1).
Figure 1.
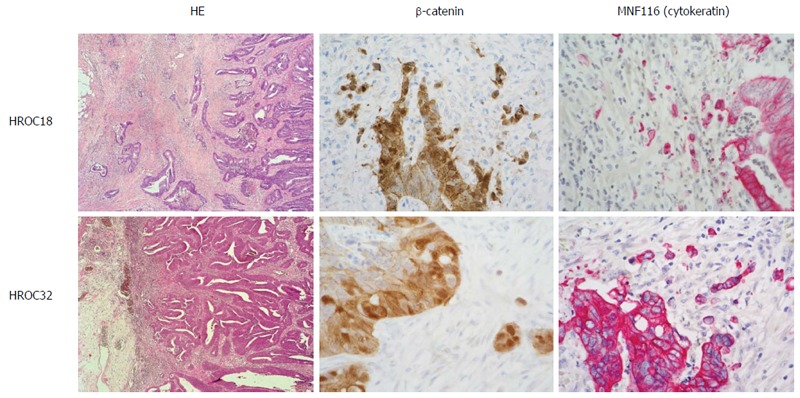
Representative histology of original patients’ tumors HROC18 and HROC32. HE histology, β-catenin, and cytokeratin (MNF116) staining as described in material and methods. Original magnification × 40.
For cell line establishment, in vitro and in vivo approaches were combined as previously described[18]. With this method, two individual patient-derived cell lines could be established (HROC18 and HROC32). Furthermore, a xenopatient-derived cell line from HROC32 was also obtained. In the case of HROC18, the obtained tumor material was insufficient for xenografting in addition to primary cell line establishment. All three lines exhibited stable outgrowth as defined by passaging > 50 times. Hence, three cell lines obtained from two individual tumor patients were available for subsequent analysis. For clarity, patient-derived cell lines were marked with the suffix P and the xenograft-derived cell line with suffix X.
Morphology and viral contamination
As determined by phase contrast microscopy, cells adhered tightly to the cell culture flask. Morphologically, all cell lines showed an epithelial-like phenotype without contaminating fibroblasts. HROC18 cells proliferated as polygonal cell clusters in shape with more regular dimensions (Figure 2, upper panel). Patient-derived HROC32 and their matched xenopatient-derived cells proliferated as tightly-packed multi-cellular islands (Figure 2, middle and low panels) that did not grow to complete confluence. Of note, no apparent morphological differences were observed between these two lines, indicative for the same dominant clone growing out of the initial tumor culture. In all cases, morphology did not change during long-term cell culture (up to 60 passages). As determined by semi-quantitative PCR, cell lines were found to be free of mycoplasma (Figure 3A) or other potential contaminants like human polyoma viruses (SV40 and JC/BK, data not shown).
Figure 2.
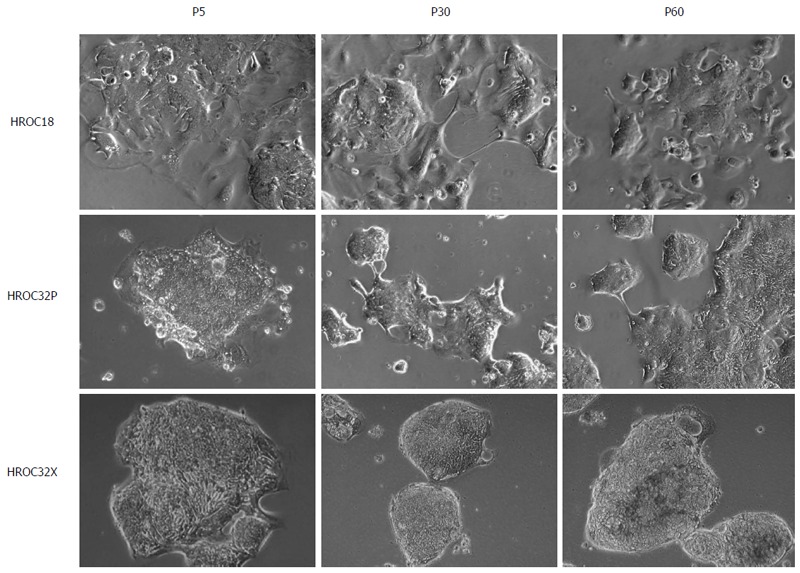
Light microscopy of tumor cell lines after direct establishment (P5), and following short- (P30) and long-term in vitro culture (P60). Cell lines were established directly from patients’ tumor material and following xenografting in murine recipients as described in material and methods. Original magnification × 100.
Figure 3.
