

Significance
Coenzyme A (CoA) is a small-molecular-weight thiol that plays a central role in cellular metabolism. We have discovered a novel, phylogenetically conserved class of enzymes that reduce S-nitroso-CoA (SNO-CoA) and thereby regulate protein S-nitrosylation. These denitrosylases, identified as alcohol dehydrogenase 6 (Adh6) in yeast and aldo-keto reductase 1A1 in mammals, may be analogized to deacetylases, which regulate CoA-mediated protein acetylation. In yeast, Adh6 (previously without ascribed cellular function) regulates endogenous protein S-nitrosylation (heretofore unknown) including function-altering S-nitrosylation that impacts CoA-related metabolism. Thus, our findings establish a novel role for CoA in protein S-nitrosylation (operating through SNO-CoA), which is governed by specific enzymes. This mechanism may regulate the influence of nitric oxide on cellular metabolism in health and disease.
Keywords: denitrosylase, S-nitrosylation, Adh6, AKR1A1, denitrosylation
Abstract
Coenzyme A (CoA) mediates thiol-based acyl-group transfer (acetylation and palmitoylation). However, a role for CoA in the thiol-based transfer of NO groups (S-nitrosylation) has not been considered. Here we describe protein S-nitrosylation in yeast (heretofore unknown) that is mediated by S-nitroso-CoA (SNO-CoA). We identify a specific SNO-CoA reductase encoded by the alcohol dehydrogenase 6 (ADH6) gene and show that deletion of ADH6 increases cellular S-nitrosylation and alters CoA metabolism. Further, we report that Adh6, acting as a selective SNO-CoA reductase, protects acetoacetyl–CoA thiolase from inhibitory S-nitrosylation and thereby affects sterol biosynthesis. Thus, Adh6-regulated, SNO-CoA–mediated protein S-nitrosylation provides a regulatory mechanism paralleling protein acetylation. We also find that SNO-CoA reductases are present from bacteria to mammals, and we identify aldo-keto reductase 1A1 as the mammalian functional analog of Adh6. Our studies reveal a novel functional class of enzymes that regulate protein S-nitrosylation from yeast to mammals and suggest that SNO-CoA–mediated S-nitrosylation may subserve metabolic regulation.
S-nitrosylation, a phylogenetically conserved posttranslational modification of proteins that mediates transduction across a broad spectrum of cellular signaling pathways, involves the covalent addition of NO groups to Cys thiols to generate S-nitrosothiols (SNOs) (1). There is increasing evidence that S-nitrosylation is regulated enzymatically (2, 3). One highly conserved enzyme implicated in regulating S-nitrosylation is represented by S-nitroso-glutathione (GSNO) reductase (GSNOR), which metabolizes the low-molecular-weight SNO, GSNO, by using reducing equivalents from NADH (4). Because many S-nitrosylated proteins (SNO-proteins) are in equilibrium with GSNO, GSNOR plays a major role in regulating protein S-nitrosylation/denitrosylation (4–6).
Coenzyme A (CoA) is an abundant, low-molecular-weight thiol that plays an essential role in cells through involvement in >100 reactions of intermediary metabolism (7, 8). Although CoA can be S-nitrosylated in vitro (9), endogenous S-nitrosylation of CoA has not been reported, and a role for S-nitroso-CoA (SNO-CoA) in protein S-nitrosylation has not been considered. We wondered whether an enzymatic activity might be involved in regulating the abundance of SNO-CoA and thereby protein S-nitrosylation/denitrosylation (analogous to regulation by GSNOR). We focused initially on an experimentally tractable model eukaryote, the yeast Saccharomyces cerevisiae.
Results
Adh6 Is a SNO-CoA Reductase in Yeast.
In extracts of yeast, NADPH, but not NADH, oxidation was greatly enhanced in the presence of SNO-CoA (Fig. 1 A and B), consistent with the operation of an NADPH-specific SNO-CoA reductase. Addition of SNO-CoA to yeast lysates led to the S-nitrosylation of multiple proteins as demonstrated by the SNO-Resin Assisted Capture (SNO-RAC) method (10), and coaddition of NADPH (but not NADH) markedly diminished SNO-protein formation (Fig. 1C). Thus, in yeast, SNO-CoA can serve as a source of NO groups for protein S-nitrosylation that may be regulated by NADPH-dependent SNO-CoA reductase activity. SNO-CoA–metabolizing activity was purified from yeast to homogeneity, as assessed by SNO-CoA–dependent NADPH consumption, and identified as the NADPH-dependent enzyme alcohol dehydrogenase 6 (Adh6; product of the ADH6 gene) (Fig. 1D, Fig. S1 A and B, and Table S1), a member of the cinnamyl alcohol dehydrogenase family (11, 12) with no previously known physiological roles or substrates. NADPH-dependent catabolism of SNO-CoA by Adh6 was confirmed directly with isolated, recombinant Adh6 (Fig. S3A). CoA–sulfinamide was identified by mass spectrometry (MS) as the major stable product of SNO-CoA metabolism (Fig. 2A and Fig. S2 A and B), confirming a reductase mechanism that produces an S-(N-hydroxy)-CoA intermediate (Fig. S2D). Kinetic analysis with SNO-CoA as substrate gave a Km of 180.5 ± 16.8 μM, an estimated kcat of 2,596.5 ± 110.7 min−1 (Fig. 2B and Fig. S3B), and a stoichiometry with cosubstrate NADPH of 1:1 (Fig. 2C). The catalytic efficiency (kcat/Km) of Adh6 (for substrate SNO-CoA) compares closely with that of microbial GSNOR (for substrate GSNO) (4), supporting physiological relevance (and as for GSNOR, a relatively high Km is consistent with a homeostatic functional role). Importantly, Adh6 was specific for SNO-CoA vs. GSNO or S-nitroso-cysteine (CysNO), oxidized CoA or CoA–glutathione mixed disulfide (Fig. 2D and Fig. S4 A and B). Adh6 is the principal source of SNO-CoA–metabolizing activity, because genetic deletion of ADH6 (adh6Δ) resulted in ∼80% decrease in SNO-CoA–consuming activity in lysates, whereas deletion of closely homologous ADH7 (Fig. S5A) had no effect (Fig. 2E).
