

Abstract
AKI is a common clinical condition associated with a number of adverse outcomes. More timely diagnosis would allow for earlier intervention and could improve patient outcomes. The goal of early identification of AKI has been the primary impetus for AKI biomarker research, and has led to the discovery of numerous novel biomarkers. However, in addition to facilitating more timely intervention, AKI biomarkers can provide valuable insight into the molecular mechanisms of this complex and heterogeneous disease. Furthermore, AKI biomarkers could also function as molecular phenotyping tools that could be used to direct clinical intervention. This review highlights the major studies that have characterized the diagnostic and prognostic predictive power of these biomarkers. The mechanistic relevance of neutrophil gelatinase–associated lipocalin, kidney injury molecule 1, IL-18, liver-type fatty acid–binding protein, angiotensinogen, tissue inhibitor of metalloproteinase-2, and IGF-binding protein 7 to the pathogenesis and pathobiology of AKI is discussed, putting these biomarkers in the context of the progressive phases of AKI. A biomarker-integrated model of AKI is proposed, which summarizes the current state of knowledge regarding the roles of these biomarkers and the molecular and cellular biology of AKI.
Keywords: acute renal failure, pathophysiology of renal disease and progression, renin-angiotensin system
The Search for a Kidney Troponin
Despite numerous clinical trials, AKI remains a cause of significant morbidity and mortality for which there is no effective intervention (1). Barriers to translating the successes of animal studies to efficacy in human clinical trials include the heterogeneity of the patient population with AKI with respect to the underlying etiology of renal injury and patient comorbidities, the complexity of the pathogenesis and pathobiology of AKI, and the late timing of the enrollment and initiation of experimental interventions (1,2). Delayed intervention has been attributed to the deficiency of serum creatinine as an AKI biomarker and is the primary impetus for the discovery of novel AKI biomarkers. The goal of most AKI biomarker research has been the discovery of a “kidney troponin”, a sensitive and specific early marker of renal injury.
After more than a decade of intensive research effort, several novel AKI biomarkers have been identified, including neutrophil gelatinase–associated lipocalin (NGAL), kidney injury molecule 1 (KIM-1), cystatin C, IL-18, and liver-type fatty acid–binding protein (L-FABP). Unfortunately, the clinical use of novel AKI biomarkers is unclear, because large, prospective, multicenter trials, such as the Translational Research Investigating Biomarker Endpoints in AKI (TRIBE-AKI) (n=1530) study failed to show troponin-like diagnostic performance (3–6). The recent SAPPHIRE study (n=728) could represent an important moment in AKI biomarker research (7). Using a two-stage study, Kashani et al. (7) discovered and validated two novel AKI biomarkers, tissue inhibitor of metalloproteinase-2 (TIMP-2) and IGF-binding protein 7 (IGFBP7), which predicted the onset of severe AKI (defined as Kidney Disease Improving Global Outcomes [KDIGO] stage 2 or 3) within 12 hours with significantly greater accuracy than NGAL, KIM-1, IL-18, L-FABP, or cystatin C, having areas under the receiver-operator characteristic curve (AUCs) of 0.79 and 0.76, respectively. The product of TIMP-2×IGFBP7 had an AUC of 0.8 (7). TIMP-2 and IGFBP7 are promising early-diagnostic biomarkers, although they will need substantial additional investigation to define their clinical role.
Although the accuracy of AKI biomarkers is often compared with that of troponins, there are fundamental differences in the pathobiology of AKI and an acute myocardial infarction, and, consequently, biomarkers of AKI are qualitatively different from troponins. In contrast to the straightforward injury mechanism of myocardial infarction, AKI is not a single disease entity, but a clinical syndrome that can be the result of myriad renal insults, and it is often multifactorial. Thus, it seems unlikely that a single AKI biomarker would achieve troponin-like diagnostic accuracy. However, novel kidney injury biomarkers, such as NGAL, KIM-1, IL-18, L-FABP, angiotensinogen, TIMP-2, and IGFBP7, reflect physiologic and pathophysiologic processes in the injured tissue, whereas serum troponin levels represent the release of preformed protein from injured cardiac myocytes. Mechanistically relevant biomarkers have the potential to improve our understanding of complex heterogeneous diseases, improving our ability to predict prognosis and response to therapy. For example, the field of oncology has been revolutionized by the use of biomarkers to predict response to therapy, and many antineoplastic drugs target biomarkers themselves. Similarly, AKI biomarkers have made significant contributions to our understanding of the molecular underpinnings of AKI, and, like cancer biomarkers, they could also have use as molecular phenotyping tools that facilitate the identification of patients who could benefit from a specific intervention, even a biomarker-targeted intervention.
The purpose of this review is to highlight the mechanistic relevance of novel AKI biomarkers as well as the potential for biomarker-directed therapy for AKI. We also briefly discuss the prognostic significance of novel AKI biomarkers. We have framed our discussion using the model of the clinical phases of AKI first proposed by Sutton et al. (8) and later adapted to postcardiac surgery AKI by Rosner and Okusa (9). Using time-course data from the TRIBE-AKI study, we propose a model for integrating novel AKI biomarkers into the current understanding of the progression of AKI through these clinical phases (4–6).
NGAL
NGAL is a widely expressed 25-kD protein of the lipocalin family. After ischemic or nephrotoxic kidney injury, intrarenal NGAL is dramatically upregulated at the transcript and protein levels (10–12). Elevated NGAL protein is detectable in the urine as early as 3 hours after injury, and in vivo data have suggested the thick ascending limb and the collecting duct as the sites of intrarenal NGAL production, although proximal tubule cells secrete NGAL in vitro in response to ATP depletion (11,13,14). Although the concentration of urine NGAL peaks approximately 6 hours after injury, there is some evidence of sustained elevation in its concentration as long as 5 days postinjury (5,6,15). Plasma NGAL also increases after AKI as a result of increased hepatic production, and NGAL is filtered by the glomerulus and taken up by the proximal tubule in a megalin-dependent manner (13,14,16). Thus, both urine and plasma NGAL can potentially exert an effect on the intrarenal molecular and cellular events that occur during AKI, and both have been used to predict the onset and course of AKI.
All known functions of NGAL depend on its ability to bind to iron-siderophore complexes. The canonical function of NGAL is as a bacteriostatic agent in the innate immune response, where it sequesters iron-siderophore complexes and prevents their uptake by bacterial pathogens (17,18). However, NGAL has also been implicated in iron-trafficking in the developing renal epithelium (19). During nephrogenesis, it is secreted by the ureteric bud and induces tubulogenesis in the metanephric mass, and importantly, it must be loaded with an iron-siderophore complex to do so (20). Elegant studies performed in rodent models of AKI have shown that parenteral administration of iron–siderophore-loaded NGAL (holo-NGAL) protects the kidney from ischemia-reperfusion injury and can attenuate the severity of the injury if administered 1 hour after reperfusion (13,21). Although the underlying mechanism is not fully understood, treatment with holo-NGAL decreased the number of apoptotic tubular epithelial cells and increased the number of proliferating epithelial cells (13,21). Holo-NGAL also upregulated the renoprotective enzyme heme oxygenase–1 (13).