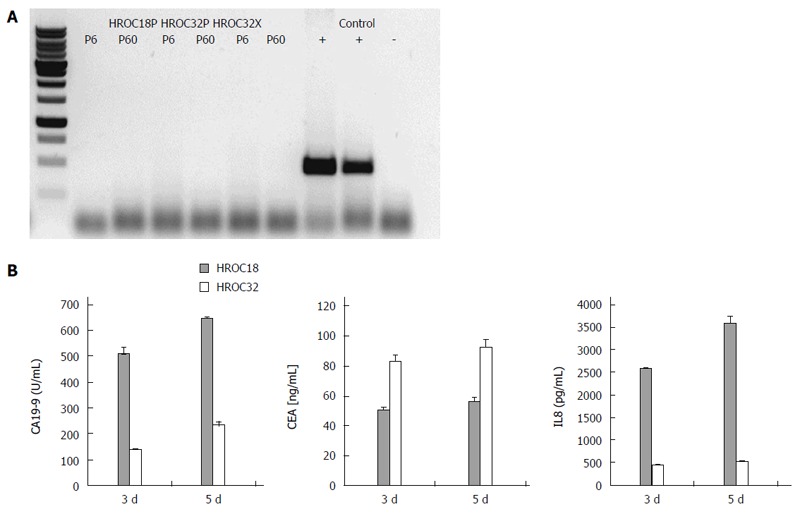
Mycoplasma polymerase chain reaction, ploidy status, and secretion profile of tumor cells. A: Displayed are the results for 16S-rRNA-gene-based polymerase chain reaction for mycoplasma. All three lines were found free of contaminating mycoplasma. Samples amplified in the absence of tumor DNA served as a negative control, whereas a highly contaminated cell line was applied as a positive control, run in duplicates; B: Cytokine secretion pattern as determined by enzyme-linked immunosorbent assays. Cytokine concentrations were determined by comparison with a standard curve generated from serial dilutions of individual standards. Quantitative analysis of carcinoembryonic antigen (CEA), carbohydrate antigen 19-9 (CA19-9), and interleukin (IL) 8 secretion after three and five days of culture, respectively. Results show the mean ± SD of three independent experiments.
Growth kinetics, ploidy analysis, and phenotype
Next, doubling times were determined over a culture period of seven days. Experiments revealed no significant differences between individual cell lines. Doubling times were 32 ± 10.6 h for HROC32P, 33.8 ± 5.4 h for HROC32X, and 35 ± 7.2 h for HROC18 cells. Cells did not grow faster after serial passages (data not shown).
Their epithelial origin was confirmed by high epithelial cell adhesion molecule expression (EpCAM; each > 98%; Table 2). More detailed phenotypic characterization revealed comparable expression levels for the transferrin receptor CD71, but heterogeneous expression of adhesion and co-stimulation markers (CD54, CD58, and CD102). All cell lines expressed MHC class I and were additionally found to be HLA-A2 positive (Table 2). MHC class II expression was absent on all lines, but could be induced by IFN-γ pre-treatment (up to 80%; Table 3).
Table 2.
Flow cytometric phenotyping of colorectal carcinoma cell lines and major histocompatibility complex expression with and without interferon-γ pre-treatment
| Antigen | HROC18P | HROC32P |
| CD58 | 3.6% | 16.4% |
| CD71 | 38.5% | 39.4% |
| CD50 | 15.1% | 20.3% |
| CD54 | 8.9% | 27.0% |
| CD102 | 22.4% | 11.8% |
| CD326 | 88.1% | 83.0% |
| CD24 | 9.5% | 0.6% |
| CD44 | 52.3% | 41.4% |
| CD66acde | 3.3% | 13.3% |
| CD15 | 75.1% | 46.7% |
| CD55 | 1.0% | 6.8% |
| HLA-A2 | 98.6% | 96.5% |
| MHC class I | ||
| - IFN-γ | 99.0% | 99.0% |
| + IFN-γ | 99.0% | 99.0% |
| MHC class II | ||
| - IFN-γ | 0.0% | 2.5% |
| + IFN-γ | 77.0% | 80.0% |
Data are given from one representative experiment. CD: Cluster of differentiation; IFN: Interferon; HLA: Human leukocyte antigen; MHC: Major histocompatibility complex.
Table 3.
Human leukocyte antigen: Typing of colorectal carcinoma cell lines
|
HLA class I |
HLA class I |
|||||||||
| DNA |
A1 |
B1 |
Cw1 |
DRB11 |
DQB11 |
|||||
| 1 allele | 2 allele | 1 allele | 2 allele | 1 allele | 2 allele | 1 allele | 2 allele | 1 allele | 2 allele | |
| HROC18 | 01 | 02 | 08 | 39 | 07 | - | 03 | 13 | 02 | 03 |
| HROC32P | 01 | 02 | 08 | 51 | 07 | 14 | 03 | 11 | 02 | 03 |
The number of the alleles. Information on both alleles is provided; in case of homozygosity the “second” allele is marked by “-“. HLA: Human leukocyte antigen.
Secretion profile
Secretion of the CRC tumor markers CA19-9 and CEA, as well as chemotactic and pro-angiogenic factor IL8, was determined from supernatants collected at different times of culture (Figure 3B). HROC18 and HROC32 secreted detectable levels of both tumor markers, with HROC18 showing high CA19-9, but low CEA secretion. On the other hand, the secretory picture of HROC32 was reversed, with high CEA and quite low CA19-9 levels. IL8 was predominantly detected in HROC18-derived supernatants. IL10 was not detectable at all.
Molecular signature, DNA fingerprint, and identification of novel tumor suppressor genes
First, DNA fingerprint was performed to verify genetic identity with the patients. A panel of highly polymorphic STR DNA markers covering four validated STR loci on different chromosomal locations was included into this analysis. Using this approach, identity with the patient was confirmed for all three cell lines even at late passage, hence cross-contamination with other (tumor) cells could be ruled out. Additionally, mutational profiling was done for a series of frequently affected tumor suppressor/proto-oncogenes (i.e., TP53, APC, KRAS, and BRAF). Examinations were done on all three cell lines and compared to the parental tumor. Notably, no differences were found in any of the matched samples (Table 4). HROC18 and HROC32 showed characteristic molecular features associated with the spStd type. HROC18 displayed TP53 and APC gene mutations. HROC32 cells presented with wild type APC, but mutations in the TP53 and KRAS genes. In line with the characteristic molecular signature, no methylation in CIMP-sensitive promoters, as traced by MethyLight, was found. Similarly, no BRAF or PTEN mutations were detected.
