

Abstract
The principal cell of the kidney collecting duct is one of the most highly regulated epithelial cell types in vertebrates. The effects of hormonal, autocrine, and paracrine factors to regulate principal cell transport processes are central to the maintenance of fluid and electrolyte balance in the face of wide variations in food and water intake. In marked contrast with the epithelial cells lining the proximal tubule, the collecting duct is electrically tight, and ion and osmotic gradients can be very high. The central role of principal cells in salt and water transport is reflected by their defining transporters—the epithelial Na+ channel (ENaC), the renal outer medullary K+ channel, and the aquaporin 2 (AQP2) water channel. The coordinated regulation of ENaC by aldosterone, and AQP2 by arginine vasopressin (AVP) in principal cells is essential for the control of plasma Na+ and K+ concentrations, extracellular fluid volume, and BP. In addition to these essential hormones, additional neuronal, physical, and chemical factors influence Na+, K+, and water homeostasis. Notably, a variety of secreted paracrine and autocrine agents such as bradykinin, ATP, endothelin, nitric oxide, and prostaglandin E2 counterbalance and limit the natriferic effects of aldosterone and the water-retaining effects of AVP. Considerable recent progress has improved our understanding of the transporters, receptors, second messengers, and signaling events that mediate principal cell responses to changing environments in health and disease. This review primarily addresses the structure and function of the key transporters and the complex interplay of regulatory factors that modulate principal cell ion and water transport.
Keywords: renal physiology, collecting duct, principal cell, epithelial cell
Introduction
The principal cell is one of two major epithelial cell types in what is often referred to as the aldosterone-sensitive distal nephron, comprising the connecting segment through the collecting duct. Ion and water transport in this part of the nephron are highly regulated by a wide variety of stimuli, including hormones, autocrine and paracrine factors, osmotic conditions, and physical factors. The principal cell is central to salt and water transport, as reflected by its defining transporters—the epithelial sodium channel (ENaC) and the aquaporin 2 (AQP2) water channel.
In humans, by the time tubular fluid reaches the aldosterone-sensitive distal nephron under physiologic conditions, virtually all amino acids, glucose, bicarbonate, and other nonwaste organic solutes have been removed and water volume has been reduced to approximately 10% of that of glomerular filtrate. Thus, the absolute level of transport of critical ions and water in this nephron segment is markedly lower than in most upstream segments; however, the variability of transport rates is markedly higher. Ion and osmotic gradients between the tubule lumen and the interstitium are also more variable, and are frequently much higher than in other regions. A future article in this series will address the integrated tubule physiology and compare characteristics of different segments. The coordinated regulation of ENaC by aldosterone, and AQP2 by arginine vasopressin (AVP) in principal cells is essential for the control of plasma Na+ and K+ concentrations, extracellular fluid volume, and BP in most vertebrates (1). In addition to these essential hormones, other hormones, autacoids, and mechanical factors influence Na+, K+, and water homeostasis. This review primarily addresses the regulation of Na+, K+, and water transport in principal cells.
Control of Principal Cell Ion Transport
Figure 1 shows the integrated transport of Na+ and K+ in a typical principal cell, emphasizing the regulatory events that control ENaC (the principal pathway for apical Na+) and the renal outer medullary K+ (ROMK) channel (the principal pathway for apical exit of K+). Aldosterone is the primary hormonal regulator of both Na+ and K+ transport, as addressed further below. ENaC is the primary target of regulation, and its stimulation by aldosterone affects both Na+ reabsorption and K+ secretion. Cl− transport is not shown in Figure 1 because it is not a simple function of principal cells; rather, Cl− transport is a function of both principal and intercalated cells, as well as the paracellular pathway (2,3).
Figure 1.

Hormonal regulation of ion transport in a typical principal cell. Principal cells respond to a variety of stimuli to control Na+ and K+ transport. Aldosterone has the most pronounced effect. It acts through the MR to increase expression of the serine-threonine kinase SGK1. Other hormones, including insulin, regulate SGK1 activity through the master kinase phosphatidylinositide 3-kinase (PI3K). SGK1 phosphorylates a variety of proteins; Nedd4-2 is shown, which is an ENaC inhibitor (see the text). SGK1 phosphorylation triggers interaction of Nedd4-2 with 14-3-3 proteins, and hence inhibition, by reducing its internalization and degradation. ENaC surface expression and activity are governed by a multiprotein ERC, whose assembly at the plasma membrane is orchestrated by the aldosterone-induced small chaperone GILZ (not shown), and the scaffold protein CNK3. Electrogenic Na+ reabsorption via ENaC is balanced by K+ secretion through ROMK and Cl− reabsorption through multiple pathways (not shown). The apical surface expression and activity of ROMK are positively regulated by SGK1 through phosphorylation of WNK4. Similar to ENaC, ROMK assembly at the luminal membrane is dictated by a multiprotein complex facilitated by interactions with the scaffold proteins NHERF-1 and NHERF-2. The driving force that sets the electrochemical gradient for principal cell Na+ and K+ transport is the basolateral Na+-K+-ATPase. c-Src, cellular homologue of the v-src gene of the Rous sarcoma virus; CFTR, cystic fibrosis transmembrane conductance regulator; ENaC, epithelial Na+ channel; ERC, ENaC regulatory complex; INS, insulin; IRS, insulin receptor substrate; MR, mineralocorticoid receptor; Nedd4-2, neural precursor cell–expressed developmentally downregulated gene 4-2; PDK1, phosphoinositide-dependent kinase-1; PH, pleckstrin homology; ROMK, renal outer medullary K+; SGK1, serum- and glucocorticoid-regulated kinase 1; WNK, with no lysine kinase.
This review discusses the key Na+ and K+ pathways operative in principal cells and their regulation, and also describes Cl− transport.
ENaC
Basic Biology of ENaC
ENaC mediates apical entry of Na+ into principal cells, and constitutes the rate-limiting step for transepithelial Na+ transport in the aldosterone-sensitive distal nephron. As is often the case for the rate-limiting step in any pathway, ENaC is also the primary locus of transepithelial Na+ transport regulation (Figure 1). Like all channels, it does not directly couple Na+ transport to the movement of any other ion or solute. Hence, unlike many upstream transporters such as the Na+-H+ exchanger isoform-3 (NHE3), Na+-K+-2Cl− cotransporter (NKCC2), and Na+-Cl− cotransporter (NCC), it does not participate in secondary active transport (4). However, because ENaC mediates electrogenic Na+ transport, it increases the driving force for K+ secretion via K+ channels, such as ROMK (expressed in principal cells, see below) (5) and BK channels (expressed in both principal and intercalated cells). It also enhances H+ secretion by adjacent intercalated cells, as well as Cl− reabsorption via a variety of pathways; a future review in this series will address these topics, along with BK channels, in detail.
ENaC comprises three distinct, but structurally related, subunits (α, β, and γ) (6). Based on the homotrimeric subunit arrangement in the crystal structure of the acid-sensing ion channel (7), an ENaC relative, it is currently thought that ENaC is a heterotrimer (8). Although this issue is not fully resolved, considerable insight into ENaC function was gained by performing a comparison with the acid-sensing ion channel (Figure 2). ENaC is highly selective for Na+ (and Li+) over other ions (most notably K+), and is highly sensitive to the K+-sparing diuretic amiloride. Selectivity is mediated by a signature Gly/Ser-X-Ser sequence, which is adjacent to the amiloride binding site (Figure 2D). ENaC activity and cell surface expression are regulated by a variety of hormonal, autocrine, paracrine, and nonhormonal signals. These regulatory signals are integrated to produce appropriate levels of Na+ transport to meet physiologic demands. Recent evidence suggests that this signal integration requires ENaC at the plasma membrane to be organized into a large (1.2 MDa) multiprotein termed the ENaC regulatory complex (ERC), which includes both positive (e.g., serum- and glucocorticoid-regulated kinase 1 [SGK1]) and negative (e.g., neural precursor cell–expressed developmentally downregulated gene 4-2 [Nedd4-2]) regulators. Regulatory molecules within the ERC interact with the cytoplasmic domains of ENaC, which are absent in current models of the ENaC structure (Figure 2). The formation and stability of the complex requires an aldosterone-induced chaperone (GILZ1) and a scaffold protein (CNK3) (9,10), which keep the complex together by stimulating interactions among multiple proteins (Figure 1). It is interesting to note that CNK3, like many scaffolds involved in stabilizing membrane expression of transport proteins, has a PDZ (PSD-95/DLG-1/ZO-1) domain (1). ROMK membrane stability requires another PDZ domain protein, sodium-proton exchanger regulatory factor (NHERF) (both isoforms NHERF-1 and NHERF-2 have been implicated) (11).
Figure 2.

