

Significance
RNA-binding motif protein 38 (Rbm38) is a target of the p53 family and modulates p53 expression via mRNA translation. However, the biological function of Rbm38 and the role of the p53-Rbm38 loop in tumor suppression have not been studied in vivo. Here, we show that mice deficient in Rbm38 exhibit hematopoietic defects and are susceptible to spontaneous tumors and accelerated aging. Furthermore, we show that Rbm38 is critical for p53-mediated radiosensitivity and tumor suppression. Together, our results suggest that Rbm38 is necessary for normal hematopoiesis and for suppressing accelerated aging and tumorigenesis. In addition, the p53-Rbm38 axis might be explored for extending longevity and for tumor suppression.
Keywords: Rbm38, p53, aging, hematopoiesis, tumor suppression
Abstract
RNA-binding motif protein 38 (Rbm38), also called RNPC1 [RNA-binding region (RNP1, RRM) containing 1], is a target of the p53 family and modulates p53 expression via mRNA translation. To investigate the biological function of Rbm38 in vivo, we generated an Rbm38-null mouse model. We showed that mice deficient in Rbm38 exhibit signs of accelerated aging and are prone to hematopoietic defects and spontaneous tumors. To determine the biological significance of the p53-Rbm38 loop, we showed that Rbm38 deficiency enhances accumulation of p53 induced by ionizing radiation (IR) and sensitizes mice to IR-induced lethality in a p53-dependent manner. Most importantly, Rbm38 deficiency markedly decreases the tumor penetrance in mice heterozygous for p53 via enhanced p53 expression. Interestingly, we found that Rbm38 deficiency shortens the life span of, and promotes lymphomagenesis in, mice deficient in p53. These results provide genetic evidence that Rbm38 is necessary for normal hematopoiesis and for suppressing accelerated aging and tumorigenesis. Thus, the p53-Rbm38 axis might be explored for extending longevity and for tumor suppression.
RNA-binding proteins (RBPs) are master regulators of RNA biogenesis and metabolism (1). Consistent with these crucial functions, RBPs are found to be altered in many human diseases, such as neurological disorders and muscular atrophy (2). Studies also suggest that RBPs form a complex network with oncoproteins and tumor suppressors and thus have profound impacts on tumor development and progression (3, 4).
Rbm38, also called RNPC1, encodes an RBP and is expressed as two isoforms, Rbm38 with 239 aa and Rbm38b with 121 aa. Both Rbm38 and Rbm38b contain one RNA recognition motif (RRM), which shares a sequence similarity with the ones found in Musashi, HuR, and nucleolin (5). Rbm38 is known to interact with its target mRNAs and regulate their expression via mRNA stability. For example, Rbm38 is necessary for mRNA stability of p21, p73, GDF15, and HuR transcripts but suppresses mRNA stability of p63 and Mdm2 transcripts (5–10). Rbm38 is also found to regulate alternative splicing of the EPB41 and FGFR2 genes (11, 12). Likewise, SUP-12, the Rbm38 ortholog in Caenorhabditis elegans, regulates alternative splicing of the ADF and cofilin genes (13–15). Additionally, Rbm38 is found to repress p53 mRNA translation via interaction with eIF4E on p53 mRNA (16). Interestingly, phosphorylation of Rbm38 at serine 195 by GSK3 abolishes its interaction with eIF4E and converts Rbm38 from a repressor to an activator of p53 mRNA translation (17). Furthermore, Rbm38 is found to modulate p53 activity by relieving microRNA-mediated repression of several p53 targets, including p21, DDIT4, LATS2, and Rbm38, itself (18).
The biological function of Rbm38 is implicated in the cell cycle control, differentiation, and senescence (5, 16, 19). Consistently, altered expression of Rbm38 is found in many types of cancers. For example, Rbm38 overexpression is found in breast-cancer patients with poor prognosis (20, 21) and is associated with malignant transformation of colorectal adenoma to carcinoma (22, 23). We show that Rbm38 is overexpressed in canine lymphoma, which is correlated with decreased expression of p53 (16). However, a study also showed that Rbm38 is down-regulated in breast cancer via promoter hypermethylation (18). In addition, Rbm38 is found to be a target of E2F1, and overexpression of E2F1 and Rbm38 is associated with increased survival in patients diagnosed with ovarian cancer, breast cancer, and glioblastoma (24). Thus, it seems that Rbm38 may have two opposing functions during cancer development and thus further studies are needed to elucidate the underlying mechanisms.
To investigate the biological functions of Rbm38 in vivo, we generated and characterized Rbm38-deficient mice. We found that Rbm38-null mice exhibit accelerated aging phenotypes and are prone to hematopoietic defects and spontaneous tumors. Moreover, we showed that the Rbm38 deficiency leads to p53-dependent radiosensitivity and reduces tumor penetrance in p53 heterozygous mice. Furthermore, we showed that Rbm38 cooperates with p53 to suppress lymphomagenesis.
Results
Mice Deficient in Rbm38 Exhibit Accelerated Aging-Related Phenotypes.