Table 4.
Comparative molecular characterization of patients’ tumor and corresponding colorectal carcinoma cell lines
|
TP53 |
APC |
KRAS |
||||||||
| Tumor-ID | Exon 5 | Exon 6 | Exon 7 | Exon 8 | Cd1 | Cd2 | Cd12 | Cd13 | B-rafV600 | |
| HROC18 | Patient tumor | wt | wt | wt | mut | mut | wt | wt | wt | wt |
| Cell line | wt | wt | wt | mut | mut | wt | wt | wt | wt | |
| HROC32 | Patient tumor | wt | wt | wt | mut | wt | wt | mut | wt | wt |
| P-Cell line | wt | wt | wt | mut | wt | wt | mut | wt | wt | |
| X-Cell line | wt | wt | wt | mut | wt | wt | mut | wt | wt | |
wt: Wild type; mut: Mutated; Cd: Codon.
More detailed analysis of potentially novel tumor suppressor genes identified individual differences between both tumor models. In detail, expression patterns of MBNL1, IMMP2L, FHIT, WWOX, and MACROD2 were examined by endpoint PCR. Alterations were detected as heterozygous point mutations, as demonstrated for FHIT (HROC18) and WWOX. IMMP2L and MACROD2 were homozygously deleted in both tumor models. MBNL1 showed no alteration with normal expression values.
Finally, we performed a very high resolution genomic analysis employing SNP Array 6.0. In this analysis, instability on the chromosomal level was confirmed. Both cell lines presented with complex aberrations (Figure 4).
Figure 4.
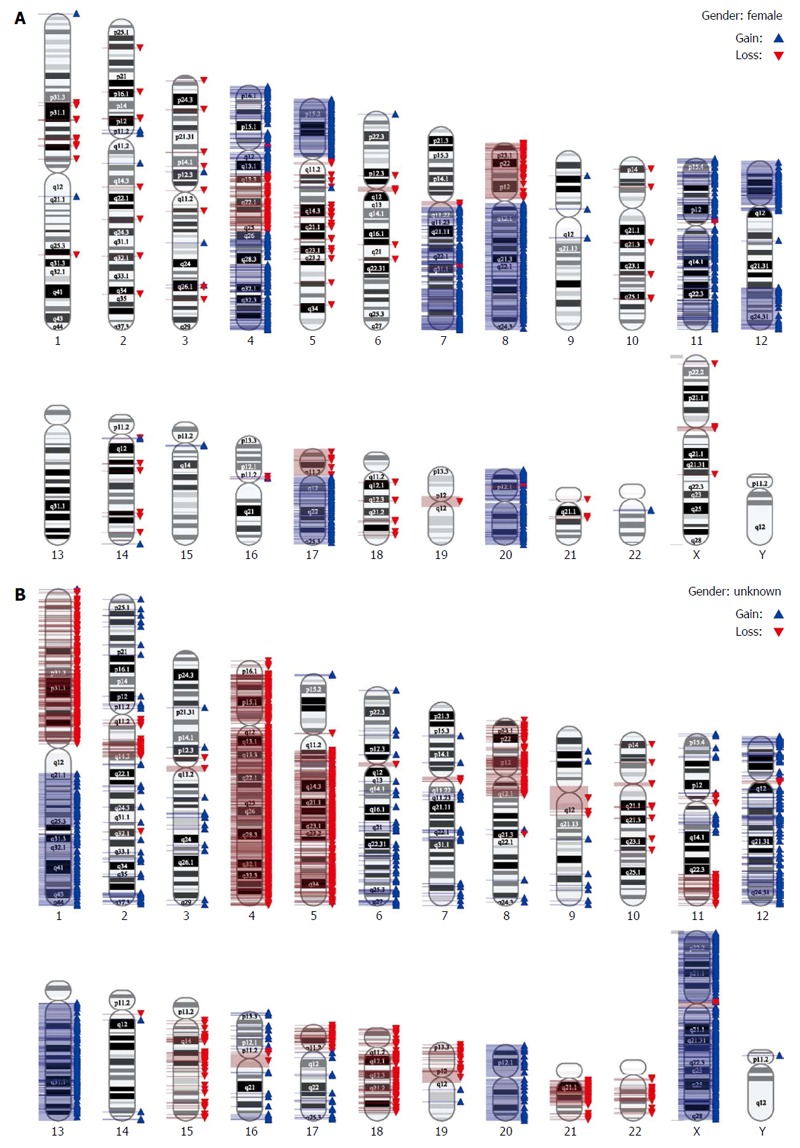
Single nucleotide polymorphism array 6.0 for assessment of chromosomal instability in cell lines. Analysis was performed according to manufacturer’s instructions. A: HROC18 cells; B: HROC32P cells.