Important questions regarding the function of NGAL during AKI and its potential use in the treatment of renal injury remain unanswered. First, studies describing the effects of holo-NGAL have all used a bacterial iron-loaded siderophore, but their results suggest the existence of an endogenous siderophore, which NGAL requires for its function. Indeed, work by Bao et al. (22) showed that NGAL sequesters iron through its interaction with a class of metabolites known as catechols, which are abundant in the urine. The discovery of this relationship opens the door for investigation of therapies that modulate the interaction of endogenous NGAL, catechols, and iron. Second, animal studies investigating the prophylactic and therapeutic effects of holo-NGAL were only designed to evaluate the severity of renal injury at a 24-hour time point and did not investigate the effect of NGAL treatment on recovery from AKI (13,21). Whereas the renoprotective and antiapoptotic effects of NGAL neatly explain the observed attenuation of renal injury, augmented epithelial cell proliferation observed after treatment with holo-NGAL suggests that NGAL enhances the ability of the damaged kidney to recover from injury. Such a role is congruent with studies that have reported that elevation in urine NGAL is sustained for up to 5 days after renal injury (5,6,15).
KIM-1
KIM-1 is a 38.7-kD transmembrane protein that contains extracellular mucin and Ig domains (23). Basal expression of KIM-1 is low in the normal kidney. However, it is upregulated after ischemia-reperfusion injury, and KIM-1 protein can be localized to proliferating dedifferentiated epithelial cells of the proximal tubule 48 hours after injury (23). Importantly, the extracellular component of KIM-1 is constitutively shed from the membrane in a matrix metalloproteinase (MMP)–dependent manner (24). Although the functional importance of KIM-1 shedding is unclear, it is ostensibly the mechanism underlying the increase in urinary KIM-1 observed after renal ischemic or toxic injury (25,26).
Contrary to what its name would suggest, KIM-1 is more than a marker of renal injury, and it seems to have a functional role in the molecular and cellular biology of AKI. KIM-1 expression has been shown to confer a phagocytic phenotype on cultured primary kidney cells by its function as a phosphatidylserine receptor, which allows it to promote the phagocytosis of apoptotic bodies and necrotic debris (27). Therefore, it has been proposed that KIM-1 could play a role in renal recovery and tubular regeneration after AKI (27). Such a role seems congruent with the late timing of peak changes (2–3 days after injury) in urine KIM-1 concentration during AKI (4,26). Pharmacologic interventions that enhance the effect of KIM-1 could potentially benefit patients by facilitating effective clearance of debris from the injured tubules. The inhibition of KIM-1 shedding is a logical target, because it would increase the amount of membrane-bound KIM-1 on surviving epithelial cells. Accelerated shedding of KIM-1 from renal tubular epithelial cells is mediated by MMP-3, which could be inhibited to increase the amount of membrane-bound KIM-1 and potentially augment the clearance of debris from the tubule (28). Additionally, because accelerated KIM-1 shedding is thought to be driven by p38 mitogen-activated protein kinase signaling in response to the production of growth factors involved in cell proliferation and recovery, the urine KIM-1 concentration could potentially be used to differentiate between the extension phase and the maintenance and recovery phases of AKI (29). Thus, KIM-1 could be used to direct interventions specific to these phases. For example, a low KIM-1 concentration (or one that is rising but has not yet peaked) would suggest that a patient could still benefit from therapies designed to attenuate injury, whereas a high concentration might indicate that a patient would benefit from therapies designed to enhance renal recovery, such as those that target mitochondrial dysfunction and enhance mitochondrial biogenesis, a process thought to be critical to the repair of the damaged renal epithelium (30).
Unfortunately, studies evaluating the prognostic use of KIM-1 have reported only modest results (31,32). However, because increased urinary KIM-1 can indicate either injury or the repair response to injury, concentration of KIM-1 by itself may not be able to discriminate with high accuracy between AKI, which will proceed to severe AKI, and injury, which will recover. Therefore, combinations of KIM-1 with other injury markers may be highly useful. In support of this hypothesis, a study of 32 urine biomarkers in AKI after cardiac surgery found that urinary KIM-1 concentrations had relatively poor correlation with other injury biomarkers and that the combination of IL-18 and KIM-1 had the best predictive ability to predict severe AKI (AUC=0.93) (32).
IL-18
IL-18 is a 22-kD proinflammatory cytokine, the amount of which increases in the kidney after ischemia-reperfusion injury, glycerol injection, and cisplatin-induced renal injury in a caspase-1–dependent manner (33–35). IL-18 is translated as a procytokine that must be subsequently cleaved by caspase-1 as part of the function of a multiprotein complex called the inflammasome, a maturation pathway that it shares with IL-1β (36). A central component of the inflammasome is the pyrin domain containing members of the NOD-like receptor (NLR) family of proteins, which induces an inflammatory response on binding a variety of ligands (36). The NLR ligands can be broadly grouped into two classes: pathogen-associated molecular patterns, of which LPS is the most well known, and damage-associated molecular patterns released from damaged and dying cells (36,37). The binding of these molecules to an NLR results in the recruitment of scaffold proteins and proteolytic enzymes and ultimately, caspase-1 activation, allowing caspase-1 to proteolytically activate IL-1β and IL-18 before their secretion (36). Mature IL-18 promotes inflammation by signaling through the IL-18 receptor–α/accessory protein–like heterodimer (38). It promotes acute injury through the NF-κB pathway, inducing the upregulation of other inflammatory mediators, including TNF-α and iNOS, and the chemokines MCP-1 and MIP-2, which drive intrarenal infiltration by macrophages and neutrophils (35).
IL-18 is an attractive target for biomarker-directed therapy of AKI, because this cytokine seems to play a prominent role in the inflammatory processes that exacerbate renal injury during the extension phase of AKI. Targeted therapies that disrupt the IL-18–signaling axis itself could prove efficacious, because work done in animal models of AKI has consistently shown that doing so attenuates renal injury (33–35,37,39,40). An engineered IL-18–binding protein would be a natural approach, because this protein antagonizes IL-18 (41). Indeed, pretreatment with exogenous IL-18–binding protein has been shown to be renoprotective in an ischemia-reperfusion injury model of AKI (35). Experiments to test the hypothesis that postinjury administration of an IL-18 antagonist attenuates renal injury have not been reported in the literature. Because urine IL-18 is elevated within the first 6 hours after renal injury but does not peak until after 12–18 hours, anti–IL-18 treatment would likely need to be initiated within the first 6 hours after renal injury. Additionally, urine IL-18 concentration after a renal insult could be predictive of response to anti–IL-18 therapy, a hypothesis that would need to be tested if an IL-18 antagonist were to prove promising in preclinical and clinical studies. For example, urine IL-18 concentration >60 pg/ml at 0–6 hours after cardiac surgery has been shown to have a sensitivity of 54% and specificity of 82% in adults for prediction of severe AKI (5). It is plausible that patients with urine IL-18 concentrations above this cutoff would benefit from anti–IL-18 therapy, whereas those with lower IL-18 concentrations would not.
L-FABP
The L-FABP gene is expressed in the renal cortex, and the 14-kD L-FABP protein can be localized predominantly in the proximal tubule (42,43). Elevated urinary L-FABP is detectable immediately postoperatively in patients undergoing cardiac surgery who go on to develop AKI, and it peaks within 6 hours (4,44). L-FABP is a renoprotective protein. In addition to its canonical function, which is to bind to and promote the metabolism of long-chain and very long–chain fatty acids, L-FABP also has antioxidant properties (45–47). The human L-FABP gene contains a hypoxia-inducible factor 1α response element, and consequently, L-FABP gene expression is induced by hypoxia (48,49). In renal transplant recipients, immediate post-transplant urinary L-FABP excretion is strongly correlated with ischemic time (49). The mechanistic import of L-FABP in AKI has been investigated using a transgenic mouse model in which the genomic DNA of the human L-FABP gene, including its promoter region, was integrated into the mouse genome, an elegant model considering that the mouse kidney does not express L-FABP, whereas it is expressed in the human renal cortex (42,50). Using this model, Yamamoto et al. (49) showed that human L-FABP expression reduces the severity of renal ischemia-reperfusion injury. It has been proposed that L-FABP binds lipid peroxides and promotes their redistribution from the cytosol to the tubular lumen, thereby protecting tubular epithelial cells from the damage caused by these reactive oxygen species (49).
