

Abstract
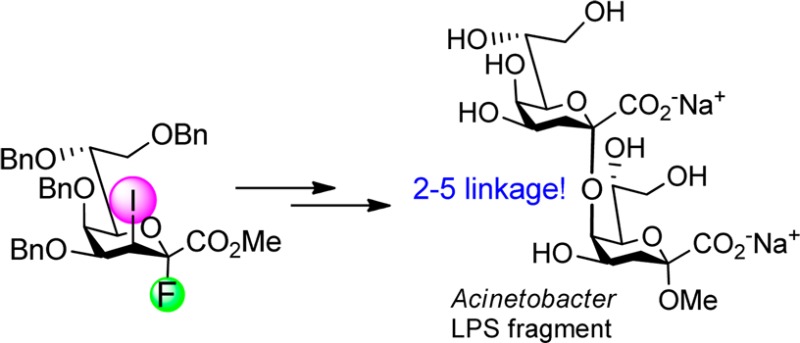
Resistance of bacterial pathogens toward antibiotics has revived interest in lipopolysaccharide (LPS) motifs as potential therapeutic targets. The LPS of several pathogenic Acinetobacter strains comprises a 4,5-branched Kdo trisaccharide containing an uncommon (2→5)-linkage. In this contribution the first stereoselective glycosylation method for obtaining an α-Kdo-(2→5)-α-Kdo disaccharide in good yield is highlighted. The synthetic approach used for accessing this linkage type will allow for future studies of the immunoreactivity associated with this unique bacterial Kdo inner core structure.
Lipopolysaccharides (LPS) are found in the outer membrane of the Gram-negative bacterial cell wall and comprise the conserved Lipid A which is noncovalently anchored to the cell wall via sugar-bound fatty acids. This lipophilic part is further linked to the core region followed by a species-specific polysaccharide, termed O-antigen.1 The common bacterial sugar 3-deoxy-d-manno-oct-2-ulosonic acid (Kdo) is a fundamental component of the LPS core domain, being present in a structurally conserved α-(2→4)-linked Kdo disaccharide unit.2 This basic disaccharide is elongated at position 5 of the proximal Kdo unit by various additional core sugars such as l-glycero-d-manno-heptose, glucose or mannose, resulting in branched structures.3 Investigations of LPS from different Acinetobacter strains, however, have revealed the presence of branched Kdo tetra- and trisaccharides containing an unusual α-(2→5) inter-Kdo linkage. In particular, several A. baumannii strains as well as the A. radioresistans strain S13 have been found to harbor an α-Kdo-(2→5)[α-Kdo-(2→4)]α-Kdo trisaccharide fragment in the inner core region.4a−4d An α-(2→5)-linked Kdo disaccharide was also detected in Campylobacter lari strain ATCC 35221.4eAcinetobacter strains are known to be responsible for severe nosocomial infections which are difficult to treat due to a pronounced resistance of the bacteria against antimicrobial drugs.5
While various routes toward (2→4)-, (2→8)-, and also (2→7)- interconnected Kdo disaccharides have been reported,6−8 a synthetic access toward the (2→5)-disaccharide via glycosylation has not been achieved thus far. The chemical glycosylation of Kdo glycosyl acceptors at position 5 is hampered by low reactivity due to the axial orientation of the C-5 hydroxyl group, the steric hindrance exerted by the side chain unit, and the deactivating effect of the C-1 ester group. Likewise, Kdo donors (e.g. 1, Scheme 1) usually suffer from considerable degradation during glycosylation reactions since they are prone to elimination reactions that produce glycal esters such as 2.2c,9 The elimination is also observed for conformationally constrained Kdo donors equipped with a 4,5-isopropylidene group.6c,6d In addition, Kdo donors show low anomeric selectivity and reduced reactivity for glycoside formation due to the deactivating effect of the carboxylic group adjacent to the anomeric center. The challenge met in the construction of a Kdo-(2→5)-Kdo linkage is reflected in the complete regioselectivity observed during coupling of Kdo donors with 4,5-unprotected Kdo diol acceptors, leading to O-4 substituted products only.6,10 Also, previous attempts in the author’s laboratory using a peracetylated Kdo phosphite donor and 4-O-protected Kdo acceptor derivatives did not produce any disaccharides.11 Notably, however, glycosylations of the 5-OH group of 4-O-protected Kdo substrates with aldopyranosyl donors (e.g., heptosyl, glucosyl, mannosyl donors) have been successfully implemented, illustrating the difficulty in forming a ketosidic bond to the sterically hindered 5-OH group of a Kdo acceptor.12
Scheme 1. Kdo Donors (LG = Leaving Group) and Glycal Ester Formation during Glycosylation.