Structural model of the ENaC extracellular domains and pore. The model represents a hypothetical α subunit trimer and was built on the basis of sequence homology to ASIC1 and functional data (8,122). Sequence conservation among ENaC subunits suggests that the α, β, and γ subunits adopt similar folds. The intracellular domains, accounting for 25%–30% of each subunit’s mass, are absent from current models of ENaC structure. These domains contain essential sites for regulatory interactions (e.g., Nedd4-2 and CNK3) and modification (e.g., ubiquitinylation and phosphorylation). (A) Surface representation showing the spatial arrangement of the three subunits with the approximate location of outer and inner borders of the lipid bilayer. (B) One subunit is represented as a ribbon diagram showing the five extracellular domains and transmembrane α-helices labeled as indicated. (C) Close-up of the finger domain (from the model in B), highlighting the peripheral location of the furin cleavage sites. (D) Close-up of the pore highlighting the likely permeation pathway as well as sites implicated in amiloride binding and permeant ion discrimination. ASIC, acid-sensing ion channel; TM, transmembrane.
Although the stable presence of ENaC at the apical membrane requires the ERC, its activity at the cell surface requires proteolytic cleavage at specific sites within the extracellular loops of the α and γ subunits to liberate embedded inhibitory tracts (12) (Figure 2). Under physiologic conditions, this effect appears to be mediated by furin and a secondary membrane-resident protease. Furin is a proprotein convertase that resides primarily in the trans-Golgi network and processes proteins transiting through the biosynthetic pathway. Furin increases ENaC open probability (i.e., the percentage of time the channel spends open) and, hence, net Na+ transport. It is unclear at this time whether the furin-mediated cleavage is an important locus of regulation or is a device to keep the channel from being turned on prematurely (see below). However, it is increasingly clear that ENaC cleavage plays a pathophysiologic role in the Na+ retention associated with the nephrotic syndrome. The serine protease plasmin cleaves the ENaC γ subunit and activates the channel (13). Plasmin is not present in the tubule lumen under normal conditions; however, in the setting of proteinuria (as seen in the nephrotic syndrome), plasminogen is filtered by the glomerulus and can be converted to plasmin by urokinase, which is present within the tubular lumen (13). In the context of glomerular proteinuria, plasmin-dependent ENaC activation may contribute to Na+ retention, and edema or hypertension (14).
Animals or humans with decreased ENaC function have severe disorders of Na+ wasting and K+ retention. Increased channel activity (or excess aldosterone) results in hypertension and K+ wasting (15), as seen with the heritable disorder Liddle’s syndrome. The first identified Liddle mutation resulted in a premature translation stop in the β subunit (16), leaving the Na+ pore intact but deleting intracellular target sites for inhibitory control mechanisms (16). Other mutations that cause variable degrees of hyperactivation of the channel were also identified. On the basis of these observations, it was suggested that mild increases in ENaC activity could act in concert with other signaling defects in the pathogenesis of essential hypertension (17).
Hormonal Regulation of ENaC
Renin-Angiotensin-Aldosterone System.
Aldosterone is central to the normal regulation of Na+ and K+ handling by principal cells, and, hence, to the control of ion concentrations, extracellular fluid volume, and BP in all land mammals (18). The effects of aldosterone on ion transport in principal cells are mediated by the mineralocorticoid receptor (MR). The MR, which is almost certainly the only receptor for aldosterone in principal cells, is an intracellular hormone-regulated transcription factor that triggers coordinated changes in the expression of numerous genes. The key ones required for regulating Na+ transport encode either transporters themselves, or regulatory proteins that control transporter abundance or activity. The bulk of evidence supports the view that the central effect of aldosterone is to increase ENaC apical membrane density approximately 2- to 5-fold (18,19) (Figure 1), but effects on channel open probability may also contribute. The ENaC α subunit gene itself is an important target; however, its transcription is stimulated somewhat slowly (over approximately 6 hours). Similarly, aldosterone stimulates transcription of both the α and β subunits of the Na+-K+-ATPase, but also relatively slowly (20) (see below). In fact, most of the aldosterone-induced increase in Na+ transport occurs within the first 3 hours and is primarily mediated by rapidly stimulated genes that encode regulatory proteins, not by increases in transporter gene expression per se.
The best characterized of the ENaC regulatory proteins is the serine-threonine kinase SGK1 (21). Aldosterone acts through the MR to rapidly increase SGK1 gene transcription and, thus, the SGK1 protein level (Figure 1). Importantly, SGK1 must also be activated by two phosphorylation events that control its inherent activity (22). These activating phosphorylations are regulated by other hormones, including insulin and possibly angiotensin II (AngII) (23). Activated SGK1 then phosphorylates and regulates various targets, most notably the ubiquitin ligase Nedd4-2 (24), which post-translationally modifies proteins by covalently adding a ubiquitin group to specific lysines. Nedd4-2 inhibits ENaC by ubiquitinylating the channel, triggering its internalization from the plasma membrane and ultimately its degradation (25). SGK1 phosphorylates and inhibits Nedd4-2, and this double negative (inhibiting the inhibitor) results in ENaC accumulation at the apical plasma membrane (26). Importantly, the above-mentioned Liddle mutation disrupts interaction between Nedd4-2 and ENaC, as if aldosterone were always present (18). In fact, aldosterone levels are suppressed in Liddle’s syndrome, and hence it is a form of pseudohyperaldosteronism. It is also interesting to note that SGK1 indirectly regulates ENaC gene expression through effects on the activities of Dot1a and Af9 (27).
SGK1 acts in other cell types to regulate a variety of other transporters, including NHE3 (proximal tubule and thick ascending limb), NKCC2 (thick ascending limb), and NCC (distal convoluted tubule). It is particularly worth noting recent evidence supporting the idea that SGK1 regulation of NCC in distal convoluted tubule proceeds, at least in part, through regulation of Nedd4-2, in a manner similar to how the SGK1–Nedd4-2 module regulates ENaC in principal cells (28). On the other hand, SGK1 also regulates ROMK in principal cells (29), although this effect is likely less important than its effects on ENaC (see below). Small molecule inhibitors of SGK1 have been shown to have antihypertensive effects in animals; however, there have been no clinical trials (30). An area of considerable recent interest is the convergent regulation by AngII and aldosterone of ENaC and other transporters such as NCC (31).
Atrial Natriuretic Peptide.
Maintaining cardiorenal homeostasis by regulating fluid volume is an important aspect of the cardioprotective properties of atrial natriuretic peptide (ANP). ANP achieves these effects by regulating multiple renal processes, as will be addressed in other reviews in this series. It acts through the membrane-bound natriuretic peptide receptor-A, which triggers generation of the second messenger cGMP (32) to stimulate a variety of downstream targets including cGMP-dependent protein kinases and cGMP-gated ion channels (33). In principal cells, ANP inhibits ENaC, which contributes to its natriuretic properties (34). It also regulates BP by inhibiting renin secretion (35) and aldosterone production from the adrenal gland by downregulating the steroidogenic acute regulatory protein (36).
Insulin.
The physiologic role for insulin in the control of Na+ transport has remained obscure; however, its pathophysiologic effects are clear. Kidney tubule Na+ transport remains insulin sensitive, even as other tissues and processes become resistant (37). Insulin stimulates NHE3-dependent Na+ transport in the proximal tubule (38), whereas it stimulates ENaC in principal cells. As insulin levels rise to maintain normal glucose concentration, the retained sensitivity of Na+ transport results in excessive Na+ reabsorption in insulin-resistant states. Because neither Na+ nor BP participates in the feedback loop to inhibit islet β-cell insulin secretion, the principal cell becomes an “unwilling accomplice” in the ensuing salt-sensitive hypertension (39). Further contributing to this problem, the vasodilatory effects of insulin are blunted in insulin-resistant states, and hence only the prohypertensive effects remain (40).
AVP.
In addition to its critical role in regulating principal cell water transport (see below), AVP also has important effects to stimulate ENaC. These effects are mediated substantially by cAMP, which activates cAMP-dependent kinase (protein kinase A [PKA]). PKA can phosphorylate and inhibit Nedd4-2 in a manner similar to that of SGK1 (41,42). This direct effect is complicated by variable effects of AVP on the renin-angiotensin-aldosterone system (RAAS) hormonal axis, either to stimulate or suppress renin, depending on extracellular fluid volume status and AVP blood level (42). Importantly, despite stimulation of ENaC (which itself should raise the serum Na+ concentration), the net effect of AVP is to lower Na+ concentration. However, under conditions of high sodium consumption and low water intake (raising AVP levels, while lowering aldosterone), AVP may contribute to salt-sensitive hypertension through its effects to stimulate ENaC. It was suggested that AVP stimulation of ENaC might play a physiologically beneficial role in overall water conservation when both water and salt intake are low, although this effect would blunt its ability to lower serum Na+ concentration (42). This suggestion harkens back to the clinical maxim that “the body defends volume above all else.”
Paracrine and Autocrine Regulation of ENaC and Na+ Transport in the Collecting Duct
Hormones are key regulators of principal cell Na+ transport; however, another key aspect of this regulation involves local factors. In particular, autocrine (acting on the same cell) and paracrine (acting on neighboring cells) factors play an important role in the modulation of principal cell Na+ transport. Physical factors, such as tubule fluid flow, can also play a role. Figure 3 summarizes these effects.
Figure 3.
