

Abstract
Background and Purpose
Accumulation of amyloid-β in cerebral blood vessels occurs in familial and sporadic forms of cerebral amyloid angiopathy and is a prominent feature of Alzheimer disease. However, the functional correlates of the vascular pathology induced by cerebral amyloid angiopathy and the mechanisms involved have not been fully established.
Methods
We used male transgenic mice expressing the Swedish, Iowa, and Dutch mutations of the amyloid precursor protein (Tg-SwDI) to examine the effect of cerebral amyloid angiopathy on cerebrovascular structure and function. Somatosensory cortex cerebral blood flow was monitored by laser-Doppler flowmetry in anesthetized Tg-SwDI mice and wild-type littermates equipped with a cranial window.
Results
Tg-SwDI mice exhibited reductions in cerebral blood flow responses to whisker stimulation, endothelium-dependent vasodilators, or hypercapnia at 3 months when compared with wild-type mice, whereas the response to adenosine was not attenuated. However, at 18 and 24 months, all cerebrovascular responses were markedly reduced. At this time, there was evidence of cerebrovascular amyloid deposition, smooth muscle fragmentation, and pericyte loss. Neocortical superfusion with the free radical scavenger manganic(I–II)meso-tetrakis(4-benzoic acid) porphyrin rescued endothelium-dependent responses and functional hyperemia completely at 3 months but only partially at 18 months.
Conclusions
Tg-SwDI mice exhibit a profound age-dependent cerebrovascular dysfunction, involving multiple regulatory mechanisms. Early in the disease process, oxidative stress is responsible for most of the vascular dysfunction, but with advancing disease structural alterations of the vasomotor apparatus also play a role. Early therapeutic interventions are likely to have the best chance to counteract the deleterious vascular effects of cerebral amyloid angiopathy.
Keywords: amyloid-β, cerebral blood flow, cerebrovascular disorder
Cerebral amyloid angiopathy (CAA) is characterized by accumulation of amyloid-β in cerebral arteries, arterioles, and capillaries and is most commonly associated with Alzheimer disease.1–3 CAA can also occur independently of Alzheimer disease, either as a familial form resulting from gene mutations or as a sporadic form observed in the elderly.1,4,5 CAA is associated with damage to the vessels wall, resulting in ischemic or hemorrhagic strokes, microinfarcts, and microhemorrhages.6,7 However, CAA can also alter the regulation of the cerebral circulation leading to vascular insufficiency and increased susceptibility to white matter damage and cognitive impairment.8–10 The mechanisms of these functional alterations and their relationships to the structural damage to cerebral blood vessels remain to be defined.
Transgenic mice (Tg-SwDI), expressing low levels of human amyloid-β precursor protein harboring the Swedish K670N/M671L and vasculotropic Dutch/Iowa E693Q/D694N mutations in neurons, develop extensive cerebral microvascular amyloid deposition especially in cerebral arterioles and capillaries.11 These mice have elevated levels of amyloid-β in brain and exhibit amyloid plaques or amyloid accumulation in blood vessels starting at 6 months.11 The CAA is associated with an age-dependent structural alteration of the cerebral vasculature, neuroinflammation, astrogliosis, and microgliosis.12,13 Cerebrovascular function is also altered in these mice. Thus, the increase in cerebral blood flow (CBF) evoked by neural activity (functional hyperemia), a key mechanism that matches the metabolic needs of active neurons with the delivery of energy substrates via blood flow, and the ability of brain endothelial cells to control CBF are markedly impaired.14 However, because previous studies were conducted in 3-month-old Tg-SwDI mice before amyloid deposition, the relationships linking vascular dysfunction, amyloid accumulation, and vascular damage have not been established. Furthermore, the mechanisms of the cerebrovascular dysfunction in Tg-SwDI mice remain to be defined. Growing evidence suggests that amyloid-β exerts its detrimental vascular effects through reactive oxygen species (ROS).15–19 However, the contribution of vascular oxidative stress to the cerebrovascular impairments observed in Tg-SwDI mice remains unclear.
Therefore, we sought to investigate the cerebrovascular dysfunction in Tg-SwDI mice and to correlate such dysfunction with the development of vascular damage and with the potential for rescue by ROS scavengers. We found that Tg-SwDI mice exhibit a profound cerebrovascular dysfunction, the magnitude of which is age dependent and is greatest when vascular damage develops. At this age, counteraction of oxidative stress no longer reverses the vascular dysfunction. The findings provide mechanistic insight into the alterations in cerebrovascular regulation induced by CAA and suggest that early intervention is needed to counteract its devastating vascular effects.
Materials and Methods
Mice
All procedures were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medical College. Experiments were performed in Tg-SwDI male mice at 3, 18, and 24 months on a C57BL/6 background.11,14 Age-matched C57BL/6 male mice were used as wild-type (WT) controls.