Recently, we have developed the acetylated 3-iodo-Kdo fluoride donor 3(13) with a stereodirecting group at position 3. This donor has proven itself capable of α-stereoselective glycosylation reactions to provide several α-Kdo homomers related to Chlamydia LPS in high yield. Additionally, the elimination side reaction toward glycal 2 could be significantly suppressed and we surmised that these properties might provide access to the α-(2→5) Kdo disaccharide motif. For the design of an appropriate Kdo glycosyl acceptor, three prerequisites for protecting groups were foreseen: (a) activating groups at remote positions to compensate for the intrinsically low reactivity of HO-5, (b) exclusion of silyl groups that would eventually be cleaved by fluoride species formed during the glycosylation step, and (c) stability toward Bronsted acids. The latter requirement resulted from a previous observation that addition of triethylamine abolished the glycosylation capability of donor 3 (thus, Bronsted acid formed after HF cleavage seems to be the active species involved in the coupling step). Since the disaccharide should serve as a ligand in STD-NMR and crystallographic studies, a methyl aglycon was selected.13 Thus, the 4,7,8-tri-O-benzyl protected Kdo glycosyl acceptor derivative 11 was prepared starting from the previously described 7,8-O-carbonyl derivative 4(13) (Scheme 2).
Scheme 2. Synthesis of the 4,7,8-Tri-O-benzyl Kdo Acceptor (11).

Reaction of 4 with acetone in the presence of TMSOTf yielded the 4,5-O-isopropylidene derivative 5 in 95% yield after silica gel filtration. Treatment of 5 with sodium methoxide in anhydrous methanol liberated the 7-OH and 8-OH groups (97% of 6(14)), which were subsequently O-benzylated using sodium hydride and BnBr in dry DMF. Thus, a mixture of the methyl ester 7 and the benzyl ester 8 was obtained in a total yield of 79%. Cleavage of the 4,5-O-acetonide by treatment of the mixture of 7 and 8 with para-toluenesulfonic acid in methanol yielded a mixture of the two esters 9 and 10 after filtration through silica gel. Treatment of the mixture with dibutyl tin oxide and subsequent conversion of the tin acetals with BnBr in the presence of tetra-n-butylammonium iodide (TBAI) in DMF afforded a 52% yield of methyl ester 11 and 8% of benzyl ester 12 (over 2 steps) after chromatography. The latter product was finally quantitatively transformed into the methyl ester 11 by treatment with stoichiometric amounts of sodium methoxide in MeOH.