To understand the biological function of Rbm38 in vivo, conventional Rbm38-null mice were generated by intercrossing cre transgenic mice (EIIa-cre) with Rbm38fl/fl mice, which were generated using the cre-loxP system (16). The loss of Rbm38 mRNA was confirmed in Rbm38−/− mouse embryonic fibroblasts (MEFs) by RT-PCR (Fig. S1A). We found that Rbm38-null mice were viable and readily obtained at Mendelian ratios without any apparent defects in survival or fertility. To investigate the function of Rbm38 in vivo, we monitored a cohort of wild-type (WT) (n = 17) and Rbm38−/− (n = 23) mice throughout their life span. Interestingly, for the first year, Rbm38−/− mice were indistinguishable from WT control mice. However, by 18 mo, most of the Rbm38−/− mice exhibited weight loss and ulcerated skin lesions compared with WT mice (Fig. 1A and Fig. S1B). In addition, Rbm38−/− mice had a shorter life span than WT mice. The median survival time for tumor-free Rbm38−/− mice (n = 11) was 94 wk compared with 121 wk for tumor-free WT mice (n = 14) (Fig. 1B). The maximal life span was 128 wk for an Rbm38−/− mouse but 144 wk for a WT mouse (Fig. 1B). Pairwise comparison revealed a significant difference in survival between these two groups (P = 0.006 by log-rank test). To determine a potential cause associated with these phenotypes, age- and sex-matched WT and Rbm38−/− mice were used for gross necropsies to determine the size and weight of various organs. Indeed, we found a significant reduction of adipose tissue in Rbm38−/− mice compared with WT mice. The relative percentage of gonadal fat vs. body weight was 1.8% for WT mice, but only 0.4% for Rbm38−/− mice (Fig. 1C). H&E staining showed small adipocytes in Rbm38−/− mice compared with WT mice (Fig. S1C). Thus, the reduction of adipose tissue is most likely responsible for the loss of weight in Rbm38−/− mice. Moreover, because Rbm38−/− mice showed hunchback, the degree of spinal curvature was measured by microcomputed tomography (micro-CT) scan. We found pronounced lordokyphosis in Rbm38−/− mice compared with age- and sex-matched WT mice (Fig. 1D and Fig. S1D). The average spine angle was reduced from 103 degrees in WT mice (n = 3) to 76 degrees in Rbm38−/− mice (n = 4) (Fig. S1E).
Fig. 1.

Mice deficient in Rbm38 exhibit accelerated aging-related phenotypes. (A) Representative photograph of 18-mo-old WT and Rbm38−/− mice. (B) Kaplan–Meyer survival curves of WT (n = 14) and Rbm38−/− (n = 11) mice (P = 0.006 by log-rank test). (C) Relative percentage of gonadal fat pad weight relative to the whole body weight in WT (n = 3) and Rbm38−/− mice (n = 3). Bars represent means ± SD. (D) Representative Micro-CT images from age- and sex-matched WT and Rbm38−/− mice. Rbm38−/− mouse displays a pronounced lordokyphosis (curvature of the spine) phenotype. (E) SA-β-gal staining of the liver tissue from age- and sex-matched WT and Rbm38−/− mice. (F) The SA-β-gal–stained liver tissues shown in E were sectioned and then counterstained with nuclear fast red. (G) The level of Rbm38, p16Ink4a, and GAPDH was determined in the liver tissues of 85-wk-old or 128-wk-old WT and Rbm38−/− mice by RT-PCR. (H) The level of Rbm38, p53, and actin was determined in the spleen and thymus of WT and Rbm38−/− mice.
Mice deficient in Rbm38 exhibit signs of short lifespan, reduced body fat, and lordokyphosis, which resembles some aspects of accelerated aging. Thus, the expression of senescence-associated-β-galactosidase (SA-β-gal), a widely used biomarker for aging, was examined in the tissues from WT and Rbm38−/− mice at various ages. We found that young Rbm38−/− mice (17 wk of age) showed slightly increased SA-β-gal staining in the kidney but not in the liver compared with WT mice (Fig. S1F). However, a significant increase in SA-β-gal activity was detected in the livers from old Rbm38−/− mice (85 and 128 wk of age) compared with WT mice (Fig. 1 E and F). We also found that the level of p16Ink4a transcript, another aging biomarker (25), was increased in the livers of old Rbm38−/− mice (85 and 128 wk of age) (Fig. 1G). Additionally, we showed that p16Ink4a transcript was not altered by Rbm38 deficiency in MEFs (Fig. S1G), suggesting that p16Ink4a is not a target of Rbm38. Furthermore, because p53 has a profound impact on aging and Rbm38 represses p53 translation (16, 26), we examined p53 expression in both WT and Rbm38-null mice. We found that the basal level of p53 was increased in the spleen and thymus of Rbm38−/− mice compared with WT mice (Fig. 1H). Together, these results indicate that enhanced p53 activation may contribute to the accelerated aging mediated by Rbm38 deficiency.
Mice Deficient in Rbm38 Are Prone to Hematopoietic Defects.
To determine other potential pathological defects in Rbm38−/− mice, we performed gross necropsies on WT and Rbm38−/− mice and noticed that the spleens were consistently enlarged (splenomegaly) in Rbm38−/− mice compared with WT mice. Indeed, splenomegaly was found in Rbm38−/− mice at 2 mo of age and became much pronounced in aged Rbm38−/− mice (Fig. 2A). Histological examination revealed that, compared with WT mice, the spleen structure of Rbm38−/− mice was altered with a striking expansion of the white pulp (Fig. 2B). In addition, the spleens of Rbm38−/− mice showed extensive extramedullary hematopoiesis (EMH), with an increased number of megakaryocytes (Fig. S2A). Similarly, EMH was found in the livers of Rbm38−/− mice (Fig. S2B). Indeed, ∼78% of RBM38−/− mice showed splenic EMH compared with 47% of older WT mice (Tables S1 and S2). Likewise, ∼26% of Rbm38−/− mice showed EMH in the livers compared with 12% of WT mice (Tables S1 and S2). Because the EMH is likely to be a compensatory mechanism due to bone-marrow dysfunction, we examined the bone marrows of WT and Rbm38−/− mice. We found that, compared with WT mice, the bone marrow of Rbm38−/− mice exhibited decreased erythropoiesis (erythroid hypoplasia), with relatively increased ratio of myeloid vs. erythroid lineages (Fig. 2C and Fig. S2C). In contrast, the percentage of hematocrit was decreased in Rbm38−/− mice (30.5 ± 6.3) compared with WT mice (43.4 ± 4.1) (Fig. 2D). Similarly, the level of hemoglobin was decreased in Rbm38−/− mice (8.6 ± 1.7 g/dL) compared with WT mice (13.6 ± 1.4 g/dL) (Fig. 2D), suggesting that Rbm38−/− mice are anemic. Together, these data suggest that mice deficient in Rbm38 are prone to hematopoietic defects.
