

Significance
The domestication of the horse revolutionized warfare, trade, and the exchange of people and ideas. This at least 5,500-y-long process, which ultimately transformed wild horses into the hundreds of breeds living today, is difficult to reconstruct from archeological data and modern genetics alone. We therefore sequenced two complete horse genomes, predating domestication by thousands of years, to characterize the genetic footprint of domestication. These ancient genomes reveal predomestic population structure and a significant fraction of genetic variation shared with the domestic breeds but absent from Przewalski’s horses. We find positive selection on genes involved in various aspects of locomotion, physiology, and cognition. Finally, we show that modern horse genomes contain an excess of deleterious mutations, likely representing the genetic cost of domestication.
Keywords: ancient DNA, horse domestication, Przewalski’s horse, positive selection, cost of domestication
Abstract
The domestication of the horse ∼5.5 kya and the emergence of mounted riding, chariotry, and cavalry dramatically transformed human civilization. However, the genetics underlying horse domestication are difficult to reconstruct, given the near extinction of wild horses. We therefore sequenced two ancient horse genomes from Taymyr, Russia (at 7.4- and 24.3-fold coverage), both predating the earliest archeological evidence of domestication. We compared these genomes with genomes of domesticated horses and the wild Przewalski’s horse and found genetic structure within Eurasia in the Late Pleistocene, with the ancient population contributing significantly to the genetic variation of domesticated breeds. We furthermore identified a conservative set of 125 potential domestication targets using four complementary scans for genes that have undergone positive selection. One group of genes is involved in muscular and limb development, articular junctions, and the cardiac system, and may represent physiological adaptations to human utilization. A second group consists of genes with cognitive functions, including social behavior, learning capabilities, fear response, and agreeableness, which may have been key for taming horses. We also found that domestication is associated with inbreeding and an excess of deleterious mutations. This genetic load is in line with the “cost of domestication” hypothesis also reported for rice, tomatoes, and dogs, and it is generally attributed to the relaxation of purifying selection resulting from the strong demographic bottlenecks accompanying domestication. Our work demonstrates the power of ancient genomes to reconstruct the complex genetic changes that transformed wild animals into their domesticated forms, and the population context in which this process took place.
The domestication of the horse had a far-reaching impact on the sociopolitical and economic trajectories of human societies (1). It not only provided meat and milk (2) but also enabled the development of continent-sized nomadic empires, by transforming warfare and allowing for the rapid spread of goods and information over long distances. However, despite the characterization of the genome of modern horses (3), an understanding of the genetic processes underlying horse domestication is still lacking. In other organisms, such an understanding is usually achieved by comparing the genomes of domesticated species and their wild relatives (4–6), but this approach is not directly applicable to horses. Recent genome-wide analyses have shown that Przewalski’s horse, the last truly wild horse population remaining today, is not the direct ancestor of domesticated horses (7, 8). Instead, it likely represents a sister population that separated from the ancestral population of domesticated horses some 38–72 kya (9). This date significantly predates not only the widely accepted date for the beginning of horse domestication, ca. 5.5 kya (2), but also the earliest potential evidence for horse domestication, ca. 7.5 kya (10). In addition, the current Przewalski’s horse population descends from a captive stock consisting of only 13 founder individuals (7). This severe demographic bottleneck, together with inbreeding resulting from unequal contributions from different captive lineages, likely caused a substantial loss of the diversity once present in Przewalski’s horses. As a result, no modern horse population can fully represent the genetic diversity ancestral to the modern, domesticated gene pool (11–13).
Ancient DNA allows tracking of past population histories through time, accessing the gene pools of wild animals predating domestication and exploring genetic variation that has been lost in extant populations. Recovery of ancient DNA, coupled with low-throughput gene candidate analyses, has previously been used to investigate changes in the genetic diversity of horses over time. This approach revealed coat color variation as one early result of domestication, by showing that the selection of multiple alleles driving diverse coloration patterns was already ongoing in the Early Bronze Age (14). It has also revealed a loss of Y-chromosomal haplotypes on both the Przewalski’s and domestic horse lineages (12).
Using next-generation sequencing, the complete genome sequence of ancient individuals can now be deciphered (15), with qualities rivaling the qualities of modern genomes (16–18). We characterized complete genome sequences of two ancient horse specimens predating the earliest evidence of horse domestication to reveal the population context in which horse domestication took place. We compared the genomic information of these specimens with genomic information of six domestic horses, representing five breeds, and one Przewalski’s horse (9), ranging from a 7.4- to 32.7-fold average depth of coverage. We also included the domestic donkey as an outgroup, which represents a sister lineage of modern horses and shares a most common recent ancestor with horses 4.0–4.5 Mya ago (9). This comparison enabled us to reconstruct the relationships between wild and domesticated horses, and to explore the genetic mechanisms underlying the behavioral, physiological, and other biological changes that accompanied horse domestication.
Results and Discussion
Genome-Wide Sequencing of Two Ancient Wild Horses.
From 29 permafrost-preserved horse bones previously screened for DNA preservation (9), we selected two samples, CGG10022 and CGG10023, suitable for genome sequencing (SI Appendix, section S1). These two samples were excavated from the Russian Siberian permafrost of the Taymyr Peninsula (Fig. 1A) and radiocarbon-dated to the Late Pleistocene [42,692 ± 891 calibrated y before present (cal BP) and 16,099 ± 192 cal BP, respectively], clearly placing them before the onset of domestication (2, 10). Because our previous genetic characterization of these samples was limited to a 1.8- to 0.2-fold depth of coverage (9), we generated additional sequence data, achieving a final coverage of 24.3-fold and 7.4-fold for CGG10022 and CGG10023, respectively. Ratios of X-chromosomal and autosomal coverages indicated that CGG10022 is a mare, whereas CGG10023 is a stallion.
Fig. 1.
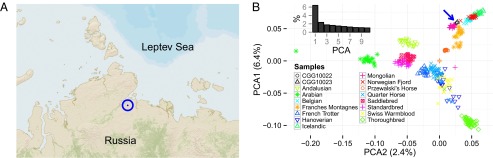
Sampling location and PCA. (A) Geographic origin of ancient horse remains (73.046° N, 109.708° E). (B) Analysis is based on array data for ∼54,000 SNPs. The blue arrow indicates the position of CGG10022 and CGG10023. The smaller bar plot indicates the relative variance contribution of the 10 first principal components.
The recovered DNA sequences exhibited signatures of postmortem DNA damage typical of ancient DNA, with preferential fragmentation at purines and inflated rates of cytosine deamination at single-stranded overhangs (19, 20). Additionally, we found an ∼10-bp periodicity in the size of ancient DNA library inserts for fragments shorter than ∼150 bp. This periodicity is reminiscent of the nucleosomal protection patterns characterized for DNA sequences from other fossil material (21). Finally, the microbial metagenome of both samples was dominated by Actinobacteria, as observed for permafrost soils (22, 23) and other Arctic horse bones (24). Altogether, these characteristics support the data as ancient and authentic.