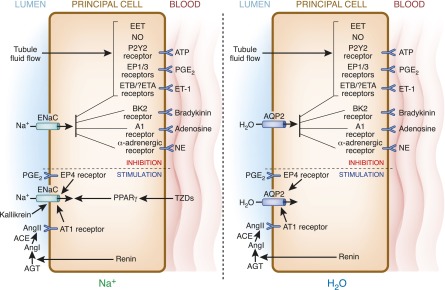
Autocrine and paracrine regulation of collecting duct principal cell ENaC and AQP2. Much commonality exists in regulation of ENaC (left) and AQP2 (right). Flow stimulates ATP, PGE2, and ET-1, which act on their cognate receptors to inhibit Na and water reabsorption. Similarly, bradykinin, adenosine, and NE act on their receptors to inhibit ENaC and AQP2. Flow-stimulated EET uniquely inhibits Na, but not water, transport. Compared with the wide variety of inhibitors, relatively few autocrine or paracrine factors stimulate ENaC and/or AQP2 activity. Renin, ultimately via AngII, as well as PGE2 binding to EP4 receptors, are potentially capable of augmenting principal cell Na and water transport. TZDs (via PPARγ) and kallikrein (via cleavage of an autoinhibitory domain in ENaC) may increase Na reabsorption. See the text for more detailed descriptions of each regulatory factor. ACE, angiotensin-converting enzyme; AGT, angiotensinogen; Ang, angiotensin; AQP, aquaporin; EET, eicosataetranoic acid; EP, PGE receptor; ET, endothelin; NE, norepinephrine; NO, nitric oxide; PPARγ, peroxisome proliferator–activated receptor-γ; TZD, thiazolidinedione.
Adrenergic Nerves.
The findings of close apposition of sympathetic nerve extensions and principal cells (43), as well as collecting duct expression of β- and α-adrenergic receptors, suggests that principal cell function can be modulated by catecholamines released by efferent renal sympathetic nerves. The nature of such regulation is unclear because differing results have been obtained depending upon the species studied and the agonist utilized. It is likely that adrenergic effects on principal cells are complex, depending upon whether β- or α-adrenergic receptors are activated, the specific cAMP-regulated pathway affected (urea, Na, or water), and the hormonal milieu. In general, α-adrenergic receptor activation in the collecting duct appears to inhibit agonist-induced cAMP accumulation (43,44); however, much more clarification is needed.
Prostaglandins and Cytochrome P450 Metabolites.
AA in the collecting duct can be metabolized by cyclooxygenases to prostaglandins (PGs) and by cytochrome P450 epoxygenase to form various epoxyeicosatrienoic acids (EETs). The collecting duct produces relatively large amounts of PGs and particularly PGE2 (45,46). The collecting duct expresses three PG receptors (EP1, EP3, and EP4). Collecting duct PGE2 production is stimulated by a variety of factors that generally exert natriuretic and diuretic effects, including ATP, EETs, endothelin (ET)-1, shear stress, and others (45,47). The effects of PGE2 on collecting duct Na+ transport are complex. In the absence of agonists, PGE2 may augment cAMP-dependent Na+ reabsorption in the collecting duct, most likely through stimulation of EP4 receptors. However, in the presence of agonists, PGE2 inhibits collecting duct Na+ transport. Because agonists are virtually always present in vivo, and as confirmed using knockout of EP receptors, the primary physiologic effect of PGE2 on the collecting duct is natriuretic. Activation of EP1 and EP3 receptors in the collecting duct reduces adenylyl cyclase–dependent cAMP production. Finally, PGE2 inhibits collecting duct Na+ reabsorption through a calcium-dependent mechanism. Taken together, these findings suggest that nonsteroidal anti-inflammatory drug–induced Na+ retention is caused, at least in part, by inhibition of collecting duct PGE2.
Although species differences may exist with regard to the specific EET stereoisomer involved, EETs have been repeatedly shown to inhibit ENaC activity (48–51). The physiologic relevance of EETs in the control of collecting duct Na+ transport is largely unknown; however, it is likely that EETs interact with other signaling systems, including mediating adenosine inhibition of ENaC (52) and stimulating PGE2 formation (50). Finally, cortical collecting duct EET formation is stimulated by elevated tubule fluid flow, potentially promoting Na+ excretion.
Tubule Fluid Flow.
Increased collecting duct tubule fluid flow occurs in natriuretic states. The increase in collecting duct tubule fluid flow, at least in the setting of salt loading, results from increased GFR and reduced solute reabsorption by nephron segments proximal to the collecting duct. ENaC is mechanosensitive and is activated by shear stress [reviewed by Kashlan and Kleyman (53)]. Such flow-stimulated ENaC activity would be counterproductive during salt loading; hence, several mechanisms appear to exist to inhibit flow-stimulated ENaC activity. Increases in intracellular or extracellular Na+ concentration inhibit ENaC activity. In addition, luminal collecting duct shear stress stimulates a variety of factors that can reduce Na+ transport, including 11-EET and 12-EET (54), ATP (55), nitric oxide (NO) (56), PGE2 (47), and ET-1 (57).
Kinins.
The connecting segment and cortical collecting duct are the major sites of renal tissue kallikrein (TK) expression [reviewed by Eladari et al. (58)]. TK cleaves kininogen to yield lysyl-BK, which is cleaved by N-aminopeptidase to form BK. BK released by the collecting duct has the potential to interact with BK type 2 (BK2) receptors expressed by the collecting duct. BK activation of BK2 receptors inhibits ENaC activity (59). TK per se may regulate ENaC independent of BK (58). The addition of TK to the lumen side of the cortical collecting duct increases ENaC activity associated with increased cleavage of ENaCγ (i.e., removal of the autoinhibitory peptide within ENaCγ). Finally, the BK system should be considered in the context of angiotensin-converting enzyme inhibitors (ACEIs), which are well known to inhibit BK degradation, thereby leading to increased BK levels. This elevation of BK could, in principle, enhance the effect of ACEIs to inhibit ENaC, over and above what angiotensin receptor blockers, which do not inhibit BK degradation, can do. However, the clinical relevance of this difference between ACEIs and angiotensin receptor blockers remains controversial.
NO.
Collecting duct NO production is stimulated by tubule fluid flow, ATP, and ET-1 [reviewed by Hyndman and Pollock (60)]. NO reduces ENaC activity (61,62), possibly via reduction of AVP-stimulated cAMP accumulation [reviewed by Ortiz and Garvin (62)]. In addition, NO may mediate ATP inhibition of collecting duct ROMK activity (63). Collecting duct NO is likely of physiologic significance because collecting duct–specific knockout of NO synthase 1 causes salt-sensitive hypertension (64).
Peroxisome Proliferator–Activated Receptors.
Activation of the peroxisome proliferator–activated receptor-γ by thiazolidinediones is well known to cause Na+ retention. Mice with principal cell–specific knockout of peroxisome proliferator–activated receptor-γ are resistant to thiazolidinedione-induced Na+ retention (65,66). In addition, other noncollecting duct mechanisms may be involved (67).
Adenosine.
Adenosine is derived from ATP and cAMP metabolism. Salt loading increases renal interstitial adenosine; relatively large amounts of adenosine are found in the inner medulla [reviewed by Rieg and Vallon (68)]. Activation of adenosine A1 receptors inhibits AVP-dependent cAMP-stimulated ENaC activity in the collecting duct.
ATP.
Collecting duct cells release ATP into the lumen in response to tubule flow. In addition, dietary salt increases urinary ATP and its metabolites [reviewed by Vallon and Rieg (55)]. The collecting duct lumen expresses low levels of ectonucleotidases, facilitating ATP regulation of collecting duct function. The collecting duct expresses luminal P2Y2 purinergic receptors, the activation of which inhibits ENaC (55,69,70). P2Y2 regulation of ENaC is physiologically relevant in that P2Y2 knockout prevents high-Na diet–induced decreases in ENaC activity. Finally, ATP activation of P2Y2 may inhibit collecting duct ROMK activity (63).
ET.
The collecting duct is the major source of ET-1 in the kidney and may produce more ET-1 than any other cell type in the body [reviewed by Kohan et al. (71)]. ET-1 production by the collecting duct is stimulated by luminal shear stress, luminal Na+ delivery, and other factors (57,71). In general, collecting duct ET-1 synthesis is increased by extracellular fluid volume expansion. Collecting duct–derived ET-1 can act in an autocrine manner on basolateral ET receptors to regulate Na+ transport. The collecting duct expresses relatively high levels of ETB receptors and low levels of ETA receptors. Activation of collecting duct ETB receptors inhibits Na+ transport in the cortical collecting duct; this effect is mediated by several signaling systems, including NO (71,72). The physiologic significance of the collecting duct ET system was demonstrated in studies with principal cell-specific knockout of ET-1 or ET receptors. The absence of principal cell ET-1 or both ET receptors causes marked salt-sensitive hypertension (73,74). These findings may be clinically relevant in that ET receptor antagonists, which are currently approved for treatment of pulmonary hypertension and are being studied for the treatment of diabetic nephropathy, can cause significant fluid retention (75). It is possible that this is mediated, at least in part, by blockade of collecting duct ET receptors.
Renin.
Renin is synthesized and secreted into the tubule lumen by collecting duct cells [reviewed by Navar et al. (76)]. Luminal renin may bind to prorenin receptors on intercalated cells, enhancing its catalytic activity and modulating intercalated cell function. Because angiotensinogen and angiotensin-converting enzymes are present in the distal nephron lumen, secreted renin may stimulate luminal AngII formation leading to enhanced ENaC activity. Because collecting duct renin production is markedly increased by circulating AngII and in the setting of diabetes mellitus, this distal nephron renin system may be of pathophysiologic significance.