General Surgical Procedures
As described in detail previously,14,15,17,18 mice were anesthetized with isoflurane (1%–2%), followed by urethane (750 mg/kg, IP) and α-chloralose (50 mg/kg, IP). A femoral artery was cannulated for recording of arterial pressure and collection of blood samples to analyze physiological variables. Arterial blood pressure, blood gases, and rectal temperature were monitored and controlled (Tables I and II in the online-only Data Supplement).
Monitoring of CBF
A small opening was drilled through the parietal bone, the dura was removed, and the site was superfused with a modified Ringer’s solution (37°C; pH 7.3–7.4).14,15,17,18 Relative CBF was continuously monitored at the site of superfusion with a laser-Doppler flow probe (Vasamedic).
Detection of ROS by Dihydroethidine Fluoromicrography
ROS production was monitored using dihydroethidine (Molecular Probes) fluoromicrography, as previously described.16–18 Dihydroethidine (2 μmol/L) was topically superfused on the somatosensory cortex for 60 minutes. In some experiments, dihydroethidine was followed by superfusion with the ROS scavenger manganic(I–II)meso-tetrakis(4-benzoic acid) porphyrin (MnTBAP; 100 μmol/L; Calbiochem) for 30 minutes. At the end of the superfusion, the brain was removed, sectioned (thickness, 20 μm), and processed for detection of ROS-dependent fluorescence.16–18 ROS data are expressed as relative fluorescence units.
Measurement of Brain Amyloid-β
Methods for determination of brain amyloid-β levels have been described.11,14 Briefly, cerebral hemispheres were sonicated and centrifuged, and amyloid-β1–40 or amyloid-β1–42 concentrations (pg/mg) were determined using a sandwich ELISA assay.
Assessment of CAA, Smooth Muscle Fragmentation, and Pericytes
Anesthetized mice were perfused transcardially with heparinized saline, followed by 4% (wt/vol) paraformaldehyde.18,20 Brains were removed, postfixed, and sectioned (thickness, 40 μm). Free-floating sections were randomly selected and processed for the labeling as described below. The specificity of the labeling was ensured by omitting the primary antibody or by preabsorption with the antigen. Images were acquired using a confocal laser scanning microscope (Leica) from somatosensory cortex underlying the cranial window (0.38 to −1.94 mm from Bregma). Brain sections from Tg-SwDI and WT mice were processed in a blinded fashion under identical conditions and imaged using identical settings.
For assessment of CAA, brain sections were first incubated with an antibody against the basement membrane marker collagen IV (Col IV; 1:500, rabbit; Abcam) followed by cyanine dye 5-conjugated anti-rabbit secondary IgG (1:200; Jackson ImmunoResearch), and then stained with 0.05% (wt/vol) thioflavin-S and imaged using the appropriate filters.20
Quantification of amyloid-β–associated fragmentation of smooth muscle cells is detailed previously.20 Briefly, brain sections were incubated with an antibody directed against the smooth muscle marker α-actin (1:8000, mouse; Sigma) for 48 hours. After washing, sections were labeled with donkey cyanine dye 5-conjugated anti-mouse IgG (1:200; Jackson ImmunoResearch) and then double-labeled with 0.05% (wt/vol) thioflavin-S. Neocortical arterioles (n=30–50 per group) positive for both amyloid-β and thioflavin-S, ranging in diameter from 20 to 100 μm, were randomly imaged by confocal microscopy (×60). The smooth muscle fragmentation was quantified by counting the number of α-actin fragments of each arteriole using ImageJ and was expressed as the fragmentation index: 100−[(1/number of α-actin fragments)×100].
For pericyte identification, sections were processed for immunofluorescence with anti-rabbit Col IV (1:500; Abcam) and the pericyte marker platelet-derived growth factor receptor-β (10 μg/mL; R&D Systems). Sections were then incubated with donkey fuorescein isothiocyanate–conjugated anti-rabbit or cyanine dye 5-conjugated anti-goat IgG. Images were obtained with a confocal microscope, and platelet-derived growth factor receptor-β+ cell bodies and Col IV+ vessels were counted in random fields using ImageJ and presented as number of Col IV+ vessels or platelet-derived growth factor receptor-β+ cell bodies/mm2.
Experimental Protocol for CBF Experiments
CBF recordings were started after arterial pressure, and blood gases were in a steady state (Tables I and II in the online-only Data Supplement). To study the increase in CBF produced by neural activity, the whiskers were mechanically stimulated for 60 s. The endothelium-dependent vasodilators, acetylcholine (10 μmol/L; Sigma), bradykinin (50 μmol/L; Sigma), and the calcium ionophore A23187 (3 μmol/L; Sigma), were superfused over the cortex, and the resulting changes in CBF were monitored. CBF responses to the NO donor S-nitroso-N-acetylpenicillamine (50 μmol/L; Sigma), adenosine (400 μmol/L; Sigma), or hypercapnia (Pco2; 50–60 mm Hg) were also tested.21,22 In some studies, responses were tested before and 30 minutes after superfusion with Ringer containing vehicle or MnTBAP (100 μmol/L).