Relationship Between Wild and Domesticated Horses.
To investigate the genomic processes of horse domestication, we must first understand the evolutionary relationships between domesticated horses, Przewalski’s horses, and the two newly sequenced ancient horses (SI Appendix, section S2). The mitochondrial genome sequences of the two ancient horses clustered within the extensive diversity present in modern horses, as previously reported (9, 12, 25). The Y-chromosome haplotype reconstructed for CGG10023 was distinct from those Y-chromosome haplotypes present in the modern horses, supporting previous hypotheses that domestication was associated with a significant loss of Y-chromosomal diversity (12, 26).
Inferring the phylogenetic relationships of ancient and modern horses from their nuclear genomes is challenging because of the recombination of the nuclear DNA. In particular, diversity of the nuclear genome is unlikely to be reciprocally monophyletic among subpopulations, a phenomenon called incomplete lineage sorting, which results in support for different topologies across loci (27). We therefore used several approaches and different nuclear markers to infer the phylogenetic relationships and to estimate the amount of gene flow between ancient and modern horses.
Principal component analysis (PCA) of ∼54,000 SNPs present in the EquineSNP50 (Illumina) assay placed the two ancient horses close to a group of geographically and morphologically disparate domesticated breeds (Belgian, Franches-Montagnes, Icelandic, Mongolian, and Norwegian Fjord; Fig. 1B). However, the EquineSNP50 assay is heavily affected by ascertainment bias toward a subset of modern breeds, making PCA results difficult to interpret. We therefore performed two independent phylogenetic analyses based on larger subsets of the nuclear data, whole-exome sequences and all polymorphic sites, to reconstruct the most likely relationships between ancient and modern horses.
The average tree topology inferred from the coding sequences of the whole exome is shown in Fig. 2A. In this phylogeny, the two ancient horses cluster together outside of the diversity of all living horses. Within living horses, domesticated horses form a monophyletic clade that is a sister to Przewalski’s horse. Next, we inferred patterns of population splits using TreeMix applied to the genome-wide polymorphisms identified in our dataset, representing ∼4,200,000 SNPs called for each sample. This analysis produced the same average topology as in Fig. 2A. We therefore assumed that this topology best reflected the relationships between ancient and living horse populations.
Fig. 2.
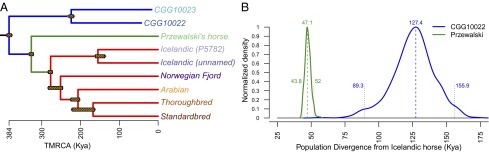
Horse phylogenetic relationships and population split times. (A) Maximum likelihood chronogram for horses based on whole-exome sequences and rooted using the domestic donkey as an outgroup. Most nodes received 100% bootstrap support, except for the Thoroughbred-Standardbred and Norwegian Fjord-Arabian-Thoroughbred-Standardbred clades, which showed 67% and 70% support, respectively. Maximum likelihood estimates for the TMRCA of each clade (x axis with the unit of kya), providing upper boundaries for population split times, were obtained using r8s. Yellow hatched bars indicate the 95% confidence interval derived from the dating of 100 bootstrap pseudoreplicates. (B) Lower boundaries for population split times. The posterior distributions shown correspond to analyses restricting mutations to transversions and considering CGG10022 and Przewalski’s horse, comparing these genomes with that of the Icelandic (unnamed) horse. The dashed and dotted lines indicate the mode and the 95% credibility interval, respectively. Additional analyses performed with other combinations of sequence data provided similar estimates (SI Appendix, section S2.8.2).
To place the horse evolutionary history in a chronological context, we estimated the divergence time between the ancestors of the ancient horse population and ancestors of the modern horse populations, as well as the divergence time between the ancestors of Przewalski’s horses and domesticated horses. We first calibrated the whole-exome phylogeny using the time to the most recent common ancestor (TMRCA) of Equus (donkey vs. horses) at 4.0–4.5 Mya (9) and tip dates for ancient samples, assuming that the combined evolution of protein-coding genes follows a molecular clock (Fig. 2A). Because the estimated TMRCA of two populations predates the actual population split event, this estimate provides upper boundaries of the population split times. Next, we used F-statistics and coalescent simulations to estimate population split times directly between the ancestors of Przewalski’s horses and domesticated horses, under the assumption of no gene flow between populations (Fig. 2B and SI Appendix, Table S20). When considering the split between the ancient population and the ancestors of modern domesticated horses, where we identified significant amounts of gene flow (below), this method underestimates the population split time, and therefore provides a lower boundary for the population split time estimate (SI Appendix, section 2.8.2). Here, we disregarded sites containing transitions, because transitions may represent DNA damage-related sequencing errors. Our analyses indicated that the ancient horses from Taymyr split from the common ancestors of domesticated and Przewalski’s horses at least 127–159 kya (credible interval = 89–211 kya; SI Appendix, Table S20). We further refined this estimate using ∂a∂i analyses to allow for gene flow (SI Appendix, Table S27) and found that the population time split occurred 169 kya. This timing predates the second-to-last interglacial, a time period during which warmer climatic conditions prevailed and grasslands contracted (28), and is associated with an ∼40% demographic decrease for horses, as inferred by pairwise sequential Markovian coalescent (PSMC) analyses (Fig. 3A) (29). The common ancestor of Przewalski’s horses diverged from the common ancestor of domesticated horses at least 43–52 kya (confidence interval = 41–70 kya; SI Appendix, Table S20), in line with previous results (9). This time period coincides with an ∼60% demographic decline for horses inferred using PSMC analyses (Fig. 3A), potentially resulting from ongoing fragmentation of horse habitats around that time.
Fig. 3.
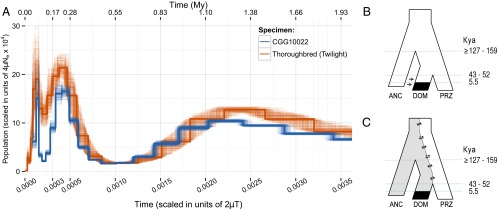
PSMC demographic inference and population models. (A) Demographic profile of the horse lineage over the past 2 My as inferred from bootstrapped PSMC analyses. (B) Population model assuming admixture between the descending population of ancient horses and the population that gave rise to domesticated horses [split times in kya (Kyr)]. ANC, ancient horse population; DOM, population of domesticated horses; PRZ, Przewalski’s horse population. (C) Alternative population model assuming the presence of ancestral population structure in these populations. Lower boundaries for population split times are derived from approximate Bayesian computation and display the full range of modes observed across the analyses performed (SI Appendix, Table S20).