Migration of tumor lines
A scratch wound healing assay was performed, with cell migration upon wounding being assessed over a period of 9 d. As can be taken from Figure 5, HROC18 cells migrated within five days throughout the initial wound (migration speed: 34.8 μm/d). By contrast, HROC32P cells exhibited lower levels of migration (Figure 5), finally taking 9 d to close the wound (migration speed: 24.8 μm/d).
Figure 5.
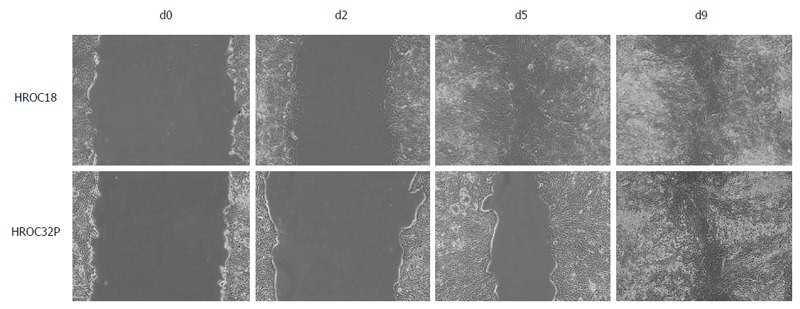
Wound healing assays were performed with HROC18 and HROC32P cells, respectively. Representative images of migrating tumor cells around the wound area at d0, d2, d5, and d9 after wounding are shown. d: Day.
In vitro drug response
Drug response testing on individual cell lines has become increasingly important due to the large heterogeneity of individual tumors in terms of therapy response. Hence, a series of clinically approved and potentially novel substances was included into in vitro analysis (Table 5). Responsiveness was examined following two 72-hour cycles, closely resembling the clinical regimen.
Table 5.
IC50 values of antitumor drugs evaluated for colorectal carcinoma cell lines
| Cell line |
IC50 |
||||||||
| 5-FU (μg/mL) | Cisplatin (μg/mL) | Irinotecan (μmol/L) | Gemcitabine (μmol/L) | Paclitaxel1 | Trastuzumab2 | Erlotinib (μmol/L) | Nilotinib (μmol/L) | Rapamycin (μmol/L) | |
| HROC18 | 0.48 ± 0.28 | 1.58 ± 0.29 | 7.88 ± 2.26 | 3.35 ± 1.18 | Resistant | Resistant | 12.21 ± 5.4 | 7.55 ± 1.77 | 0.08 ± 0.04 |
| HROC32P | 0.15 ± 0.07 | 0.65 ± 0.40 | 0.15 ± 0.07 | 3.90 ± 2.16 | Resistant | Resistant | 1.40 ± 1.01 | 5.47 ± 3.13 | 0.14 ± 0.14 |
| HROC32X | 1.97 ± 1.95 | 1.30 ± 0.46 | 3.83 ± 1.40 | 5.90 ± 1.06 | Resistant | Resistant | 0.87 ± 0.15 | 5.5 ± 3.1 | 0.13 ± 0.06 |
Range 0.9 nmol/L: 60 mmol/L;
Range: 310 ng/mL: 20 μg/mL. Values are given as mean, resulting from at least three independent experiments each performed in triplicates. 5-FU: 5-Fluorouracil.
Generally, HROC18 and HROC32 (P + X) cells responded to clinically approved CRC drugs (i.e., 5-FU, cisplatin, and irinotecan). HROC18 cells showed the highest sensitivity towards 5-FU (IC50 value: 0.48 ± 0.28 μmol/L). HROC32X tended to be more resistant against these drugs than its parental line (HROC32P), especially following irinotecan treatment (IC50 values: 3.83 ± 1.40 μmol/L vs 0.15 ± 0.07 μmol/L). Of note, all lines were susceptible towards targeted therapies. Among these therapeutics, the mammalian target of rapamycin (mTOR) inhibitor rapamycin (IC50 value of each cell line: < 0.20 μmol/L) was most potent. Nilotinib, an inhibitor of the DDR, KIT, and PDGFR receptor kinases, showed IC50 values between 5.4 and 7.5 μmol/L, and erlotinib, a selective EGFR inhibitor, inhibited proliferation with IC50 values in the range of 0.87-12.2 μmol/L. By contrast, taxol did not show significant growth inhibition at all, which is in line with the recent literature in CIN tumors[28]. Likewise, trastuzumab had no impact on cellular growth.
In vivo tumorigenicity
To evaluate in vivo growth behavior, mice were subcutaneously implanted with 5 × 106 patient-derived HROC18 and HROC32 tumor cells, respectively. Growth kinetics were different between these two cells, with HROC32P growing more rapidly than HROC18 (Figure 6). Thirty-five days after injection, HROC32 tumors became apparent and continued to grow until the experimental endpoint (day 77, tumor diameter: 10.2 ± 1.9 mm). By contrast, HROC18 tumors presented with low tumorigenic potential and tumor sizes suspended at 6.2 ± 0.3 mm in diameter (day 77). The addition of Matrigel to the tumor cell suspension did not improve tumor growth (data not shown).
Figure 6.