Fig. 2.

Mice deficient in Rbm38 are prone to hematopoietic defects. (A) Representative photographs of spleens from 2-mo-old (Left) and 18-mo old (Right) WT and Rbm38−/− mice. (B) Representative images of H&E-stained sections of spleens from WT and Rbm38−/− mice. (C) Representative images of periodic acid–Schiff (PAS)-stained sections of bone marrows from WT and Rbm38−/− mice. (D) The level of hemoglobin and the percentage of hematocrit were measured from the blood samples of WT (n = 4) and Rbm38−/− (n = 4) mice. Data are represented as mean ± SD.
Mice Deficient in Rbm38 Are Prone to Spontaneous Tumors.
Unexpectedly, we noticed that Rbm38−/− mice started to show tumor-associated phenotypes as they aged. Thus, a cohort of WT (n = 17) and Rbm38−/− mice (n = 23) were monitored for potential tumor formation over a 3-y period. The overall survival time was significantly shorter for Rbm38−/− mice compared with WT mice (Fig. S3A). We would like to mention that the tumor-free mice from this cohort were also used for survival analysis in Fig. 1B. Moribund mice of each genotype were scarified for pathological analysis. We found that 12 out of 23 Rbm38−/− mice, but only 3 out of 17 WT mice, developed spontaneous tumors (Fig. 3A and Tables S1 and S2). Statistical analysis indicated that the tumor penetrance in Rbm38−/− mice was significantly higher than that in WT mice (P = 0.023 by Fisher’s exact test). Moreover, Rbm38−/− mice developed a broader spectrum of tumors compared with WT mice. In addition to lymphoma and histiocytic sarcoma, Rbm38−/− mice developed metastatic hemangiosarcoma (Fig. 3B) and hepatoma (Fig. 3C). Furthermore, to confirm the diagnoses of histiocytic sarcoma and lymphoma, immunohistochemsitry was performed using antibodies against B220 (B-cell marker), CD3 (T-cell marker), and F4/80 (macrophage marker). We showed that the histiocytic sarcoma was positive for F4/80 but not for B220 and CD3 (Fig. S3B). By contrast, the lymphoma was positive for B220, with background scattered reactive CD3-positive T cells, but not for F4/80 (Fig. S3C). Finally, to determine the potential mechanism whereby Rbm38 is involved in tumor suppression, RNA-seq was performed with total RNAs isolated from WT and Rbm38−/− MEFs. Interestingly, comparative analysis indicated that several immune and inflammatory genes, including TLR7, IL17D, and Tnfsf15, were highly up-regulated in Rbm38−/− MEFs, which were then confirmed by RT-PCR analysis (Fig. 3D). TLR7 is a member of the TLR family and mediates production of proinflammatory cytokines (27). IL17D, a member of the IL17 family, and Tnfsf15, also called TL1A and a member of the tumor necrosis factor (TNF) ligand superfamily, are proinflammatory cytokines (28, 29). Importantly, all three genes are involved in proinflammation, a process linking inflammation and cancer. Together, these data indicate that Rbm38 deficiency may lead to production of proinflammatory cytokines, which contribute to a tumor microenvironment and, consequently, tumorigenesis.
Fig. 3.

Mice deficient in Rbm38 are prone to spontaneous tumors. (A) Spontaneous tumor spectrum in WT (n = 17) and Rbm38−/− (n = 23) mice (P = 0.023 by Fisher’s exact test). (B) H&E-stained sections of hemangiosarcoma in the liver of an Rbm38−/− mouse at 20× (Left) and 200× (Right). (C) H&E-stained sections of hepatoma in the liver of an Rbm38−/− mouse at 20× (Left) and 200× (Right). (D) The level of IL17D, Tnfsf15, and TLR7 transcripts was determined from WT and Rbm38−/− MEFs by RT-PCR analysis.
Loss of Rbm38 Sensitizes Mice to IR-Induced Lethality in a p53-Dependent Manner.
We showed previously that Rbm38 is a p53 target and forms a feedback regulatory loop with p53 by modulating p53 mRNA translation (16). Thus, to investigate the biological significance of the p53-Rbm38 loop in vivo, we determined whether Rbm38 has any effect on p53-mediated radiosensitivity. To address this, we examined whether Rbm38 regulates p53 expression in response to γ-irradiation. Specifically, WT and Rbm38−/− mice were exposed to 4 gray of whole-body γ-irradiation. Four hours postirradiation, the level of p53 protein was examined in radiosensitive tissues, including spleen and thymus. We found that the level of p53 protein induced by IR was markedly higher in Rbm38−/− mice than that in WT mice (Fig. 4A). Similarly, the level of γ-H2A.X was increased by Rbm38 deficiency (Fig. 4A). Next, we determined whether loss of Rbm38 has any effect on radiosensitivity. To address this, a cohort of WT (n = 29) and Rbm38−/− (n = 38) mice at the age of 6–8 wk were exposed to 8 gray of whole-body γ-irradiation and monitored daily for survival. We found that the median survival time was 13 d for Rbm38−/− mice and 20 d for WT mice, suggesting that loss of Rbm38 leads to enhanced radiosensitivity (Fig. 4B). Pairwise comparison indicated that the difference in survival time between WT and Rbm38−/− mice was statistically significant (P < 0.001 by log-rank test). Furthermore, to determine whether p53 is responsible for the increased radiosensitivity in Rbm38−/− mice, a cohort of p53−/− (n = 10) and Rbm38−/−; p53−/− (n = 14) mice was generated and subjected to 8 gray of whole-body γ-irradiation. Like p53−/− mice, all Rbm38−/−; p53−/− mice survived for at least 34 d after γ-irradiation (Fig. 4B). Together, these data suggest that loss of Rbm38 enhances radiosensitivity in a p53-dependent manner.