Na-K-ATPase
Ultimately, the primary driving force for principal cell Na+ reabsorption and K+ secretion is provided by the Na+-K+-ATPase. As in other nephron segments and, indeed, most epithelia, Na+-K+-ATPase is targeted exclusively to the basolateral membrane of principal cells. Its activity and membrane density are regulated by hormones, such as aldosterone, AVP, and insulin, as well as nonhormonal factors, such as intracellular [Na+] and tonicity (77). However, in the control of Na+ balance, most notably by aldosterone, the Na+-K+-ATPase is not the major target of regulation. As addressed below, the bulk of evidence supports the conclusion that ENaC is the primary site of regulation. This is an important detail for clinicians to be aware of, because it is relevant to commonalities among hypertensive diseases, such as Liddle syndrome, apparent mineralocorticoid excess, and primary aldosteronism, as well as to the treatment of salt-sensitive essential hypertension.
ROMK
A predominant role of the RAAS is to regulate ENaC (18); however, other principal cell transporters, such as ROMK, are also regulated by aldosterone (acting through the MR) (78). ROMK is a critical, but not the sole, mediator of K+ transport in principal cells (Figure 1). Like ENaC, it is a cation channel. ROMK is highly selective for K+ over Na+, and is inhibited by Ba2+ (79). Some of the effects of the RAAS on ROMK may be the result of regulation of with no lysine kinases (WNKs) (2) that, in addition to inhibiting NKCC2 and NCC in the thick ascending limb and distal convoluted tubule, respectively, also inhibit K channels in principal cells (80). Interconnections between RAAS and WNK signaling have been identified, both through effects of AngII and effects of aldosterone, at least in part, mediated by SGK1 (78,81). Assembly and trafficking of ROMK to the cell surface is dictated by a multiprotein complex facilitated by PDZ interactions with scaffold proteins NHERF-1 and NHERF-2 (82). Increased dietary potassium causes a large increase in apical expression of ROMK in the distal convoluted tubule-2, connecting tubule, and collecting duct but not in distal convoluted tubule-1, indicating that multiple regulatory mechanisms are involved in dietary K–regulated ROMK channel function in the distal nephron (83). Recent studies suggest that Cullin3-RING (really interesting new gene) ligases that contain Kelch-like 3 (components of an E3 ubiquitin ligase complex) target ubiquitinylation of WNK4 and thereby regulate WNK4 levels, which, in turn, regulate levels of ROMK (84). Interestingly, ROMK variants with decreased channel activity have been associated with resistance to hypertension, suggesting that ROMK may also be a determinant of BP control in the general population (85). Hypertension resistance variants of ROMK were found to have decreased channel function as a result of increased sensitivity to the inhibitory effects of a G protein–coupled receptor, which stimulates phosphatidylinositol 4,5-bisphosphate hydrolysis (86).
Cl− Transport
There are three contributory pathways for Cl− transport. First, in principal cells, Cl− can be reabsorbed or secreted (depending on electrochemical gradient) via the cystic fibrosis transmembrane regulator. Cl− basolateral transport is through a Cl− channel of the ClC family. Second, Cl− also moves through a paracellular pathway, at least in part via tight junction claudins (2). Finally, Cl− is transported through intercalated cells (3). Fascinating new data support the idea that, in intercalated cells, the ability of the receptor for aldosterone (the MR) to respond to aldosterone is controlled by phosphorylation (87). This effect influences the ability of these cells to increase Cl− transport in response to aldosterone, and allows the collecting duct to shift from mediating primarily Na+-K+ exchange to mediating both Na+-K+ exchange and Na+-Cl+ cotransport.
Control of Water Transport in Principal Cells
Overview of Collecting Duct Water Transport
The primary mechanism by which water is reabsorbed in the principal cell is through AVP stimulation of AQP2 expression and accumulation in the luminal plasma membrane (Figure 4). AVP binding to its type 2 receptor (V2R) in the basolateral membrane of principal cells induces a cAMP signaling cascade, which leads rapidly to translocation of AQP2 from intracellular vesicles to the apical membrane. Tubule water enters the cell through AQP2, traverses the cytosol, and exits to the interstitium via AQP3 and AQP4, with water movement ultimately being driven by the tubule lumen–interstitium osmotic gradient (88,89). Inactivating mutations in V2R or AQP2 genes lead to nephrogenic diabetes insipidus, a disorder characterized by polyuria and, consequently, polydipsia (90,91) [reviewed by Robben et al. (92) and Loonen et al. (93)]. Overstimulation of water retention is found in the syndrome of inappropriate release of the antidiuretic hormone and in activating mutations of the V2R, which both lead to hyponatremia with normovolemia.
Figure 4.

Hormonal regulation of AQP2 in the principal cell. In states of hypernatremia or hypovolemia, AVP is released from the pituitary and binds its V2R. AVP-bound V2R results in dissociation of the trimeric G protein into a GTP-bound α subunit (Gsα) and βγ subunits (Gsβ and Gsγ) of which the first activates AC to generate cAMP. Activation of PKA by cAMP increases AQP2 transcription through phosphorylated activation of the CREB transcription factor and induces translocation of AQP2 from intracellular vesicles to the apical membrane by dephosphorylating AQP2 at Ser261 (pS261) and by phosphorylating AQP2 at Ser256 (pS256), Ser264 (pS264), and Thr269 (pT269). Driven by the transcellular osmotic gradient, prourinary water will then enter the principal cell through AQP2 and exit the cell via AQP3 and AQP4 located in the basolateral membrane of these cells, thereby concentrating urine. Activation of receptors by hormones, such as PGE2, ATP, and dopamine, counteracts this action of AVP by inducing short-chain ubiquitination of AQP2 at Lys270 (K270), leading to its internalization and lysosomal targeting and degradation. AQP2 pS261 is reinstalled during this internalization process, in which PKC is a central kinase. AC, adenylate cyclase; AVP, arginine vasopressin; CREB, cAMP-responsive element-binding protein; Dop, dopamine; PKA, protein kinase A; PKC, protein kinase C; Ub, ubiquitin; V2R, vasopressin receptor type 2.
Basic Biology of AQP2
The principal cell mechanisms for regulation of water reabsorption primarily involve modulation of AQP2 abundance in the luminal plasma membrane. Basolateral AQP3 and, to a lesser extent, AQP4 are necessary for transcellular water movement in the principal cell (94,95), but these channels are mainly constitutively expressed and, therefore, serve a permissive function. By contrast, AQP2 is highly regulated in a complex manner. This involves short-term modulation through alterations in trafficking and long-term regulation through changes in protein expression.
AQP2 has at least five phosphorylation sites that appear to regulate apical membrane insertion and endocytosis [reviewed by Fenton et al. (96) and Nedvetsky et al. (97)]. PKA is the canonical cAMP-dependent mechanism for AQP2 phosphorylation; however, protein kinase G, protein kinase C, casein kinases, and other kinases may also phosphorylate AQP2 (98). In addition, several phosphatases are likely involved in the regulation of AQP2 phosphorylation and activity. AQP2 phosphorylation near the carboxy terminus at serines S256 and S269 appears to be of primary importance in trafficking to the plasma membrane. Conventionally, PKA-dependent phosphorylation at S256 was considered a priming event for S269 phosphorylation and plasma membrane insertion (99–101); however, recent studies suggest that S256 phosphorylation may be involved in trafficking of AQP2 through the endoplasmic reticulum and Golgi apparatus. Phosphorylation at S269 is important for increasing AQP2 activity by enhancing exocytosis and reducing endocytosis. AQP2 accumulates in clathrin-coated pits and is internalized in a dynamin-dependent manner; this process may be regulated by the phosphorylation state of S256 and S269. Finally, AQP2 is highly phosphorylated at S261 without AVP, but this is reduced by AVP (102,103), suggesting that S261 may also be involved in AQP2 trafficking.
AQP2 internalization is mediated, at least in part, by an unknown ubiquitin ligase, which mediates ubiquitinylation at lysine K270 (104). K270 is essential in this process, because endocytosis of AQP2-K270R, which cannot be further ubiquitinylated, was strongly retarded (104). Relevant to this, AQP2 K63-linked ubiquitinylation is short lived and internalized AQP2 may recycle to the plasma membrane multiple times (105). This suggests that, as with many other membrane proteins, AQP2 is likely deubiquitinated before being targeted for lysosomal degradation. Although the involved ubiquitin ligase and deubiquitinating enzyme are not known, initial studies have identified candidates for further research (106). It is unlikely that this ubiquitin ligase is Nedd4-2; however, the parallels with ENaC regulation are striking.
Another important mechanism for regulation of principal cell water transport is through control of total cell AQP2 levels (i.e., long-term regulation of AQP2 expression) [see the excellent review by Radin et al. (107)]. The time frame for maximal changes in AQP2 abundance is not certain, but likely takes several days for up to 10-fold alterations to occur. Changes in principal cell AQP2 degradation or elimination (through exosomes excreted in the urine) do not account for this wide fluctuation in AQP2 abundance. Rather, it appears that transcriptional modulation of AQP2 promoter activity is important. The AQP2 promoter contains cAMP response elements as well as a number of other transcription factor consensus recognition sites.