In addition to incomplete lineage sorting, the nuclear phylogeny and relationships among lineages can be influenced by gene flow that occurs between populations after their initial divergence. We therefore tested for the presence or absence of gene flow across populations using admixture tests based on the D-statistic (30). This analysis revealed a statistically significant excess of shared derived polymorphisms between the ancient and domesticated horses in the following quartets (Przewalski’s, Domesticated; Ancient, Donkey), where “Domesticated” represents any of the six domesticated horse genomes included in this study (SI Appendix, section S2.7). This excess of shared derived mutations suggests the presence of gene flow between the population ancestral to domesticated horses and the population descended from the one to which our ancient horses belonged, as confirmed by the ∂a∂i analyses, which also showed statistically significant support for population models including migration over models without migration (SI Appendix, section S2.9).
The D-statistics revealed that no domesticated horse shares more derived alleles than any other domesticated horse with the ancient horses when examining quartets, including two domesticated horses, one ancient horse, and the domestic donkey (Domesticated1, Domesticated2; Ancient, Donkey). This observation suggests that both ancient horses are equidistant to all domesticated breeds investigated here, and that admixture predated the divergence of modern breeds and occurred between the population ancestral to domesticated horses and the population to which our ancient horses belonged. Alternatively, this admixture could have occurred at comparable levels in all domesticated breeds after they diverged. The large majority of the quartets including two domesticated horses, the Przewalski’s horse, and the domestic donkey (Domesticated1, Domesticated2; Przewalski’s, Donkey) were nonsignificant. These results suggested an absence of a detectable admixture between the domesticated and Przewalski’s horse populations after their initial divergence, in line with previous results (9).
Overall, our analyses suggest two possible models to explain horse evolutionary history before domestication (Fig. 3 B and C). Both models suggest some levels of genetic structure among Eurasian horse populations during the Late Pleistocene, which was not apparent in the temporal and geographic distribution of mitochondrial haplotypes (31). In the first model (Fig. 3B), the population including our ancient horses and their descendants first separated from common ancestors of domesticated horses and Przewalski’s horses, and later admixed with the population ancestral to domesticated horses after it diverged from the Przewalski’s horse population. This admixture could have occurred before domestication or during the early stages of the domestication process, following restocking from the wild as previously suggested (13, 32, 33). Following the methods of Durand et al. (30) and Cahill et al. (34), and assuming instantaneous admixture, we can estimate that at least 12.9–17.8% of the genomic variation present in domesticated horse genomes, and potentially as much as 29.4–60.7%, could result from such predomestication admixture or postdomestication restocking (SI Appendix, section S2.7.3 and Table S19). In the second model (Fig. 3C), population structure was present among Late Pleistocene horses in Eurasia, with the ancient horse population originating, albeit earlier, from a similar background to the ancestral population of domesticated horses, whereas Przewalski’s horses derived from a different background. The data presented in this paper do not, however, allow us to select between these two possible models.
Our analyses reveal that a substantial fraction of the genome-wide variation that is present in domesticated horses is not present in Przewalski’s horses. These genetic differences may have been exacerbated by the recent bottleneck, and consequent loss of diversity, within Przewalski’s horses. We found the presence of highly homozygous tracks indicative of inbreeding within the Przewalski’s horse genome (Fig. 4A and SI Appendix, Fig. S36), as would be expected given the founder effect resulting from only 13 individuals (7). Such tracks were also found among domesticated horses but were almost absent from the ancient genomes (Fig. 4A and SI Appendix, Figs. S36–S38). Together, the distinct evolutionary histories of the lineages leading to domesticated horses and Przewalski’s horses and the recent loss of variation within the Przewalski’s horse population limit what can be learned about horse domestication using only genomic data from living horses. Thus, the ancient horse genomes offer an unprecedented opportunity to investigate changes associated with domestication.
Fig. 4.
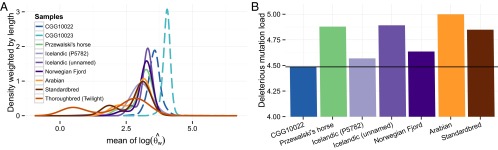
Inbreeding and genetic load of predomesticated vs. modern breeds. (A) Genome-wide distribution of heterozygosity estimates for genomic tracks, calculated as the logarithm of the Watterson estimators, averaged across the track and weighted by its length. Transitions were excluded when calculating the Watterson estimators. Genomics tracks are defined as continuous regions of similar Watterson estimator values. The bimodal distributions observed for the modern horses, but not for CGG10022 and CGG10023, are indicative of inbreeding in these individuals. Dashed lines indicate ancient individuals. (B) Additive deleterious mutation load estimated from coding sequences of modern breeds and the high-coverage predomesticated sample (CGG10022), excluding the Thoroughbred Twilight and CGG10023.
Genetic Changes Associated with Horse Domestication.
By comparing genetic information of ancient and modern domesticated horses, we could pinpoint the genetic changes associated with domestication shared by all modern breeds. This methodology contrasts with scans for outlier genomic regions among modern breeds (35), which can, at best, identify genes that are specific to breeds, in the absence of knowledge of the predomesticated genetic background and sources of variation.
We first screened the ancient genomes for 50 SNPs associated with known Mendelian traits in modern horses. We observed the reference allele at most loci, including the reference alleles associated with diseases and coat color variation. Exceptions included the ZFAT gene, which is associated with variation in wither height (36), for which both ancient individuals carried a mixture of reference and alternative alleles. Additionally, we found that CGG10023 carried the alternative allele at ACN9 homologue (S. cerevisiae; ACN9), CKM, and COX4/1, all of which are associated with increased winning performance in modern race horses (37, 38). For ACN9, the alternative allele was also observed in CGG10022. In contrast, the short-distance, performance-enhancing allele for the “speed gene,” MSTN, (39) was not observed in either of the predomesticated horses. These results suggest that some genetic variants associated with the desirable phenotypes of elite Thoroughbreds have existed in the horse population for at least ∼16,000–43,000 y. The presence of these SNPs in the ancient horses implies that selection for these traits did not occur on de novo mutations but on existing variation, which was either contributed to the domesticated stock by the ancient population through admixture or was cosegregating in both the ancestors of the ancient horses and the ancestors of the domesticated horses after their split.
We next used four complementary methods to scan the genomes of modern and predomesticated horses for regions that have undergone positive selection during domestication (Fig. 5 and SI Appendix, section S4). Despite exploiting signatures displaying different statistical properties, these methods individually detected genes previously identified as undergoing selection in domesticated horses, such as KITLG (9) and melanocortin 1 receptor (MC1R) (40), providing important validation of the approach as a whole. We focused on a conservative set of 125 loci that were identified by at least two of the above methods so as to limit method-specific bias and narrow the list of candidates. Annotations showed genes involved in gastrointestinal and neurological diseases; endocrine system disorders; metabolism; perception; bone and limb morphogenesis; growth; and the development of the muscular, cardiovascular, and nervous systems.
Fig. 5.