Fig. 4.
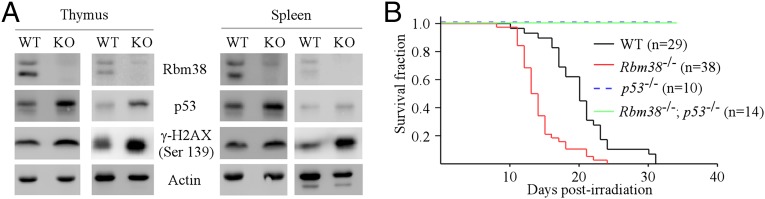
Loss of Rbm38 sensitizes mice to IR-induced lethality in a p53-dependent manner. (A) WT and Rbm38−/− mice were exposed to 4 gray of whole body γ-irradiation. Four hours postirradiation, lysates from spleen and thymus were collected, and the level of Rbm38, p53, phospho-γ-H2AX, and actin was determined. (B) Kaplan–Meier survival curves of WT (n = 29), Rbm38−/− (n = 38), p53−/−(n = 10), and Rbm38−/−; p53−/− (n = 14) mice after 8 gray of whole-body γ-irradiation.
Loss of Rbm38 Significantly Reduces Tumor Penetrance in p53 Heterozygous Mice.
p53 is the most commonly mutated gene in human cancer, and p53+/− mice are tumor-prone due to haploinsufficiency (30–32). Thus, we asked whether Rbm38 deficiency has an effect on tumor formation in a p53 heterozygous background. To address this hypothesis, we first showed that p53 expression was increased in Rbm38−/−; p53+/− MEFs compared with that in p53+/− MEFs (Fig. 5A). Next, we determined whether the increased expression of p53 by Rbm38 deficiency plays a role in tumor suppression in p53 heterozygous mice. In this regard, a cohort of p53+/− (n = 24) and Rbm38−/−; p53+/− (n = 21) mice was generated and monitored for their survival time, tumor penetrance, and spectrum. Although the median survival time for Rbm38−/−; p53+/− mice (70 wk) was longer than that for p53+/− mice (64 wk), the difference in survival time was not statistically significant (P = 0.371 by log-rank test) (Fig. 5B). Additionally, the tumor spectrum in Rbm38−/−; p53+/− mice exhibited minor difference compared with that in p53+/− mice (Table S3). Histopathological analysis indicated that the most frequent tumors for p53+/− and Rbm38−/−; p53+/− mice were sarcomas and lymphomas, consistent with previous reports (30, 31). Most importantly, we found that the tumor penetrance was significantly decreased in RBM38−/−; p53+/− mice compared with p53+/− mice (Fig. 5C and Tables S4 and S5). Indeed, 9 out of 21 Rbm38−/−; p53+/− mice, but only 1 out of 24 p53+/− mice, did not succumb to tumors. Statistical analysis indicated that the difference in tumor penetrance between these two groups was highly significant (P = 0.00221 by Fisher’s exact test). We would like to mention that most of the tumor-free Rbm38−/−; p53+/− mice displayed nonmalignant lesions, such as splenic follicular hyperplasia, steatosis in livers (Fig. 5C and Table S5), and extensive inflammation in various organs. These lesions may be the cause of death for tumor-free Rbm38−/−; p53+/− mice. Together, these data indicate that loss of Rbm38 reduces tumor penetrance in p53 heterozygous mice.
Fig. 5.

Loss of Rbm38 significantly reduces the tumor penetrance in p53-heterozygous mice. (A) The level of Rbm38, p53, and actin was determined in p53+/− and Rbm38−/−; p53+/− MEFs. (B) Kaplan–Meyer survival curves of p53+/− (n = 24) and Rbm38−/−; p53+/− (n = 21) mice (P = 0.371 by log-rank test). (C) Comparison of tumor-free mice between a cohort of p53+/− and Rbm38−/−; p53+/− mice.
Loss of Rbm38 Shortens the Life Span of, and Facilitates Lymphomagenesis in, p53-Null Mice.
p53-null mice are prone to spontaneous tumors (30, 33, 34). In addition, we showed that Rbm38−/− mice develop spontaneous tumors (Fig. 3A). Thus, we asked whether mice deficient in both p53 and Rbm38 are highly prone to tumor formation. To address this hypothesis, a cohort of p53−/− (n = 24) and Rbm38−/−; p53−/− (n = 19) mice was generated and monitored for their survival, tumor penetrance, and spectrum. The tumor penetrance in Rbm38−/−; p53−/− mice did not significantly differ from that in p53−/− mice (89% vs. 100%; P = 0.199 by Fisher’s exact test). However, two Rbm38−/−; p53−/− mice did not die from tumor-associated disease. We also found that the tumor latency was shorter in Rbm38−/−; p53−/− mice than that in p53−/− mice. The tumor onset was 15 wk for a Rbm38−/−; p53−/− mouse compared with 18 wk for a p53−/− mouse (Fig. 6A and Tables S6 and S7). The longest lifespan was 29 wk for a Rbm38−/−; p53−/− mouse compared with 34 wk for a p53−/− mouse (Fig. 6A and Tables S6 and S7). The median survival time was 19 wk for Rbm38−/−; p53−/− mice compared with 25 wk for p53−/− mice (Fig. 6A). The difference in survival time between these two groups was statistically significant (P = 0.006 by log-rank test). Furthermore, the tumor spectrum in Rbm38−/−; p53−/− mice was significantly different from the one in p53−/− mice (Table S8). Unlike p53−/− mice, Rbm38−/−; p53−/− mice did not develop hibernoma or hemangioma. Moreover, the frequency of lymphomas was significantly increased in Rbm38−/−; p53−/− mice than that in p53−/− mice (94.1% vs. 60.6%; P = 0.010 by Fisher’s exact test). By contrast, the sarcoma incidence was significantly decreased in Rbm38−/−; p53−/− mice compared with p53−/− mice (5.8% vs. 30.3%, P = 0.042 by Fisher’s exact test). Furthermore, tumor burden was markedly reduced in Rbm38−/−; p53−/− mice compared with p53−/− mice (Tables S6 and S7). 33.3% of p53−/− mice whereas none of Rbm38−/−; p53−/− mice developed more than one primary tumors. Together, these data suggest that Rbm38 deficiency shortens the life span of, and promotes lymphomagenesis in, p53-null mice.
