

Significance
The DNA damage response (DDR) maintains genomic integrity following DNA damage to prevent cancer and developmental disorders. The DDR operates in part through controlling localization of factors to chromatin. Here, we detail an interaction between the DDR protein NBS1 and TCOF1, a nucleolar protein mutated in Treacher Collins syndrome that regulates ribosomal DNA transcription. We show that NBS1 relocalizes to nucleoli after DNA damage in a manner dependent on TCOF1 and independent on the NBS1-associated protein MRE11. This process is regulated by casein kinase II and ATM, two protein kinases that phosphorylate TCOF1 and promote NBS1 nucleolar localization. Our work identifies TCOF1 as a DDR factor that cooperates with NBS1 to preserve genomic stability after DNA damage.
Keywords: DNA damage response, nucleolus, protein phosphorylation, ATM, CK2
Abstract
The signal transduction pathway of the DNA damage response (DDR) is activated to maintain genomic integrity following DNA damage. The DDR promotes genomic integrity by regulating a large network of cellular activities that range from DNA replication and repair to transcription, RNA splicing, and metabolism. In this study we define an interaction between the DDR factor NBS1 and TCOF1, a nucleolar protein that regulates ribosomal DNA (rDNA) transcription and is mutated in Treacher Collins syndrome. We show that NBS1 relocalizes to nucleoli after DNA damage in a manner dependent on TCOF1 and on casein kinase II and ATM, which are known to modify TCOF1 by phosphorylation. Moreover, we identify a putative ATM phosphorylation site that is required for NBS1 relocalization to nucleoli in response to DNA damage. Last, we report that TCOF1 promotes cellular resistance to DNA damaging agents. Collectively, our findings identify TCOF1 as a DDR factor that could cooperate with ATM and NBS1 to suppress inappropriate rDNA transcription and maintain genomic integrity after DNA damage.
The faithful conservation of genomic information is an essential process for cell survival and for preventing malignant transformation (1). To maintain genomic integrity, DNA has to be protected from damage either spontaneously induced or generated by environmental sources, including ionizing radiation or chemical agents. The DNA damage response (DDR) is a signal transduction network that is activated to maintain genomic integrity after DNA damage (1, 2). A principal component of the DDR is the ATM kinase, which is primarily activated by the presence of DNA double-strand breaks (DSBs).
DSBs are deleterious DNA lesions that can lead to cell death if unresolved. DSBs are fixed either by joining the two DNA ends together by nonhomologous end joining (NHEJ) or by homology-directed repair mediated by homologous recombination (HR) (3). The regulation of DSB end-processing represents a key step in the choice between NHEJ and HR. Whereas NHEJ occurs with minimal end-processing, extensive resection of DNA ends and formation of single-stranded DNA regions is required for the initiation of HR (4, 5).
NBS1 is a critical component of the heterotrimeric MRE11-RAD50-NBS1 (MRN) complex, which plays a central role in the repair of DSBs through the activation of the DDR and the initiation of HR. After binding and stabilizing DSB ends, the MRN complex recruits ATM and the mediator protein MDC1 to the break site through their direct interaction with NBS1. MDC1 subsequently associates with the phosphorylated histone variant H2AX (γH2AX) locally to amplify the ATM signaling cascade at DSBs (6–9). Direct interaction with NBS1 also promotes the recruitment of the DNA repair factor CtIP to DSB ends by the MRN complex, where it promotes end resection to initiate HR (10). The importance of NBS1 to the maintenance of genomic integrity is further highlighted by the predisposition to growth defects, craniofacial abnormalities, and B-cell lymphomas of patients with Nijmegen breakage syndrome, who carry biallelic mutations in NBS1 (2). In addition, mutations in the subunits of the MRN complex have also been linked to familial breast cancer (1).
To fully understand how NBS1 prevents genetic disorders and cancer, it is important to have a comprehensive view of the multiple functions of NBS1 and define all NBS1-associated factors. Here we identify TCOF1, a nucleolar protein that regulates ribosomal RNA transcription and is mutated in the craniofacial syndrome Treacher Collins, as an interactor of NBS1. We show that NBS1 colocalizes with TCOF1 in the nucleolus transiently after DNA damage in a manner dependent on TCOF1 and on ATM and casein kinase II (CK2), which are kinases known to phosphorylate TCOF1. Our experiments identify TCOF1 as a DDR factor that cooperates with NBS1 in the DNA damage response.