Fig. 1.

Identification of Adh6 as the NADPH-dependent SNO-CoA reductase in yeast. (A) SNO-CoA–dependent NADPH consumption in yeast (S. cerevisiae). Extracts (800 µg/mL) were incubated with 100 μM SNO-CoA and 100 μM NADPH, and NADPH consumption (absorbance at 340 nm) was monitored continuously. (B) SNO-CoA–metabolizing activity in yeast extracts requires NADPH and not NADH. Extracts were incubated with 200 μM SNO-CoA and 100 μM NADPH or NADH and monitored continuously for 1 min. Data are presented as mean ± SD; n = 3. (C) SNO-CoA–mediated protein S-nitrosylation. Representative Coomassie-stained SDS/PAGE gel displaying SNO-proteins isolated by SNO-RAC following incubation of yeast lysate for 10 min with SNO-CoA (60 μM) alone or in combination with NADPH or NADH (100 μM). Ascorbate (Asc) was omitted from the SNO-RAC assay as a specificity control. Results are representative of three independent experiments. (D) Isolation and identification of SNO-CoA reductase. Representative Coomassie-stained SDS/PAGE gel corresponding to the five-step chromatographic purification scheme detailed in Table S1, which yielded from a crude extract (lane 1) 2,826-fold enrichment of NADPH-dependent SNO-CoA reductase activity identified by MS as Adh6 (lane 6).
Fig. 2.

Characterization of the yeast SNO-CoA reductase, Adh6. (A) CoA-sulfinamide was identified by MS as the major stable product of SNO-CoA reduction by purified Adh6 (see Fig. S2 A, B, and D for product analysis). (B) Kinetic analysis of SNO-CoA reductase activity of purified Adh6. (C) Stoichiometry of NADPH:SNO-CoA in Adh6-catalyzed SNO-CoA reduction. Sequential additions of 87 μM NADPH to an excess of SNO-CoA led to consumption of 79–85 μM (mean 82 ± 3 μM; n = 6 additions) of SNO-CoA, demonstrating a stoichiometry of 1:1. Results shown are representative of two independent experiments. (D) Specificity of Adh6 for SNO-CoA. Purified Adh6 (Table S1 and Fig. 1D) (20 nM) was incubated with NADPH (100 µM) and SNO-CoA, GSNO, or CysNO (100 µM), and NADPH consumption was measured over time. (E) Adh6 is the principal source of NADPH-dependent SNO-CoA reductase activity. Activity was assayed in lysates from WT yeast and adh6Δ, adh7Δ, and adh6Δadh7Δ yeast. Data are presented as mean ± SD; n = 3.
SNO-CoA–Mediated Protein S-Nitrosylation Is Regulated by Adh6.
In yeast, GSNOR-regulated denitrosylation of SNO-proteins (coupled to metabolism of GSNO) protects against nitrosative stress imposed by exogenous NO, as demonstrated by enhanced susceptibility to nitrosative challenge in GSNOR-null cells (4, 13). In contrast, deletion of ADH6 did not affect the growth response to nitrosative stress (Fig. S5B), suggesting distinct functions for SNO-CoA and GSNO. To reveal possible roles for SNO-CoA—and in particular to explore a possible role for SNO-CoA in metabolic signaling—we used a MS-based approach to identify substrates of SNO-CoA–mediated, Adh6-regulated protein S-nitrosylation. We treated lysates of wild-type (WT) yeast with SNO-CoA (which is not cell-permeable) and treated intact WT and adh6Δ yeast with the cell-permeable S-nitrosylating agent, S-nitroso-cysteine ethyl ester (EtCysNO). NO groups originating from EtCysNO will distribute among intracellular SNOs (14), forming SNO-CoA (Fig. S6), and as shown in Fig. 3A and detailed below, there is substantial overlap between the sets of proteins S-nitrosylated by EtCysNO or SNO-CoA. SNO-proteins were captured by SNO-RAC (10), and tryptic peptides were quantified by isobaric tags for relative and absolute quantification (iTRAQ) and liquid chromatography-coupled tandem MS (LC-MS/MS) (detailed in SI Materials and Methods).