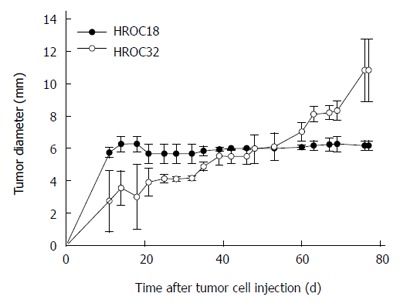
In vivo tumorigenicity. Female NMRI Foxn1nu mice were challenged subcutaneously with 5 × 106 tumor cells. Monitoring of tumor growth was performed for a total of 80 d. Both cell lines exhibited low tumorigenic potential in vivo, as can be depicted from the tumor growth curve. Values are given as mean tumor diameter ± SD.
Next, experiments were performed in NOD-SCID mice. Due to a lack of NK cells, these mice graft human cells better than Foxn1nu mice. Once again, low or absent tumor growth was observed for HROC18 cells, confirming their low tumorigenic potential in vivo (data not shown).
DISCUSSION
Identifying subsets of patients who will benefit from a particular (targeted) therapy requires improved integration of pre-clinical models and cutting-edge clinical examination. In this case, individual tumor models have become increasingly important research tools[29-32]. Such models helped to identify the large morphologic and molecular heterogeneity that exists among individual tumor cases; yet these findings have contributed to a better understanding of CRC biology and guided the way for individualized pre-clinical drug testing. Hence, this study was conducted to establish novel CRC cell lines obtained from patients with primary spStd type tumors. Tumors were resected at early and late stages, thereby representing two different models that expand our collection on individual tumor models. This collection includes primary material (frozen and paraffin-embedded tumors, normal tissue, and lymphocytes) together with xenografts, patient-derived and xenopatient-derived cell lines, as well as matched B-LCLs[18,33,34].
Cell lines generated in this study were directly obtained from patient tumor material, thereby perfectly reflecting the original molecular signature of the tumor. For culturing primary tumor cells, a simple, yet rapid, method described by Maletzki et al[18] was applied. This method was found to be reliable and highly reproducible, yielding an approximately 10% success rate of primary tumor cell line establishment, which is comparable to that reported by others[35,36]. In order to improve cell line establishment and obtain more individual tumor models for functional comparative studies, tumor fragments were additionally engrafted in immunodeficient mice without prior in vitro exposure[35,37]. With this approach, up to 60%-80% success can be achieved (own unpublished data). Cell lines established following xenografting often show even better in vitro growth behavior than their parental counterpart cell line. They even adequately recapitulate molecular hallmarks of parental tumors and exhibit good in vivo tumorigenic potential, which is crucial for therapeutic interventions. However, there are also some limitations that must be kept in mind: (1) xenopatient-derived cell lines undergo a kind of in vivo preselection and may thus not represent the full tumor heterogeneity; (2) due to selective pressure, growth behavior, and consequently drug response, may differ; and (3) in vivo xenografting is very time-consuming, lasting up to 6 mo with follow-up cell line establishment potentially adding another year.
In this study, HROC32 cells gave rise to in vitro growth after xenografting, and consequently three cell lines were available from two individual tumor cases. The genetic identity with the patient was verified for these lines by means of STR fingerprinting.
All three lines showed characteristic molecular features associated with the spStd type (i.e., complex chromosomal aberrations, TP53 gene mutations, and absence of GpG island promoter methylation). In line with our previous study on microsatellite instable tumor cell lines, molecular profiles matched between cell lines (both X and P) and the parental counterpart[18]. KRAS, a well-established predictive biomarker for EGFR-targeted therapies[38], was mutated only in the late stage CRC case (HROC32). Moreover, this tumor harbored a mutation in codon 12 that is associated with worse outcome and early relapse after chemotherapy compared to codon 13 or other KRAS gene mutations[39].
Confirming their origin, cell lines were deliberated as epithelial tumor cells (> 80% EpCAM+) with heterogeneous expression of adhesion molecules (CD15, CD54, CD58, and CD102) and tumor antigens (CD66acde). Transferrin receptor CD71 was highly expressed on tumor cells, indicative for a high proliferative index. Due to their natural immunosuppressive behavior, co-stimulatory molecules (CD80 and CD86) were not detectable at all, while MHC class I-expression was preserved. HROC18 and HROC32 cells were additionally found to be HLA-A2 positive. This provides a ready basis for subsequent immunological studies aiming at the identification of immunogenic epitopes from novel candidate antigens. Given the fact that about half of Caucasians and Asians are HLA-A2 positive, this finding is of particular relevance.
Common characteristics for adaptation to cell culture conditions include changes in morphology towards a rather undifferentiated phenotype and an accelerated rate of growth[18,40]. However, both parameters remained unchanged in HROC18 and HROC32 (X + P) lines, at least over 60 passages. Directional cell migration, as determined by a classical wound healing assay[41], was quite individual, with HROC18 cells migrating faster than HROC32 cells. Since these findings may give a hint towards in vivo growth behavior and has potential consequences for the efficacy of antineoplastic drugs in vivo, the tumorigenic potential was studied. However, tumorigenicity was always found to be low. Together, these findings are indicative for outgrowth of one dominant cell clone already in the initial in vivo and in vitro culture.