Data Analysis
Mice treated with MnTBAP were randomly assigned to the vehicle or treatment group. Histological and biochemical analyses were performed in a blinded fashion. Data are expressed as mean±SEM. Two-group comparisons were analyzed by the 2-tailed t test. Multiple comparisons were evaluated by the ANOVA and Tukey test. Differences were considered statistically significant for P<0.05.
Results
Age-Dependent Alterations in Cerebrovascular Function in Tg-SwDI Mice
First, we examined key regulatory responses of the cerebral circulation in WT and Tg-SwDI mice at different ages. In WT mice, the increase in CBF elicited by neural activity (whisker stimulation) and by the endothelium-dependent vasoactive agents, acetylcholine and bradykinin, but not A23187, were attenuated at 18 and 24 months (Figure 1A and 1B). In contrast, CBF responses evoked by the smooth muscle relaxants adenosine or the NO donor S-nitroso-N-acetylpenicillamine were not significantly attenuated, and the response to hypercapnia was attenuated only at 24 months (Figure 1C). In Tg-SwDI mice, CBF responses to whisker stimulation, hypercapnia, endothelium-dependent agents, including A23187, and S-nitroso-N-acetylpenicillamine were already reduced at 3 months (Figure 1A–1C). However, the response to adenosine was not attenuated (Figure 1C). With aging (18 and 24 months), cerebrovascular responses were suppressed further in Tg-SwDI mice, and the response to adenosine, which was intact at 3 months, was markedly attenuated (Figure 1C).
Figure 1.
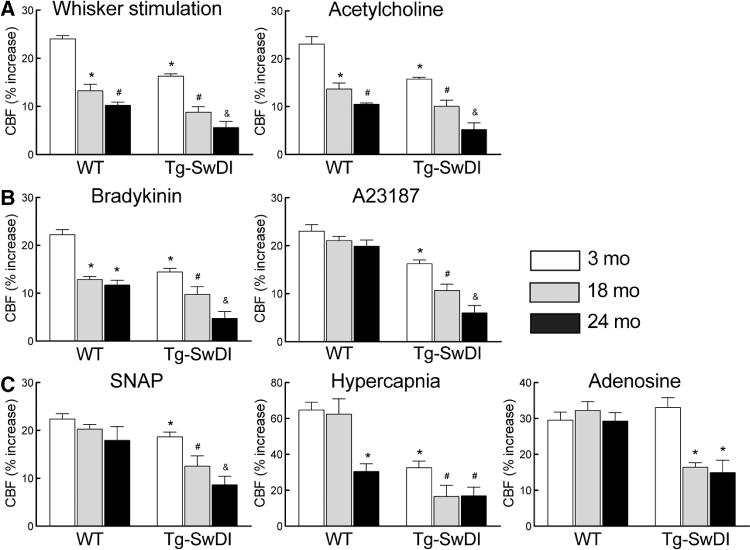
Age-dependent attenuation of cerebral blood flow (CBF) responses in transgenic mice expressing the Swedish, Iowa, and Dutch mutations of the amyloid precursor protein (Tg-SwDI). Increase in CBF produced by whisker stimulation or acetylcholine (A), bradykinin or A23187 (B), S-nitroso-N-acetylpenicillamine (SNAP), hypercapnia, and adenosine (C) in 3-, 18-, and 24-month-old Tg-SwDI and wild-type (WT) mice. *P<0.05 from 3-month-old WT, #P<0.05 from 3- or 18-month-old WT or Tg-SwDI mice, and &P<0.05 from 18-month-old Tg-SwDI mice, ANOVA and Tukey test; n=5 per group.
Age-Dependent Alterations in Cerebrovascular Structure in Tg-SwDI Mice
Next, we examined the cerebrovascular damage induced by amyloid-β accumulation in Tg-SwDI mice. Brain levels of amyloid-β1–40 and amyloid-β1–42 were already elevated in 3-month-old Tg-SwDI mice (Figure 2A), but there was no amyloid deposition in the somatosensory cortex or its blood vessels, as previously reported.11,14 Amyloid-β levels increased dramatically at 18 and 24 months (Figure 2A), and at 18 months there were abundant amyloid deposits in cerebral arterioles and capillaries (Figure 2B and 2C). Concurrent with the amyloid deposition, microvessels developed a more irregular appearance and smooth muscle cells became fragmented (Figure 2B and 2C). Because pericytes are susceptible to the cytotoxic effects of amyloid-β,20,23,24 we examined their number and morphology using platelet-derived growth factor receptor-β as a marker in the somatosensory cortex. In 3-month-old WT and Tg-SwDI mice, pericytes had a normal appearance and number20 (Figure 3A). However, at 18 months, pericytes were reduced in number and their processes were shorter (Figure 3A–3C). No differences in the number of vascular profiles were observed in WT and Tg-SwDI mice at 18 months when compared with those at 3 months (Figure 3C).
Figure 2.