Fig. 3.
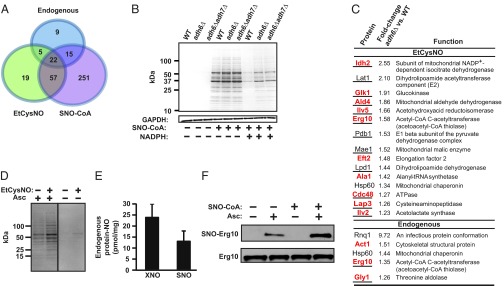
Adh6 regulates protein S-nitrosylation mediated by SNO-CoA. (A) A Venn diagram illustrates the relationships between the sets of SNO proteins identified in intact WT yeast under basal growth conditions (endogenous SNO proteins) or following treatment with EtCysNO, a cell-permeable S-nitrosylating agent, and in lysates treated with SNO-CoA. (B) Adh6-regulated protein S-nitrosylation. Representative Coomassie-stained SDS/PAGE gel illustrating SNO-proteins isolated by SNO-RAC following treatment of WT yeast lysates with SNO-CoA (in the presence or absence of NADPH) and the regulation by Adh6 of protein S-nitrosylation. Results are representative of three independent experiments. (C) SNO-CoA–mediated protein S-nitrosylation in situ. (Upper) SNO-proteins exhibiting enhanced S-nitrosylation in adh6Δ vs. WT yeast following treatment with EtCysNO. (Lower) Endogenous SNO proteins showing enhanced S-nitrosylation in adh6Δ vs. WT yeast under basal growth conditions. Shared targets identified following treatment of lysates with SNO-CoA are indicated in red, and metabolic enzymes are underlined. n = 3; P < 0.05 by Student t test; relative standard deviation < 35%. (D) Isolation of endogenous and in-situ-formed SNO-proteins. Representative Coomassie-stained SDS/PAGE gel illustrating endogenous SNO-proteins isolated by SNO-RAC from WT yeast (untreated) as well as SNO-proteins formed in situ (EtCysNO). Shared targets are shown in A. (Note that the demonstration of endogenous SNO-proteins in D but not B reflects different amounts of total protein used in the SNO-RAC assay (4 vs. 1 mg, respectively). Results are representative of three independent experiments. (E) Endogenous protein-bound NO. Total protein-bound NO (XNO; which includes FeNO and SNO) and SNO were quantified in WT yeast extracts by mercury-coupled photolysis-chemiluminescence. Data are presented as mean ± SEM (n = 7). (F) Endogenous S-nitrosylation of thiolase and its enhanced S-nitrosylation by SNO-CoA. Results of a representative analysis by SNO-RAC of endogenous and SNO-CoA–induced S-nitrosylation of Erg10. Data are representative of two independent experiments.
Treatment of lysates with SNO-CoA (60 µM, 10 min) resulted in the identification of 345 SNO-proteins (Fig. 3A and Dataset S1). SNO-CoA–induced protein S-nitrosylation was greatly attenuated in WT lysates by the addition of NADPH, and this attenuation was partially eliminated in adh6Δ lysates (Fig. 3B), confirming regulation by Adh6 of SNO-CoA–mediated protein S-nitrosylation. Similarly, treatment of intact cells with EtCysNO (100 µM, 2 h) resulted in the identification of 103 SNO-proteins (Fig. 3C and Dataset S2), and iTRAQ analysis revealed that ADH6 deletion resulted in significantly enhanced S-nitrosylation of 15 of those proteins (Fig. 3C, Upper). Notably, 10 of 15 proteins exhibiting Adh6-dependent enhanced S-nitrosylation after EtCysNO treatment of intact cells were identified as SNO-CoA substrates in lysates (Fig. 3C, Upper; Datasets S1 and S2). The majority of substrates for Adh6-regulated S-nitrosylation were identified as metabolic enzymes (Fig. 3C), including Erg10 (acetoacetyl-CoA thiolase) (Fig. 3C and Fig. S7A), which plays a key role in CoA-dependent sterol biosynthesis (15), as well as several enzymes influencing acyl-CoA levels (see below). We further confirmed that the enhanced S-nitrosylation of Erg10 in adh6Δ yeast did not reflect changes in Erg10 abundance (Fig. S7B).
Endogenous S-Nitrosylation and Regulation by Adh6 of Erg10 Activity.