In many CRC patients, therapy resistance and subsequent relapse occurs early after initial objective response[42]. In terms of targeted therapies, it was found that single cells resistant to any targeted agent are already present at the start of therapy[43]. In most cases, these cells clonally expand once the therapy is applied, causing tumor recurrence. Recently, a mathematical model describing the evolutionary dynamics of lesions in response to treatment was developed that attempts to predict the efficacy of drugs and certain combinations[43]. Hence, a large individual variability is actually found even in molecular closely-matched tumor samples[25,44]. Therefore, maintenance of the original tumors’ molecular signature is in demand for determining (initial) drug sensitivity experimentally and for predicting response. Since this condition is completely fulfilled in our freshly established cancer cell lines, a large panel of chemotherapeutics and targeted drugs was tested. In principle, cell lines were susceptible towards commonly used chemotherapeutics (i.e., 5-FU, cisplatin, and irinotecan)[24], while the targeted therapeutics[45] evaluated did not show significant growth inhibition, they were indicative for intrinsic resistance (of the in vitro outgrown cell clone). Of note, HROC18 cells were generally more resistant than HROC32 cells (both X and P), underscoring the necessity of personalized therapy. A rather unexpected, yet very interesting, finding was the responsiveness towards rapamycin. Rapamycin, produced by Streptomyces hygroscopicus, was the first mTOR inhibitor discovered that has been defined as a “longevity enhancer and cancer preventative agent” in TP53 gene deficiency[46]. Though evidence from the literature indicates the resistance of CRCs to the antitumorigenic effects of rapamycin, we still observed significant growth inhibition in our cell lines. The underlying resistance mechanism is supposed to be mediated by feedback activation of PI3K/Akt and Ras-MAPK signaling and, accordingly, cell survival[46]. Recently, the addition of sorafenib has been found to abrogate rapamycin resistance in cells carrying oncogenic KRAS and PIK3CA[47]. Hence, this combination may deliver another option for selected patients and underscores the role of mTOR inhibitors as combinatorial agents in cancer therapy despite their so far limited clinical success[48].
In principle, an ideal preclinical tumor model is of low passage, shows close similarity to its parental tumor (≥ 95%), harbors specific genomic alterations of interest (for targeted approaches), and fulfils more practical issues like good in vitro and in vivo growth characteristics. Our collection of individual tumor models meets these criteria; hence these models provide a virtually unlimited source of tumor material for predicting treatment and response. This will help to improve the ability for personalizing tumor therapy.
ACKNOWLEDGMENTS
The authors kindly thank Dr. Dirk Koczan for performing the SNP 6.0 analysis of HROC18 and HROC32 cell lines. We also thank the Institute of Clinical Chemistry and Laboratory Medicine for performing carcinoembryonic antigen and carbohydrate antigen 19-9 measurements from cell culture supernatants. Finally, we thank Novartis AG for supplying us with small molecule inhibitors for in vitro studies.
COMMENTS
Background
The chromosomal instability (CIN+) subtype occurs in 65%-70% of all sporadic colorectal cancer (CRC). Preclinical validation of novel biomarkers that allow drug response testing and prediction is ideally being performed in patient-derived, individual tumor models. However, ultra-low passage and well-characterized CRC models are still rare.
Research frontiers
Individual tumor models provide excellent tools for both basic and translational research. Even more importantly, they are a prime choice for the development of personalized tumor therapies.
Innovations and breakthroughs
In this study, the authors describe the successful establishment of two different tumor models showing characteristic features associated with the CIN+ phenotype. Due to their individual character, morphology, phenotype, and drug response were quite heterogeneous. This substantially extends the collection of individual tumor models, which include all known CRC subtypes. By establishing and comprehensively characterizing obtained tumor models, useful instruments for subsequent development of individualized tumor therapies are supplied.
Applications
Patient-derived tumor models constitute a virtually unlimited source of tumor material, both for further analyzing their biological characteristics and for predicting drug response. This will help to improve the ability for personalizing tumor therapy in the near future.
Terminology
Chromosomal instability: A typical feature of most solid cancers involving structural and numerical alterations of the normal chromosome numbers. Ultra-low passage tumor cell lines: Culture of primary (patient-derived) tumor cells which grow continuously, but with only a low number of cell divisions outside the body. This guarantees very low or absent molecular changes in comparison to the parental tumor.
Peer review
This descriptive study represents the methodology and characterization of CRC cell lines both from patient material and xenografts of original patients’ tumors. By applying a large number of methods for analyzing tumor biology, genetic alterations, and chemosensitivity, the authors present an interesting way for generating individual tumor models.