Results
TCOF1 Is an NBS1 Interactor.
To characterize in detail the mechanisms by which NBS1 operates in the DDR, we sought to identify factors associated with NBS1 in human cells. To this end, we expressed in human embryonic kidney HEK 293T-REx cells a cDNA coding for human NBS1 fused to an HA tag and then performed anti-HA immunoprecipitation of NBS1 protein complexes following treatment with ionizing radiation (IR). Protein complexes were then identified by mass spectrometry and further analyzed using CompPASS software, which assigns to each protein a normalized weighted D (NWD) score dependent on protein abundance, frequency, and reproducibility of the interactions (11). In addition to known NBS1 interactors, such as the ATR kinase and the DDR mediator MDC1, we were able to identify the TCOF1 protein (also known as Treacle) as a potential component of the NBS1 protein complex (Fig. 1). The association between NBS1 and TCOF1 was confirmed following immunoprecipitation of HA-tagged TCOF1 protein complexes from HEK 293T-REx cells after IR (Fig. 1B). TCOF1 encodes a nucleolar protein that is mutated in Treacher Collins syndrome (TCS), an autosomal dominant disorder that causes hypoplasia of the facial bones, hearing loss, and cleft palate in 1 in 50,000 children (12). TCOF1 interacts with RNA polymerase I and the transcription factor UBF to promote ribosomal DNA (rDNA) transcription (13). Consistent with this role, we identified subunits of RNA polymerase I (POLR1A, B and E) as components of TCOF1 complexes (Fig. 1B).
Fig. 1.
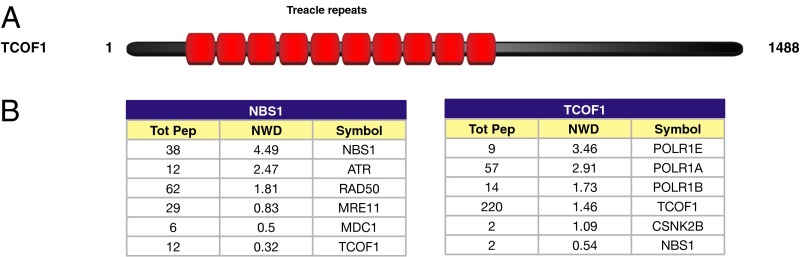
Characterization of the protein complexes of NBS1 and TCOF1. (A) Schematic representation of TCOF1 protein. Treacle repeats are indicated in red. (B) Lists of NBS1 or TCOF1 interactors identified after mass spectrometry and CompPASS analyses of NBS1 or TCOF1 protein complexes isolated from HEK 293T-REx cells after IR. The total number of peptides (Tot Pep) and the NWD score are indicated as in ref. 11.
NBS1 Localizes to Nucleoli After DNA Damage in a TCOF1-Dependent Manner.
To determine whether NBS1 colocalizes with TCOF1 after DNA damage, human osteosarcoma U2OS cells were transfected with HA-tagged NBS1 and stained with an antibody against the HA tag. As shown in Fig. 2 and Fig. S1A, we observed that, following treatments with IR or with the cross-linking agent cisplatin, a portion of the NBS1 protein pool relocates into the nucleolus, where it colocalizes with TCOF1. NBS1 localizes transiently to the nucleolus between 5 and 30 min after IR in a TCOF1-dependent manner, as indicated by the abrogation of NBS1 nucleolar staining after siRNA-dependent depletion of TCOF1 (Fig. 2 C and D). Similar data were also obtained for endogenous NBS1 expressed in U2OS cells (Fig. S2A). Unlike NBS1, MRE11 did not exhibit localization to nucleoli after DNA damage, suggesting that the nucleolar localization of NBS1 is MRE11-independent (Fig. S1B). Furthermore, other DDR components, such as MDC1 and 53BP1, did not colocalize with TCOF1 in nucleoli after IR treatment, as shown in Fig. S2B.
Fig. 2.