Stimulators of AQP2
AVP.
AVP is the major regulator of AQP2 trafficking and expression in principal cells. AVP exerts multiple effects on cell signaling of the principal cell, foremost of which is stimulation of adenylyl cyclase–dependent cAMP accumulation and activation of PKA. Another cAMP-activated mediator, Epac, was recently implicated in mediating AQP2 phosphorylation (96,97). AVP increases intracellular [Ca2+] as well as activates Akt (protein kinase B) via the phosphatidylinositide 3-kinase and inhibition of extracellular signal–regulated kinases 1 and 2; some of these processes are likely cAMP dependent. The net effect is that AVP can modulate phosphorylation AQP2 through a variety of protein kinases and likely also through regulation of phosphatases. AVP also reduces AQP2 internalization through decreasing ubiquitinylation as well as depolymerization of the actin cytoskeleton. AVP increases AQP2 protein expression primarily through enhanced AQP2 gene transcription and, to a lesser extent, through reduced AQP2 degradation.
Interstitial Osmolality.
Interstitial osmolality per se may regulate AQP2 abundance. AVP-deficient Brattleboro rats start concentrating their urine when water deprived, or when made hypertonic, which all lead to the generation of a hypertonic interstitium (108,109). The tonicity-responsive enhancer-binding protein (TonEBP) may be involved in the hypertonicity response, because its expression is upregulated with hypertonicity (110), inactivating mutations in the tonicity-responsive element in the AQP2 promoter reduced AQP2 expression in mpkCD cells (111), and TonEBP knockout mice or mice transgenic for dominant-negative TonEBP showed decreased AQP2 abundance (112,113).
Other Stimulators.
AngII, via angiotensin II type 1 receptors, and aldosterone have been shown to increase AQP2 abundance in mpkCCD cells (114,115), suggesting that the RAAS can control principal cell water reabsorption. In agreement with this, recent studies found that mice with collecting duct–specific angiotensin II type 1 receptor knockout had reduced urinary concentrating ability associated with reduced AVP-stimulated cAMP accumulation (116). ANP was also suggested to increase principal cell water reabsorption because ANP increased AQP2 phosphorylation at S256 and its translocation to the plasma membrane in inner medullary collecting duct cells (117,118). This, however, is controversial, because others showed that ANP antagonizes AVP-mediated water permeability by decreasing AQP2 phosphorylation and enhancing AQP2 retrieval from the apical membrane (119).
Autocrine and Paracrine Inhibitors of AQP2
Similar to regulation of principal cell Na+ transport, several autocrine and paracrine factors modulate principal cell water reabsorption. The previous section on Na+ transport provides details regarding the cell sources and regulation of expression of these factors. In the following, we specifically address these factors in the context of modulation of principal cell water transport.
PGs and Cytochrome P450 Metabolites.
As for Na+ reabsorption in the collecting duct, PGE2 may increase cAMP-dependent water reabsorption via binding to EP4 receptors. In the presence of agonists, PGE2 inhibits collecting duct water transport through stimulation of EP1 and EP3 receptors leading to inhibition of cAMP production, induction of AQP2 retrieval from the plasma membrane, and Rho-dependent actin depolymerization with resultant inhibition of AQP2 translocation to the plasma membrane (45,46).
Kinins.
BK released by the collecting duct can bind to collecting duct BK2 receptors and inhibit AQP2 trafficking to the plasma membrane (120). Whether this is physiologically relevant remains to be determined.
NO.
The effects of NO/cGMP on principal cell water transport are controversial, with results varying depending upon the model and conditions studied. NO has been reported by some, but not all, to inhibit collecting duct water transport, the latter through reduction of AVP-stimulated cAMP accumulation [reviewed by Ortiz and Garvin (62)]. By contrast, NO/cAMP has been described to increase AQP2 plasma membrane expression, particularly in in vitro studies (98).
Adenosine.
Adenosine activation of A1 receptors inhibits AVP-dependent cAMP-stimulated water reabsorption in the collecting duct [reviewed by Rieg and Vallon (68)]. Because caffeine is shown to exert a diuretic effect through antagonism of the adenosine A1 receptor, this effect must be the result of nonprincipal cell actions.
ATP.
Water loading may increase collecting duct ATP release by increasing cell swelling caused by decreased extracellular tonicity (55). Activation of apical collecting duct P2Y2 receptors inhibits water reabsorption by reducing AVP-stimulated cAMP and increasing PGE2 production.
ET.
ET-1 activation of ETB receptors inhibits AVP-stimulated cAMP accumulation and water reabsorption by the collecting duct. Mice with principal cell–specific knockout of ET-1 demonstrate enhanced water retention and increased cAMP accumulation in response to AVP (121).
Summary
The principal cell is arguably the most highly regulated cell type in the kidney tubules, if not in all mammalian epithelia. Because the regulated transport of ions and water is of vital importance to the role that this tubule cell plays in kidney function and homeostasis, we have addressed the basic transport functions of the principal cell primarily from the standpoint of regulation. The central role of two different hormone-transporter pairs was emphasized: aldosterone and ENaC for control of ion transport, and AVP and AQP2 for control of water transport. However, the complex regulation of principal cell function is underscored by the panoply of hormonal, autocrine, paracrine, and physical factors that regulate its activities. A few examples are as follows: ATP, PGE2, and ET, which inhibit both Na+ and water transport; AngII, which stimulates both Na+ and water transport; and insulin, which selectively stimulates Na+ transport. Under normal physiologic conditions, these diverse regulators exert their effects in various combinations to provide context-appropriate integrated responses to different environmental conditions.
Understanding the signaling and transport mechanisms that underlie principal cell function is valuable to practicing clinicians, both because it enhances our appreciation of the kidney and its role in homeostasis, and because it provides a foundation for greater depth and flexibility in our approach to diagnosis and treatment of the wide array of fluid and electrolyte disorders we encounter.
Disclosures
None.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1.Palmer LG, Frindt G: Na+ and K+ transport by the renal connecting tubule. Curr Opin Nephrol Hypertens 16: 477–483, 2007 [DOI] [PubMed] [Google Scholar]
- 2.Hou J, Rajagopal M, Yu AS: Claudins and the kidney. Annu Rev Physiol 75: 479–501, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schuster VL, Stokes JB: Chloride transport by the cortical and outer medullary collecting duct. Am J Physiol 253: F203–F212, 1987 [DOI] [PubMed] [Google Scholar]
- 4.Palmer LG, Patel A, Frindt G: Regulation and dysregulation of epithelial Na+ channels. Clin Exp Nephrol 16: 35–43, 2012 [DOI] [PubMed] [Google Scholar]
- 5.Wang WH, Giebisch G: Regulation of potassium (K) handling in the renal collecting duct. Pflugers Arch 458: 157–168, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC: Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367: 463–467, 1994 [DOI] [PubMed] [Google Scholar]
- 7.Jasti J, Furukawa H, Gonzales EB, Gouaux E: Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature 449: 316–323, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Kashlan OB, Kleyman TR: ENaC structure and function in the wake of a resolved structure of a family member. Am J Physiol Renal Physiol 301: F684–F696, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soundararajan R, Melters D, Shih IC, Wang J, Pearce D: Epithelial sodium channel regulated by differential composition of a signaling complex. Proc Natl Acad Sci U S A 106: 7804–7809, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soundararajan R, Ziera T, Koo E, Ling K, Wang J, Borden SA, Pearce D: Scaffold protein connector enhancer of kinase suppressor of Ras isoform 3 (CNK3) coordinates assembly of a multiprotein epithelial sodium channel (ENaC)-regulatory complex. J Biol Chem 287: 33014–33025, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoo D, Flagg TP, Olsen O, Raghuram V, Foskett JK, Welling PA: Assembly and trafficking of a multiprotein ROMK (Kir 1.1) channel complex by PDZ interactions. J Biol Chem 279: 6863–6873, 2004 [DOI] [PubMed] [Google Scholar]