In yeast, as in bacteria, NO is generated by respiratory enzymes that reduce nitrite and/or nitrate [i.e., NO is produced in the absence of a nitric oxide synthase (NOS) (1, 16)]. However, endogenous protein S-nitrosylation has not been described in yeast, and in general the role of yeast NO is unknown. Our analysis revealed constitutive protein S-nitrosylation in yeast under basal conditions with both SNO-RAC (Fig. 3 C, lower list, and D) and mercury-coupled photolysis/chemiluminescence (Fig. 3E) and identified 51 endogenous SNO proteins (Dataset S2). Notably, 37 of 51 endogenous substrates were also identified as targets of exogenous SNO-CoA (Datasets S1 and S2). In addition, five endogenous SNO-proteins exhibited enhanced basal S-nitrosylation in the absence of Adh6 (Fig. 3C, lower list, and Dataset S2), and three of these substrates were among the set identified as targets of exogenous SNO-CoA, including Erg10 thiolase (Fig. S7C). Acetoacetyl-CoA thiolase has also been identified by proteomic analysis as an endogenous SNO-protein in mammals (17, 18). Both endogenous and exogenous SNO-CoA–mediated S-nitrosylation of Erg10 were confirmed directly by SNO-RAC analysis of untreated extracts and extracts treated with SNO-CoA (60 µM, 10 min) (Fig. 3F and Fig. S7 A and C). Nitrite-dependent NO production by yeast mitochondria is enhanced under hypoxic conditions (16). Supplementation of intact yeast with nitrite (100 µM) under hypoxia to enhance endogenous NO production led to the progressive accumulation of SNO-proteins as assessed by photolysis/chemiluminescence (Fig. 4A), and these increases in endogenous SNO-protein levels were substantially greater in adh6Δ vs. WT yeast (Fig. 4B). Thus, collectively, these data support a role for SNO-CoA in endogenous protein S-nitrosylation in yeast that is regulated by Adh6 and indicate that SNO-CoA–mediated, Adh6-regulated protein S-nitrosylation is coupled to endogenous NO production.
Fig. 4.

S-nitrosylation by SNO-CoA inhibits acetoacetyl-CoA thiolase (Erg10)-dependent metabolism. (A) Endogenous SNO-protein formation in yeast grown under hypoxia in the presence of nitrite. WT yeast were grown for the indicated times in the presence or absence of 100 µM nitrite. SNO levels in lysates were quantified by mercury-coupled photolysis chemiluminescence. Data are presented as mean ± SEM (n = 4). *P < 0.05 (by Student t test). (B) Adh6 regulates endogenous SNO-protein synthesis in yeast. Cells were grown under hypoxia for 24 h in the presence of 0, 100, or 500 µM nitrite, and SNO-protein levels in lysates were quantified as in A. Data are presented as mean ± SEM (n = 5). *P < 0.05 (by Student t test). (C) Erg10 activity is inhibited by Adh6-regulated, SNO-CoA–mediated S-nitrosylation. Erg10 activity was assayed in extracts of WT or adh6Δ yeast grown under hypoxia with or without 100 µM nitrite (as in A and B) or treated at normoxia with EtCysNO (100 µM). Data are mean ± SD (n = 3). *P < 0.05 vs. untreated; †P < 0.05 adh6Δ vs. WT by ANOVA. (D and E) Selective inhibition of Erg10 by SNO-CoA vs. GSNO (D) or succinyl-CoA (E). Erg10 activity was assayed in extracts of WT yeast in the presence or absence of SNO-CoA (D and E), GSNO (D), or succinyl-CoA (E). (F–H) Effects of enhanced S-nitrosylation on the metabolic profile of the mevalonate pathway. Midlog phase yeast were untreated or treated with 100 µM (F) or 500 µM (G and H) EtCysNO for 2, and metabolites (mevalonate, CoA, and acetyl-CoA) were measured as described in SI Materials and Methods. Data are mean ± SD (n = 5 or 6). *P < 0.05 (by Student t test in F and G and by ANOVA in H, normalized with respect to culture turbidity). Note in F that erg10–DAmP yeast exhibits ∼50% of WT thiolase activity (Fig. S7D). (I) Schematic summary of the potential role of SNO-CoA–mediated protein S-nitrosylation in regulation of Erg10-dependent metabolism. Erg10 converts acetyl-CoA to free CoA and acetoacetyl-CoA, a precursor via 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) in mevalonate biosynthesis. S-nitrosylation of Erg10 by SNO-CoA, regulated by Adh6 acting as a SNO-CoA reductase, inhibits thiolase activity, resulting in decreased levels of mevalonate and may contribute to diminished levels of CoA and increased levels of acetyl-CoA. Altered levels of CoA and acetyl-CoA may reflect actions of Adh6-regulated SNO-CoA at additional loci (FAox, fatty acid β-oxidation; PDC, pyruvate dehydrogenase multienzyme complex). SNO-CoA reductase thus acts as a cognate denitrosylase for substrates of SNO-CoA by direct analogy to GSNOR, which acts as a denitrosylase for protein substrates of GSNO.
To illustrate regulation of metabolism through endogenous SNO-CoA–mediated protein S-nitrosylation, we focused on Erg10 (identified as a substrate for Adh6-regulated S-nitrosylation by both endogenous and exogenous SNO-CoA; Fig. 3C and F and Fig. S7 A and C). Nitrite supplementation of hypoxic yeast cultures to enhance endogenous S-nitrosylation resulted in inhibition of acetoacetyl-CoA thiolase activity (Fig. 4C), and this inhibition was significantly greater in adh6Δ vs. WT cells (Fig. 4C), implicating SNO-CoA. Treatment of normoxic cultures with EtCysNO (100 µM) revealed similar regulation of thiolase activity by Adh6 (Fig. 4C), specifically implicating SNO-CoA in the inhibitory effects of EtCysNO (recall that Adh6 does not metabolize CysNO; Fig. 2D). Moreover, we verified in yeast extracts and with purified protein (IC50 = 4 µM) that SNO-CoA potently inhibited Erg10 thiolase activity (Fig. 4 D and E), whereas, notably, neither GSNO (Fig. 4D) nor succinyl–CoA (a CoA analog) (Fig. 4E) had a significant effect. That is, thiolase is selectively inhibited by SNO-CoA, and, thus, the inhibition of thiolase that is coupled to endogenous NO production or exogenous nitrosative stress (and regulated by the SNO-CoA reductase Adh6) is likely to be selectively mediated by SNO-CoA.