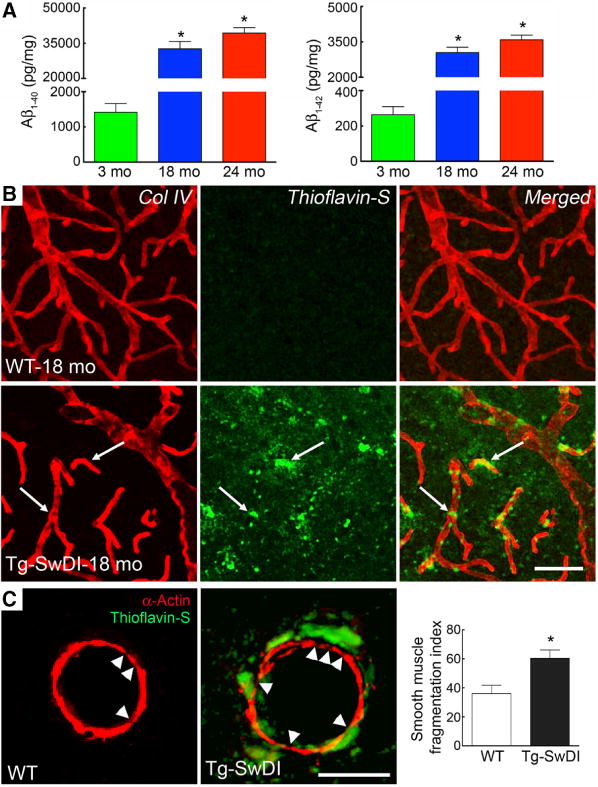
Amyloid-β (Aβ) increases age dependently and accumulates in neocortical vessels, leading to smooth muscle damage in transgenic mice expressing the Swedish, Iowa, and Dutch mutations of the amyloid precursor protein (Tg-SwDI). A, The brain concentration of Aβ increases age dependently (*P<0.05, ANOVA and Tukey test; n=5 per group). B, Aβ (thioflavin-S) accumulates in collagen IV (Col IV)-positive somatosensory cortex capillaries. C, Aβ (thioflavin-S) accumulates around α-actin+ cerebral arteries in 18-month-old Tg-SwDI mice when compared with wild-type (WT) mice. The number of smooth muscle cell fragments, expressed as fragmentation index, is increased in Tg-SwDI mice when compared with age-matched WT mice (*P<0.05, t test; n=30–50 arterioles/group). Scale bar, 50 μm in B and 20 μm in C.
Figure 3.
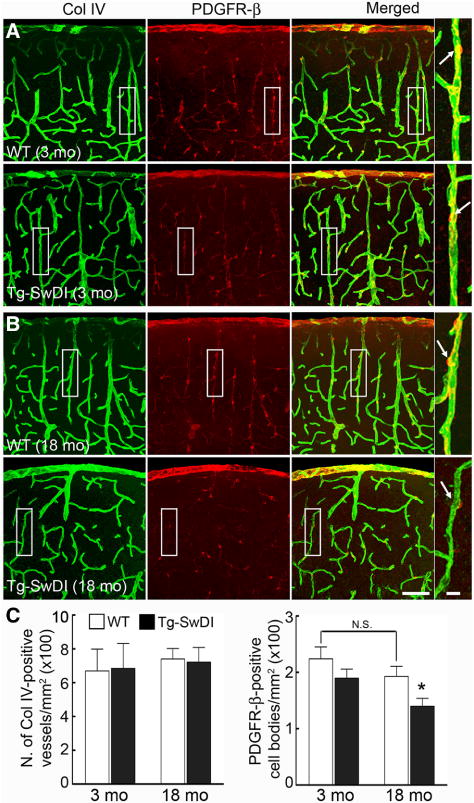
Pericyte morphology is altered in 18-month-old Tg-SwDI mice. A and B, Representative images of pericytes, identified by platelet-derived growth factor receptor-β (PDGFR-β) immunostaining (red), and microvessels, identified by the basement membrane marker collagen VI (Col IV; green), in 3- and 18-month-old transgenic mice expressing the Swedish, Iowa, and Dutch mutations of the amyloid precursor protein (Tg-SwDI) and age-matched wild-type (WT) mice. C, In Tg-SwDI mice, the number of Col IV–positive vessels is comparable between groups, but pericyte cell bodies are reduced. *P<0.05 from 3-month-old Tg-SwDI and 18-month-old WT, ANOVA and Tukey test; n=5 per group. Scale bar, 75 μm in all panels, except for the enlarged merged images (10 μm).
Role of Oxidative Stress in the Cerebrovascular Dysfunction
ROS have been implicated in the cerebrovascular effects of aging and amyloid-β.16,18,21 Therefore, we examined whether ROS production was increased in the brains of Tg-SwDI mice and if so, whether the cerebrovascular dysfunction could be rescued by the treatment with the ROS scavenger MnTBAP. In Tg-SwDI mice, ROS production in somatosensory cortex was higher than that of WT mice, an effect observed both at 3 and at 18 months (Figure 4A). MnTBAP attenuated the ROS-dependent fluorescence both in WT and in Tg-SwDI mice (Figure 4A). However, in WT mice, the cerebrovascular dysfunction was completely reversed by MnTBAP at all ages; but in Tg-SwDI mice, complete rescue was observed only at 3 months (Figure 4B). In 18-month-old Tg-SwDI mice, MnTBAP improved the CBF response to whisker stimulation and acetylcholine but had no statistically significant effect on responses to adenosine and hypercapnia (Figure 4B and 4C).