NBS1 nucleolar localization after DNA damage. (A) Representative pictures of U2OS cells expressing HA-NBS1 stained with antibodies against the HA tag (green) or TCOF1 (red) with or without IR treatment (10 Gy). Images of cells with merged green and red signals are indicated. (B) Quantification of the percentage of U2OS cells expressing HA-NBS1 that exhibit nucleolar staining of NBS1 with or without IR treatment (10 Gy) as in A. (C) Representative pictures of U2OS cells expressing HA-NBS1 stained as in A treated with control or TCOF1 siRNAs with IR treatment (10 Gy). Images of cells with merged green and red signals are indicated. (D) Western blotting showing the levels of TCOF1 knockdown obtained with two TCOF1 siRNAs. (E) Quantification of the percentage of U2OS cells expressing HA-NBS1 that exhibit nucleolar staining of NBS1 after treatment with TCOF1 siRNAs and IR.
NBS1 Localization to Nucleoli After IR Is Dependent on Casein Kinase II and ATM Kinase Activity.
Previous experiments have shown that the association between NBS1 and many of its interactors, such as MDC1 and CtIP, is mediated by NBS1’s FHA domain, which binds to protein motifs phosphorylated by CK2 (14–16). TCOF1 harbors 10 highly repetitive regions, known as treacle repeats, containing conserved CK2 sites and is a known CK2 substrate (17, 18). Consistent with these observations, we identified a CK2 subunit CSNK2B as part of a TCOF1 complex (Fig. 1B). It has been previously shown that CK2 is required for the association between NBS1 and MDC1 (15, 16). To test whether CK2 is required for the recruitment of NBS1 to the nucleolus after IR, we treated cells with siRNAs against the CK2 subunits α and α′ before irradiation (Fig. 3A). Following this treatment, we observed that the recruitment of NBS1 to the nucleolus was strongly impaired, suggesting that CK2 is required for the nucleolar localization of NBS1 after DNA damage (Fig. 3 A and B).
Fig. 3.

NBS1 localization to nucleoli after CK2 depletion or ATM inhibition. (A) Representative images of U2OS cells expressing HA-NBS1 treated with control or siRNAs targeting the CK2 subunits α and α′ (CK2α and CK2α′) and stained as in Fig. 2 after IR treatment (10 Gy). (B) Quantification of the percentage of U2OS cells expressing HA-NBS1 that exhibit nucleolar staining treated as in A. (C) Representative images of U2OS cells expressing HA-NBS1 with or without ATM inhibitor (10 μM) and stained as in Fig. 2 after IR treatment (10 Gy). (D) Quantification of the percentage of U2OS cells expressing HA-NBS1 that exhibit nucleolar staining treated as in C.
TCOF1 has previously been shown to be phosphorylated by the ATM kinase in response to IR (19). To test whether ATM kinase activity could regulate the recruitment of NSB1 to the nucleolus, U2OS cells were incubated with an ATM kinase inhibitor during and after IR. As indicated in Fig. 3, treatment with the ATM kinase inhibitor KU-55933 abrogated the recruitment of NBS1 to the nucleolus after IR, indicating that ATM activity is required for the colocalization of NBS1 with TCOF1 in the nucleolus after DNA damage (Fig. 3 C and D).
TCOF1 S1199 Is Required for the Nucleolar Localization of TCOF1 After IR.
ATM and ATR kinases phosphorylate their substrates on SQ or TQ motifs (1). We have previously shown that TCOF1 is phosphorylated by ATM/ATR kinases on an SQ site located at amino acids 1410–1411 of TCOF1 (19). To determine the function of this phosphorylation event, we mutated serine 1410 to alanine. Following expression of wild-type and S1410A mutant HA-TCOF1, we observed that the S1410A mutant was proficient for nucleolar localization of NBS1 after IR (Fig. S3). The C-terminal region of TCOF1 contains two additional SQ sites at amino acids 1199–1200 and 1216–1217 that are highly conserved among mammals (Fig. 4A). To examine the role of these SQ sites, we generated HA-TCOF1 SQ to AQ mutants and expressed them in U2OS cells. As shown in Fig. 4 B and C, the TCOF1 S1199A mutant displayed defective localization of NBS1 to nucleoli after IR, whereas the TCOF1 S1216A mutant exhibited a phenotype similar to wild-type TCOF1 in this assay. These experiments suggest that ATM could potentially phosphorylate TCOF1 to recruit or retain NBS1 at nucleolar sites after DNA damage.
Fig. 4.