The glycosylation reaction of acceptor 11 with the acetylated donor 3 in dry CH2Cl2 containing ground molecular sieves (3 Å) and 2 equiv of BF3·Et2O did not afford the disaccharide. Instead, unreacted acceptor 11 was isolated, while donor 3 degraded very slowly over a period of several hours. Thus, in order to enhance the reactivity of the glycosyl donor, the armed analogue 16 (Scheme 3) was prepared. De-O-acetylation of glycal ester 2(15) and benzylation of the resulting tetraol 13 using NaH/BnBr in dry DMF provided 14(7c) in 77% isolated yield. Notably, destruction of excessive NaH for the workup was performed, after dilution with anhydrous CH2Cl2 at 0 °C, by addition of anhydrous MeOH followed by quick extraction and purification. Thus, degradation of the reactive glycal 14 and ester saponification could be avoided. Iodoacetoxylation using N-iodosuccinimide (NIS) in acetic acid was performed at ambient temperature and afforded the 2,3-trans-diaxial product 15 in 83% yield.16 Direct conversion of the anomeric acetate, with an optimized amount of hydrogen fluoride pyridine complex at −5–0 °C, afforded the α-fluoride 16 (JH-3eq,F-ax 5.1 Hz) selectively and in excellent yield (96%).7b Increased amounts of HF-pyridine and temperatures above 0 °C, however, resulted in epimerization of the 3-iodo substituent, and an inseparable mixture of the desired d-glycero-d-talo donor 16 and the d-glycero-d-galacto-epimer 17 was obtained.
Scheme 3. Preparation of Fluoride Donor (16) and Model Glycosylation.
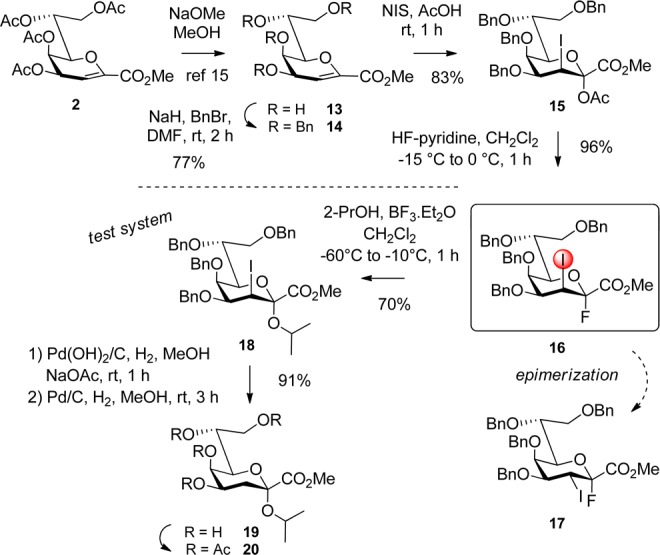
To evaluate its glycosylation properties, donor 16 was converted in 70% yield into the 2-propyl glycoside 18 using 2 equiv of 2-propanol under BF3·Et2O-promotion in CH2Cl2. TLC-analysis of the glycosylation mixture indicated a pronounced reactivity of 16, which was already activated at −60 °C. In comparison, no reaction of acetylated donor 3 with 2-propanol had been observed below ambient temperature.13 Dehalogenation was performed by catalytic hydrogenation using Pd(OH)2/C (20%) in the presence of NaOAc in MeOH; it was followed by workup (extraction with sodium thiosulfate and water to remove iodo impurities). The ensuing debenzylation over Pd/C (10%) gave 19 in 91% yield (2 steps). Notably, dehalogenation also led to partial cleavage of the benzyl groups. O-Acetylation of 19 afforded the known glycoside 20, and comparison with published data confirmed the assignment of the α-anomeric configuration.13
When applying these optimized glycosylation conditions to the coupling of donor 16 with 4,7,8-tri-O-benzyl acceptor 11, trace amounts of product 21 could be isolated (Scheme 4). Furthermore, large amounts of unreacted acceptor 11 and hydrolyzed donor were present. Surprisingly, 1H NMR analysis revealed the presence of a second 2→5-linked disaccharide bearing an equatorially oriented 3-iodo substituent resulting from epimerization. Optimization of the glycosylation conditions, however, allowed for suppression of the 3-iodo-epimerization when toluene was used as the solvent. Still, the isolated yield was quite low (∼30%). On the one hand, at low temperature (−40 °C), incomplete conversion of acceptor 11 was observed, while at elevated temperatures (0 °C) extensive degradation of the product occurred. Stability tests of the isolated product 21 revealed that it was unstable toward BF3·Et2O at temperatures above −10 °C. However, working at high dilution and keeping the temperature between −40 °C to −10 °C (warming up to −10 °C was, however, essential) afforded 21 in a good yield (71%) as the α-anomer only. In addition, only very minor formation of the glycal ester 14 was observed and no β-anomeric product was detected. For workup, BF3·Et2O had to be neutralized by dropwise addition of diluted triethylamine at low temperature followed by quick extraction and purification to secure a high isolated yield. This procedure was reproducible without the need for a large excess of donor 16 (only 1.5 equiv were used).