- 12.Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong Q, Carattino MD, Johnson JP, Stockand JD, Kleyman TR: Epithelial sodium channels are activated by furin-dependent proteolysis. J Biol Chem 279: 18111–18114, 2004 [DOI] [PubMed] [Google Scholar]
- 13.Passero CJ, Mueller GM, Rondon-Berrios H, Tofovic SP, Hughey RP, Kleyman TR: Plasmin activates epithelial Na+ channels by cleaving the gamma subunit. J Biol Chem 283: 36586–36591, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Svenningsen P, Bistrup C, Friis UG, Bertog M, Haerteis S, Krueger B, Stubbe J, Jensen ON, Thiesson HC, Uhrenholt TR, Jespersen B, Jensen BL, Korbmacher C, Skøtt O: Plasmin in nephrotic urine activates the epithelial sodium channel. J Am Soc Nephrol 20: 299–310, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warnock DG, Rossier BC: Renal sodium handling: The role of the epithelial sodium channel. J Am Soc Nephrol 16: 3151–3153, 2005 [DOI] [PubMed] [Google Scholar]
- 16.Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, Gill JR, Jr, Ulick S, Milora RV, Findling JW, Canessa CM, Rossier BC, Lifton RP: Liddle’s syndrome: Heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell 79: 407–414, 1994 [DOI] [PubMed] [Google Scholar]
- 17.Soundararajan R, Pearce D, Hughey RP, Kleyman TR: Role of epithelial sodium channels and their regulators in hypertension. J Biol Chem 285: 30363–30369, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pearce D, Bhalla V, Funder JW, Stokes JB: Aldosterone regulation of ion transport. In: The Kidney, edited by Brenner BM, Philadephia, Saunders Elsevier, 2012 [Google Scholar]
- 19.Frindt G, Ergonul Z, Palmer LG: Surface expression of epithelial Na channel protein in rat kidney. J Gen Physiol 131: 617–627, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verrey F, Schaerer E, Zoerkler P, Paccolat MP, Geering K, Kraehenbuhl JP, Rossier BC: Regulation by aldosterone of Na+,K+-ATPase mRNAs, protein synthesis, and sodium transport in cultured kidney cells. J Cell Biol 104: 1231–1237, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soundararajan R, Wang J, Melters D, Pearce D: Glucocorticoid-induced Leucine zipper 1 stimulates the epithelial sodium channel by regulating serum- and glucocorticoid-induced kinase 1 stability and subcellular localization. J Biol Chem 285: 39905–39913, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu M, Wang J, Jones KT, Ives HE, Feldman ME, Yao LJ, Shokat KM, Ashrafi K, Pearce D: mTOR complex-2 activates ENaC by phosphorylating SGK1. J Am Soc Nephrol 21: 811–818, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCormick JA, Bhalla V, Pao AC, Pearce D: SGK1: A rapid aldosterone-induced regulator of renal sodium reabsorption. Physiology (Bethesda) 20: 134–139, 2005 [DOI] [PubMed] [Google Scholar]
- 24.Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Münster C, Chraïbi A, Pratt JH, Horisberger JD, Pearce D, Loffing J, Staub O: Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. EMBO J 20: 7052–7059, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kabra R, Knight KK, Zhou R, Snyder PM: Nedd4-2 induces endocytosis and degradation of proteolytically cleaved epithelial Na+ channels. J Biol Chem 283: 6033–6039, 2008 [DOI] [PubMed] [Google Scholar]
- 26.Bhalla V, Soundararajan R, Pao AC, Li H, Pearce D: Disinhibitory pathways for control of sodium transport: Regulation of ENaC by SGK1 and GILZ. Am J Physiol Renal Physiol 291: F714–F721, 2006 [DOI] [PubMed] [Google Scholar]
- 27.Zhang W, Xia X, Reisenauer MR, Rieg T, Lang F, Kuhl D, Vallon V, Kone BC: Aldosterone-induced Sgk1 relieves Dot1a-Af9-mediated transcriptional repression of epithelial Na+ channel alpha. J Clin Invest 117: 773–783, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ronzaud C, Loffing-Cueni D, Hausel P, Debonneville A, Malsure SR, Fowler-Jaeger N, Boase NA, Perrier R, Maillard M, Yang B, Stokes JB, Koesters R, Kumar S, Hummler E, Loffing J, Staub O: Renal tubular NEDD4-2 deficiency causes NCC-mediated salt-dependent hypertension. J Clin Invest 123: 657–665, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lang F, Huang DY, Vallon V: SGK, renal function and hypertension. J Nephrol 23[Suppl 16]: S124–S129, 2010 [PMC free article] [PubMed] [Google Scholar]
- 30.Ackermann TF, Boini KM, Beier N, Scholz W, Fuchss T, Lang F: EMD638683, a novel SGK inhibitor with antihypertensive potency. Cell Physiol Biochem 28: 137–146, 2011 [DOI] [PubMed] [Google Scholar]
- 31.van der Lubbe N, Zietse R, Hoorn EJ: Effects of angiotensin II on kinase-mediated sodium and potassium transport in the distal nephron. Curr Opin Nephrol Hypertens 22: 120–126, 2013 [DOI] [PubMed] [Google Scholar]
- 32.Tremblay J, Gerzer R, Vinay P, Pang SC, Béliveau R, Hamet P: The increase of cGMP by atrial natriuretic factor correlates with the distribution of particulate guanylate cyclase. FEBS Lett 181: 17–22, 1985 [DOI] [PubMed] [Google Scholar]
- 33.Hayek S, Nemer M: Cardiac natriuretic peptides: From basic discovery to clinical practice. Cardiovasc Ther 29: 362–376, 2011 [DOI] [PubMed] [Google Scholar]
- 34.Guo LJ, Alli AA, Eaton DC, Bao HF: ENaC is regulated by natriuretic peptide receptor-dependent cGMP signaling. Am J Physiol Renal Physiol 304: F930–F937, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gambaryan S, Wagner C, Smolenski A, Walter U, Poller W, Haase W, Kurtz A, Lohmann SM: Endogenous or overexpressed cGMP-dependent protein kinases inhibit cAMP-dependent renin release from rat isolated perfused kidney, microdissected glomeruli, and isolated juxtaglomerular cells. Proc Natl Acad Sci U S A 95: 9003–9008, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kudo T, Baird A: Inhibition of aldosterone production in the adrenal glomerulosa by atrial natriuretic factor. Nature 312: 756–757, 1984 [DOI] [PubMed] [Google Scholar]
- 37.Rocchini AP: Obesity hypertension, salt sensitivity and insulin resistance. Nutr Metab Cardiovasc Dis 10: 287–294, 2000 [PubMed] [Google Scholar]
- 38.Baum M: Insulin stimulates volume absorption in the rabbit proximal convoluted tubule. J Clin Invest 79: 1104–1109, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reaven GM: The kidney: An unwilling accomplice in syndrome X. Am J Kidney Dis 30: 928–931, 1997 [DOI] [PubMed] [Google Scholar]
- 40.Muniyappa R, Quon MJ: Insulin action and insulin resistance in vascular endothelium. Curr Opin Clin Nutr Metab Care 10: 523–530, 2007 [DOI] [PubMed] [Google Scholar]
- 41.Snyder PM, Olson DR, Kabra R, Zhou R, Steines JC: cAMP and serum and glucocorticoid-inducible kinase (SGK) regulate the epithelial Na(+) channel through convergent phosphorylation of Nedd4-2. J Biol Chem 279: 45753–45758, 2004 [DOI] [PubMed] [Google Scholar]
- 42.Bankir L, Bichet DG, Bouby N: Vasopressin V2 receptors, ENaC, and sodium reabsorption: A risk factor for hypertension? Am J Physiol Renal Physiol 299: F917–F928, 2010 [DOI] [PubMed] [Google Scholar]
- 43.Loesch A, Unwin R, Gandhi V, Burnstock G: Sympathetic nerve varicosities in close apposition to basolateral membranes of collecting duct epithelial cells of rat kidney. Nephron, Physiol 113: 15–21, 2009 [DOI] [PubMed] [Google Scholar]
- 44.Wallace DP, Reif G, Hedge AM, Thrasher JB, Pietrow P: Adrenergic regulation of salt and fluid secretion in human medullary collecting duct cells. Am J Physiol Renal Physiol 287: F639–F648, 2004 [DOI] [PubMed] [Google Scholar]
- 45.Breyer MD, Breyer RM: Prostaglandin E receptors and the kidney. Am J Physiol Renal Physiol 279: F12–F23, 2000 [DOI] [PubMed] [Google Scholar]
- 46.Boone M, Deen PM: Physiology and pathophysiology of the vasopressin-regulated renal water reabsorption. Pflugers Arch 456: 1005–1024, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flores D, Liu Y, Liu W, Satlin LM, Rohatgi R: Flow-induced prostaglandin E2 release regulates Na and K transport in the collecting duct. Am J Physiol Renal Physiol 303: F632–F638, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Capdevila J, Wang W: Role of cytochrome P450 epoxygenase in regulating renal membrane transport and hypertension. Curr Opin Nephrol Hypertens 22: 163–169, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pavlov TS, Ilatovskaya DV, Levchenko V, Mattson DL, Roman RJ, Staruschenko A: Effects of cytochrome P-450 metabolites of arachidonic acid on the epithelial sodium channel (ENaC). Am J Physiol Renal Physiol 301: F672–F681, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sakairi Y, Jacobson HR, Noland TD, Capdevila JH, Falck JR, Breyer MD: 5,6-EET inhibits ion transport in collecting duct by stimulating endogenous prostaglandin synthesis. Am J Physiol 268: F931–F939, 1995 [DOI] [PubMed] [Google Scholar]