L-FABP could be used to guide clinical intervention in patients at risk for AKI and has already been approved as a diagnostic test for use in Japan. First, urinary L-FABP can be thought of as an index marker of renal hypoxia and susceptibility to additional insult. For example, elevated preprocedure urinary L-FABP concentration has been shown to predict contrast-induced nephropathy (51). Similarly, a study in the intensive care unit (ICU) population found that urinary L-FABP at the time of ICU admission predicted development of AKI within 1 week (AUC=0.7), suggesting that patients with elevated baseline L-FABP values are at greater risk of AKI (52). Therefore, L-FABP measurement could be used to identify high-risk patients and minimize their exposure to renal insults. Second, because L-FABP is renoprotective, strategies that increase its expression or potentiate its effects could benefit patients who will undergo a predictable renal insult. For example, L-FABP gene expression is upregulated by peroxisome proliferator–activated receptor-α (PPAR-α). PPAR-α agonists have been used in animal models to attenuate cisplatin-induced AKI and renal ischemia-reperfusion injury (53–55). This finding could represent a potentially fruitful avenue of investigation for novel interventions for AKI, although recent observational studies have reported an increased risk of AKI associated with the use of fibrates, which are potent PPAR-α agonists (56,57).
Angiotensinogen
Urinary angiotensinogen has been recently described as a novel prognostic biomarker of AKI, although it has not yet been investigated as an early diagnostic biomarker. In patients with AKI after cardiac surgery and patients with AKI secondary to nonsurgical causes, elevated urinary angiotensinogen is associated with progression to higher stages of AKI as well as hard outcomes, such as increased length of hospital stay, need for RRT, and death (58,59). In patients with AKI after cardiac surgery, elevated urinary angiotensinogen had an AUC of 0.75 for prediction of progression from Acute Kidney Injury Network (AKIN) stage 1 to the composite end-point AKIN stage 3 or death (60). In the critically ill AKI population, angiotensinogen was a strong predictor of worsening of AKI (AUC=0.77) and also predicted the composite end-point need for RRT or death (AUC=0.73) (59). It is also noteworthy that one study found that elevated angiotensinogen is associated with elevated urinary renin and that patients with concomitantly elevated concentrations of these urine biomarkers were at very high risk (80.4% positive predictive value) of progressing from mild to severe AKI (60).
The findings regarding the prognostic significance of angiotensinogen have strong implications for the molecular and cellular mechanisms underlying renal injury in AKI and could lead to novel interventional approaches. Activation of the renin-angiotensin system (RAS) has long been recognized as an important contributor to chronic renal injury. Urinary angiotensinogen has been proposed as a marker of intrarenal RAS activity, and it is predictive of progression of CKD (61–63). The prognostic significance of urinary angiotensinogen as an AKI biomarker is strongly suggestive of a role for RAS activation in modulating the severity of AKI. Animal models of AKI have shown that intrarenal angiotensin II increases after renal ischemia-reperfusion injury, whereas the concentration of angiotensin 1–7, the molecular counterbalance to angiotensin II, decreases in kidney tissue (64,65). An unexplored area is the potential therapeutic benefit of shunting the RAS pathway from activation of angiotensin II receptor type 1 by angiotensin II to activation of the Mas receptor by angiotensin 1–7. Pretreatment with angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, and aliskiren has been shown to mitigate renal ischemia-reperfusion injury in experimental models, primarily by reduction in postreperfusion inflammation (66–68). Additionally, one study reported a reduction in the severity of AKI at 48 hours postreperfusion when captopril was continuously administered through an intraperitoneal osmotic micropump implanted immediately after renal ischemia-reperfusion injury.
The aforementioned data suggest that pharmacologic inhibition of the RAS could potentially be used in the treatment of AKI, although the optimal timing of intervention remains unclear. Such a proposition contradicts the conventional wisdom that RAS inhibitors should be discontinued because of the potential of these drugs to exacerbate AKI, since they cause vasodilation of the efferent arteriole and decrease GFR. However, a reduction in GFR is not equivalent to more severe renal injury. On the contrary, a recent study by Coca et al. (69) highlighted the need to differentiate structural AKI, which involves renal parenchymal damage, from functional AKI, which only represents a decrease in GFR. This study showed that, although angiotensin-converting enzyme inhibitor and angiotensin receptor blocker use increases the incidence of AKI after cardiac surgery, the drugs are not associated with an increase in renal injury markers (69). Therefore, future studies that investigate the therapeutic role of RAS inhibition in patients with AKI should not have GFR-related outcomes only but also, harder outcomes, such as development of CKD at 90 days, need for RRT, and death.
Inhibition of the RAS could also have long-term benefits for patients with AKI. AKI that is superimposed on CKD accelerates CKD progression, and even in the absence of CKD, AKI is associated with an increased risk of ESRD (70,71). Given the clearly defined role for the RAS in CKD progression, it is plausible that RAS activation not only modulates the severity of an episode of AKI but also mediates progression to chronic disease. In support of this hypothesis, a study performed using a rat model of AKI and chronic kidney injury shows that spironolactone treatment reduced progression to CKD (72). However, other RAS inhibitors have not been investigated. Importantly, given the predictive power of urinary angiotensinogen for CKD progression, measurement of urinary angiotensinogen could play a role in identifying patients with AKI who are likely to develop CKD and could potentially benefit from RAS blockade.
TIMP-2
TIMP-2 was identified as a novel AKI biomarker by Kashani et al. (7) in a two-stage, multicenter study (n=522 in stage 1; n=728 in stage 2), in which they tested the ability of 340 proteins, including known AKI biomarkers, to predict development of AKI in the ICU population (including patients after cardiac surgery). TIMP-2 was a strong predictor of development of KDIGO stage 2 or 3 AKI within 12 hours (AUC=0.79) (7). The investigators proposed that the diagnostic performance of TIMP-2 is derived from its MMP-independent role in inducing G1 cell cycle arrest after an ischemic insult, which prevents ensuing cell death (7). The supporting data come from an in vitro study of human microvascular endothelial cells, which showed that TIMP-2 binds to α3β1-integrin to induce a Shp-1–mediated increase in the synthesis of the cyclin-dependent kinase inhibitor p27Kip1, resulting in G1 cell cycle arrest (73).
However, the role of TIMP-2 in AKI could be more complex. For instance, studies in the same cell line have shown that TIMP-2 binding to α3β1-integrin also leads to Shp-1–mediated inactivation of tyrosine kinase–type growth factor receptors (74,75). Furthermore, in contradistinction to its antiproliferative effects, TIMP-2 can, in some instances, promote cellular proliferation (76). The role of TIMP-2 in nephrogenesis is particularly pertinent to its putative role in AKI. TIMP-2 is secreted by the ureteric bud in response to paracrine metanephric signals (77). It exerts antiapoptotic and proproliferative effects on the metanephric mesenchyme through an MMP-independent mechanism; TIMP-2 also negatively regulates ureteric bud branching, which seems to be caused by its function as an MMP inhibitor (77). Finally, TIMP-2 is implicated in activation of MMP-2, an enzyme that facilitates renal recovery after ischemia-reperfusion injury. Therefore, the literature supports both renoprotective and prorecovery roles for TIMP-2. Given the divergent potential mechanisms of TIMP-2 in AKI, it is currently unclear how this biomarker may be used for mechanistic understanding and therapeutic modulation of AKI, and additional studies using conditional knockouts and pharmacologic inhibitors of TIMP-2 are needed to better define its mechanistic role in renal injury.