Erg10 is a critical component of the CoA-based mevalonate pathway for sterol biosynthesis; yeast lacking Erg10 are mevalonate auxotrophs (15). To establish a functional corollary of thiolase inhibition by SNO-CoA, we carried out metabolomic analyses in WT vs. adh6Δ yeast, focusing on components of CoA-based metabolism, including mevalonate. Metabolomic analysis demonstrated that treatment with EtCysNO, under conditions used to demonstrate SNO-CoA–mediated Erg10 S-nitrosylation (Fig. 3 A and C), resulted in a significant decrease of mevalonate levels in adh6Δ yeast, but not in WT yeast (Fig. 4F), consistent with inhibition of Erg10 activity by SNO-CoA (Fig. 4I). As a measure of the effect of Erg10 inhibition by SNO-CoA on mevalonate biosynthesis, mevalonate levels in EtCysNO-treated adh6Δ yeast were similar to the levels observed in yeast harboring the erg10–DAmp gene (Fig. 4F), which codes for a form of Erg10 that exhibits ∼50% reduction in thiolase activity (Fig. S7D). In addition, metabolomic analysis revealed that treatment with EtCysNO resulted in large decreases in free CoA (Fig. 4G), consistent with formation of SNO-CoA (Fig. S6 A–G) (which escapes detection at physiological concentrations, as is the case for other unstable, short-lived SNOs, including GSNO and CysNO; note that SNO-CoA is not regenerated to CoA by Adh6). However, whereas levels of precursor CoA and downstream product mevalonate were decreased, levels of acetyl-CoA were increased, and this increase was significantly larger in adh6Δ vs. WT cells (Fig. 4H). Metabolic blockade of acetyl-CoA utilization although Erg10, which would also contribute to decreased levels of CoA, may play a role in enhancement of acetyl-CoA levels (Fig. 4I). However, it is well known that acetyl-CoA generation is governed in large part by fatty acid (β-)oxidation, which is enhanced by S-nitrosylation (19), and by the pyruvate dehydrogenase multienzyme complex, and we identified some components of those mechanisms as SNO-proteins (Datasets S1 and S2), including components of the pyruvate dehydrogenase complex that exhibited Adh6-regulated S-nitrosylation (Fig. 3C and Dataset S2). Thus, Adh6-regulated S-nitrosylation may potentially signal through modification of multiple components of CoA-related metabolic pathways. Finally, it is important to note that constitutive levels of acetyl-CoA were increased significantly in adh6Δ vs. WT yeast in the absence of exogenous SNO (Fig. 4H), indicating directly metabolic regulation by endogenous SNO-CoA. Adh6 is thus a newly discovered SNO-CoA reductase that influences CoA metabolism in yeast.
AKR1A1 Is a SNO-CoA Reductase in Mammals.
Adh6 is unique to yeast (phylum Ascomycetes). However, the generality of our findings is indicated by our discovery of NADPH-dependent SNO-CoA reductase activity across a phylogenetic spectrum from bacteria to mammals (Fig. 5 A and B; see also Fig. 6D). As in yeast, addition of SNO-CoA to lysates of mouse tissues led to the S-nitrosylation of multiple proteins, and coaddition of NADPH (but not NADH) markedly diminished SNO-protein formation (Fig. 5C). Using an approach identical in all significant respects to that used in the analysis of Adh6 in yeast, we purified (from bovine liver and kidney) the mammalian SNO-CoA reductase and identified it as aldo-keto reductase 1A1 (AKR1A1) (Fig. 5D and Figs. S2C and S8 A and B). AKR1A1 is the founding member of the AKR superfamily, and orthologs are present across the vertebrate phylum (20). However, the physiological role of AKR1A1 remains unknown, with the exception of a role in vitamin C synthesis that has been demonstrated in the mouse (21), although many mammals, including humans, do not synthesize vitamin C. MS analysis demonstrated that the reductive mechanism (involving hydride transfer; Fig. 6A and Fig. S2D) and product (CoA-sulfinamide; Fig. S2C) of AKR1A1 operating as a SNO-CoA reductase were identical to those of yeast Adh6. Kinetic analysis, with SNO-CoA as substrate, gave a Km of 20.5 ± 1.8 μM and an estimated kcat of 627 ± 23.76 min−1 (Fig. 6B and Fig. S3 C and D), and, as for Adh6, a stoichiometry with cosubstrate NADPH of 1:1 (Fig. 6C). AKR1A1 and Adh6 are evolutionarily unrelated and are therefore functional analogs. The existence of evolutionarily unrelated functional analogs in yeast and mammals indicates strongly that SNO-CoA reductase activity is biologically significant.