Scheme 4. Glycosylation and Deprotection towards the α-(2→5) Kdo Disaccharide (24).

Broad signal shapes in the 1H NMR spectrum (600 MHz, in CDCl3) of 21 complicated the analysis and assignments. This line broadening suggests a high steric load and a rigid structure of 21. Thus, the sample was subjected to high-temperature measurements17 in d8-toluene to confirm the presence of the (2→5) linkage. Indeed, the HMBC spectrum showed a cross-correlation between H-5 and C-2′ (Figure 1) and the assignment was further supported by a NOESY signal between H-5 and H-6′ (see Supporting Information).
Figure 1.

HMBC spectrum (d8-tol, 323 K) of disaccharide 21.
The dehalogenation/debenzylation sequence of 21 using Pd(OH)2/C-NaOAc and Pd/C in MeOH under an atmosphere of H2 yielded an impure product, which was O-acetylated and purified by normal-phase HPLC providing a mixture of 22 together with the 1′→4 lactone 23 in 61% yield (3 steps).
NMR characterization of 22 supported the assignment of the α-anomeric configuration as indicated by the characteristic chemical shifts of H-3′ax and H-3′eq and the low-field shift of H-4′.18 Analysis of the side products revealed that partial cleavage of the glycosidic bond had occurred. This was further supported by LC-MS studies showing that glycoside hydrolysis (and lactone formation) had already occurred during the treatment with Pd(OH)2/C-NaOAc. Deacetylation of the ∼10:1 mixture of 22 and 23 using sodium methoxide and subsequent methyl ester hydrolysis with aq NaOH afforded 24 containing a trace impurity. To obtain the pure target disaccharide, the mixture of 22 and 23 was partially separated by normal-phase HPLC, providing a pure fraction of 22 (1.2 mg from 9.9 mg of a 10:1 mixture of 22/23, 12%), which was globally deprotected to afford the α-(2→5)-linked Kdo disaccharide 24.
The NMR spectroscopic data for 24 (recorded in D2O) were in close agreement with the related Kdo units present in the branched Kdo-tetrasaccharide.4a−4d The proton signals of H-4′ and H-6′ of Kdo II (see Scheme 4) were substantially shifted to lower field (4.26 and 3.90 ppm, respectively), presumably due to the deshielding effect of the O-4 group of Kdo I. Similarly, the equatorial proton H-3′eq at the distal Kdo II unit was also shifted to lower field (2.24 ppm), which is most likely attributable to the fixed orientation of the lone pairs at O-5 of the glycosidic bond. These effects have been published for the branched Kdo tetrasaccharide isolated from A. baumannii strain NCTC 10303.4a Since these features are also seen in the α-(2→5)-linked disaccharide, the additional, lateral α-(2→4)-linked Kdo unit has thus only a limited impact on the NMR characteristics of the α-(2→5)-unit (except for 13C NMR shift differences at C-4 and C-5 for the branched Kdo moiety, respectively). Carbon-5 of Kdo I revealed a significant shift to low field (74.2 ppm), confirming the site of attachment of the second Kdo unit (which was again proven by an HMBC correlation from H-5 to carbon C-2′).