IGFBP7
IGFBP7, also known as IGFBP-related protein 1, mac25, angiomodulin, tumor-derived adhesion factor, and prostacyclin-stimulating factor, is a ubiquitously expressed 29-kD protein that is primarily known as a tumor suppressor and regulator of cellular senescence (78). IGFBP7, like TIMP-2, was unknown as an AKI biomarker until the study published by the SAPPHIRE investigators. This study reported that elevated urine IGFBP7 predicted the onset of KDIGO stage 2 or 3 AKI within 12 hours of sample collection (AUC=0.76) (7). A subsequent smaller study (n=52) of patients in the ICU has reported that, on the day of AKI diagnosis, elevated urinary IGFBP7 outperforms urine NGAL as a predictor of nonresolution of AKI within 7 days (79).
It has been proposed that injured tubular epithelial cells secrete IGFBP7, which attenuates renal injury by the induction of G1 cell cycle arrest in nearby surviving cells through upregulation of p21 and p53 expression (7). This hypothesis is grounded in data from in vitro studies of colon and breast cancer cell lines, which have shown the capacity of IGFBP7 to induce G1 cell cycle arrest and senescence, as well as a study of melanocyte and melanoma cell lines, which has shown that IGFBP7 regulates proliferation and senescence (80,81). These studies have reported that the effects of IGFBP7 are mediated by increased expression of p21 and p53 as well as inhibition of mitogen-activated protein kinase signaling (80–82). However, it is also plausible that elevated IGFBP7 could have a deleterious effect on the injured kidney, because IGFBP7 is an IGF-1 receptor antagonist (83). IGF-1 improves renal perfusion and increases GFR. Therefore, elevated IGFBP7 could alter renal hemodynamics in such a way that exacerbates renal injury.
The role of IGFBP7 as an IGF-1 antagonist could have therapeutic implications. In an animal model of AKI, administration of IGF-1 5 hours after injury attenuated the severity of AKI and promoted renal recovery by increasing the number of proliferating proximal tubule epithelial cells (84). A recombinant human IGF-1 was tested as an AKI therapeutic in a randomized controlled clinical trial (85). Unfortunately, the trial failed to show a benefit, and its failure has been attributed to poorly timed intervention and IGF-1–associated hypotension (2). However, elevated intrarenal IGFBP7 could have contributed to the failure of this trial by antagonizing the renal protective effects of the intervention. Future studies could investigate the potential benefits of using IGFBP7 to predict response to IGF-1 therapy (for instance, using urinary IGFBP7 concentration to titrate IGF-1 dosage).
Integrating Novel Biomarkers into Existing Conceptual Models of AKI
Vigorous research efforts have failed to deliver a kidney troponin for the diagnosis of AKI. However, novel biomarkers of AKI, specifically NGAL, KIM-1, IL-18, L-FABP, angiotensinogen, TIMP-2, and IGFBP7, are qualitatively different from cardiac troponin and more akin to cancer biomarkers, in that they are reflective of the molecular and cellular events that occur throughout the clinical phases of AKI. To illustrate how biomarker kinetics relate to the molecular and cellular events that are known to occur throughout the clinical course of AKI, we plotted the median biomarker concentration at each time point from the recent TRIBE-AKI study and superimposed these trends on the previously described phases of AKI (Figure 1) (4–6,8,9). Although this approach is limited in that it is on the basis of kinetics from a single human study of a single etiology of AKI, the TRIBE-AKI study is the largest prospective study for which biomarker kinetics data are available, and the cardiac surgery population is ideal for the study of the AKI biomarker kinetics, because the timing of the injury is known. As a corollary to these data, we propose the biomarker-integrated model of AKI (Figure 2); it shows how novel AKI biomarkers are molecular determinants of the course of AKI by framing them in the context of the clinical phases of AKI, which were proposed by Sutton et al. (8) and later adapted by Rosner and Okusa (9).
Figure 1.

Changes in AKI biomarker concentration over time after renal injury. Data from the Translational Research Investigating Biomarker Endpoints in AKI (TRIBE-AKI) study in adult patients undergoing cardiac surgery were used, and the relationship between the stages of AKI proposed by Sutton et al. (4,5,8) and the time course of changes in biomarker concentration that occur after renal injury is shown. The relative change in median biomarker concentration at each time point was plotted against time. To create this plot, the time point with the highest median value was set to 100%, and all other time points were converted to a percentage of this maximum. pNGAL, plasma neutrophil gelatinase–associated lipocalin; uIL-18, urine IL-18; uKIM-1, urine kidney injury molecule 1; uL-FABP, urine liver-type fatty acid–binding protein; uNGAL, urine neutrophil gelatinase-associated lipocalin.
Figure 2.
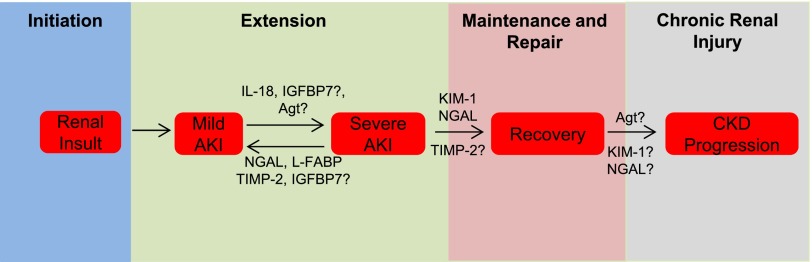
Biomarker integrated model of AKI. This figure shows how the novel AKI biomarkers neutrophil gelatinase–associated lipocalin (NGAL), kidney injury molecule 1 (KIM-1), IL-18, liver-type fatty acid binding protein (L-FABP), angiotensinogen (Agt), tissue inhibitor of metalloproteinase-2 (TIMP-2), and IGF-binding protein 7 (IGFBP7) contribute to the different clinical phases of AKI and progression to chronic renal injury. Phases are shown as colored boxes, and specific events within each phase are denoted by text boxes. After an initiating renal insult, such as ischemia-reperfusion injury, proinjury biomarker IL-18 promotes additional renal injury and the development of severe AKI by upregulating the inflammatory response during the extension phase. Additionally, although the precise mechanistic import of urinary Agt is unknown, urinary Agt concentration is considered a marker of activation of the intrarenal renin-angiotensin system (RAS). Inhibition of the RAS has been shown to attenuate the severity of AKI in animal models. Agt may contribute to renal injury during the extension phase by promoting RAS activation. In contrast, the renoprotective biomarkers NGAL and L-FABP mitigate renal injury by antiapoptotic (NGAL) and antioxidant (L-FABP) effects during this phase. Putative cell cycle arrest biomarkers TIMP-2 and IGFBP7 could also attenuate injury during the extension phase, although some of the literature suggest that IGFBP7 could promote injury. During the maintenance and repair phases, KIM-1 promotes renal recovery by conferring a phagocytic phenotype to surviving, proliferating renal tubular epithelial cells, allowing these cells to clean up debris from the injury and facilitating tissue remodeling. Similarly, NGAL increases cellular proliferation, which is necessary for repopulation of the denuded tubular epithelium. TIMP-2 may also promote recovery by promoting proliferation of surviving tubular epithelial cells. Finally, although the mechanistic link between AKI and progression to CKD and ESRD is unknown, the RAS is likely involved. Thus, elevated Agt could predict chronicity of renal injury or even promote it.