Localization of NBS1 to nucleoli after expression of TCOF1 mutants. (A) Sequence alignment of TCOF1 regions containing putative ATM SQ phosphorylation sites from different species. The putative ATM phosphorylation sites are in red. (B) Representative images of U2OS cells expressing HA-TCOF1 wild-type or mutant proteins stained with antibodies against NBS1 (green) and the HA tag (red) after IR treatment (10 Gy). (C) Quantification of the percentage of U2OS cells expressing HA-TCOF1 wild-type or mutant proteins that exhibit nucleolar staining treated as in B.
A Small Fraction of TCOF1 Is Recruited to Sites of DNA Damage After Laser Microirradiation.
TCOF1 is predominantly localized in the nucleolus. To determine whether TCOF1 localization could be affected by DNA damage, we conducted UV laser microirradiation on U2OS cells expressing HA-TCOF1. Interestingly, we observed that a small fraction of TCOF1 localized to the DNA damage sites generated by the UV laser outside of the nucleolus (Fig. S4A).
TCOF1 has at least six different isoforms in mammalian cells. To determine whether the localization of TCOF1 to DNA damage sites differs among TCOF1 isoforms, we expressed in U2OS cells the TCOF1 isoform c [National Center for Biotechnology Information (NCBI) reference sequence NM_001008657.2], which displays pan-nuclear localization due to the absence of the nucleolar signal located at the C terminus of the protein. As shown in Fig. S4B, the TCOF1 isoform c exhibited a very clear recruitment to DNA damage sites induced by laser stripes. These observations suggest that TCOF1 could also have a role in the DNA damage response outside of the nucleolus.
TCOF1 Protects Cells from DNA Damage.
Many proteins that associate with NBS1 are known to protect cells from DNA damage. To test whether TCOF1 is required for survival in response to DNA damage, GFP-labeled U2OS cells treated with control or TCOF1 siRNAs were mixed with unlabeled U2OS cells. The relative survival of green vs. unlabeled U2OS cells after IR (5 Gy) or cisplatin (0.25 μM) treatment was then measured by FACS cytometry (20). As shown in Fig. 5, depletion of TCOF1 with two independent siRNAs led to sensitivity to IR and cisplatin treatment, indicating that TCOF1 protects cells from DNA damage.
Fig. 5.

Cellular response to DNA damaging agents after TCOF1 depletion. Cell competition assays between control U2OS green cells and U2OS cells treated with control or TCOF1 siRNAs after IR (5 Gy) or cisplatin (CIS; 0.25 μM) treatment.
Discussion
Our study has identified the nucleolar protein TCOF1 as a DDR factor required for the recruitment of NBS1 to nucleoli after DNA damage. NBS1 contains FHA and BRCT motifs, which form a supramodular phosphobinding interface (14, 21). It has been previously shown that the FHA motif of NBS1 interacts with CK2 phosphorylation sites of MDC1 (15, 16). Given that TCOF1 is a CK2 substrate and that CK2 is required for NBS1 localization to nucleoli, we propose that TCOF1 and MDC1 interact with NBS1 in a similar manner through the binding of CK2 phosphorylation sites to the FHA motif of NBS1; this would suggest that TCOF1 and MDC1 are mutually exclusive interactors of NBS1. Indeed, we observed that MDC1 does not localize to the nucleolus together with NBS1 and TCOF1. Similar findings were reported by Larsen et al. (22) during the preparation of our manuscript.
In addition to CK2, the ATM kinase was identified by us to regulate the localization of NBS1 to nucleoli after DNA damage. We previously found TCOF1 to be phosphorylated on S1410 by ATM/ATR after DNA damage (19). However, here we find that phosphorylation of this site is not required for the nucleolar localization of NBS1 after DNA damage. Instead, we identified S1199, an SQ site previously found to be phosphorylated on TCOF1 in the absence of exogenous DNA damage (23, 24), as a putative ATM site that is required for NBS1 localization to nucleoli in response to DNA damage. Based on these observations, we propose that the phosphorylation of S1199 might be recognized by NBS1 phosphobinding motifs, which could then promote the association with NBS1 after DNA damage. The recognition of TCOF1 ATM phosphorylation sites could be mediated by the NBS1 BRCT motif. Indeed, BRCT motifs are known to bind sites phosphorylated by ATM or ATR (25). Altogether, our data suggest that ATM and CK2 phosphorylation sites may cooperate to mediate NBS1 localization to TCOF1.