- 51.Wei Y, Lin DH, Kemp R, Yaddanapudi GS, Nasjletti A, Falck JR, Wang WH: Arachidonic acid inhibits epithelial Na channel via cytochrome P450 (CYP) epoxygenase-dependent metabolic pathways. J Gen Physiol 124: 719–727, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wei Y, Sun P, Wang Z, Yang B, Carroll MA, Wang WH: Adenosine inhibits ENaC via cytochrome P-450 epoxygenase-dependent metabolites of arachidonic acid. Am J Physiol Renal Physiol 290: F1163–F1168, 2006 [DOI] [PubMed] [Google Scholar]
- 53.Kashlan OB, Kleyman TR: Epithelial Na(+) channel regulation by cytoplasmic and extracellular factors. Exp Cell Res 318: 1011–1019, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun P, Liu W, Lin DH, Yue P, Kemp R, Satlin LM, Wang WH: Epoxyeicosatrienoic acid activates BK channels in the cortical collecting duct. J Am Soc Nephrol 20: 513–523, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vallon V, Rieg T: Regulation of renal NaCl and water transport by the ATP/UTP/P2Y2 receptor system. Am J Physiol Renal Physiol 301: F463–F475, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cai Z, Xin J, Pollock DM, Pollock JS: Shear stress-mediated NO production in inner medullary collecting duct cells. Am J Physiol Renal Physiol 279: F270–F274, 2000 [DOI] [PubMed] [Google Scholar]
- 57.Lyon-Roberts B, Strait KA, van Peursem E, Kittikulsuth W, Pollock JS, Pollock DM, Kohan DE: Flow regulation of collecting duct endothelin-1 production. Am J Physiol Renal Physiol 300: F650–F656, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Eladari D, Chambrey R, Peti-Peterdi J: A new look at electrolyte transport in the distal tubule. Annu Rev Physiol 74: 325–349, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zaika O, Mamenko M, O’Neil RG, Pochynyuk O: Bradykinin acutely inhibits activity of the epithelial Na+ channel in mammalian aldosterone-sensitive distal nephron. Am J Physiol Renal Physiol 300: F1105–F1115, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hyndman KA, Pollock JS: Nitric oxide and the A and B of endothelin of sodium homeostasis. Curr Opin Nephrol Hypertens 22: 26–31, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Helms MN, Yu L, Malik B, Kleinhenz DJ, Hart CM, Eaton DC: Role of SGK1 in nitric oxide inhibition of ENaC in Na+-transporting epithelia. Am J Physiol Cell Physiol 289: C717–C726, 2005 [DOI] [PubMed] [Google Scholar]
- 62.Ortiz PA, Garvin JL: Role of nitric oxide in the regulation of nephron transport. Am J Physiol Renal Physiol 282: F777–F784, 2002 [DOI] [PubMed] [Google Scholar]
- 63.Lu M, MacGregor GG, Wang W, Giebisch G: Extracellular ATP inhibits the small-conductance K channel on the apical membrane of the cortical collecting duct from mouse kidney. J Gen Physiol 116: 299–310, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hyndman KA, Boesen EI, Elmarakby AA, Brands MW, Huang P, Kohan DE, Pollock DM, Pollock JS: Renal collecting duct NOS1 maintains fluid-electrolyte homeostasis and blood pressure. Hypertension 62: 91–98, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guan Y, Hao C, Cha DR, Rao R, Lu W, Kohan DE, Magnuson MA, Redha R, Zhang Y, Breyer MD: Thiazolidinediones expand body fluid volume through PPARgamma stimulation of ENaC-mediated renal salt absorption. Nat Med 11: 861–866, 2005 [DOI] [PubMed] [Google Scholar]
- 66.Zhang H, Zhang A, Kohan DE, Nelson RD, Gonzalez FJ, Yang T: Collecting duct-specific deletion of peroxisome proliferator-activated receptor gamma blocks thiazolidinedione-induced fluid retention. Proc Natl Acad Sci U S A 102: 9406–9411, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pavlov TS, Imig JD, Staruschenko A: Regulation of ENaC-mediated sodium reabsorption by peroxisome proliferator-activated receptors. PPAR Res 2010: 703735, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rieg T, Vallon V: ATP and adenosine in the local regulation of water transport and homeostasis by the kidney. Am J Physiol Regul Integr Comp Physiol 296: R419–R427, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pochynyuk O, Bugaj V, Rieg T, Insel PA, Mironova E, Vallon V, Stockand JD: Paracrine regulation of the epithelial Na+ channel in the mammalian collecting duct by purinergic P2Y2 receptor tone. J Biol Chem 283: 36599–36607, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wildman SS, Marks J, Turner CM, Yew-Booth L, Peppiatt-Wildman CM, King BF, Shirley DG, Wang W, Unwin RJ: Sodium-dependent regulation of renal amiloride-sensitive currents by apical P2 receptors. J Am Soc Nephrol 19: 731–742, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kohan DE, Rossi NF, Inscho EW, Pollock DM: Regulation of blood pressure and salt homeostasis by endothelin. Physiol Rev 91: 1–77, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bugaj V, Mironova E, Kohan DE, Stockand JD: Collecting duct-specific endothelin B receptor knockout increases ENaC activity. Am J Physiol Cell Physiol 302: C188–C194, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ahn D, Ge Y, Stricklett PK, Gill P, Taylor D, Hughes AK, Yanagisawa M, Miller L, Nelson RD, Kohan DE: Collecting duct-specific knockout of endothelin-1 causes hypertension and sodium retention. J Clin Invest 114: 504–511, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ge Y, Bagnall A, Stricklett PK, Webb D, Kotelevtsev Y, Kohan DE: Combined knockout of collecting duct endothelin A and B receptors causes hypertension and sodium retention. Am J Physiol Renal Physiol 295: F1635–F1640, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Barton M, Kohan DE: Endothelin antagonists in clinical trials: Lessons learned. Contrib Nephrol 172: 255–260, 2011 [DOI] [PubMed] [Google Scholar]
- 76.Navar LG, Kobori H, Prieto MC, Gonzalez-Villalobos RA: Intratubular renin-angiotensin system in hypertension. Hypertension 57: 355–362, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vinciguerra M, Mordasini D, Vandewalle A, Feraille E: Hormonal and nonhormonal mechanisms of regulation of the NA,K-pump in collecting duct principal cells. Semin Nephrol 25: 312–321, 2005 [DOI] [PubMed] [Google Scholar]
- 78.McCormick JA, Ellison DH: The WNKs: Atypical protein kinases with pleiotropic actions. Physiol Rev 91: 177–219, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ho K, Nichols CG, Lederer WJ, Lytton J, Vassilev PM, Kanazirska MV, Hebert SC: Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature 362: 31–38, 1993 [DOI] [PubMed] [Google Scholar]
- 80.Kahle KT, Wilson FH, Leng Q, Lalioti MD, O’Connell AD, Dong K, Rapson AK, MacGregor GG, Giebisch G, Hebert SC, Lifton RP: WNK4 regulates the balance between renal NaCl reabsorption and K+ secretion. Nat Genet 35: 372–376, 2003 [DOI] [PubMed] [Google Scholar]
- 81.Hoorn EJ, Nelson JH, McCormick JA, Ellison DH: The WNK kinase network regulating sodium, potassium, and blood pressure. J Am Soc Nephrol 22: 605–614, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yoo D, Flagg TP, Olsen O, Raghuram V, Foskett JK, Welling PA: Assembly and trafficking of a multiprotein ROMK (Kir 1.1) channel complex by PDZ interactions. J Biol Chem 279: 6863–6873, 2004 [DOI] [PubMed] [Google Scholar]
- 83.Wade JB, Fang L, Coleman RA, Liu J, Grimm PR, Wang T, Welling PA: Differential regulation of ROMK (Kir1.1) in distal nephron segments by dietary potassium. Am J Physiol Renal Physiol 300: F1385–F1393, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shibata S, Zhang J, Puthumana J, Stone KL, Lifton RP: Kelch-like 3 and Cullin 3 regulate electrolyte homeostasis via ubiquitination and degradation of WNK4. Proc Natl Acad Sci U S A 110: 7838–7843, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ji W, Foo JN, O’Roak BJ, Zhao H, Larson MG, Simon DB, Newton-Cheh C, State MW, Levy D, Lifton RP: Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet 40: 592–599, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fang L, Li D, Welling PA: Hypertension resistance polymorphisms in ROMK (Kir1.1) alter channel function by different mechanisms. Am J Physiol Renal Physiol 299: F1359–F1364, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shibata S, Rinehart J, Zhang J, Moeckel G, Castañeda-Bueno M, Stiegler AL, Boggon TJ, Gamba G, Lifton RP: Mineralocorticoid receptor phosphorylation regulates ligand binding and renal response to volume depletion and hyperkalemia. Cell Metab 18: 660–671, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ishibashi K, Sasaki S, Fushimi K, Yamamoto T, Kuwahara M, Marumo F: Immunolocalization and effect of dehydration on AQP3, a basolateral water channel of kidney collecting ducts. Am J Physiol 272: F235–F241, 1997 [DOI] [PubMed] [Google Scholar]