Fig. 6.
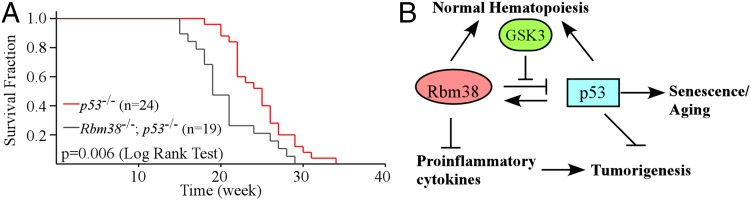
Loss of Rbm38 shortens the life span of, and facilitates lymphomagenesis in, p53-null mice. (A) Kaplan–Meyer survival curves of p53−/− (n = 24) and Rbm38−/−; p53−/− (n = 19) mice (P = 0.006 log-rank test). (B) A proposed model for the role of the p53-Rbm38 loop in aging, normal hematopoiesis, and tumor suppression.
Discussion
In this study, Rbm38- and p53-deficient mice models were used to characterize the biological function of Rbm38 and the role of the p53-Rbm38 loop in aging and tumor suppression. We showed that Rbm38 plays an instrumental role in aging, normal hematopoiesis, and tumor suppression. Moreover, we showed that Rbm38 deficiency enhances radiosensitivity in mice, which can be rescued by loss of p53. Furthermore, we showed that Rbm38 deficiency reduces tumor penetrance in p53 heterozygous mice but promotes lymphomagenesis in p53-null mice. Based on these findings, a model for the role of the p53-Rbm38 axis in aging, normal hematopoiesis, and tumor suppression is proposed and presented in Fig. 6B.
The Role of Rbm38 in Normal Hematopoiesis.
According to a previous report (11) and the Human Protein Atlas (35), Rbm38 is highly expressed in hematopoietic organs, such as spleen, thymus, and bone marrow. The expression profile suggests that Rbm38 plays a role in hematopoiesis. Indeed, we found that Rbm38−/− mice display severe hematopoietic defects, including chronic normocytic anemia, decreased marrow erythropoiesis, increased ratio of myeloid vs. erythroid, increased EMH in the spleen and liver, and splenomegaly (Fig. 2 and Fig. S2). Of note, EMH is considered as a physiologic, compensatory mechanism for insufficient bone marrow hematopoiesis. The excessive EMH observed in Rbm38−/− mice may be indicative of dysregulation of hematopoietic stem cells and pathological alterations in bone marrow niche microenvironment. Additionally, the increased ratio of myeloid vs. erythroid in bone marrow in the context of persistent anemia suggests that there is a delayed or depressed erythropoietic response in Rbm38−/− mice. Interestingly, a recent study indicates that Rbm38 plays a role in the late stage of erythroid differentiation by regulating alternative splicing of EPB41 (11), which may contribute to the increased ratio of myeloid vs. erythroid in the bone marrow of Rbm38−/− mice. Moreover, most tumors from Rbm38−/− mice, including lymphoma and histiocytic sarcoma, are of hematopoietic origin, suggesting that the hematopoietic defects induced by Rbm38 deficiency may contribute to the hematopoietic malignancy. Thus, further studies are needed to address the potential link of hematopoietic defects and malignancy mediated by Rbm38 deficiency.
The Role of RBM38 in Aging and Tumorigenesis.
In our study, we showed that Rbm38 deficiency results in accelerated aging, including short life span, weight loss with reduced adipose tissue, and lordokyphosis (Fig. 1 B–D and Fig. S1 B–E). Consistent with these aging phenotypes, both the activity of SA-β-gal and the level of p16Ink4A transcript were increased in the livers of old Rbm38−/− mice (85 and 128 wk of age) compared with WT mice (Fig. 1 E–G). Furthermore, we showed that the p53 level is increased in Rbm38-null mice compared with WT mice under normal and IR-induced conditions (Figs. 1H and 4A). It is likely that the increased level of p53 in Rbm38-deficient mice drives apoptosis or cellular senescence and consequently causes tissue atrophy and degeneration, leading to aging. Although the underlying mechanism is not fully understood, we tentatively suggest that Rbm38 prevents aging by repressing p53-dependent apoptosis and/or senescence. In agreement with this hypothesis, we showed previously that Rbm38 deficiency leads to premature senescence in MEFs in a p53-dependent manner (16). Notably, similar to Rbm38−/− mice, both p53+/m and p44+/+ mice express a hyperactive p53 and exhibit signs of premature aging (36, 37). By contrast, two other mice models, the “super p53” mice and the MDM2 hypomorphic mice, express high levels of WT p53 but do not exhibit premature aging phenotypes (38, 39). Thus, p53 may promote aging depending on its regulation or activity. Alternatively, Rbm38 may prevent aging by regulating targets other than p53. For example, Rbm38 is able to stabilize both p21 and TAp73 transcripts (5, 7), both of which are known to have an impact on aging (40, 41). Nevertheless, further studies are warranted to determine the underlying mechanism whereby Rbm38 prevents aging via p53-dependent and -independent processes.