NBS1 does not form classical foci in the nucleolus as observed in nonnucleolar DNA. Instead, NBS1 appears to fully colocalize with TCOF1-bound chromatin, which comprises a large fraction of nucleolar DNA. Although speculative, the extensive colocalization of NBS1 with TCOF1 in nucleoli suggests the existence of a possible spreading mechanism for NBS1 on TCOF1 akin to MDC1–NBS1 spreading on H2AX. It is known that phosphorylation of H2AX on S139 by ATM near DSBs triggers the initial association of the MDC1–NBS1–ATM complex, which then further phosphorylates H2AX, thereby promoting a feed-forward loop that results in the spreading of the MDC1–NBS1–ATM complex for several megabases around DSBs (7–9). A similar phenomenon could exist for the TCOF1–NBS1–ATM complex in nucleoli, where TCOF1 phosphorylation by CK2 and ATM could promote the binding of the NBS1–ATM complex to TCOF1 and the subsequent propagation of TCOF1 phosphorylation, thus allowing the NBS1–ATM complex to extensively coat nucleolar chromatin through TCOF1 association.
Previous experiments have indicated that ribosomal RNA transcription is transiently inhibited after DNA damage through ATM and NBS1 to maintain genomic instability (26); this would facilitate the repair of DNA lesions by preventing the collision between the transcriptional and DNA repair machineries. Based on our observation that TCOF1 is required for the ATM-dependent recruitment of NBS1 to nucleoli after DNA damage, we suggest that TCOF1 could be a component of the ATM- and NBS1-dependent pathway that blocks ribosomal DNA transcription after DNA damage, as suggested also by Larsen et al. (22). It will be critical in the future to determine whether defective localization of NBS1 to nucleoli results in increased instability of ribosomal DNA repeats.
The craniofacial abnormalities caused by TCOF1 mutations have been shown to depend on hyperactivation of p53-dependent apoptotic processes in neural crest progenitors during craniofacial development (27). The observation that mutations in RNA polymerase I can cause TCS suggests that the increased cell death observed in TCS patients could result from impaired rDNA transcription and consequent dysfunctional ribosomal biogenesis. However, excessive p53 activation in TCS may alternatively be consistent with increased and/or unresolved DNA damage in TCS cells due to defective regulation of RNA polymerase I transcription after DNA damage or to potential functions for TCOF1 in nonnucleolar DNA repair. It will therefore be important to test the possibility that defective DNA repair may contribute to TCS pathophysiology. Similar to TCS, the Nijmegen breakage syndrome displays craniofacial abnormalities (1). Future experiments will be needed to determine how defects in NBS1 nucleolar localization contribute to the craniofacial abnormalities displayed by Nijmegen breakage syndrome patients.
Together, our experiments have identified TCOF1 as a DDR factor that recruits NBS1 to nucleoli after DNA damage and have suggested a potential role of alterations of the DNA damage response in the pathogenesis of TCS.
Materials and Methods
Antibodies.
Rabbit polyclonal anti-TCOF1 (1:1,000; Proteintech, 11003-1-AP1), anti-GAPDH (1:2,000; Santa Cruz, sc-25778), and mouse monoclonal anti-HA (1:1,000; Covance, HA.11) antibodies were used in Western blot experiments.
DNA Clones.
The TCOF1 clone corresponding to NCBI NM_001135243.1 was cloned by Gateway recombination into pENTRD-TOPO to generate pENTRD-TOPO-TCOF1. The Gateway pENTRD-TOPO-TCOF1-S1199A, pENTRD-TOPO-TCOF1-S1216A, and pENTRD-TOPO-TCOF1-S1410A were generated by site-directed mutagenesis of pENTRD-TOPO-TCOF1. The TCOF1 isoform c corresponding to NCBI NM_001008657.2, the NBS1 clone corresponding to NCBI AK312410 and the MRE11 clone corresponding to NCBI NM_005591 were cloned by Gateway recombination into pDONR223. pENTRD-TOPO-TCOF1, pENTRD-TOPO-TCOF1-S1199A, pENTRD-TOPO-TCOF1-S1216A, pENTRD-TOPO-TCOF1-S1410A, pDONR223-NBS1, and pDONR223-MRE11 were used in Gateway recombination reactions with pMSCV-FLAG-HA (28) to generate pMSCV-FLAG-HA-TCOF1; pMSCV-FLAG-HA-TCOF1-S1199A, pMSCV-FLAG-HA-TCOF1-S1216A, and pMSCV-FLAG-HA-TCOF1-S1410A; pMSCV-FLAG-HA-NBS1; and pMSCV-FLAG-HA-MRE11. pDONR223-TCOF1 isoform c was recombined with the Gateway vector pHAGE-HA to generate pHAGE-HA-TCOF1 isoform c.