Figure 4.
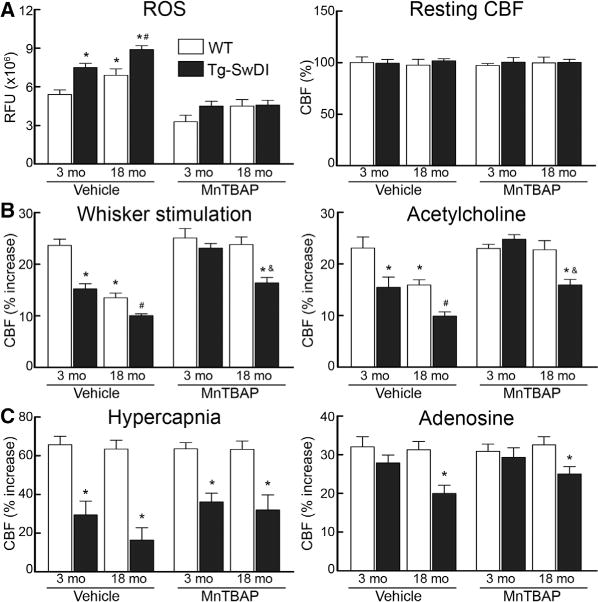
Manganic(I–II)meso-tetrakis(4-benzoic acid) porphyrin (MnTBAP) suppresses reactive oxygen species (ROS) production but fails to rescue cerebrovascular responses in aged transgenic mice expressing the Swedish, Iowa, and Dutch mutations of the amyloid precursor protein (Tg-SwDI). Effect of neocortical superfusion of MnTBAP or vehicle on ROS production or resting cerebral blood flow (CBF; A) and on the increase in CBF elicited by whisker stimulation or acetylcholine (B), hypercapnia or adenosine (C) in 3- and 18-month-old wild-type (WT) and Tg-SwDI mice. *P<0.05 from 3-month-old WT, #P<0.05 from 3-month-old Tg-SwDI mice, and &P<0.05 from vehicle-treated 18-month-old Tg-SwDI mice; ANOVA and Tukey test; n=5 per group. LDU indicates laser-Doppler perfusion units; and RFU, relative fluorescence unit.
Discussion
Novel Findings of the Study
The present study describes several novel findings. First, we demonstrated that Tg-SwDI mice, a model of CAA, exhibit age-dependent cerebrovascular dysfunction, which manifest itself first with alterations in neurovascular coupling, hypercapnia, and endothelium-dependent vasodilatation, and then progresses in older mice to a global vasomotor dysfunction involving all responses independently of their mechanism. Second, we found that the global cerebrovascular dysfunction in older Tg-SwDI mice is associated with marked alterations in the vasomotor apparatus, involving damage to smooth muscle cells and loss of pericytes. Third, we demonstrated that the cerebrovascular dysfunction could be rescued by short-term treatment with an ROS scavenger at 3 months, and that these beneficial effects were less evident in older mice, when vascular damage sets in. These observations collectively indicate that, in the absence of CAA, the cerebrovascular dysfunction induced by amyloid-β is fully reversible by counteracting oxidative stress, except for the response to hypercapnia. With the development of CAA, vascular damage occurs and the dysfunction broadens to involve all the CBF responses studied and is no longer reversible by ROS scavengers.
Exclusion of Potential Sources of Artifacts
The findings of the present study cannot result from differences in the physiological variables of the mice because arterial pressure, blood gases, and body temperature were monitored and did not differ among the groups studied. Differences in the genetic background of the mice are unlikely to play a role in the findings because Tg-SwDI mice were studied longitudinally at different ages, and mice were congenic with the C57BL/6 background, the strain used as WT control. Furthermore, the rescue of the responses by MnTBAP in 3-month-old Tg-SwDI mice cannot be attributed to a nonspecific enhancement of baseline responses because this agent did not affect resting CBF or the magnitude of CBF increases in WT mice.
Tg-SwDI Mice Have a Profound Impairment in Cerebrovascular Function
We found that functional hyperemia, endothelium-dependent responses, and CBF responses to hypercapnia were altered in Tg-SwDI mice starting at 3 months. The involvement of hypercapnia is at variance with the vascular dysfunction observed in 3-month-old Tg2576 mice in which the hypercapnic response is not altered.15,18 Although the reasons for this difference remain to be established, the findings suggest a broad impairment in the mechanisms of the regulation of the cerebral circulation in Tg-SwDI mice, which is more pronounced than that observed in Tg2576 mice. One possibility is that amyloid-β with the Dutch–Iowa mutations produced by Tg-SwDI is more vasotoxic than WT amyloid-β produced by Tg2576, also leading to alterations of the hypercapnic vasodilatation. Furthermore, it remains to be established whether the dysfunction is related to amyloid-β–induced enhancement of constrictor mechanisms,25 which may antagonize the effect of the vasodilatatory stimuli. Additional studies are required to examine these outstanding questions.