Footnotes
Supported by German Cancer Aid, No. 108919
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 23, 2014
First decision: June 18, 2014
Article in press: July 26, 2014
P- Reviewer: Feng ZL, Kim SJ, Teresa Valenti M S- Editor: Gou SX L- Editor: Rutherford A E- Editor: Liu XM
References
- 1.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 2.Jass JR. Molecular heterogeneity of colorectal cancer: Implications for cancer control. Surg Oncol. 2007;16 Suppl 1:S7–S9. doi: 10.1016/j.suronc.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 3.Ostwald C, Linnebacher M, Weirich V, Prall F. Chromosomally and microsatellite stable colorectal carcinomas without the CpG island methylator phenotype in a molecular classification. Int J Oncol. 2009;35:321–327. [PubMed] [Google Scholar]
- 4.Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007;50:113–130. doi: 10.1111/j.1365-2559.2006.02549.x. [DOI] [PubMed] [Google Scholar]
- 5.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 6.Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM, vanTuinen P, Ledbetter DH, Barker DF, Nakamura Y, et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science. 1989;244:217–221. doi: 10.1126/science.2649981. [DOI] [PubMed] [Google Scholar]
- 7.Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995;268:1336–1338. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 8.Thiagalingam S, Lengauer C, Leach FS, Schutte M, Hahn SA, Overhauser J, Willson JK, Markowitz S, Hamilton SR, Kern SE, et al. Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat Genet. 1996;13:343–346. doi: 10.1038/ng0796-343. [DOI] [PubMed] [Google Scholar]
- 9.Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle. 2004;3:1221–1224. doi: 10.4161/cc.3.10.1164. [DOI] [PubMed] [Google Scholar]
- 10.Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 11.Velculescu VE. Defining the blueprint of the cancer genome. Carcinogenesis. 2008;29:1087–1091. doi: 10.1093/carcin/bgn096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loeb LA, Loeb KR, Anderson JP. Multiple mutations and cancer. Proc Natl Acad Sci USA. 2003;100:776–781. doi: 10.1073/pnas.0334858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 15.Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology. 2010;138:2059–2072. doi: 10.1053/j.gastro.2009.12.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freeman DJ, Juan T, Reiner M, Hecht JR, Meropol NJ, Berlin J, Mitchell E, Sarosi I, Radinsky R, Amado RG. Association of K-ras mutational status and clinical outcomes in patients with metastatic colorectal cancer receiving panitumumab alone. Clin Colorectal Cancer. 2008;7:184–190. doi: 10.3816/CCC.2008.n.024. [DOI] [PubMed] [Google Scholar]
- 17.Wadlow RC, Hezel AF, Abrams TA, Blaszkowsky LS, Fuchs CS, Kulke MH, Kwak EL, Meyerhardt JA, Ryan DP, Szymonifka J, et al. Panitumumab in patients with KRAS wild-type colorectal cancer after progression on cetuximab. Oncologist. 2012;17:14. doi: 10.1634/theoncologist.2011-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maletzki C, Stier S, Gruenert U, Gock M, Ostwald C, Prall F, Linnebacher M. Establishment, characterization and chemosensitivity of three mismatch repair deficient cell lines from sporadic and inherited colorectal carcinomas. PLoS One. 2012;7:e52485. doi: 10.1371/journal.pone.0052485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poupon MF, Arvelo F, Goguel AF, Bourgeois Y, Jacrot M, Hanania N, Arriagada R, Le Chevalier T. Response of small-cell lung cancer xenografts to chemotherapy: multidrug resistance and direct clinical correlates. J Natl Cancer Inst. 1993;85:2023–2029. doi: 10.1093/jnci/85.24.2023. [DOI] [PubMed] [Google Scholar]
- 20.Fielding LP, Arsenault PA, Chapuis PH, Dent O, Gathright B, Hardcastle JD, Hermanek P, Jass JR, Newland RC. Clinicopathological staging for colorectal cancer: an International Documentation System (IDS) and an International Comprehensive Anatomical Terminology (ICAT) J Gastroenterol Hepatol. 1991;6:325–344. doi: 10.1111/j.1440-1746.1991.tb00867.x. [DOI] [PubMed] [Google Scholar]
- 21.Ogino S, Nosho K, Kirkner GJ, Kawasaki T, Meyerhardt JA, Loda M, Giovannucci EL, Fuchs CS. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut. 2009;58:90–96. doi: 10.1136/gut.2008.155473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masibay A, Mozer TJ, Sprecher C. Promega Corporation reveals primer sequences in its testing kits. J Forensic Sci. 2000;45:1360–1362. [PubMed] [Google Scholar]
- 23.Klier U, Maletzki C, Klar E, Linnebacher M. Generation of highly pure fusions of colorectal carcinoma and antigen-presenting cells. Langenbecks Arch Surg. 2010;395:365–371. doi: 10.1007/s00423-010-0598-1. [DOI] [PubMed] [Google Scholar]
- 24.Petrelli F, Borgonovo K, Cabiddu M, Ghilardi M, Lonati V, Maspero F, Sauta MG, Beretta GD, Barni S. FOLFIRI-bevacizumab as first-line chemotherapy in 3500 patients with advanced colorectal cancer: a pooled analysis of 29 published trials. Clin Colorectal Cancer. 2013;12:145–151. doi: 10.1016/j.clcc.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 25.Salgado M, Reboredo M, Mendez JC, Quintero G, Pellón ML, Romero C, Jorge M, Montes AF, Valladares-Ayerbes M, Ramos M, et al. Gemcitabine and capecitabine as third- or later-line therapy for refractory advanced colorectal cancer: a retrospective study. Anticancer Res. 2013;33:4089–4096. [PubMed] [Google Scholar]