Cell Culture and RNAi.
The human osteosarcoma cell line U2OS and human embryonic kidney fibroblast cell line HEK 293T-REx were maintained in McCoy’s or DMEM, respectively, supplemented with 10% (vol/vol) FBS. Stable U2OS cell lines expressing HA-TCOF1; HA-TCOF1-S1199A, HA-TCOF1-S1216A, HA-TCOF1-S1410A; HA-NBS1; HA-MRE11; and HA-TCOF1 isoform c were obtained after puromycin selection of cells infected with retroviruses or lentiviruses generated from the vectors pMSCV-FLAG-HA-TCOF1; pMSCV-FLAG-HA-TCOF1-S1199A, pMSCV-FLAG-HA-TCOF1-S1216A, and pMSCV-FLAG-HA-TCOF1-S1410A; pMSCV-FLAG-HA-NBS1; pMSCV-FLAG-HA-MRE11; and pHAGE-HA-TCOF1 isoform c. Stable HEK 293T-REx cells expressing HA-TCOF1 and HA-NBS1 were obtained after infection with retroviruses generated from the pMSCV-FLAG-HA-TCOF1 and pMSCV-FLAG-HA-NBS1 vectors.
TCOF1 siRNAs (Invitrogen; Stealth siRNA HS110575 and HS110577), CK2α and CK2α′ siGenome siRNA pools (Dharmacon; M-003475-03-0005 and M-004752-00-0005) were used to transfect U2OS cells.
Immunofluorescence.
Parental U2OS cells and U2OS cells expressing HA-TCOF1 wild-type and mutant proteins, HA-TCOF1 isoform c, HA-NBS1, or HA-MRE11 were stained with rabbit polyclonal anti-MDC1 (1:100; Abcam, ab11169), anti-NBS1 (1:100; Novus Biologicals, NB100-143), anti-TCOF1 (1:100; Proteintech, 11003-1-AP1), anti-53BP1 (1:1,000; Bethyl Laboratories, A300-272A), or mouse monoclonal anti-HA (1:1,000; Covance, HA.11), anti-γH2AX (1:500; Millipore, JBW301), anti-TCOF1 (1:100; Santa Cruz Biotechnology, sc-374536) antibodies. For NBS1 localization experiments, U2OS cells expressing HA-NBS1 or HA-TCOF1 wild-type and mutant proteins were treated with 10 Gy irradiation with or without the ATM inhibitor KU-55933 (10 μM) and then fixed with 4% formaldehyde 15 min after irradiation. Following permeabilization with 0.2% Triton, U2OS cells were stained with the antibodies indicated above. Similar experiments were conducted after cisplatin (1 μM) treatment for 6 h. The percentage of cells with NBS1 nucleolar localization was determined as the average value of three or more replicates. Microirradiation experiments were performed as previously described (29), and U2OS cells expressing HA-TCOF1 and HA-TCOF1 isoform c were fixed 5 min after irradiation.
Protein Purification and Mass Spectrometry.
Stable HEK 293T-REx cells were subjected to doxycycline treatment to induce the expression of HA-TCOF1 and HA-NBS1 and purification of HA-TCOF1 and HA-NBS1 protein complexes after IR treatment (10 Gy) was conducted as previously described (29). Mass spectrometry and CompPASS analysis was performed as reported (11, 29).
DNA Damage Sensitivity Assays.
Cell competition assays were performed as previously described (28). In particular, U2OS cells were transfected with control or TCOF1 siRNAs and then mixed with GFP expressing U2OS cells as reported (28). Cells were then treated with a single IR dose (5 Gy) or with cisplatin (0.25 μM) for 16 h, and the ratio of uncolored to GFP expressing U2OS cells was determined by flow cytometric analysis after 7 d.
Supplementary Material
Acknowledgments
We thank members of the S.J.E. and A.C. laboratories for comments and advice on this project. This work was supported by National Institutes of Health Grants GM44664 (to S.J.E.) and AG011085 (to J.-W.H.). S.J.E. is an Investigator with the Howard Hughes Medical Institute.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1422488112/-/DCSupplemental.