In the first phase (initiation), the proximal insult occurs, and there is a precipitous fall in the GFR (8,9). In the extension phase, GFR continues to fall because of worsening injury from renal hemodynamic alterations and inflammation and apoptosis and necrosis of tubular epithelial cells (8,9). It is during the extension phase that AKI transitions from mild to severe, and it may be promoted by the proinjury marker IL-18, which peaks in this phase (5). Data from animal models clearly show that IL-18 exacerbates renal injury through its proinflammatory effects. Although kinetic data are not available, we hypothesize that elevated angiotensinogen also promotes renal injury in the acute extension phase and drives the progression to chronic renal injury, because activation of RAS is known to promote renal injury. IGFBP7 may also contribute to injury through its effect as an IGF-1 antagonist. In contrast to IL-18 and angiotensinogen, increases in renoprotective NGAL and L-FABP during the extension phase likely represent a response to a kidney insult, because these biomarkers attenuate renal injury through antiapoptotic and antioxidant mechanisms. TIMP-2 and IGFBP7, recently discovered markers of early AKI, have also been proposed to play a renoprotective role, although the precise mechanism underlying the diagnostic value of these biomarkers is just beginning to be elucidated. The maintenance phase is characterized by stabilization of GFR at its nadir and proliferation and migration of surviving tubular epithelial cells to repair the damaged epithelium (4,5,8,9). The final phase (repair) involves the re-establishment of normal cellular and physiologic function. These final two phases do not begin until 2–3 days after the initiating renal injury, but they last >1 week (8,9). Unfortunately, time course data on AKI biomarker concentration throughout the duration of these phases have not been collected. However, sustained elevation of NGAL and L-FABP suggests that they are also somehow involved in renal recovery during the maintenance and repair phases of AKI. Although such a role has not been suggested for L-FABP, NGAL is known to have proliferative effects on tubular epithelial cells, important components of the complex process of tubular regeneration. Similarly, KIM-1, which peaks in its urine concentration at 48 hours after injury just before the onset of the maintenance phase, has been proposed as a prorecovery biomarker, because it enhances the phagocytosis of apoptotic bodies and cellular debris, implicating this biomarker in wound-healing.
Finally, the link between AKI and CKD is beginning to be elucidated, and it is plausible that novel AKI biomarkers play a role in driving the transition from acute to chronic renal injury (71). As we have noted, angiotensinogen has already been established as a biomarker of CKD progression, and urinary angiotensinogen is believed to reflect activation of the intrarenal RAS, a known driver of CKD progression (61–63). We hypothesize that elevated urinary angiotensinogen during AKI could promote the progression to CKD. Similarly, others have noted in transcriptomic analysis that KIM-1 and NGAL expression remain high in the later stages of AKI, and it has been proposed that sustained elevation of these biomarkers could represent ongoing kidney injury and a transition to chronic renal disease (86).
Disclosures
J.L.A. and J.M.A. are named inventors on a patent application filed by the Medical University of South Carolina for the use of urinary angiotensinogen as a kidney disease biomarker.
Acknowledgments
This manuscript was supported by the Department of Veterans Affairs and National Institutes of Health Grant R01-DK080234.
The contents do not necessarily represent the views of the Department of Veterans Affairs or the US Government.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1.Palevsky PM, Molitoris BA, Okusa MD, Levin A, Waikar SS, Wald R, Chertow GM, Murray PT, Parikh CR, Shaw AD, Go AS, Faubel SG, Kellum JA, Chinchilli VM, Liu KD, Cheung AK, Weisbord SD, Chawla LS, Kaufman JS, Devarajan P, Toto RM, Hsu CY, Greene T, Mehta RL, Stokes JB, Thompson AM, Thompson BT, Westenfelder CS, Tumlin JA, Warnock DG, Shah SV, Xie Y, Duggan EG, Kimmel PL, Star RA: Design of clinical trials in acute kidney injury: report from an NIDDK workshop on trial methodology. Clin J Am Soc Nephrol 7: 844–850, 2012 [DOI] [PubMed] [Google Scholar]
- 2.Jo SK, Rosner MH, Okusa MD: Pharmacologic treatment of acute kidney injury: why drugs haven’t worked and what is on the horizon. Clin J Am Soc Nephrol 2: 356–365, 2007 [DOI] [PubMed] [Google Scholar]
- 3.Endre ZH, Pickering JW, Walker RJ, Devarajan P, Edelstein CL, Bonventre JV, Frampton CM, Bennett MR, Ma Q, Sabbisetti VS, Vaidya VS, Walcher AM, Shaw GM, Henderson SJ, Nejat M, Schollum JB, George PM: Improved performance of urinary biomarkers of acute kidney injury in the critically ill by stratification for injury duration and baseline renal function. Kidney Int 79: 1119–1130, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parikh CR, Thiessen-Philbrook H, Garg AX, Kadiyala D, Shlipak MG, Koyner JL, Edelstein CL, Devarajan P, Patel UD, Zappitelli M, Krawczeski CD, Passik CS, Coca SG, TRIBE-AKI Consortium : Performance of kidney injury molecule-1 and liver fatty acid-binding protein and combined biomarkers of AKI after cardiac surgery. Clin J Am Soc Nephrol 8: 1079–1088, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parikh CR, Coca SG, Thiessen-Philbrook H, Shlipak MG, Koyner JL, Wang Z, Edelstein CL, Devarajan P, Patel UD, Zappitelli M, Krawczeski CD, Passik CS, Swaminathan M, Garg AX, TRIBE-AKI Consortium : Postoperative biomarkers predict acute kidney injury and poor outcomes after adult cardiac surgery. J Am Soc Nephrol 22: 1748–1757, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parikh CR, Devarajan P, Zappitelli M, Sint K, Thiessen-Philbrook H, Li S, Kim RW, Koyner JL, Coca SG, Edelstein CL, Shlipak MG, Garg AX, Krawczeski CD, TRIBE-AKI Consortium : Postoperative biomarkers predict acute kidney injury and poor outcomes after pediatric cardiac surgery. J Am Soc Nephrol 22: 1737–1747, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kashani K, Al-Khafaji A, Ardiles T, Artigas A, Bagshaw SM, Bell M, Bihorac A, Birkhahn R, Cely CM, Chawla LS, Davison DL, Feldkamp T, Forni LG, Gong MN, Gunnerson KJ, Haase M, Hackett J, Honore PM, Hoste EA, Joannes-Boyau O, Joannidis M, Kim P, Koyner JL, Laskowitz DT, Lissauer ME, Marx G, McCullough PA, Mullaney S, Ostermann M, Rimmelé T, Shapiro NI, Shaw AD, Shi J, Sprague AM, Vincent JL, Vinsonneau C, Wagner L, Walker MG, Wilkerson RG, Zacharowski K, Kellum JA: Discovery and validation of cell cycle arrest biomarkers in human acute kidney injury. Crit Care 17: R25, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sutton TA, Fisher CJ, Molitoris BA: Microvascular endothelial injury and dysfunction during ischemic acute renal failure. Kidney Int 62: 1539–1549, 2002 [DOI] [PubMed] [Google Scholar]