It seems intriguing that RBM38−/− mice develop spontaneous tumors (Fig. 3) because one would expect tumor resistance in these mice owing to the increased expression of p53. However, we would like to mention that the spontaneous tumors observed in Rbm38−/− mice do not necessarily contradict the finding that p53 levels are increased in Rbm38−/− mice (Figs. 1H and 4A). First, Rbm38 deficiency may result in tumorigenesis independently of p53 because loss of Rbm38 promotes lymphomagenesis in p53−/− mice (Fig. 6). Second, p53 activity is known to be progressively declined during the aging process (42). It is likely that the increased p53 expression in old Rbm38−/− mice is not sufficient for tumor suppression. In support of this notion, most Rbm38−/− mice develop tumors between 18 and 24 mo. Third, we showed previously that loss of Rbm38 in MEFs leads to premature senescence in a p53-dependent manner (16). Interestingly, senescence is suggested to function as a double-edged sword that suppresses tumor formation at the early stage of life but may promote tumorigenesis later via the senescence-associated secretory phenotype (SASP) (43). The SASP refers to an increase in the mRNA levels and secretion of several growth factors, cytokines, and chemokines. In support of this notion, we showed that several proinflammatory cytokines are up-regulated in Rbm38−/− MEFs (Fig. 3D), which may contribute to tumor development. Thus, the implication and significance of these proinflammatory cytokines in Rbm38-mediated senescence, aging, and cancer are worth further investigation. Notably, both accelerated aging and tumorigenesis observed in Rbm38−/− mice are reminiscent of those observed in the Brca1Δ11/Δ11; p53 +/− mice (44) and p53S18A knockin mice (45) although the underlying mechanism may be different.
We would like to mention that, according to The Cancer Genome Atlas (TCGA) research network (cancergenome.nih.gov), Rbm38 is frequently overexpressed in human cancers, such as lymphoma and breast and colorectal carcinomas, suggesting that Rbm38 may function as an oncogene. Although these data seem contradictory with the ones from our mouse model, it is likely that Rbm38 may have positive and negative impacts on tumorigenesis by regulating different signaling pathways. Of particular interest, because Rbm38 represses both WT and mutant p53 mRNA translation (16), Rbm38 deficiency may have two opposing functions on tumorigenesis dependent on the genetic status of p53. Thus, further studies are needed to determine how and why Rbm38 possesses two opposing roles in tumor development.
The Role of the p53-RBM38 Loop in Tumor Suppression.
In our study, we found that the effect of Rbm38 loss on tumor formation differs widely among p53+/+, p53+/−, and p53−/− mice. In a p53+/+ background, mice deficient in Rbm38 have shorter lifespan and develop spontaneous tumors, mostly lymphoma (Fig. 3). In a p53+/− background, Rbm38 deficiency does not have an effect on the overall survival and the tumor spectrum (Fig. 5B and Table S3). Instead, Rbm38−/−; p53+/− mice are less prone to spontaneous tumors compared with p53+/− mice (Fig. 5C). These results suggest that, depending on the p53 level, Rbm38 may act positively or negatively during tumor development. We postulate that, in a p53+/+ background, p53 level is sufficient to prevent tumor formation. Thus, the increased p53 expression by Rbm38 deficiency may contribute to accelerated aging, which promotes tumor formation at a late stage of life as discussed in The Role of RBM38 in Aging and Tumorigenesis. However, in a p53+/− background, these mice are tumor prone due to haploinsufficiency of p53. Therefore, the increased p53 expression by Rbm38 deficiency may contribute to tumor suppression. Indeed, several other mouse models, including the MDM2 hypomorphic and super Ink4a/ARF mice, express an increased level of p53 and are resistant to spontaneous tumor formation (38, 46). Notably, although Rbm38−/−; p53+/− mice are less tumor prone, they have similar lifespan as p53+/− mice (Fig. 5B), which is likely due to the pathological defects mediated by Rbm38 deficiency, including hematopoietic defects and accelerated aging. Furthermore, in a p53−/− background, loss of Rbm38 significantly shortens the life span and promotes lymphomagenesis (Fig. 6), suggesting that loss of Rbm38 promotes tumor formation independently of p53. We speculate that loss of p53 leads to accumulation of malignant lymphoma cells, which are further exacerbated by additional loss of Rbm38. We also observed that, upon Rbm38 deletion, the tumor burden and sarcoma incidence are significantly reduced in p53−/− mice (Tables S6–S8). The reduced tumor burden and sarcoma incidence may simply be due to the shortened life span for Rbm38−/−; p53−/− mice, which die too early to develop other tumors. However, it remains possible that loss of RBM38 may suppress sarcoma or other precursor tumor cells in p53-null mice. Overall, these data suggest that there is a complex reciprocal regulation between p53 and Rbm38 in vivo. Thus, further studies with conditional p53 and Rbm38 knockout mouse models are warranted to address the role of the p53-Rbm38 axis in aging and tumor suppression.
In conclusion, the genetically engineered mouse models described in this study reveal the physiologic functions of Rbm38, especially in aging, normal hematopoiesis, and tumor suppression. In addition, our data suggest that Rbm38 is an ideal molecule to fine-tune the level of p53 and that the p53-Rbm38 axis may be targeted for cancer management.