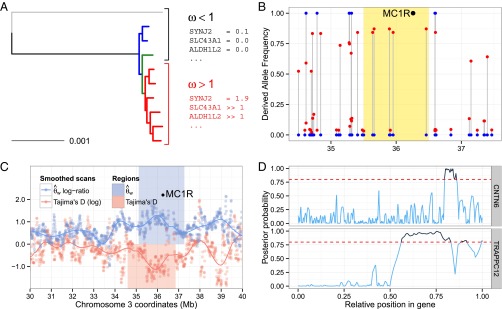
Scans for positive selection. (A) Detection of selection by an increased and nonneutral ω-ratio, which represents the rate of nonsynonymous over synonymous mutations in the clade containing the domesticated horses (red). Examples include the SYNJ2, SLC43A1, and ALDH1L2 genes. (B) Detection of selection by a deficit in the observed rate of derived alleles in CGG10022 (blue) vs. the expected rate of derived alleles (red) among 32 modern breeds (represented here by the Belgian horse); the position of MC1R within the region under selection, indicated in yellow, is signified by a black dot. (C) Detection of selection defined by regions associated with decreased genetic diversity in domesticated horses relative to predomesticated horses [indicated by an increased log-ratio of the Watterson estimator (θw)] and showing potential deviation from neutrality (indicated by decreased Tajima’s D values). Such a simultaneous decrease in genetic diversity and deviation from neutrality is exemplified by the region overlapping the MC1R gene. (D) Detection of selection defined by a hidden Markov model that identifies long genomic tracks with a high probability (≥0.80) of carrying advantageous mutations cosegregating within the six modern breeds.
Of the 125 genes undergoing positive selection during domestication, a subset is related to the differentiation, organization, and contraction of skeletal muscles, including ACTA1, C-SKI, MYBPC1, and delta-sarcoglycan (SGCD). Defects in these genes are associated with several forms of myopathies (41), including limb-girdle muscular dystrophy 2F, which causes limb musculature wasting and locomotory troubles in juvenile individuals. Additionally, many selected genes are involved in balance and motor coordination, including VRK1 and TCTN1, with defects associated with underdevelopment of the cerebellum, motor dysfunction and muscle hypotonia (42, 43), and CNTN6, loss of which causes motor coordination and equilibrium impairment in KO mice (44). This set of genes also includes a member of the collagen protein family, COL22A1, which localizes to myotendinous and articular junctions (45). Taken together, these observations reveal that genes influencing muscles, joints, balance, and locomotion have represented important targets of selection during horse domestication.
The cardiac system appears to be another key domestication target, with multiple related genes showing evidence for positive selection, including acyl-CoA dehydrogenase family, member 8 (ACAD8), B-raf proto-oncogene (BRAF), fanconi anemia, complementation group A (FANCA), SGCD, and calcium channel, voltage-dependent, L type, alpha 1D subunit (CACNA1D). Defects in these genes are associated with cardiomyopathy, cardiac malformation, sinoatrial node dysfunction, arrhythmia, and bradycardia (46). We also identify genes involved in electrolyte metabolism and homeostasis, including NR3C2, SCPEP1, WNK2, and CACNA1D, with important direct (47, 48) and indirect (49) physiological consequences for blood pressure regulation. These genes may have enabled the adaptation of the equine physiology to the increased energetic demands following their use in transportation, racing, chariotry, and agriculture.
Interestingly, our scans also identify genes associated with syndromes resulting in facial dysmorphy, skeletal dysplasia with severe growth retardation, and malformed or shortened limbs, including ACSF3, beta 1,3-galactosyltransferase-like (B3GALTL), N-acetylglucosamine-1-phosphate transferase, alpha and beta subunits (GNPTAB), nipped-B homolog (Drosophila; NIPBL), and POP1, as well as ACAD8, BRAF, and FANCA, which are also involved in cardiac pathologies (50–53). These genes may have been essential in skeletal and bone remodeling processes, possibly in relation to size shifts documented in the horse archeological record as early as the Late Bronze Age (54).
Finally, a large subset of genes is linked to brain development, including neural growth (NINJ1, NTM), axon and glial guidance (MATN2, DCC, ASTN1), synapse plasticity (DLGAP1), neurogenesis (ALK, NUMB) and a variety of neurological disorders. The latter include B3GALTL, GNPTAB, NIPBL, and voltage-dependent anion-selective channel protein 1 (VDAC1), which are associated with mental and psychomotor retardation and learning disability. A second set of genes is associated with behavioral syndromes, including JPH3, which is involved in Huntington disease-like 2 and is associated with movement impairment and chorea, as well as subcortical dementia (55). The voltage-dependent channel VDAC1 is involved in the behavioral fear response, and GRID1 is associated with social behavior and schizophrenia (56). CACNA1D has also been associated with schizophrenia (57) and has been reported in selection scans of cattle (58). These findings may enable characterization of the mechanisms facilitating the emergence of learning capabilities, social interactions, and behavioral responses compatible with human interactions, which are hallmarks of animal domestication (59).
In addition to the 125 genes considered above, many of the genes identified as associated with domestication by only one of the four methods are potentially interesting and worthy of further research. These genes include synaptojanin 2 (SYNJ2), which has been detected with PAML [phylogenetic analysis by maximum likelihood; false discovery rate (FDR) = 5%] and represents a longevity gene candidate associated with variation in levels of agreeableness and cognitive abilities (60, 61). Such changes could have been instrumental in the development of tameness and increased social interactions with humans.
Genetic Load as the Cost of Domestication.
Previous scans of domesticated genomes have revealed an accumulation of deleterious mutations in rice (62, 63), tomatoes (64), and dogs (4). This phenomenon has been termed the “cost of domestication” and is proposed to be driven by the repetitive bottlenecks associated with domestication, which reduce the efficiency of purifying selection (4).
We estimated the deleterious mutation load before and after domestication using genomic evolutionary rate profiling (GERP) scores to measure the strength of purifying selection (65) (SI Appendix, section S3). We disregarded CGG10023, due to insufficient genome coverage and high error rate, and Twilight, which represents the Thoroughbred horse used for generating the EquCab2.0 assembly and is therefore expected to show a strong deficit of derived alleles. We found significantly higher deleterious mutation loads in the genomes of the modern domesticated horses (Fig. 4B), where a higher proportion of sites under strong purifying selection were affected by mutations (SI Appendix, Fig. S30). The higher mutational load in domesticated horses is consistent with both the cost of domestication hypothesis and the recent major demographic bottleneck detected in horses (9) (SI Appendix, section S2.10). We note a striking difference in the estimated mutational loads for the two Icelandic genomes, which does not have an impact on the significance of our tests, however, and is likely explained by the different methodology used for generating the P5782 genome (66).
The observed difference in deleterious mutation loads between ancient and domesticated horses could also reflect the difference in their inbreeding levels (Fig. 4A and SI Appendix, Fig. S38). This is because the presence of a heterozygous mutation at a highly conserved site, which is assumed to be deleterious, implies a lower mutational load than a similar homozygous mutation. Consequently, long homozygosity tracts and a deficit of heterozygous sites resulting from inbreeding would inflate the estimated genetic load. We therefore made the calculation of mutational loads robust to inbreeding by conditioning the analyses on homozygous sites. We found that sites under especially strong selective constraint (GERP ≥ 4) remained enriched for mutations in the genomes of the domesticated horses relative to CGG10022 (P = 0.033). This relative enrichment suggests that our findings are not simply due to inbreeding but, instead, support the cost of domestication for horses. Interestingly, we also found that the deleterious mutation load in the genome of the wild Przewalski’s horse was similar to the deleterious mutation load observed in domesticated horses (Fig. 4B), which may result from their recent population collapse (7).