- 9.Rosner MH, Okusa MD: Acute kidney injury associated with cardiac surgery. Clin J Am Soc Nephrol 1: 19–32, 2006 [DOI] [PubMed] [Google Scholar]
- 10.Supavekin S, Zhang W, Kucherlapati R, Kaskel FJ, Moore LC, Devarajan P: Differential gene expression following early renal ischemia/reperfusion. Kidney Int 63: 1714–1724, 2003 [DOI] [PubMed] [Google Scholar]
- 11.Mishra J, Ma Q, Prada A, Mitsnefes M, Zahedi K, Yang J, Barasch J, Devarajan P: Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol 14: 2534–2543, 2003 [DOI] [PubMed] [Google Scholar]
- 12.Mishra J, Mori K, Ma Q, Kelly C, Barasch J, Devarajan P: Neutrophil gelatinase-associated lipocalin: a novel early urinary biomarker for cisplatin nephrotoxicity. Am J Nephrol 24: 307–315, 2004 [DOI] [PubMed] [Google Scholar]
- 13.Mori K, Lee HT, Rapoport D, Drexler IR, Foster K, Yang J, Schmidt-Ott KM, Chen X, Li JY, Weiss S, Mishra J, Cheema FH, Markowitz G, Suganami T, Sawai K, Mukoyama M, Kunis C, D’Agati V, Devarajan P, Barasch J: Endocytic delivery of lipocalin-siderophore-iron complex rescues the kidney from ischemia-reperfusion injury. J Clin Invest 115: 610–621, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmidt-Ott KM, Mori K, Kalandadze A, Li JY, Paragas N, Nicholas T, Devarajan P, Barasch J: Neutrophil gelatinase-associated lipocalin-mediated iron traffic in kidney epithelia. Curr Opin Nephrol Hypertens 15: 442–449, 2006 [DOI] [PubMed] [Google Scholar]
- 15.Mishra J, Dent C, Tarabishi R, Mitsnefes MM, Ma Q, Kelly C, Ruff SM, Zahedi K, Shao M, Bean J, Mori K, Barasch J, Devarajan P: Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet 365: 1231–1238, 2005 [DOI] [PubMed] [Google Scholar]
- 16.Hvidberg V, Jacobsen C, Strong RK, Cowland JB, Moestrup SK, Borregaard N: The endocytic receptor megalin binds the iron transporting neutrophil-gelatinase-associated lipocalin with high affinity and mediates its cellular uptake. FEBS Lett 579: 773–777, 2005 [DOI] [PubMed] [Google Scholar]
- 17.Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK: The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell 10: 1033–1043, 2002 [DOI] [PubMed] [Google Scholar]
- 18.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A: Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 432: 917–921, 2004 [DOI] [PubMed] [Google Scholar]
- 19.Yang J, Goetz D, Li JY, Wang W, Mori K, Setlik D, Du T, Erdjument-Bromage H, Tempst P, Strong R, Barasch J: An iron delivery pathway mediated by a lipocalin. Mol Cell 10: 1045–1056, 2002 [DOI] [PubMed] [Google Scholar]
- 20.Li JY, Ram G, Gast K, Chen X, Barasch K, Mori K, Schmidt-Ott K, Wang J, Kuo HC, Savage-Dunn C, Garrick MD, Barasch J: Detection of intracellular iron by its regulatory effect. Am J Physiol Cell Physiol 287: C1547–C1559, 2004 [DOI] [PubMed] [Google Scholar]
- 21.Mishra J, Mori K, Ma Q, Kelly C, Yang J, Mitsnefes M, Barasch J, Devarajan P: Amelioration of ischemic acute renal injury by neutrophil gelatinase-associated lipocalin. J Am Soc Nephrol 15: 3073–3082, 2004 [DOI] [PubMed] [Google Scholar]
- 22.Bao G, Clifton M, Hoette TM, Mori K, Deng SX, Qiu A, Viltard M, Williams D, Paragas N, Leete T, Kulkarni R, Li X, Lee B, Kalandadze A, Ratner AJ, Pizarro JC, Schmidt-Ott KM, Landry DW, Raymond KN, Strong RK, Barasch J: Iron traffics in circulation bound to a siderocalin (Ngal)-catechol complex. Nat Chem Biol 6: 602–609, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ichimura T, Bonventre JV, Bailly V, Wei H, Hession CA, Cate RL, Sanicola M: Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J Biol Chem 273: 4135–4142, 1998 [DOI] [PubMed] [Google Scholar]
- 24.Bailly V, Zhang Z, Meier W, Cate R, Sanicola M, Bonventre JV: Shedding of kidney injury molecule-1, a putative adhesion protein involved in renal regeneration. J Biol Chem 277: 39739–39748, 2002 [DOI] [PubMed] [Google Scholar]
- 25.Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV: Kidney Injury Molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney Int 62: 237–244, 2002 [DOI] [PubMed] [Google Scholar]
- 26.Ichimura T, Hung CC, Yang SA, Stevens JL, Bonventre JV: Kidney injury molecule-1: a tissue and urinary biomarker for nephrotoxicant-induced renal injury. Am J Physiol Renal Physiol 286: F552–F563, 2004 [DOI] [PubMed] [Google Scholar]
- 27.Ichimura T, Asseldonk EJ, Humphreys BD, Gunaratnam L, Duffield JS, Bonventre JV: Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J Clin Invest 118: 1657–1668, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lim AI, Chan LY, Lai KN, Tang SC, Chow CW, Lam MF, Leung JC: Distinct role of matrix metalloproteinase-3 in kidney injury molecule-1 shedding by kidney proximal tubular epithelial cells. Int J Biochem Cell Biol 44: 1040–1050, 2012 [DOI] [PubMed] [Google Scholar]
- 29.Zhang Z, Humphreys BD, Bonventre JV: Shedding of the urinary biomarker kidney injury molecule-1 (KIM-1) is regulated by MAP kinases and juxtamembrane region. J Am Soc Nephrol 18: 2704–2714, 2007 [DOI] [PubMed] [Google Scholar]
- 30.Funk JA, Schnellmann RG: Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am J Physiol Renal Physiol 302: F853–F864, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hall IE, Coca SG, Perazella MA, Eko UU, Luciano RL, Peter PR, Han WK, Parikh CR: Risk of poor outcomes with novel and traditional biomarkers at clinical AKI diagnosis. Clin J Am Soc Nephrol 6: 2740–2749, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arthur JM, Hill EG, Alge JL, Lewis EC, Neely BA, Janech MG, Tumlin JA, Chawla LS, Shaw AD, SAKInet Investigators : Evaluation of 32 urine biomarkers to predict the progression of acute kidney injury after cardiac surgery. Kidney Int 85: 431–438, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melnikov VY, Ecder T, Fantuzzi G, Siegmund B, Lucia MS, Dinarello CA, Schrier RW, Edelstein CL: Impaired IL-18 processing protects caspase-1-deficient mice from ischemic acute renal failure. J Clin Invest 107: 1145–1152, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Homsi E, Janino P, de Faria JB: Role of caspases on cell death, inflammation, and cell cycle in glycerol-induced acute renal failure. Kidney Int 69: 1385–1392, 2006 [DOI] [PubMed] [Google Scholar]