Materials and Methods
Mutant Mice.
Rbm38-conditional knockout mice (on a pure C57BL/6 background) were previously generated using standard gene-targeting techniques in embryonic stem cells (16). The resulting allele contains two loxP sites flanking exon 1. Mice with targeted allele were then bred with Cre transgenic mice (EIIa-Cre) (The Jackson Laboratory) to generate straight Rbm38 knockout mice with exon 1 deletion. The p53+/− mice (on a C57BL/6 background) were purchased from The Jackson Laboratory. Rbm38+/− mice were crossed with p53+/− mice to generate double heterozygous mice. The latter were intercrossed to generate Rbm38−/−; p53+/− and Rbm38−/−; p53−/− mutant mice. The p53+/− mice were also intercrossed to generate p53+/− and p53−/− mice, respectively. All animals and use protocols were approved by the University of California at Davis Institutional Animal Care and Use Committee (Animal Protocol 18216).
Supplementary Material
Acknowledgments
We thank Drs. Lihong Qi and Philip H. Kass for help with statistical analysis, Drs. Neil E. Hubbard and Olulanu Aina for help with histological and immunohistochemistry (IHC) analyses, Dr. Kent Lloyd and the Mouse Biology Program at the University of California, Davis (UCD), for generating the RBM38-deficient mouse model, UCD Center for Molecular and Genomic Imaging for performing micro-CT scan, UCD Comparative Pathology Laboratory for blood sample analysis, and Drs. Huaijun Zhou and Ying Wang for analyzing RNA-seq data. This study was supported in part by NIH Grants CA076069, CA081237, and CA121137.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1415607112/-/DCSupplemental.
References
- 1.Dreyfuss G, Kim VN, Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol. 2002;3(3):195–205. doi: 10.1038/nrm760. [DOI] [PubMed] [Google Scholar]
- 2.Lukong KE, Chang KW, Khandjian EW, Richard S. RNA-binding proteins in human genetic disease. Trends Genet. 2008;24(8):416–425. doi: 10.1016/j.tig.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 3.Kim MY, Hur J, Jeong S. Emerging roles of RNA and RNA-binding protein network in cancer cells. BMB Reports. 2009;42(3):125–130. doi: 10.5483/bmbrep.2009.42.3.125. [DOI] [PubMed] [Google Scholar]
- 4.Wurth L. Versatility of RNA-binding proteins in cancer. Comp Funct Genomics. 2012;2012:178525. doi: 10.1155/2012/178525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shu L, Yan W, Chen X. RNPC1, an RNA-binding protein and a target of the p53 family, is required for maintaining the stability of the basal and stress-induced p21 transcript. Genes Dev. 2006;20(21):2961–2972. doi: 10.1101/gad.1463306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yin T, Cho SJ, Chen X. RNPC1, an RNA-binding protein and a p53 target, regulates macrophage inhibitory cytokine-1 (MIC-1) expression through mRNA stability. J Biol Chem. 2013;288(33):23680–23686. doi: 10.1074/jbc.M113.480186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan W, Zhang J, Zhang Y, Jung YS, Chen X. p73 expression is regulated by RNPC1, a target of the p53 family, via mRNA stability. Mol Cell Biol. 2012;32(13):2336–2348. doi: 10.1128/MCB.00215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho SJ, Jung YS, Zhang J, Chen X. The RNA-binding protein RNPC1 stabilizes the mRNA encoding the RNA-binding protein HuR and cooperates with HuR to suppress cell proliferation. J Biol Chem. 2012;287(18):14535–14544. doi: 10.1074/jbc.M111.326827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu E, Zhang J, Chen X. MDM2 expression is repressed by the RNA-binding protein RNPC1 via mRNA stability. Oncogene. 2013;32(17):2169–2178. doi: 10.1038/onc.2012.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J, Xu E, Chen X. Regulation of Mdm2 mRNA stability by RNA-binding protein RNPC1. Oncotarget. 2013;4(8):1121–1122. doi: 10.18632/oncotarget.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heinicke LA, et al. The RNA binding protein RBM38 (RNPC1) regulates splicing during late erythroid differentiation. PLoS ONE. 2013;8(10):e78031. doi: 10.1371/journal.pone.0078031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell. 2009;33(5):591–601. doi: 10.1016/j.molcel.2009.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohno G, et al. Muscle-specific splicing factors ASD-2 and SUP-12 cooperatively switch alternative pre-mRNA processing patterns of the ADF/cofilin gene in Caenorhabditis elegans. PLoS Genet. 2012;8(10):e1002991. doi: 10.1371/journal.pgen.1002991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuroyanagi H, Ohno G, Mitani S, Hagiwara M. The Fox-1 family and SUP-12 coordinately regulate tissue-specific alternative splicing in vivo. Mol Cell Biol. 2007;27(24):8612–8621. doi: 10.1128/MCB.01508-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anyanful A, et al. The RNA-binding protein SUP-12 controls muscle-specific splicing of the ADF/cofilin pre-mRNA in C. elegans. J Cell Biol. 2004;167(4):639–647. doi: 10.1083/jcb.200407085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, et al. Translational repression of p53 by RNPC1, a p53 target overexpressed in lymphomas. Genes Dev. 2011;25(14):1528–1543. doi: 10.1101/gad.2069311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang M, Zhang J, Chen X, Cho SJ, Chen X. Glycogen synthase kinase 3 promotes p53 mRNA translation via phosphorylation of RNPC1. Genes Dev. 2013;27(20):2246–2258. doi: 10.1101/gad.221739.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Léveillé N, et al. Selective inhibition of microRNA accessibility by RBM38 is required for p53 activity. Nat Commun. 2011;2:513. doi: 10.1038/ncomms1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miyamoto S, Hidaka K, Jin D, Morisaki T. RNA-binding proteins Rbm38 and Rbm24 regulate myogenic differentiation via p21-dependent and -independent regulatory pathways. Genes Cells. 2009;14(11):1241–1252. doi: 10.1111/j.1365-2443.2009.01347.x. [DOI] [PubMed] [Google Scholar]