Fig. 5.
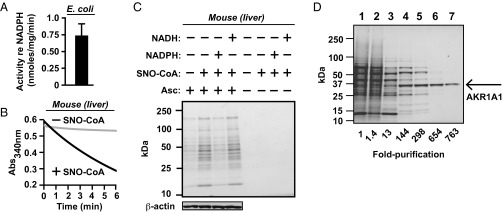
Identification of aldo-keto reductase 1A1 (AKR1A1) as the NADPH-dependent SNO-CoA reductase in mammals. (A and B) Conservation of SNO-CoA reductase activity from bacteria to mammals. NADPH-dependent SNO-CoA metabolizing activity in extracts from Escherichia coli (A) and mouse liver (B) is shown. Conditions in A were similar to Fig. 1B. In B, 160 µg/mL liver extract was incubated with 100 μM SNO-CoA and 100 μM NADPH. Note that high levels of basal NADH consumption (diaphorase activity) in E. coli under aerobic conditions prevented assessment of SNO-CoA dependence. (C) SNO-CoA–mediated protein S-nitrosylation. Representative Coomassie-stained SDS/PAGE gels displaying SNO proteins isolated by SNO-RAC following incubation of mouse liver extract (1 mg/mL in 1 mL of reaction volume) for 10 min with SNO-CoA (60 μM) alone or in combination with NADPH or NADH (100 μM). Ascorbate (Asc) was omitted from the SNO-RAC assay as a specificity control. (D) SNO-CoA reductase isolation. Representative SDS/PAGE gel corresponding to the six-step chromatographic purification scheme detailed in Table S1, which yielded from a crude extract (lane 1) 763-fold enrichment of SNO-CoA reductase activity identified by MS as AKR1A1 (lane 7).
Fig. 6.
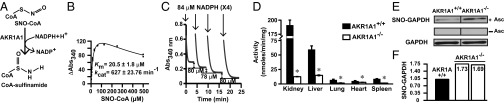
Characterization of the mammalian SNO-CoA reductase AKR1A1. (A) CoA-sulfinamide was identified by MS as the major stable product of SNO-CoA reduction by purified AKR1A1 (see Fig. S2 A, C, and D for product analysis). (B) Kinetic analysis of SNO-CoA reductase activity of purified AKR1A1. (C) Stoichiometry of NADPH:SNO-CoA in AKR1A1-catalyzed SNO-CoA reduction. Sequential additions of 84 μM NADPH to an excess of SNO-CoA led to consumption of 75–82 μM (mean 79 ± 3 μM; n = 8 additions) SNO-CoA, demonstrating a stoichiometry of 1:1. Results shown are representative of two independent experiments. (D) SNO-CoA reductase activity in AKR1A1 knockout animals. NADPH-dependent SNO-CoA reductase activity across various tissues from WT or AKR1A1−/− mice is shown. Extracts were incubated with 100 µM NADPH and 0 or 200 µM SNO-CoA. Values are from three WT (filled) and three AKR1A1−/− (open) mice. *P < 0.05 (Student t test). (E and F) Regulation of endogenous protein S-nitrosylation by AKR1A1. Analysis of S-nitrosylated GAPDH (SNO-GAPDH) in kidney extracts of AKR1A1+/+ and AKR1A1−/− mice is shown. Changes in SNO-GAPDH levels were determined by SNO-RAC coupled to Western blotting (E; n = 2) or to iTRAQ-based MS (F; n = 2).
Transgenic mice bearing an unconditional knockout of AKR1A1 were generated (Fig. S8 C and D) to demonstrate that AKR1A1 is the predominant source of NADPH-dependent SNO-CoA reductase activity in mammalian tissue (Fig. 6D and Fig. S8E), and these results were confirmed by immunodepletion of AKR1A1 from tissue extracts (Fig. S8F). To illustrate regulation by AKR1A1 of endogenous SNO-CoA–mediated protein S-nitrosylation, we focused as an exemplar on glyceraldehyde 3-phosphate dehydrogenase (GAPDH). GAPDH is best characterized among the multitude of mammalian metabolic enzymes that are regulated by S-nitrosylation, which includes both loss- and gain-of-function roles for SNO-GAPDH in metabolic regulation (22), metabolic inflammation (23), and glycolysis (24). Further, GAPDH is among the set of SNO-proteins in which S-nitrosylation is not regulated through coupling to GSH/GSNO (i.e., is independent of GSNOR) (25) and that we find is mediated effectively by SNO-CoA (Dataset S1). Analysis by SNO-RAC in AKR1A1-null and WT tissues, followed by either Western blotting (Fig. 6E) or iTRAQ-MS (Fig. 6F), showed that SNO-GAPDH levels are enhanced substantially in the absence of AKR1A1. Thus, SNO-CoA is likely a principal low-molecular-weight SNO that exists in equilibrium with SNO-GAPDH to regulate GAPDH S-nitrosylation/denitrosylation, with AKR1A1 serving to control that equilibrium.