Conclusions
Many wild progenitors of domesticated animals have gone extinct or have experienced massive demographic bottlenecks (67), making them poor surrogates for the gene pool from which domesticated animals arose. Our results showcase how ancient genomes can reveal the genetic background in which domestication took place, thereby illuminating the molecular changes underlying domestication processes. Genome-wide information from time series recapitulating major domestication stages will be essential to understand fully the complex origins of the diversity of domesticated species that surround us today.
Methods
Detailed descriptions of samples and methods are provided in SI Appendix.
Sequencing of Ancient Samples.
A total of three and eight new indexed Illumina DNA libraries were built for CGG10022 and CGG10023, respectively. Libraries were prepared based on previously reported procedures (9, 24, 68), and contamination was monitored through mock extractions and amplification blanks. All controls were negative. Libraries were sequenced on Illumina platforms at the Danish National High-Throughput DNA Sequencing Center. The sequencing data generated in this study for CGG10022 and CGG10023 are available from the European Nucleotide Archive, using accession no. PRJEB7537.
Read Processing, Mapping, and Genotyping.
Reads were processed from trimming to genotyping using the PALEOMIX pipeline (69). Mapping and genotyping was done separately for the nuclear genome (plus chrUn) and for the mitochondrial genome. Default settings were used, except that a minimum mapping quality of 25 was required, the seed option was disabled, and uncollapsed paired-ended reads were excluded for ancient samples, given the expectation that ancient DNA templates are predominantly short-sized.
Metagenomic Analyses and Postmortem Damage.
Cytosine deamination rates and fragmentation patterns were plotted using mapDamage2.0 (70), based on 100,000 randomly selected alignments against EquCab2.0. Metagenomic analyses was carried out using MetaPhlan (71) implemented in the PALEOMIX pipeline, as previously described (69). Shotgun reads were mapped using Bowtie2, using end-to-end mode and default parameters, and taxon abundances were examined in the statistical environment R, version 3.0.1 (72), as described previously (24, 69).
Mitochondrial Phylogeny and Dating.
Mitochondrial genomes were typed following mapping against the horse reference mitochondrial genome (Genbank accession no. NC_001640), using a majority rule, and requiring three or more independent unique reads showing base qualities ≥30. We partitioned the alignment into ribosomal RNA; tRNA; a control region; and the first, second, and third codon positions for coding DNA sequences (CDSs) and followed previously described procedures (9) to perform Bayesian analyses in MrBayes (73) and Beast (74). In Beast, we used radiocarbon dates and/or stratigraphic context information for tip calibration and selected a Bayesian Skyline model assuming a log-uncorrelated relaxed clock (7.764 ≤ log Bayes Factor ≤ 44.985) to reconstruct the past demographic profile of horses.
Y-Chromosome Haplotype.
Y-chromosome haplotypes were recovered by aligning reads against previously identified Y-chromosome contigs (12, 75), first against the contigs alone and then remapped against the full nuclear genome, including the Y-chromosome contigs, to control for repetitive regions. Mapping and genotyping were as described above, except that a minimum depth of 4 and a maximum depth of 50 were used when filtering SNPs. Median joining networks (76) were obtained using Network v4.612.
PCA.
PCA was carried out using SNPs overlapping with the genomic coordinates covered by the equine SNP array for nine Przewalski’s horses, as well as 14 (8) and 32 (40) domestic breeds, representing a total of 348 and 729 individuals, respectively. Individual genotypes were converted into PLINK format (77) and further analyzed using “smartpca” of EIGENSOFT 4.0 (78). The two ancient samples were combined using a Procrustes transformation as implemented by the “proc” function of the Comprehensive R Archive Network (CRAN) library vegan (79).
Whole-Exome Phylogeny.
Phylogenetic inference was carried out using a partitioned (codon positions 1–2 and 3) supermatrix of 20,384 protein-coding genes from Ensembl v72 (80) following quality filtering, using the longest transcript for each gene. One hundred bootstrap pseudoreplicate alignments were generated from the supermatrix, and parsimony starting trees were generated using Randomized Axelerated Maximum Likelihood (RAxML) v7.3.2 (81) for both the original supermatrix and the bootstrap supermatrices. Phylogenetic inference was carried out for each supermatrix using Exascale Maximum Likelihood (ExaML) v1.0.2 (sco.h-its.org/exelixis/software.html) under the “GAMMA” model of nucleotide substitutions and the starting trees generated above.
TreeMix Analyses.
Patterns of population splits and migration events were analyzed using TreeMix (82) and the intersection of SNPs passing quality filters for the ancient specimens, all modern horses, and the domestic donkey (when included). In the first analysis, each sample was considered individually. In the second analysis, breeds were grouped according to their known historical affinities and up to one migration edge was considered. Both sets of analyses were run with or without the domestic donkey and provided identical topologies.
Admixture Tests Using D-Statistics.
The D-statistic was used to examine the presence of gene flow between the ancient horse population, the Przewalski’s horse population, and the population of domesticated horses. The D-statistic was originally described by Green et al. (83), and was implemented as described in Orlando et al. (9). Because the horse reference genome EquCab2.0 was generated from the Thoroughbred (Twilight) sample, this horse was excluded from the D-statistics.
Demographic Inference Using PSMC.
PSMC (29) was performed as described previously (9), with slight modifications of the maximum coverage threshold per sample. We set up input parameters to values recommended by the developers (29): number of iterations = 25, maximum 2N0 coalescent time = 15, initial θ∕ρ = 5. 100; bootstrap pseudoreplicates were performed by splitting chromosomal sequences into shorter fragments of 500 kb and randomly selected regions, with replacement, to evaluate the spread of the PSMC reconstructions. Demographic inferences were scaled using the genome-wide substitution rate of 7.242 × 10−9 per site per generation and a generation time of 8 y (9).
Population Split Times Using F-Statistics.
The TMRCAs of major phylogenetic clades observed in the whole-exome tree represent upper boundaries for population split times. These TMRCAs were calculated for each bootstrap tree in r8s using the Langley-Fitch (LF) method and POWELL algorithm (84), by constraining the root height to 4.0–4.5 Mya (9) and fixing the dates of CGG10022 and CGG10023 at 43 kya and 16.5 kya, respectively. We also used coalescent simulations and F-statistics (83) to estimate lower boundaries of population split times. Coalescent simulations were performed using fastsimcoal2 (85) under an isolation model and a composite demographic model. This latter model was derived from the PSMC inference for times older than 10 kya. Because the PSMC approach does not provide reliable demographic information for recent times, the demographic trajectory of the past 10,000 y was derived from previous Bayesian Skyline reconstructions (11, 12). Posterior distributions of population split times were obtained by simulating over a discrete grid of possible times (every 5,000 y for the past 200,000 y) and by using F-statistics and a standard local linear regression approach within an approximate Bayesian computation framework (86).