It is likely that the attenuation in functional hyperemia and response to acetylcholine is related to a reduction in the bioavailability of NO, which participates in these response.1,26 However, the attenuation of the CBF increase evoked by A23187 and bradykinin cannot be attributed to the lack of NO because these responses are mediated by COX-1.27–29 However, the CBF response to adenosine was not affected in 3-month-old Tg-SwDI mice, indicating that the vasomotor function of smooth muscle cells was not affected at this age. However, at 18 and 24 months, the response to adenosine was attenuated, suggesting an impairment of the vasomotor apparatus. This observation is consistent with our neuropathological data, demonstrating loss of smooth muscle cell integrity and pericytes at this age. Therefore, it is likely that damage to these vasomotor cells at 18 and 24 months contributes to the worsening of functional hyperemia and endothelium-dependent responses observed at this age. Because the vascular dysfunction is present also without vascular amyloid accumulation, the data suggest that amyloid-β in soluble form is sufficient to induce vascular dysfunction. However, it would be of interest to correlate the progression of the vascular dysfunction with the development of the vascular pathology in arterioles and capillaries.
Pericytes, intramural vascular cells enriched in capillaries, have recently been implicated in CBF regulation.30 Our data demonstrating pericyte loss concomitant with a total failure of all vascular responses in aged Tg-SwDI mice are consistent with a role of these cells in cerebrovascular dysfunction. This conclusion is reinforced by the finding that the amyloid accumulation in this model occurs abundantly in capillaries.11 Pericytes in culture internalize amyloid-β, which may lead to their death,24,31 although in Tg-SwDI mice amyloid-β accumulation has been observed in microglia–macrophages but not in pericytes.23 Both pericyte loss and smooth muscle cell damage could play a role in the vascular dysfunction, but their relative contribution remains to be established.
ROS Contribution to the Cerebrovascular Dysfunction
Another major finding of this study is that acute application of the ROS scavenger MnTBAP suppressed ROS production and ameliorated vascular function. However, MnTBAP was most effective in 3-month-old mice, in which MnTBAP completely restored responses to whisker stimulation and acetylcholine. In 18-month-old mice, MnTBAP restored the response only partially. At this time, pronounced alterations in smooth muscle cell structure and pericyte number were observed. These findings suggest that the ability of acute administration of free radical scavengers to counteract the effects of CAA is greatest before the vascular damage sets in. However, we cannot rule out that a more prolonged treatment with MnTBAP or treatment with other agents could be more effective. Premenopausal female mice are resistant to the cerebrovascular dysfunction induced by angiotensin, an effect that depends on the suppression of ROS production by ovarian hormones during the estrous cycle.32,33 It remains to be determined whether such protection is observed in premenopausal Tg-SwDI female mice and is lost in the postmenopausal period.
Interestingly, MnTBAP did not rescue the response to hypercapnia even in 3-month-old Tg-SwDI mice. This observation indicates that ROS do not contribute to the effects of CAA on the response to hypercapnia. Because the vasculature is not damaged at this age,14 the findings implicate that functional alterations are distinct from those underlying the attenuation of functional hyperemia and endothelium-dependent responses, which were reversed by MnTBAP in this age group. The nature of these mechanisms remains unclear and needs further investigations.
Implications for CAA
Inasmuch as Tg-SwDI mice reflect the human disease, the present findings provide the mechanistic bases for the alterations in CBF regulation reported in patients with CAA. Furthermore, they suggest that the functional alterations induced by CAA are amenable to the treatment in the early stages of the disease process, before structural alterations of cerebral blood vessels occur. At this time, the deleterious vascular effects of amyloid are largely reversible. Unfortunately, current imaging approaches for CAA diagnosis rely on microhemorrhages, reflecting the presence of vascular damage, and early diagnosis of CAA is not yet feasible.9,34,35 A limitation of the CAA model used in the present study is that the pathology is present mainly in microvessels, which is not the case in most patients with sporadic CAA.1,9 However, a mitigating factor is that we found a similar ROS-dependent neurovascular dysfunction in aged Tg2576 mice, in which the pattern of vascular amyloid deposition resembles patients with CAA more closely.1,20 In that study, we observed that the innate immunity receptor CD36 is involved in the vascular amyloid deposition.20 It remains to be established whether CD36 has a similar role in Tg-SwDI mice.