Discussion
Our results reveal a new functional class of enzymes, SNO-CoA reductases, and establish a phylogenetically conserved role for this enzymatic machinery in the regulation of protein S-nitrosylation. Our findings include, to our knowledge, the first demonstration of endogenous S-nitrosylation in yeast and the identification of SNO-CoA as a novel signaling molecule. Endogenous SNOs in a variety of low-molecular-weight and protein forms may purvey NO bioactivity. However, assignments of individual roles for ephemeral small-molecular-weight SNOs in cellular function can only be achieved through the identification of dedicated enzymes that metabolize individual SNOs (it is important to appreciate in this regard that no enzymes have been found to metabolize NO in mammals that would allow assignment of function to NO that is independent of SNOs). Here, as in previous analyses of the role of GSNO in protein S-nitrosylation based on genetic manipulation of the GSNO-metabolizing enzyme GSNOR (4, 5), we used genetic manipulation of novel, specific SNO-CoA–metabolizing enzymes to establish a role for SNO-CoA in NO-based cellular signaling.
Our findings demonstrate that, in cells producing or otherwise exposed to NO, SNO-CoA may serve as a source of NO groups for protein S-nitrosylation and that protein S-nitrosylation regulated by SNO-CoA reductases may provide a mechanism for metabolic regulation by NO. In particular, in yeast, regulation by the dedicated SNO-CoA reductase, Adh6, of Erg10 S-nitrosylation demonstrates a previously unsuspected locus of control of sterol metabolism. Adh6 may thereby protect yeast against sterol auxotrophy caused by endogenous or exogenous (e.g., host-derived or soil) NO. More generally, the disruption of CoA homeostasis (e.g., CoA depletion) that we observed upon treatment of yeast with an S-nitrosylating agent may exemplify nitrosative stress experienced by microbes upon infection of mammalian cells, and a protective role for Adh6 may likely entail an effective mechanism to regenerate CoA from CoA-sulfinamide (Fig. 2A). The regulation in mammals by a cognate SNO-CoA reductase of GAPDH S-nitrosylation, extensively characterized as an exemplar of metabolic signaling by NO (22–24), supports the prediction that SNO-CoA may convey metabolic signals more broadly. Further, S-nitrosylated GAPDH has been shown recently to function as an S-nitrosylase and thereby to serve as an important regulator of protein acetylation within the nucleus that impacts cellular metabolism (22). Thus, our findings suggest that S-nitrosylation and acetylation provide distinct CoA-based mechanisms for posttranslational protein modification that will together exert a broad functional purview in most or all cells, with implications for both physiology and disease.
Materials and Methods
Purification of Yeast and Mammalian SNO-CoA Reductases.
SNO-CoA–metabolizing activity was purified from a crude extract of yeast cells and bovine kidneys by ammonium sulfate precipitation followed by several steps of column chromatography (SI Materials and Methods). The specific activity of NADPH-dependent SNO-CoA reduction was used to assess purification at each step.
Kinetic Parameters of Adh6 and AKR1A1.
Kinetic analysis was carried out with purified Adh6 and AKR1A1. The reactions contained a fixed concentration of NADPH (100 µM) and several concentrations of SNO-CoA. Further details are provided in SI Materials and Methods.
Metabolomic Analysis.
In extracts of WT and adh6Δ yeast (untreated and following treatment with EtCysNO), mevalonate, CoA, and acetyl-CoA were separated and quantified by GC-MS (mevalonate) or LC-MS/MS (CoA and acetyl-CoA), as described in SI Materials and Methods.
SNO-RAC Assay and iTRAQ Labeling for Quantification of Protein S-Nitrosylation.
Protein S-nitrosylation was assessed by the SNO-RAC assay (10), and differences in the profile of protein S-nitrosylation in WT vs. adh6Δ yeast (under basal growth conditions and upon treatment of cells with EtCysNO) were quantified by iTRAQ labeling (SI Materials and Methods).
Animals.
Mouse studies were approved by the Case Western Reserve University Institutional Care and Use Committee (IACUC); housing and procedures complied with the Guide for the Care and Use of Laboratory Animals (26) and the American Veterinary Medical Association guidelines on euthanasia (27).
Supplementary Material
Acknowledgments
We thank Abhijit Chakladar and Rekha Puria for help with construction of yeast mutants, Precious McLaughlin for technical assistance, and Mark Chance and Giridharan Gokulrangan for advice.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1417816112/-/DCSupplemental.