Genetic Load.
GERP scores computed from the alignment of 35 mammals to the human genome reference were used to quantify the level of evolutionary constraint at polymorphic sites (65). We converted the EquCab2.0 genomic coordinates of the horse polymorphic sites into the hg19 coordinates with the liftover tool (87), and the GERP score at each site was determined. Functional classifications into exons and nonsynonymous sites were obtained with ANNOVAR (88) applied to the Ensembl horse transcripts. Analyses were restricted to CDSs from Ensembl v72, as well as 10 bp upstream and downstream of each exon. For each polymorphic site observed in a given horse sample, we defined the ancestral state using the donkey sequence data and computed a measure of genetic load as the product of the GERP score at each site and the number of derived alleles carried by this individual at this site, averaged across sites for each individual. We considered only sites with GERP scores of −2 or greater because only those sites are considered as being under selection. We compared the distribution of genetic load measures among individuals using QQ plots and Kolmogorov–Smirnov tests.
Inbreeding Coverage.
Inbreeding was estimated following previously described methods (16) in which the proportion of mostly homozygous genomic segments is calculated; heterozygosity was estimated across the samples by calculating the individual Watterson estimator for sliding windows of 50 kb with a 10-kb step size using analysis of next-generation sequencing data (www.popgen.dk/angsd/), excluding regions where less than 90% of bases (45 kb) were covered and excluding transitions to account for the presence of postmortem DNA damage in CGG10022 and CGG10023. Segments with local changes in Watterson estimator values were estimated using the R package “changepoints,” utilizing the binary segmentation algorithm. Segment coordinates and corresponding heterozygosity estimates [mean log(Watterson estimator)] were extracted. We excluded the X-chromosome for males.
Genome-Wide Selection Scans.
First, we used branch tests as implemented in PAML to identify genes where the fixation rate of nonsynonymous mutations was significantly faster than the fixation rate of synonymous mutations in the domesticated horses (FDR = 5%). Second, we calculated Tajima’s D-statistics and the Watterson estimator to identify genomic regions where the genetic diversity decreased in the domesticated horses and significantly deviated from neutral expectations. Third, we capitalized on the recent publication of known genotypes at 54,602 SNP loci for ca. 400 horses from 32 modern domesticated breeds (8). We examined whether domesticated alleles at each locus coalesce more recently than ancient alleles, as would be expected if domestication results in a selective sweep. Finally, we developed a hidden Markov model to identify regions of the genome with longer than expected tracks in which alleles shared among the domesticated horses coalesce more recently than alleles in the predomesticated horses. Hits from each of the four methods were ranked on a scale (0, 1) separately for each analysis, and weights were calculated as the sum of ranks for each gene. We selected genes with a weight >1 as a conservative set of candidate genes.
Supplementary Material
Acknowledgments
We thank T. Brand, P. Selmer Olsen, and the laboratory technicians at the Danish National High-Throughput DNA Sequencing Centre for technical assistance. This work was supported by the Danish Council for Independent Research, Natural Sciences (FNU); the Danish National Research Foundation (DNFR94); a Marie-Curie Career Integration Grant (FP7 CIG-293845); and the International Research Group Program (IRG14-08), Deanship of Scientific Research (King Saud University, Saudi Arabia). H.J. was supported by a Marie-Curie Initial Training Network Grant (EUROTAST; FP7 ITN-290344); A.G. and L. Ermini were supported by Marie-Curie Intra-European Fellowships (FP7 IEF-299176 and FP7 IEF-302617); M.S. was supported by a Lundbeck Foundation Grant (R52-A5062); I.D., A.F., and L. Excoffier were supported by a Swiss National Science Foundation Grant (31003A-143393); M.H. was supported by a European Research Council (ERC) Consolidator Grant (310763); T.M.-B. was supported by an ERC Starting Grant (260372) and by a Ministerio de Ciencia e Innovación (MICINN) Grant (BFU2011-28549); B.S. was supported by the Packard Foundation; and D.C. was supported by start-up funds to B.S. from the University of California, Santa Cruz.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: Sequence data generated for this study (samples CGG10022 and CGG10023) have been deposited in the European Nucleotide Archive (accession no. PRJEB7537).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1416991111/-/DCSupplemental.
References
- 1.Kelekna P. The Horse in Human History. Cambridge Univ Press; Cambridge, England: 2009. [Google Scholar]
- 2.Outram AK, et al. The earliest horse harnessing and milking. Science. 2009;323(5919):1332–1335. doi: 10.1126/science.1168594. [DOI] [PubMed] [Google Scholar]
- 3.Wade CM, et al. Broad Institute Genome Sequencing Platform; Broad Institute Whole Genome Assembly Team Genome sequence, comparative analysis, and population genetics of the domestic horse. Science. 2009;326(5954):865–867. doi: 10.1126/science.1178158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cruz F, Vilà C, Webster MT. The legacy of domestication: Accumulation of deleterious mutations in the dog genome. Mol Biol Evol. 2008;25(11):2331–2336. doi: 10.1093/molbev/msn177. [DOI] [PubMed] [Google Scholar]
- 5.Groenen MA, et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature. 2012;491(7424):393–398. doi: 10.1038/nature11622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rubin CJ, et al. Strong signatures of selection in the domestic pig genome. Proc Natl Acad Sci USA. 2012;109(48):19529–19536. doi: 10.1073/pnas.1217149109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goto H, et al. A massively parallel sequencing approach uncovers ancient origins and high genetic variability of endangered Przewalski’s horses. Genome Biol Evol. 2011;3:1096–1106. doi: 10.1093/gbe/evr067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCue ME, et al. A high density SNP array for the domestic horse and extant Perissodactyla: Utility for association mapping, genetic diversity, and phylogeny studies. PLoS Genet. 2012;8(1):e1002451. doi: 10.1371/journal.pgen.1002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orlando L, et al. Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature. 2013;499(7456):74–78. doi: 10.1038/nature12323. [DOI] [PubMed] [Google Scholar]
- 10.Olsen S. Early horse domestication on the Eurasian steppe. In: Zeder M, Bradley D, Emschwiller E, Smith B, editors. Documenting Domestication: New Genetic and Archaeological Paradigms. Univ of California Press; Berkeley, CA: 2006. pp. 246–269. [Google Scholar]
- 11.Achilli A, et al. Mitochondrial genomes from modern horses reveal the major haplogroups that underwent domestication. Proc Natl Acad Sci USA. 2012;109(7):2449–2454. doi: 10.1073/pnas.1111637109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lippold S, et al. Discovery of lost diversity of paternal horse lineages using ancient DNA. Nat Commun. 2011;2:450. doi: 10.1038/ncomms1447. [DOI] [PubMed] [Google Scholar]