Conclusions
We have demonstrated that Tg-SwDI mice, which recapitulate selected features of human CAA, have a profound neurovascular dysfunction that involves not only functional hyperemia and endothelium-dependent responses but also the CBF increase induced by hypercapnia. The dysfunction is age dependent and is greater in older mice, in which profound structural and functional alterations of the cerebrovascular vasomotor apparatus (smooth muscle cells and pericytes) are observed. The ROS scavenger MnTBAP is able to rescue the alterations in functional hyperemia and endothelium-dependent responses in young mice completely, but not in older mice, in which vascular damage has occurred. The findings implicate oxidative stress in the mechanisms of neurovascular dysfunction in Tg-SwDI mice and suggest that treatments for the damaging vascular effects of amyloid are likely to be most effective if instituted early in the course of the disease.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by National Institutes of Health grants NS37853 (Dr Iadecola), NS052533 (Dr Van Nostrand), and NS41997 (Dr Carlson), by anonymous donor bequest to McLaughlin Research Institute (Dr Carlson), by the Alzheimer Association (Zenith Award to Dr Iadecola) and by the American Heart Association Grant 09SDG2060701 (Dr Park). The continued support of the Feil Family Foundation is gratefully acknowledged.
Footnotes
Guest Editor for this article was Malcolm R. Macleod, PhD, FRCP.
The online-only Data Supplement is available with this article at http://stroke.ahajournals.org/lookup/suppl/doi:10.1161/STROKEAHA.114.005179/-/DC1.
Disclosures
None.
References
- 1.Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844–866. doi: 10.1016/j.neuron.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 3.Weller RO, Boche D, Nicoll JA. Microvasculature changes and cerebral amyloid angiopathy in Alzheimer’s disease and their potential impact on therapy. Acta Neuropathol. 2009;118:87–102. doi: 10.1007/s00401-009-0498-z. [DOI] [PubMed] [Google Scholar]
- 4.Kimberly WT, Gilson A, Rost NS, Rosand J, Viswanathan A, Smith EE, et al. Silent ischemic infarcts are associated with hemorrhage burden in cerebral amyloid angiopathy. Neurology. 2009;72:1230–1235. doi: 10.1212/01.wnl.0000345666.83318.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, et al. American Heart Association Stroke Council, Council on Epidemiology and Prevention, Council on Cardiovascular Nursing, Council on Cardiovascular Radiology and Intervention, and Council on Cardiovascular Surgery and Anesthesia Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:2672–2713. doi: 10.1161/STR.0b013e3182299496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Attems J, Jellinger K, Thal DR, Van Nostrand W. Review: sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol. 2011;37:75–93. doi: 10.1111/j.1365-2990.2010.01137.x. [DOI] [PubMed] [Google Scholar]
- 7.Charidimou A, Gang Q, Werring DJ. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiatry. 2012;83:124–137. doi: 10.1136/jnnp-2011-301308. [DOI] [PubMed] [Google Scholar]
- 8.Peca S, McCreary CR, Donaldson E, Kumarpillai G, Shobha N, Sanchez K, et al. Neurovascular decoupling is associated with severity of cerebral amyloid angiopathy. Neurology. 2013;81:1659–1665. doi: 10.1212/01.wnl.0000435291.49598.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Viswanathan A, Greenberg SM. Cerebral amyloid angiopathy in the elderly. Ann Neurol. 2011;70:871–880. doi: 10.1002/ana.22516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dumas A, Dierksen GA, Gurol ME, Halpin A, Martinez-Ramirez S, Schwab K, et al. Functional magnetic resonance imaging detection of vascular reactivity in cerebral amyloid angiopathy. Ann Neurol. 2012;72:76–81. doi: 10.1002/ana.23566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, et al. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem. 2004;279:20296–20306. doi: 10.1074/jbc.M312946200. [DOI] [PubMed] [Google Scholar]
- 12.Fan R, Xu F, Previti ML, Davis J, Grande AM, Robinson JK, et al. Minocycline reduces microglial activation and improves behavioral deficits in a transgenic model of cerebral microvascular amyloid. J Neurosci. 2007;27:3057–3063. doi: 10.1523/JNEUROSCI.4371-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu F, Grande AM, Robinson JK, Previti ML, Vasek M, Davis J, et al. Early-onset subicular microvascular amyloid and neuroinflammation correlate with behavioral deficits in vasculotropic mutant amyloid betaprotein precursor transgenic mice. Neuroscience. 2007;146:98–107. doi: 10.1016/j.neuroscience.2007.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park L, Zhou P, Koizumi K, El Jamal S, Previti ML, Van Nostrand WE, et al. Brain and circulating levels of Aβ1–40 differentially contribute to vasomotor dysfunction in the mouse brain. Stroke. 2013;44:198–204. doi: 10.1161/STROKEAHA.112.670976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iadecola C, Zhang F, Niwa K, Eckman C, Turner SK, Fischer E, et al. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci. 1999;2:157–161. doi: 10.1038/5715. [DOI] [PubMed] [Google Scholar]