References
- 1.Seth D, Hausladen A, Wang YJ, Stamler JS. Endogenous protein S-nitrosylation in E. coli: Regulation by OxyR. Science. 2012;336(6080):470–473. doi: 10.1126/science.1215643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anand P, Stamler JS. Enzymatic mechanisms regulating protein S-nitrosylation: Implications in health and disease. J Mol Med (Berl) 2012;90(3):233–244. doi: 10.1007/s00109-012-0878-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benhar M, Forrester MT, Stamler JS. Protein denitrosylation: Enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol. 2009;10(10):721–732. doi: 10.1038/nrm2764. [DOI] [PubMed] [Google Scholar]
- 4.Liu L, et al. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410(6827):490–494. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 5.Liu L, et al. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116(4):617–628. doi: 10.1016/s0092-8674(04)00131-x. [DOI] [PubMed] [Google Scholar]
- 6.Whalen EJ, et al. Regulation of beta-adrenergic receptor signaling by S-nitrosylation of G-protein-coupled receptor kinase 2. Cell. 2007;129(3):511–522. doi: 10.1016/j.cell.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 7.Thauer RK, Jungermann K, Decker K. Energy conservation in chemotrophic anaerobic bacteria. Bacteriol Rev. 1977;41(1):100–180. doi: 10.1128/br.41.1.100-180.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leonardi R, Zhang YM, Rock CO, Jackowski S. Coenzyme A: Back in action. Prog Lipid Res. 2005;44(2-3):125–153. doi: 10.1016/j.plipres.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Roediger WE, Hems R, Wiggins D, Gibbons GF. Inhibition of hepatocyte lipogenesis by nitric oxide donor: Could nitric oxide regulate lipid synthesis? IUBMB Life. 2004;56(1):35–40. doi: 10.1080/15216540310001649822. [DOI] [PubMed] [Google Scholar]
- 10.Forrester MT, et al. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat Biotechnol. 2009;27(6):557–559. doi: 10.1038/nbt.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larroy C, Fernández MR, González E, Parés X, Biosca JA. Characterization of the Saccharomyces cerevisiae YMR318C (ADH6) gene product as a broad specificity NADPH-dependent alcohol dehydrogenase: Relevance in aldehyde reduction. Biochem J. 2002;361(Pt 1):163–172. doi: 10.1042/0264-6021:3610163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larroy C, Rosario Fernández M, González E, Parés X, Biosca JA. Properties and functional significance of Saccharomyces cerevisiae ADHVI. Chem Biol Interact. 2003;143-144:229–238. doi: 10.1016/s0009-2797(02)00166-7. [DOI] [PubMed] [Google Scholar]
- 13.de Jesús-Berríos M, et al. Enzymes that counteract nitrosative stress promote fungal virulence. Curr Biol. 2003;13(22):1963–1968. doi: 10.1016/j.cub.2003.10.029. [DOI] [PubMed] [Google Scholar]
- 14.Matsumoto A, Gow AJ. Membrane transfer of S-nitrosothiols. Nitric Oxide. 2011;25(2):102–107. doi: 10.1016/j.niox.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hiser L, Basson ME, Rine J. ERG10 from Saccharomyces cerevisiae encodes acetoacetyl-CoA thiolase. J Biol Chem. 1994;269(50):31383–31389. [PubMed] [Google Scholar]
- 16.Castello PR, David PS, McClure T, Crook Z, Poyton RO. Mitochondrial cytochrome oxidase produces nitric oxide under hypoxic conditions: Implications for oxygen sensing and hypoxic signaling in eukaryotes. Cell Metab. 2006;3(4):277–287. doi: 10.1016/j.cmet.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 17.Doulias PT, et al. Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc Natl Acad Sci USA. 2010;107(39):16958–16963. doi: 10.1073/pnas.1008036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zaręba-Kozioł M, Szwajda A, Dadlez M, Wysłouch-Cieszyńska A, Lalowski M. Global analysis of S-nitrosylation sites in the wild type (APP) transgenic mouse brain-clues for synaptic pathology. Mol Cell Proteomics. 2014;13(9):2288–2305. doi: 10.1074/mcp.M113.036079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doulias PT, Tenopoulou M, Greene JL, Raju K, Ischiropoulos H. Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci Signal. 2013;6(256):rs1. doi: 10.1126/scisignal.2003252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mindnich RD, Penning TM. Aldo-keto reductase (AKR) superfamily: Genomics and annotation. Hum Genomics. 2009;3(4):362–370. doi: 10.1186/1479-7364-3-4-362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gabbay KH, et al. Ascorbate synthesis pathway: Dual role of ascorbate in bone homeostasis. J Biol Chem. 2010;285(25):19510–19520. doi: 10.1074/jbc.M110.110247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kornberg MD, et al. GAPDH mediates nitrosylation of nuclear proteins. Nat Cell Biol. 2010;12(11):1094–1100. doi: 10.1038/ncb2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jia J, et al. Protection of extraribosomal RPL13a by GAPDH and dysregulation by S-nitrosylation. Mol Cell. 2012;47(4):656–663. doi: 10.1016/j.molcel.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Galli F, Rossi R, Di Simplicio P, Floridi A, Canestrari F. Protein thiols and glutathione influence the nitric oxide-dependent regulation of the red blood cell metabolism. Nitric Oxide. 2002;6(2):186–199. doi: 10.1006/niox.2001.0397. [DOI] [PubMed] [Google Scholar]
- 25.Paige JS, Xu G, Stancevic B, Jaffrey SR. Nitrosothiol reactivity profiling identifies S-nitrosylated proteins with unexpected stability. Chem Biol. 2008;15(12):1307–1316. doi: 10.1016/j.chembiol.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Committee on Care and Use of Laboratory Animals (1996) Guide for the Care and Use of Laboratory Animals (Natl Inst Health, Bethesda), DHHS Publ No (NIH) 85-23.
- 27. AVMA Guidelines for the Euthanasia of Animals, Version 2013.01 (2013)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.