- 89.Kim SW, Gresz V, Rojek A, Wang W, Verkman AS, Frøkiaer J, Nielsen S: Decreased expression of AQP2 and AQP4 water channels and Na,K-ATPase in kidney collecting duct in AQP3 null mice. Biol Cell 97: 765–778, 2005 [DOI] [PubMed] [Google Scholar]
- 90.Deen PMT, Weghuis DO, Sinke RJ, Geurts van Kessel A, Wieringa B, van Os CH: Assignment of the human gene for the water channel of renal collecting duct Aquaporin 2 (AQP2) to chromosome 12 region q12—>q13. Cytogenet Cell Genet 66: 260–262, 1994 [DOI] [PubMed] [Google Scholar]
- 91.Rosenthal W, Seibold A, Antaramian A, Lonergan M, Arthus M-F, Hendy GN, Birnbaumer M, Bichet DG: Molecular identification of the gene responsible for congenital nephrogenic diabetes insipidus. Nature 359: 233–235, 1992 [DOI] [PubMed] [Google Scholar]
- 92.Robben JH, Knoers NV, Deen PM: Cell biological aspects of the vasopressin type-2 receptor and aquaporin 2 water channel in nephrogenic diabetes insipidus. Am J Physiol Renal Physiol 291: F257–F270, 2006 [DOI] [PubMed] [Google Scholar]
- 93.Loonen AJ, Knoers NV, van Os CH, Deen PM: Aquaporin 2 mutations in nephrogenic diabetes insipidus. Semin Nephrol 28: 252–265, 2008 [DOI] [PubMed] [Google Scholar]
- 94.Ma T, Song Y, Yang B, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS: Nephrogenic diabetes insipidus in mice lacking aquaporin-3 water channels. Proc Natl Acad Sci U S A 97: 4386–4391, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chou C-L, Ma T, Yang B, Knepper MA, Verkman AS: Fourfold reduction of water permeability in inner medullary collecting duct of aquaporin-4 knockout mice. Am J Physiol 274: C549–C554, 1998 [DOI] [PubMed] [Google Scholar]
- 96.Fenton RA, Pedersen CN, Moeller HB: New insights into regulated aquaporin-2 function. Curr Opin Nephrol Hypertens 22: 551–558, 2013 [DOI] [PubMed] [Google Scholar]
- 97.Nedvetsky PI, Tamma G, Beulshausen S, Valenti G, Rosenthal W, Klussmann E: Regulation of aquaporin-2 trafficking. Handb Exp Pharmacol 190: 133–157, 2009 [DOI] [PubMed] [Google Scholar]
- 98.Bouley R, Hasler U, Lu HA, Nunes P, Brown D: Bypassing vasopressin receptor signaling pathways in nephrogenic diabetes insipidus. Semin Nephrol 28: 266–278, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Christensen BM, Zelenina M, Aperia A, Nielsen S: Localization and regulation of PKA-phosphorylated AQP2 in response to V(2)-receptor agonist/antagonist treatment. Am J Physiol Renal Physiol 278: F29–F42, 2000 [DOI] [PubMed] [Google Scholar]
- 100.van Balkom BWM, Savelkoul PJ, Markovich D, Hofman E, Nielsen S, van der Sluijs P, Deen PM: The role of putative phosphorylation sites in the targeting and shuttling of the aquaporin-2 water channel. J Biol Chem 277: 41473–41479, 2002 [DOI] [PubMed] [Google Scholar]
- 101.Fushimi K, Sasaki S, Marumo F: Phosphorylation of serine 256 is required for cAMP-dependent regulatory exocytosis of the aquaporin-2 water channel. J Biol Chem 272: 14800–14804, 1997 [DOI] [PubMed] [Google Scholar]
- 102.Hoffert JD, Nielsen J, Yu MJ, Pisitkun T, Schleicher SM, Nielsen S, Knepper MA: Dynamics of aquaporin-2 serine-261 phosphorylation in response to short-term vasopressin treatment in collecting duct. Am J Physiol Renal Physiol 292: F691–F700, 2007 [DOI] [PubMed] [Google Scholar]
- 103.Tamma G, Robben JH, Trimpert C, Boone M, Deen PM: Regulation of AQP2 localization by S256 and S261 phosphorylation and ubiquitination. Am J Physiol Cell Physiol 300: C636–C646, 2011 [DOI] [PubMed] [Google Scholar]
- 104.Kamsteeg EJ, Hendriks G, Boone M, Konings IB, Oorschot V, van der Sluijs P, Klumperman J, Deen PM: Short-chain ubiquitination mediates the regulated endocytosis of the aquaporin-2 water channel. Proc Natl Acad Sci U S A 103: 18344–18349, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Katsura T, Ausiello DA, Brown D: Direct demonstration of aquaporin-2 water channel recycling in stably transfected LLC-PK1 epithelial cells. Am J Physiol 270: F548–F553, 1996 [DOI] [PubMed] [Google Scholar]
- 106.Lee YJ, Lee JE, Choi HJ, Lim JS, Jung HJ, Baek MC, Frøkiær J, Nielsen S, Kwon TH: E3 ubiquitin-protein ligases in rat kidney collecting duct: Response to vasopressin stimulation and withdrawal. Am J Physiol Renal Physiol 301: F883–F896, 2011 [DOI] [PubMed] [Google Scholar]
- 107.Radin MJ, Yu MJ, Stoedkilde L, Miller RL, Hoffert JD, Frokiaer J, Pisitkun T, Knepper MA: Aquaporin-2 regulation in health and disease. Vet Clin Pathol 41: 455–470, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Valtin H, Edwards BR: GFR and the concentration of urine in the absence of vasopressin. Berliner-Davidson re-explored. Kidney Int 31: 634–640, 1987 [DOI] [PubMed] [Google Scholar]
- 109.Li C, Wang W, Summer SN, Cadnapaphornchai MA, Falk S, Umenishi F, Schrier RW: Hyperosmolality in vivo upregulates aquaporin 2 water channel and Na-K-2Cl co-transporter in Brattleboro rats. J Am Soc Nephrol 17: 1657–1664, 2006 [DOI] [PubMed] [Google Scholar]
- 110.Sheen MR, Kim JA, Lim SW, Jung JY, Han KH, Jeon US, Park SH, Kim J, Kwon HM: Interstitial tonicity controls TonEBP expression in the renal medulla. Kidney Int 75: 518–525, 2009 [DOI] [PubMed] [Google Scholar]
- 111.Hasler U, Jeon US, Kim JA, Mordasini D, Kwon HM, Féraille E, Martin PY: Tonicity-responsive enhancer binding protein is an essential regulator of aquaporin-2 expression in renal collecting duct principal cells. J Am Soc Nephrol 17: 1521–1531, 2006 [DOI] [PubMed] [Google Scholar]
- 112.Lam AK, Ko BC, Tam S, Morris R, Yang JY, Chung SK, Chung SS: Osmotic response element-binding protein (OREBP) is an essential regulator of the urine concentrating mechanism. J Biol Chem 279: 48048–48054, 2004 [DOI] [PubMed] [Google Scholar]
- 113.López-Capapé M, Golmayo L, Lorenzo G, Gallego N, Barrio R: Hypothalamic adipic hypernatraemia syndrome with normal osmoregulation of vasopressin. Eur J Pediatr 163: 580–583, 2004 [DOI] [PubMed] [Google Scholar]
- 114.Li C, Wang W, Rivard CJ, Lanaspa MA, Summer S, Schrier RW: Molecular mechanisms of angiotensin II stimulation on aquaporin-2 expression and trafficking. Am J Physiol Renal Physiol 300: F1255–F1261, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hasler U, Leroy V, Martin PY, Féraille E: Aquaporin-2 abundance in the renal collecting duct: New insights from cultured cell models. Am J Physiol Renal Physiol 297: F10–F18, 2009 [DOI] [PubMed] [Google Scholar]
- 116.Stegbauer J, Gurley SB, Sparks MA, Woznowski M, Kohan DE, Yan M, Lehrich RW, Coffman TM: AT1 receptors in the collecting duct directly modulate the concentration of urine. J Am Soc Nephrol 22: 2237–2246, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Silver MA: The natriuretic peptide system: Kidney and cardiovascular effects. Curr Opin Nephrol Hypertens 15: 14–21, 2006 [DOI] [PubMed] [Google Scholar]
- 118.Boone M, Kortenoeven M, Robben JH, Deen PM: Effect of the cGMP pathway on AQP2 expression and translocation: Potential implications for nephrogenic diabetes insipidus. Nephrol Dial Transplant 25: 48–54, 2010 [DOI] [PubMed] [Google Scholar]
- 119.Klokkers J, Langehanenberg P, Kemper B, Kosmeier S, von Bally G, Riethmüller C, Wunder F, Sindic A, Pavenstädt H, Schlatter E, Edemir B: Atrial natriuretic peptide and nitric oxide signaling antagonizes vasopressin-mediated water permeability in inner medullary collecting duct cells. Am J Physiol Renal Physiol 297: F693–F703, 2009 [DOI] [PubMed] [Google Scholar]
- 120.Tamma G, Carmosino M, Svelto M, Valenti G: Bradykinin signaling counteracts cAMP-elicited aquaporin 2 translocation in renal cells. J Am Soc Nephrol 16: 2881–2889, 2005 [DOI] [PubMed] [Google Scholar]
- 121.Ge Y, Ahn D, Stricklett PK, Hughes AK, Yanagisawa M, Verbalis JG, Kohan DE: Collecting duct-specific knockout of endothelin-1 alters vasopressin regulation of urine osmolality. Am J Physiol Renal Physiol 288: F912–F920, 2005 [DOI] [PubMed] [Google Scholar]
- 122.Kashlan OB, Adelman JL, Okumura S, Blobner BM, Zuzek Z, Hughey RP, Kleyman TR, Grabe M: Constraint-based, homology model of the extracellular domain of the epithelial Na+ channel α subunit reveals a mechanism of channel activation by proteases. J Biol Chem 286: 649–660, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]