- 16.Park L, Anrather J, Forster C, Kazama K, Carlson GA, Iadecola C. Abeta-induced vascular oxidative stress and attenuation of functional hyperemia in mouse somatosensory cortex. J Cereb Blood Flow Metab. 2004;24:334–342. doi: 10.1097/01.WCB.0000105800.49957.1E. [DOI] [PubMed] [Google Scholar]
- 17.Park L, Anrather J, Zhou P, Frys K, Pitstick R, Younkin S, et al. NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J Neurosci. 2005;25:1769–1777. doi: 10.1523/JNEUROSCI.5207-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, et al. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci U S A. 2008;105:1347–1352. doi: 10.1073/pnas.0711568105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tong XK, Nicolakakis N, Kocharyan A, Hamel E. Vascular remodeling versus amyloid beta-induced oxidative stress in the cerebrovascular dysfunctions associated with Alzheimer’s disease. J Neurosci. 2005;25:11165–11174. doi: 10.1523/JNEUROSCI.4031-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park L, Zhou J, Zhou P, Pistick R, El Jamal S, Younkin L, et al. Innate immunity receptor CD36 promotes cerebral amyloid angiopathy. Proc Natl Acad Sci U S A. 2013;110:3089–3094. doi: 10.1073/pnas.1300021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park L, Anrather J, Girouard H, Zhou P, Iadecola C. Nox2-derived reactive oxygen species mediate neurovascular dysregulation in the aging mouse brain. J Cereb Blood Flow Metab. 2007;27:1908–1918. doi: 10.1038/sj.jcbfm.9600491. [DOI] [PubMed] [Google Scholar]
- 22.Niwa K, Araki E, Morham SG, Ross ME, Iadecola C. Cyclooxygenase-2 contributes to functional hyperemia in whisker-barrel cortex. J Neurosci. 2000;20:763–770. doi: 10.1523/JNEUROSCI.20-02-00763.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miao J, Xu F, Davis J, Otte-Höller I, Verbeek MM, Van Nostrand WE. Cerebral microvascular amyloid beta protein deposition induces vascular degeneration and neuroinflammation in transgenic mice expressing human vasculotropic mutant amyloid beta precursor protein. Am J Pathol. 2005;167:505–515. doi: 10.1016/s0002-9440(10)62993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, Ramanathan A, et al. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat Commun. 2013;4:2932. doi: 10.1038/ncomms3932. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Niwa K, Porter VA, Kazama K, Cornfield D, Carlson GA, Iadecola C. A beta-peptides enhance vasoconstriction in cerebral circulation. Am J Physiol Heart Circ Physiol. 2001;281:H2417–H2424. doi: 10.1152/ajpheart.2001.281.6.H2417. [DOI] [PubMed] [Google Scholar]
- 26.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 27.Niwa K, Haensel C, Ross ME, Iadecola C. Cyclooxygenase-1 participates in selected vasodilator responses of the cerebral circulation. Circ Res. 2001;88:600–608. doi: 10.1161/01.res.88.6.600. [DOI] [PubMed] [Google Scholar]
- 28.Rosenblum WI. Hydroxyl radical mediates the endothelium-dependent relaxation produced by bradykinin in mouse cerebral arterioles. Circ Res. 1987;61:601–603. doi: 10.1161/01.res.61.4.601. [DOI] [PubMed] [Google Scholar]
- 29.Rosenblum WI, Nelson GH. Endothelium dependence of dilation of pial arterioles in mouse brain by calcium ionophore. Stroke. 1988;19:1379–1382. doi: 10.1161/01.str.19.11.1379. [DOI] [PubMed] [Google Scholar]
- 30.Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–427. doi: 10.1016/j.neuron.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilhelmus MM, Otte-Höller I, van Triel JJ, Veerhuis R, Maat-Schieman ML, Bu G, et al. Lipoprotein receptor-related protein-1 mediates amyloid-beta-mediated cell death of cerebrovascular cells. Am J Pathol. 2007;171:1989–1999. doi: 10.2353/ajpath.2007.070050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Capone C, Anrather J, Milner TA, Iadecola C. Estrous cycle-dependent neurovascular dysfunction induced by angiotensin II in the mouse neocortex. Hypertension. 2009;54:302–307. doi: 10.1161/HYPERTENSIONAHA.109.133249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Girouard H, Lessard A, Capone C, Milner TA, Iadecola C. The neurovascular dysfunction induced by angiotensin II in the mouse neocortex is sexually dimorphic. Am J Physiol Heart Circ Physiol. 2008;294:H156–H163. doi: 10.1152/ajpheart.01137.2007. [DOI] [PubMed] [Google Scholar]
- 34.Charidimou A, Jaunmuktane Z, Baron JC, Burnell M, Varlet P, Peeters A, et al. White matter perivascular spaces: an MRI marker in pathology-proven cerebral amyloid angiopathy? Neurology. 2014;82:57–62. doi: 10.1212/01.wnl.0000438225.02729.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yates PA, Villemagne VL, Ellis KA, Desmond PM, Masters CL, Rowe CC. Cerebral microbleeds: a review of clinical, genetic, and neuroimaging associations. Front Neurol. 2014;4:205. doi: 10.3389/fneur.2013.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.