- 26.Kozuch P, Malamud S, Wasserman C, Homel P, Mirzoyev T, Grossbard M. Phase II trial of erlotinib and capecitabine for patients with previously untreated metastatic colorectal cancer. Clin Colorectal Cancer. 2009;8:38–42. doi: 10.3816/CCC.2009.n.006. [DOI] [PubMed] [Google Scholar]
- 27.Atreya CE, Ducker GS, Feldman ME, Bergsland EK, Warren RS, Shokat KM. Combination of ATP-competitive mammalian target of rapamycin inhibitors with standard chemotherapy for colorectal cancer. Invest New Drugs. 2012;30:2219–2225. doi: 10.1007/s10637-012-9793-y. [DOI] [PubMed] [Google Scholar]
- 28.Swanton C, Tomlinson I, Downward J. Chromosomal instability, colorectal cancer and taxane resistance. Cell Cycle. 2006;5:818–823. doi: 10.4161/cc.5.8.2682. [DOI] [PubMed] [Google Scholar]
- 29.Julien S, Merino-Trigo A, Lacroix L, Pocard M, Goéré D, Mariani P, Landron S, Bigot L, Nemati F, Dartigues P, et al. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin Cancer Res. 2012;18:5314–5328. doi: 10.1158/1078-0432.CCR-12-0372. [DOI] [PubMed] [Google Scholar]
- 30.Scott CL, Becker MA, Haluska P, Samimi G. Patient-derived xenograft models to improve targeted therapy in epithelial ovarian cancer treatment. Front Oncol. 2013;3:295. doi: 10.3389/fonc.2013.00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Puig I, Chicote I, Tenbaum SP, Arqués O, Herance JR, Gispert JD, Jimenez J, Landolfi S, Caci K, Allende H, et al. A personalized preclinical model to evaluate the metastatic potential of patient-derived colon cancer initiating cells. Clin Cancer Res. 2013;19:6787–6801. doi: 10.1158/1078-0432.CCR-12-1740. [DOI] [PubMed] [Google Scholar]
- 32.Chiron M, Bagley RG, Pollard J, Mankoo PK, Henry C, Vincent L, Geslin C, Baltes N, Bergstrom DA. Differential antitumor activity of aflibercept and bevacizumab in patient-derived xenograft models of colorectal cancer. Mol Cancer Ther. 2014;13:1636–1644. doi: 10.1158/1535-7163.MCT-13-0753. [DOI] [PubMed] [Google Scholar]
- 33.Linnebacher M, Maletzki C, Ostwald C, Klier U, Krohn M, Klar E, Prall F. Cryopreservation of human colorectal carcinomas prior to xenografting. BMC Cancer. 2010;10:362. doi: 10.1186/1471-2407-10-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Available from: http://www.northgermantumorbank-crc.de/North_German_Tumor_Bank_-_CRC/Home.html.
- 35.Dangles-Marie V, Pocard M, Richon S, Weiswald LB, Assayag F, Saulnier P, Judde JG, Janneau JL, Auger N, Validire P, et al. Establishment of human colon cancer cell lines from fresh tumors versus xenografts: comparison of success rate and cell line features. Cancer Res. 2007;67:398–407. doi: 10.1158/0008-5472.CAN-06-0594. [DOI] [PubMed] [Google Scholar]
- 36.Lerescu L, Tucureanu C, Caraş I, Neagu S, Melinceanu L, Sălăgeanu A. Primary cell culture of human adenocarcinomas--practical considerations. Roum Arch Microbiol Immunol. 2008;67:55–66. [PubMed] [Google Scholar]
- 37.Seol HS, Kang HJ, Lee SI, Kim NE, Kim TI, Chun SM, Kim TW, Yu CS, Suh YA, Singh SR, et al. Development and characterization of a colon PDX model that reproduces drug responsiveness and the mutation profiles of its original tumor. Cancer Lett. 2014;345:56–64. doi: 10.1016/j.canlet.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dempke WC, Heinemann V. Ras mutational status is a biomarker for resistance to EGFR inhibitors in colorectal carcinoma. Anticancer Res. 2010;30:4673–4677. [PubMed] [Google Scholar]
- 39.Stec R, Bodnar L, Charkiewicz R, Korniluk J, Rokita M, Smoter M, Ciechowicz M, Chyczewski L, Nikliński J, Kozłowski W, et al. K-Ras gene mutation status as a prognostic and predictive factor in patients with colorectal cancer undergoing irinotecan- or oxaliplatin-based chemotherapy. Cancer Biol Ther. 2012;13:1235–1243. doi: 10.4161/cbt.21813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Danes BS, Deangelis P, Traganos F, Melamed MR, Alm T. Demonstration of altered cellular DNA content distribution in long-term colon epithelial cell lines with colon cancer genotypes. Scand J Gastroenterol. 1988;23:840–846. doi: 10.3109/00365528809090770. [DOI] [PubMed] [Google Scholar]
- 41.Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 42.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 43.Bozic I, Reiter JG, Allen B, Antal T, Chatterjee K, Shah P, Moon YS, Yaqubie A, Kelly N, Le DT, et al. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife. 2013;2:e00747. doi: 10.7554/eLife.00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sadanandam A, Lyssiotis CA, Homicsko K, Collisson EA, Gibb WJ, Wullschleger S, Ostos LC, Lannon WA, Grotzinger C, Del Rio M, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19:619–625. doi: 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Knijn N, Tol J, Punt CJ. Current issues in the targeted therapy of advanced colorectal cancer. Discov Med. 2010;9:328–336. [PubMed] [Google Scholar]
- 46.Francipane MG, Lagasse E. mTOR pathway in colorectal cancer: an update. Oncotarget. 2014;5:49–66. doi: 10.18632/oncotarget.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gulhati P, Zaytseva YY, Valentino JD, Stevens PD, Kim JT, Sasazuki T, Shirasawa S, Lee EY, Weiss HL, Dong J, et al. Sorafenib enhances the therapeutic efficacy of rapamycin in colorectal cancers harboring oncogenic KRAS and PIK3CA. Carcinogenesis. 2012;33:1782–1790. doi: 10.1093/carcin/bgs203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]