- 35.Wu H, Craft ML, Wang P, Wyburn KR, Chen G, Ma J, Hambly B, Chadban SJ: IL-18 contributes to renal damage after ischemia-reperfusion. J Am Soc Nephrol 19: 2331–2341, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anders HJ, Muruve DA: The inflammasomes in kidney disease. J Am Soc Nephrol 22: 1007–1018, 2011 [DOI] [PubMed] [Google Scholar]
- 37.Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, Eisenbarth SC, Florquin S, Flavell RA, Leemans JC, Sutterwala FS: Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A 106: 20388–20393, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheung H, Chen NJ, Cao Z, Ono N, Ohashi PS, Yeh WC: Accessory protein-like is essential for IL-18-mediated signaling. J Immunol 174: 5351–5357, 2005 [DOI] [PubMed] [Google Scholar]
- 39.Melnikov VY, Faubel S, Siegmund B, Lucia MS, Ljubanovic D, Edelstein CL: Neutrophil-independent mechanisms of caspase-1- and IL-18-mediated ischemic acute tubular necrosis in mice. J Clin Invest 110: 1083–1091, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang J, Long Q, Zhang W, Chen N: Protective effects of exogenous interleukin 18-binding protein in a rat model of acute renal ischemia-reperfusion injury. Shock 37: 333–340, 2012 [DOI] [PubMed] [Google Scholar]
- 41.Novick D, Kim SH, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M: Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity 10: 127–136, 1999 [DOI] [PubMed] [Google Scholar]
- 42.Maatman RG, Van Kuppevelt TH, Veerkamp JH: Two types of fatty acid-binding protein in human kidney. Isolation, characterization and localization. Biochem J 273[Pt 3]: 759–766, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maatman RG, van de Westerlo EM, van Kuppevelt TH, Veerkamp JH: Molecular identification of the liver- and the heart-type fatty acid-binding proteins in human and rat kidney. Use of the reverse transcriptase polymerase chain reaction. Biochem J 288[Pt 1]: 285–290, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katagiri D, Doi K, Honda K, Negishi K, Fujita T, Hisagi M, Ono M, Matsubara T, Yahagi N, Iwagami M, Ohtake T, Kobayashi S, Sugaya T, Noiri E: Combination of two urinary biomarkers predicts acute kidney injury after adult cardiac surgery. Ann Thorac Surg 93: 577–583, 2012 [DOI] [PubMed] [Google Scholar]
- 45.Atshaves BP, McIntosh AM, Lyuksyutova OI, Zipfel W, Webb WW, Schroeder F: Liver fatty acid-binding protein gene ablation inhibits branched-chain fatty acid metabolism in cultured primary hepatocytes. J Biol Chem 279: 30954–30965, 2004 [DOI] [PubMed] [Google Scholar]
- 46.Atshaves BP, McIntosh AL, Payne HR, Mackie J, Kier AB, Schroeder F: Effect of branched-chain fatty acid on lipid dynamics in mice lacking liver fatty acid binding protein gene. Am J Physiol Cell Physiol 288: C543–C558, 2005 [DOI] [PubMed] [Google Scholar]
- 47.Wang G, Gong Y, Anderson J, Sun D, Minuk G, Roberts MS, Burczynski FJ: Antioxidative function of L-FABP in L-FABP stably transfected Chang liver cells. Hepatology 42: 871–879, 2005 [DOI] [PubMed] [Google Scholar]
- 48.Noiri E, Doi K, Negishi K, Tanaka T, Hamasaki Y, Fujita T, Portilla D, Sugaya T: Urinary fatty acid-binding protein 1: An early predictive biomarker of kidney injury. Am J Physiol Renal Physiol 296: F669–F679, 200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamamoto T, Noiri E, Ono Y, Doi K, Negishi K, Kamijo A, Kimura K, Fujita T, Kinukawa T, Taniguchi H, Nakamura K, Goto M, Shinozaki N, Ohshima S, Sugaya T: Renal L-type fatty acid—binding protein in acute ischemic injury. J Am Soc Nephrol 18: 2894–2902, 2007 [DOI] [PubMed] [Google Scholar]
- 50.Kamijo-Ikemori A, Sugaya T, Matsui K, Yokoyama T, Kimura K: Roles of human liver type fatty acid binding protein in kidney disease clarified using hL-FABP chromosomal transgenic mice. Nephrology (Carlton) 16: 539–544, 2011 [DOI] [PubMed] [Google Scholar]
- 51.Nakamura T, Sugaya T, Node K, Ueda Y, Koide H: Urinary excretion of liver-type fatty acid-binding protein in contrast medium-induced nephropathy. Am J Kidney Dis 47: 439–444, 2006 [DOI] [PubMed] [Google Scholar]
- 52.Doi K, Negishi K, Ishizu T, Katagiri D, Fujita T, Matsubara T, Yahagi N, Sugaya T, Noiri E: Evaluation of new acute kidney injury biomarkers in a mixed intensive care unit. Crit Care Med 39: 2464–2469, 2011 [DOI] [PubMed] [Google Scholar]
- 53.Negishi K, Noiri E, Sugaya T, Li S, Megyesi J, Nagothu K, Portilla D: A role of liver fatty acid-binding protein in cisplatin-induced acute renal failure. Kidney Int 72: 348–358, 2007 [DOI] [PubMed] [Google Scholar]
- 54.Negishi K, Noiri E, Maeda R, Portilla D, Sugaya T, Fujita T: Renal L-type fatty acid-binding protein mediates the bezafibrate reduction of cisplatin-induced acute kidney injury. Kidney Int 73: 1374–1384, 2008 [DOI] [PubMed] [Google Scholar]
- 55.Portilla D, Dai G, Peters JM, Gonzalez FJ, Crew MD, Proia AD: Etomoxir-induced PPARalpha-modulated enzymes protect during acute renal failure. Am J Physiol Renal Physiol 278: F667–F675, 2000 [DOI] [PubMed] [Google Scholar]
- 56.Attridge RL, Linn WD, Ryan L, Koeller J, Frei CR: Evaluation of the incidence and risk factors for development of fenofibrate-associated nephrotoxicity. J Clin Lipidol 6: 19–26, 2012 [DOI] [PubMed] [Google Scholar]
- 57.Zhao YY, Weir MA, Manno M, Cordy P, Gomes T, Hackam DG, Juurlink DN, Mamdani M, Moist L, Parikh CR, Paterson JM, Wald R, Yao Z, Garg AX: New fibrate use and acute renal outcomes in elderly adults: a population-based study. Ann Intern Med 156: 560–569, 2012 [DOI] [PubMed] [Google Scholar]
- 58.Alge JL, Karakala N, Neely BA, Janech MG, Tumlin JA, Chawla LS, Shaw AD, Arthur JM, SAKInet Investigators : Urinary angiotensinogen and risk of severe AKI. Clin J Am Soc Nephrol 8: 184–193, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alge JL, Karakala N, Neely BA, Janech MG, Velez JC, Arthur JM, The SAKInet Investigators : Urinary angiotensinogen predicts adverse outcomes among acute kidney injury patients in the intensive care unit. Crit Care 17: R69, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alge JL, Karakala N, Neely BA, Janech MG, Tumlin JA, Chawla LS, Shaw AD, Arthur JM, SAKInet Investigators : Association of elevated urinary concentration of renin-angiotensin system components and severe AKI. Clin J Am Soc Nephrol 8: 2043–2052, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kobori H, Ohashi N, Katsurada A, Miyata K, Satou R, Saito T, Yamamoto T: Urinary angiotensinogen as a potential biomarker of severity of chronic kidney diseases. J Am Soc Hypertens 2: 349–354, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamamoto T, Nakagawa T, Suzuki H, Ohashi N, Fukasawa H, Fujigaki Y, Kato A, Nakamura Y, Suzuki F, Hishida A: Urinary angiotensinogen as a marker of intrarenal angiotensin II activity associated with deterioration of renal function in patients with chronic kidney disease. J Am Soc Nephrol 18: 1558–1565, 2007 [DOI] [PubMed] [Google Scholar]