- 13.Jansen T, et al. Mitochondrial DNA and the origins of the domestic horse. Proc Natl Acad Sci USA. 2002;99(16):10905–10910. doi: 10.1073/pnas.152330099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ludwig A, et al. Coat color variation at the beginning of horse domestication. Science. 2009;324(5926):485. doi: 10.1126/science.1172750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shapiro B, Hofreiter M. A paleogenomic perspective on evolution and gene function: New insights from ancient DNA. Science. 2014;343(6169):1236573. doi: 10.1126/science.1236573. [DOI] [PubMed] [Google Scholar]
- 16.Prüfer K, et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature. 2014;505(7481):43–49. doi: 10.1038/nature12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rasmussen M, et al. Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature. 2010;463(7282):757–762. doi: 10.1038/nature08835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyer M, et al. A high-coverage genome sequence from an archaic Denisovan individual. Science. 2012;338(6104):222–226. doi: 10.1126/science.1224344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brotherton P, et al. Novel high-resolution characterization of ancient DNA reveals C > U-type base modification events as the sole cause of post mortem miscoding lesions. Nucleic Acids Res. 2007;35(17):5717–5728. doi: 10.1093/nar/gkm588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Briggs AW, et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc Natl Acad Sci USA. 2007;104(37):14616–14621. doi: 10.1073/pnas.0704665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pedersen JS, et al. Genome-wide nucleosome map and cytosine methylation levels of an ancient human genome. Genome Res. 2014;24(3):454–466. doi: 10.1101/gr.163592.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fierer N, et al. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc Natl Acad Sci USA. 2012;109(52):21390–21395. doi: 10.1073/pnas.1215210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heintzman PD, Elias SA, Moore K, Paszkiewicz K, Barnes I. Characterizing DNA preservation in degraded specimens of Amara alpina (Carabidae: Coleoptera) Mol Ecol Res. 2014;14(3):606–615. doi: 10.1111/1755-0998.12205. [DOI] [PubMed] [Google Scholar]
- 24.Der Sarkissian C, et al. Shotgun microbial profiling of fossil remains. Mol Ecol. 2014;23(7):1780–1798. doi: 10.1111/mec.12690. [DOI] [PubMed] [Google Scholar]
- 25.Vilà C, et al. Widespread origins of domestic horse lineages. Science. 2001;291(5503):474–477. doi: 10.1126/science.291.5503.474. [DOI] [PubMed] [Google Scholar]
- 26.Lindgren G, et al. Limited number of patrilines in horse domestication. Nat Genet. 2004;36(4):335–336. doi: 10.1038/ng1326. [DOI] [PubMed] [Google Scholar]
- 27.Nakhleh L. Computational approaches to species phylogeny inference and gene tree reconciliation. Trends Ecol Evol. 2013;28(12):719–728. doi: 10.1016/j.tree.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Svenning J-C. A review of natural vegetation openness in north-western Europe. Biol Conserv. 2002;104(2):133–148. [Google Scholar]
- 29.Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475(7357):493–496. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Durand EY, Patterson N, Reich D, Slatkin M. Testing for ancient admixture between closely related populations. Mol Biol Evol. 2011;28(8):2239–2252. doi: 10.1093/molbev/msr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cieslak M, et al. Origin and history of mitochondrial DNA lineages in domestic horses. PLoS ONE. 2010;5(12):e15311. doi: 10.1371/journal.pone.0015311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Warmuth V, et al. Reconstructing the origin and spread of horse domestication in the Eurasian steppe. Proc Natl Acad Sci USA. 2012;109(21):8202–8206. doi: 10.1073/pnas.1111122109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lippold S, Matzke NJ, Reissmann M, Hofreiter M. Whole mitochondrial genome sequencing of domestic horses reveals incorporation of extensive wild horse diversity during domestication. BMC Evol Biol. 2011;11:328. doi: 10.1186/1471-2148-11-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cahill JA, et al. Genomic evidence for island population conversion resolves conflicting theories of polar bear evolution. PLoS Genet. 2013;9(3):e1003345. doi: 10.1371/journal.pgen.1003345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petersen JL, et al. Genetic diversity in the modern horse illustrated from genome-wide SNP data. PLoS ONE. 2013;8(1):e54997. doi: 10.1371/journal.pone.0054997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Signer-Hasler H, et al. A genome-wide association study reveals loci influencing height and other conformation traits in horses. PLoS ONE. 2012;7(5):e37282. doi: 10.1371/journal.pone.0037282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hill EW, Gu J, McGivney BA, MacHugh DE. Targets of selection in the Thoroughbred genome contain exercise-relevant gene SNPs associated with elite racecourse performance. Anim Genet. 2010;41(Suppl 2):56–63. doi: 10.1111/j.1365-2052.2010.02104.x. [DOI] [PubMed] [Google Scholar]
- 38.Gu J, et al. Association of sequence variants in CKM (creatine kinase, muscle) and COX4I2 (cytochrome c oxidase, subunit 4, isoform 2) genes with racing performance in Thoroughbred horses. Equine Vet J Suppl. 2010;(38):569–575. doi: 10.1111/j.2042-3306.2010.00181.x. [DOI] [PubMed] [Google Scholar]
- 39.Hill EW, et al. MSTN genotype (g.66493737C/T) association with speed indices in Thoroughbred racehorses. J Appl Physiol (1985) 2012;112(1):86–90. doi: 10.1152/japplphysiol.00793.2011. [DOI] [PubMed] [Google Scholar]
- 40.Petersen JL, et al. Genome-wide analysis reveals selection for important traits in domestic horse breeds. PLoS Genet. 2013;9(1):e1003211. doi: 10.1371/journal.pgen.1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nowak KJ, Ravenscroft G, Laing NG. Skeletal muscle α-actin diseases (actinopathies): Pathology and mechanisms. Acta Neuropathol. 2013;125(1):19–32. doi: 10.1007/s00401-012-1019-z. [DOI] [PubMed] [Google Scholar]
- 42.Renbaum P, et al. Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am J Hum Genet. 2009;85(2):281–289. doi: 10.1016/j.ajhg.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parisi M, Glass I. Joubert syndrome and related disorders. In: Pagon RA, et al., editors. GeneReviews(R) University of Washington; Seattle, WA: 1993. [PubMed] [Google Scholar]
- 44.Takeda Y, et al. Impaired motor coordination in mice lacking neural recognition molecule NB-3 of the contactin/F3 subgroup. J Neurobiol. 2003;56(3):252–265. doi: 10.1002/neu.10222. [DOI] [PubMed] [Google Scholar]
- 45.Koch M, et al. A novel marker of tissue junctions, collagen XXII. J Biol Chem. 2004;279(21):22514–22521. doi: 10.1074/jbc.M400536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baig SM, et al. Loss of Ca(v)1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat Neurosci. 2011;14(1):77–84. doi: 10.1038/nn.2694. [DOI] [PubMed] [Google Scholar]
- 47.Azizan EA, et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet. 2013;45(9):1055–1060. doi: 10.1038/ng.2716. [DOI] [PubMed] [Google Scholar]