- 20.Chin K, et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 2006;10(6):529–541. doi: 10.1016/j.ccr.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 21.Jenssen TK, Kuo WP, Stokke T, Hovig E. Associations between gene expressions in breast cancer and patient survival. Hum Genet. 2002;111(4-5):411–420. doi: 10.1007/s00439-002-0804-5. [DOI] [PubMed] [Google Scholar]
- 22.Carvalho B, et al. Multiple putative oncogenes at the chromosome 20q amplicon contribute to colorectal adenoma to carcinoma progression. Gut. 2009;58(1):79–89. doi: 10.1136/gut.2007.143065. [DOI] [PubMed] [Google Scholar]
- 23.Hermsen M, et al. Colorectal adenoma to carcinoma progression follows multiple pathways of chromosomal instability. Gastroenterology. 2002;123(4):1109–1119. doi: 10.1053/gast.2002.36051. [DOI] [PubMed] [Google Scholar]
- 24.Feldstein O, Ben-Hamo R, Bashari D, Efroni S, Ginsberg D. RBM38 is a direct transcriptional target of E2F1 that limits E2F1-induced proliferation. Mol Cancer Res. 2012;10(9):1169–1177. doi: 10.1158/1541-7786.MCR-12-0331. [DOI] [PubMed] [Google Scholar]
- 25.Krishnamurthy J, et al. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114(9):1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rufini A, Tucci P, Celardo I, Melino G. Senescence and aging: The critical roles of p53. Oncogene. 2013;32(43):5129–5143. doi: 10.1038/onc.2012.640. [DOI] [PubMed] [Google Scholar]
- 27.Matsushima H, Yamada N, Matsue H, Shimada S. TLR3-, TLR7-, and TLR9-mediated production of proinflammatory cytokines and chemokines from murine connective tissue type skin-derived mast cells but not from bone marrow-derived mast cells. J Immunol. 2004;173(1):531–541. doi: 10.4049/jimmunol.173.1.531. [DOI] [PubMed] [Google Scholar]
- 28.Pappu R, Ramirez-Carrozzi V, Sambandam A. The interleukin-17 cytokine family: Critical players in host defence and inflammatory diseases. Immunology. 2011;134(1):8–16. doi: 10.1111/j.1365-2567.2011.03465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meylan F, Richard AC, Siegel RM. TL1A and DR3, a TNF family ligand-receptor pair that promotes lymphocyte costimulation, mucosal hyperplasia, and autoimmune inflammation. Immunol Rev. 2011;244(1):188–196. doi: 10.1111/j.1600-065X.2011.01068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacks T, et al. Tumor spectrum analysis in p53-mutant mice. Cur Biol. 1994;4(1):1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 31.Venkatachalam S, et al. Is p53 haploinsufficient for tumor suppression? Implications for the p53+/- mouse model in carcinogenicity testing. Toxicol Pathol. 2001;29(Suppl):147–154. doi: 10.1080/019262301753178555. [DOI] [PubMed] [Google Scholar]
- 32.Kemp CJ, Wheldon T, Balmain A. p53-deficient mice are extremely susceptible to radiation-induced tumorigenesis. Nat Genet. 1994;8(1):66–69. doi: 10.1038/ng0994-66. [DOI] [PubMed] [Google Scholar]
- 33.Purdie CA, et al. Tumour incidence, spectrum and ploidy in mice with a large deletion in the p53 gene. Oncogene. 1994;9(2):603–609. [PubMed] [Google Scholar]
- 34.Donehower LA, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356(6366):215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 35.Uhlen M, et al. Towards a knowledge-based Human Protein Atlas. Nat Biotechnol. 2010;28(12):1248–1250. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- 36.Tyner SD, et al. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415(6867):45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- 37.Maier B, et al. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004;18(3):306–319. doi: 10.1101/gad.1162404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mendrysa SM, et al. Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev. 2006;20(1):16–21. doi: 10.1101/gad.1378506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.García-Cao I, et al. “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J. 2002;21(22):6225–6235. doi: 10.1093/emboj/cdf595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rufini A, et al. TAp73 depletion accelerates aging through metabolic dysregulation. Genes Dev. 2012;26(18):2009–2014. doi: 10.1101/gad.197640.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ju Z, Choudhury AR, Rudolph KL. A dual role of p21 in stem cell aging. Ann N Y Acad Sci. 2007;1100:333–344. doi: 10.1196/annals.1395.036. [DOI] [PubMed] [Google Scholar]
- 42.Feng Z, et al. Declining p53 function in the aging process: A possible mechanism for the increased tumor incidence in older populations. Proc Natl Acad Sci USA. 2007;104(42):16633–16638. doi: 10.1073/pnas.0708043104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campisi J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell. 2005;120(4):513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 44.Cao L, Li W, Kim S, Brodie SG, Deng CX. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 2003;17(2):201–213. doi: 10.1101/gad.1050003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Armata HL, Garlick DS, Sluss HK. The ataxia telangiectasia-mutated target site Ser18 is required for p53-mediated tumor suppression. Cancer Res. 2007;67(24):11696–11703. doi: 10.1158/0008-5472.CAN-07-1610. [DOI] [PubMed] [Google Scholar]
- 46.Matheu A, et al. Increased gene dosage of Ink4a/Arf results in cancer resistance and normal aging. Genes Dev. 2004;18(22):2736–2746. doi: 10.1101/gad.310304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.