- 63.Kobori H, Harrison-Bernard LM, Navar LG: Urinary excretion of angiotensinogen reflects intrarenal angiotensinogen production. Kidney Int 61: 579–585, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Allred AJ, Chappell MC, Ferrario CM, Diz DI: Differential actions of renal ischemic injury on the intrarenal angiotensin system. Am J Physiol Renal Physiol 279: F636–F645, 2000 [DOI] [PubMed] [Google Scholar]
- 65.da Silveira KD, Pompermayer Bosco KS, Diniz LR, Carmona AK, Cassali GD, Bruna-Romero O, de Sousa LP, Teixeira MM, Santos RA, Simões e Silva AC, Ribeiro Vieira MA: ACE2-angiotensin-(1-7)-Mas axis in renal ischaemia/reperfusion injury in rats. Clin Sci (Lond) 119: 385–394, 2010 [DOI] [PubMed] [Google Scholar]
- 66.Efrati S, Berman S, Hamad RA, Siman-Tov Y, Ilgiyaev E, Maslyakov I, Weissgarten J: Effect of captopril treatment on recuperation from ischemia/reperfusion-induced acute renal injury. Nephrol Dial Transplant 27: 136–145, 2012 [DOI] [PubMed] [Google Scholar]
- 67.Molinas SM, Cortés-González C, González-Bobadilla Y, Monasterolo LA, Cruz C, Elías MM, Bobadilla NA, Trumper L: Effects of losartan pretreatment in an experimental model of ischemic acute kidney injury. Nephron, Exp Nephrol 112: e10–e19, 2009 [DOI] [PubMed] [Google Scholar]
- 68.Wang Z, Liu Y, Han Y, Guan W, Kou X, Fu J, Yang D, Ren H, He D, Zhou L, Zeng C: Protective effects of aliskiren on ischemia-reperfusion-induced renal injury in rats. Eur J Pharmacol 718: 160–166, 2013 [DOI] [PubMed] [Google Scholar]
- 69.Coca SG, Garg AX, Swaminathan M, Garwood S, Hong K, Thiessen-Philbrook H, Passik C, Koyner JL, Parikh CR, TRIBE-AKI Consortium : Preoperative angiotensin-converting enzyme inhibitors and angiotensin receptor blocker use and acute kidney injury in patients undergoing cardiac surgery. Nephrol Dial Transplant 28: 2787–2799, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ishani A, Xue JL, Himmelfarb J, Eggers PW, Kimmel PL, Molitoris BA, Collins AJ: Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol 20: 223–228, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Venkatachalam MA, Griffin KA, Lan R, Geng H, Saikumar P, Bidani AK: Acute kidney injury: a springboard for progression in chronic kidney disease. Am J Physiol Renal Physiol 298: F1078–F1094, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barrera-Chimal J, Pérez-Villalva R, Rodríguez-Romo R, Reyna J, Uribe N, Gamba G, Bobadilla NA: Spironolactone prevents chronic kidney disease caused by ischemic acute kidney injury. Kidney Int 83: 93–103, 2013 [DOI] [PubMed] [Google Scholar]
- 73.Seo DW, Li H, Qu CK, Oh J, Kim YS, Diaz T, Wei B, Han JW, Stetler-Stevenson WG: Shp-1 mediates the antiproliferative activity of tissue inhibitor of metalloproteinase-2 in human microvascular endothelial cells. J Biol Chem 281: 3711–3721, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seo DW, Kim SH, Eom SH, Yoon HJ, Cho YR, Kim PH, Kim YK, Han JW, Diaz T, Wei BY, Stetler-Stevenson WG: TIMP-2 disrupts FGF-2-induced downstream signaling pathways. Microvasc Res 76: 145–151, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hoegy SE, Oh HR, Corcoran ML, Stetler-Stevenson WG: Tissue inhibitor of metalloproteinases-2 (TIMP-2) suppresses TKR-growth factor signaling independent of metalloproteinase inhibition. J Biol Chem 276: 3203–3214, 2001 [DOI] [PubMed] [Google Scholar]
- 76.Stetler-Stevenson WG: Tissue inhibitors of metalloproteinases in cell signaling: metalloproteinase-independent biological activities. Sci Signal 1: re6, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Barasch J, Yang J, Qiao J, Tempst P, Erdjument-Bromage H, Leung W, Oliver JA: Tissue inhibitor of metalloproteinase-2 stimulates mesenchymal growth and regulates epithelial branching during morphogenesis of the rat metanephros. J Clin Invest 103: 1299–1307, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhu S, Xu F, Zhang J, Ruan W, Lai M: Insulin-like growth factor binding protein-related protein 1 and cancer. Clin Chim Acta 431: 23–32, 2014 [DOI] [PubMed] [Google Scholar]
- 79.Aregger F, Uehlinger DE, Witowski J, Brunisholz RA, Hunziker P, Frey FJ, Jörres A: Identification of IGFBP-7 by urinary proteomics as a novel prognostic marker in early acute kidney injury. Kidney Int 85: 909–19, 2014 [DOI] [PubMed] [Google Scholar]
- 80.Ma Y, Lu B, Ruan W, Wang H, Lin J, Hu H, Deng H, Huang Q, Lai M: Tumor suppressor gene insulin-like growth factor binding protein-related protein 1 (IGFBP-rP1) induces senescence-like growth arrest in colorectal cancer cells. Exp Mol Pathol 85: 141–145, 2008 [DOI] [PubMed] [Google Scholar]
- 81.Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR: Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 132: 363–374, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zuo S, Liu C, Wang J, Wang F, Xu W, Cui S, Yuan L, Chen X, Fan W, Cui M, Song G: IGFBP-rP1 induces p21 expression through a p53-independent pathway, leading to cellular senescence of MCF-7 breast cancer cells. J Cancer Res Clin Oncol 138: 1045–1055, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Evdokimova V, Tognon CE, Benatar T, Yang W, Krutikov K, Pollak M, Sorensen PH, Seth A: IGFBP7 binds to the IGF-1 receptor and blocks its activation by insulin-like growth factors. Sci Signal 5: ra92, 2012 [DOI] [PubMed] [Google Scholar]
- 84.Ding H, Kopple JD, Cohen A, Hirschberg R: Recombinant human insulin-like growth factor-I accelerates recovery and reduces catabolism in rats with ischemic acute renal failure. J Clin Invest 91: 2281–2287, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hirschberg R, Kopple J, Lipsett P, Benjamin E, Minei J, Albertson T, Munger M, Metzler M, Zaloga G, Murray M, Lowry S, Conger J, McKeown W, O’shea M, Baughman R, Wood K, Haupt M, Kaiser R, Simms H, Warnock D, Summer W, Hintz R, Myers B, Haenftling K, Capra W, Pike M, Guler R: Multicenter clinical trial of recombinant human insulin-like growth factor I in patients with acute renal failure. Kidney Int 55: 2423–2432, 1999 [DOI] [PubMed] [Google Scholar]
- 86.Ko GJ, Grigoryev DN, Linfert D, Jang HR, Watkins T, Cheadle C, Racusen L, Rabb H: Transcriptional analysis of kidneys during repair from AKI reveals possible roles for NGAL and KIM-1 as biomarkers of AKI-to-CKD transition. Am J Physiol Renal Physiol 298: F1472–F1483, 2010 [DOI] [PubMed] [Google Scholar]