References
- 1.Ciccia A, Elledge SJ. The DNA damage response: Making it safe to play with knives. Mol Cell. 2010;40(2):179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol. 2003;4(6):435–445. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- 4.Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 5.Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47(4):497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 6.Lukas J, Lukas C, Bartek J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol. 2011;13(10):1161–1169. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- 7.Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421(6926):961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- 8.Stucki M, et al. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123(7):1213–1226. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 9.Lou Z, et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell. 2006;21(2):187–200. doi: 10.1016/j.molcel.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 10.Stracker TH, Petrini JH. The MRE11 complex: Starting from the ends. Nat Rev Mol Cell Biol. 2011;12(2):90–103. doi: 10.1038/nrm3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138(2):389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakai D, Trainor PA. Treacher Collins syndrome: Unmasking the role of Tcof1/treacle. Int J Biochem Cell Biol. 2009;41(6):1229–1232. doi: 10.1016/j.biocel.2008.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valdez BC, Henning D, So RB, Dixon J, Dixon MJ. The Treacher Collins syndrome (TCOF1) gene product is involved in ribosomal DNA gene transcription by interacting with upstream binding factor. Proc Natl Acad Sci USA. 2004;101(29):10709–10714. doi: 10.1073/pnas.0402492101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams RS, et al. Nbs1 flexibly tethers Ctp1 and Mre11-Rad50 to coordinate DNA double-strand break processing and repair. Cell. 2009;139(1):87–99. doi: 10.1016/j.cell.2009.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spycher C, et al. Constitutive phosphorylation of MDC1 physically links the MRE11-RAD50-NBS1 complex to damaged chromatin. J Cell Biol. 2008;181(2):227–240. doi: 10.1083/jcb.200709008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Melander F, et al. Phosphorylation of SDT repeats in the MDC1 N terminus triggers retention of NBS1 at the DNA damage-modified chromatin. J Cell Biol. 2008;181(2):213–226. doi: 10.1083/jcb.200708210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones NC, Farlie PG, Minichiello J, Newgreen DF. Detection of an appropriate kinase activity in branchial arches I and II that coincides with peak expression of the Treacher Collins syndrome gene product, treacle. Hum Mol Genet. 1999;8(12):2239–2245. doi: 10.1093/hmg/8.12.2239. [DOI] [PubMed] [Google Scholar]
- 18.Wise CA, et al. TCOF1 gene encodes a putative nucleolar phosphoprotein that exhibits mutations in Treacher Collins Syndrome throughout its coding region. Proc Natl Acad Sci USA. 1997;94(7):3110–3115. doi: 10.1073/pnas.94.7.3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsuoka S, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316(5828):1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 20.Smogorzewska A, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129(2):289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lloyd J, et al. A supramodular FHA/BRCT-repeat architecture mediates Nbs1 adaptor function in response to DNA damage. Cell. 2009;139(1):100–111. doi: 10.1016/j.cell.2009.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Larsen DH, et al. The NBS1-Treacle complex controls ribosomal RNA transcription in response to DNA damage. Nat Cell Biol. 2014;16(8):792–803. doi: 10.1038/ncb3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Courcelles M, et al. Phosphoproteome dynamics reveal novel ERK1/2 MAP kinase substrates with broad spectrum of functions. Mol Syst Biol. 2013;9:669. doi: 10.1038/msb.2013.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsu PP, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332(6035):1317–1322. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reinhardt HC, Yaffe MB. Phospho-Ser/Thr-binding domains: Navigating the cell cycle and DNA damage response. Nat Rev Mol Cell Biol. 2013;14(9):563–580. doi: 10.1038/nrm3640. [DOI] [PubMed] [Google Scholar]
- 26.Kruhlak M, et al. The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature. 2007;447(7145):730–734. doi: 10.1038/nature05842. [DOI] [PubMed] [Google Scholar]
- 27.Jones NC, et al. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat Med. 2008;14(2):125–133. doi: 10.1038/nm1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ciccia A, et al. Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol Cell. 2012;47(3):396–409. doi: 10.1016/j.molcel.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ciccia A, et al. The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev. 2009;23(20):2415–2425. doi: 10.1101/gad.1832309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.