- 48.Scholl UI, et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet. 2013;45(9):1050–1054. doi: 10.1038/ng.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rickard AJ, et al. Endothelial cell mineralocorticoid receptors regulate deoxycorticosterone/salt-mediated cardiac remodeling and vascular reactivity but not blood pressure. Hypertension. 2014;63(5):1033–1040. doi: 10.1161/HYPERTENSIONAHA.113.01803. [DOI] [PubMed] [Google Scholar]
- 50.Borck G, et al. NIPBL mutations and genetic heterogeneity in Cornelia de Lange syndrome. J Med Genet. 2004;41(12):e128. doi: 10.1136/jmg.2004.026666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tonkin ET, Wang TJ, Lisgo S, Bamshad MJ, Strachan T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat Genet. 2004;36(6):636–641. doi: 10.1038/ng1363. [DOI] [PubMed] [Google Scholar]
- 52.Glazov EA, et al. Whole-exome re-sequencing in a family quartet identifies POP1 mutations as the cause of a novel skeletal dysplasia. PLoS Genet. 2011;7(3):e1002027. doi: 10.1371/journal.pgen.1002027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aggarwal S, et al. Prenatal skeletal dysplasia phenotype in severe MLII alpha/beta with novel GNPTAB mutation. Gene. 2014;542(2):266–268. doi: 10.1016/j.gene.2014.03.053. [DOI] [PubMed] [Google Scholar]
- 54.Benecke N, Driesch AVD. Horse exploitation in the Kazakh steppes during the Eneolithic and Bronze Age. In: Levine M, Renfrew C, Boyle K, editors. Prehistoric Steppe Adaptation and the Horse. McDonald Institute for Archaeological Research; Cambridge, UK: 2003. pp. 69–82. [Google Scholar]
- 55.Schneider SA, Marshall KE, Xiao J, LeDoux MS. JPH3 repeat expansions cause a progressive akinetic-rigid syndrome with severe dementia and putaminal rim in a five-generation African-American family. Neurogenetics. 2012;13(2):133–140. doi: 10.1007/s10048-012-0318-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nenadic I, et al. Glutamate receptor δ 1 (GRID1) genetic variation and brain structure in schizophrenia. J Psychiatr Res. 2012;46(12):1531–1539. doi: 10.1016/j.jpsychires.2012.08.026. [DOI] [PubMed] [Google Scholar]
- 57.Nyegaard M, et al. CACNA1C (rs1006737) is associated with schizophrenia. Mol Psychiatry. 2010;15(2):119–121. doi: 10.1038/mp.2009.69. [DOI] [PubMed] [Google Scholar]
- 58.Qanbari S, et al. Classic selective sweeps revealed by massive sequencing in cattle. PLoS Genet. 2014;10(2):e1004148. doi: 10.1371/journal.pgen.1004148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zeder MA. The domestication of animals. J Anthropol Res. 2012;68(2):161–190. [Google Scholar]
- 60.Luciano M, et al. Longevity candidate genes and their association with personality traits in the elderly. Am J Medical Genet B Neuropsychiatr Genet. 2012;159B(2):192–200. doi: 10.1002/ajmg.b.32013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lopez LM, et al. Evolutionary conserved longevity genes and human cognitive abilities in elderly cohorts. Eur J Hum Genet. 2012;20(3):341–347. doi: 10.1038/ejhg.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lu J, et al. The accumulation of deleterious mutations in rice genomes: A hypothesis on the cost of domestication. Trends Genet. 2006;22(3):126–131. doi: 10.1016/j.tig.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 63.Nabholz B, et al. Transcriptome population genomics reveals severe bottleneck and domestication cost in the African rice (Oryza glaberrima) Mol Ecol. 2014;23(9):2210–2227. doi: 10.1111/mec.12738. [DOI] [PubMed] [Google Scholar]
- 64.Koenig D, et al. Comparative transcriptomics reveals patterns of selection in domesticated and wild tomato. Proc Natl Acad Sci USA. 2013;110(28):E2655–E2662. doi: 10.1073/pnas.1309606110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cooper GM, et al. NISC Comparative Sequencing Program Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15(7):901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Andersson LS, et al. Mutations in DMRT3 affect locomotion in horses and spinal circuit function in mice. Nature. 2012;488(7413):642–646. doi: 10.1038/nature11399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Freedman AH, et al. Genome sequencing highlights the dynamic early history of dogs. PLoS Genet. 2014;10(1):e1004016. doi: 10.1371/journal.pgen.1004016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Seguin-Orlando A, et al. Ligation bias in illumina next-generation DNA libraries: Implications for sequencing ancient genomes. PLoS ONE. 2013;8(10):e78575. doi: 10.1371/journal.pone.0078575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schubert M, et al. Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX. Nat Protoc. 2014;9(5):1056–1082. doi: 10.1038/nprot.2014.063. [DOI] [PubMed] [Google Scholar]
- 70.Jónsson H, Ginolhac A, Schubert M, Johnson PL, Orlando L. mapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics. 2013;29(13):1682–1684. doi: 10.1093/bioinformatics/btt193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Segata N, et al. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods. 2012;9(8):811–814. doi: 10.1038/nmeth.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.R-Core-Team 2013 R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna. Available at www.R-project.org.
- 73.Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17(8):754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 74.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wallner B, et al. Identification of genetic variation on the horse y chromosome and the tracing of male founder lineages in modern breeds. PLoS ONE. 2013;8(4):e60015. doi: 10.1371/journal.pone.0060015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16(1):37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- 77.Purcell S, et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2(12):e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dixon P. VEGAN, a package of R functions for community ecology. J Veg Sci. 2003;14(6):927–930. [Google Scholar]
- 80.Flicek P, et al. Ensembl 2013. Nucleic Acids Res. 2013;41(Database issue):D48–D55. doi: 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stamatakis A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 82.Pickrell JK, Pritchard JK. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012;8(11):e1002967. doi: 10.1371/journal.pgen.1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Green RE, et al. A draft sequence of the Neandertal genome. Science. 2010;328(5979):710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sanderson MJ. r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics. 2003;19(2):301–302. doi: 10.1093/bioinformatics/19.2.301. [DOI] [PubMed] [Google Scholar]
- 85.Excoffier L, Dupanloup I, Huerta-Sánchez E, Sousa VC, Foll M. Robust demographic inference from genomic and SNP data. PLoS Genet. 2013;9(10):e1003905. doi: 10.1371/journal.pgen.1003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Csilléry K, François O, Blum MGB. ABC: An R package for approximate Bayesian computation (ABC) Methods Ecol Evol. 2012;3(3):475–479. [Google Scholar]
- 87.Karolchik D, et al. The UCSC Genome Browser database: 2014 update. Nucleic Acids Res. 2014;42(Database issue):D764–D770. doi: 10.1093/nar/gkt1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang K, Li M, Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.