In conclusion, we have developed a stereoselective route toward α-(2→5)-interconnected Kdo residues using an armed 3-iodo-Kdo fluoride donor displaying high reactivity and α-anomeric stereoselectivity. The NMR data for the deprotected disaccharide corresponding to fragments of Acinetobacter LPS favorably match the assignments made for the native oligosaccharides.
Acknowledgments
Financial support of this work by the Austrian Science Fund FWF (Grant No. P-24921) and high-temperature NMR measurements by Dr. Andreas Hofinger (Department of Chemistry, University of Natural Resources and Life Sciences-Vienna) are gratefully acknowledged.
Supporting Information Available
Detailed synthetic procedures for 5–12, 14–19, and 21–24; characterization data; and NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Raetz C. R.; Whitfield C. Annu. Rev. Biochem. 2002, 71, 635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Brade H.; Galanos C.; Lüderitz O. Eur. J. Biochem. 1983, 131, 201–203. [DOI] [PubMed] [Google Scholar]; b Paulsen H.; Stiem M.; Unger F. M. Tetrahedron Lett. 1986, 27, 1135–1138. [Google Scholar]; c Unger F. M. Adv. Carbohydr. Chem. Biochem. 1983, 348, 323–387. [DOI] [PubMed] [Google Scholar]
- a Holst O. In Endotoxin in Health and Disease; Brade H., Opal S. M., Vogel S. N., Morrison D. C., Eds.; Marcel Dekker: New York, 1999; p 115. [Google Scholar]; b Holst O. FEMS Microbiol. Lett. 2007, 271, 3–11. [DOI] [PubMed] [Google Scholar]; c Holst O. In Bacterial Lipopolysaccharides; Knirel Y. A., Valvano M. A., Eds.: Springer: Wien–New York, 2011; p 21. [Google Scholar]
- a Vinogradov E. V.; Petersen B. O.; Thomas-Oates J. E.; Duus J.Ø.; Brade H.; Holst O. J. Biol. Chem. 1998, 273, 28122–28131. [DOI] [PubMed] [Google Scholar]; b Vinogradov E. V.; Duus J. Ø.; Brade H.; Holst O. Eur. J. Biochem. 2002, 269, 422–430. [DOI] [PubMed] [Google Scholar]; c Fregolino E.; Fugazza G.; Galano E.; Gargiulo V.; Landini P.; Lanzetta R.; Lindner B.; Pagani L.; Parrilli M.; Holst O.; De Castro C. Eur. J. Org. Chem. 2010, 2010, 1345–1352. [Google Scholar]; d Leone S.; Molinaro A.; Pessione E.; Mazzoli R.; Giunta C.; Sturiale L.; Garozzo D.; Lanzetta R.; Parrilli M. Carbohydr. Res. 2006, 341, 582–590. [DOI] [PubMed] [Google Scholar]; e Aspinall G. O.; Monteiro M. A.; Pang H. Carbohydr. Res. 1995, 279, 245–264. [DOI] [PubMed] [Google Scholar]
- a Dijkshoorn L.; Nemec A.; Seifert H. Nat. Rev. Microbiol. 2007, 5, 939–951. [DOI] [PubMed] [Google Scholar]; b McConnell M. J.; Actis L.; Pachón J. FEMS Microbiol. Rev. 2013, 37, 130–155. [DOI] [PubMed] [Google Scholar]
- For selected examples see:; a Paulsen H.; Krogmann C. Carbohydr. Res. 1990, 205, 31–44. [DOI] [PubMed] [Google Scholar]; b Shimoyama A.; Fujimoto Y.; Fukase K. Synlett 2011, 2359–2362. [Google Scholar]; c Yoshizaki H.; Fukuda N.; Sato K.; Oikawa M.; Fukase K.; Suda Y.; Kusumoto S. Angew. Chem., Int. Ed. 2001, 40, 1475–1480. [DOI] [PubMed] [Google Scholar]; d Shimoyama A.; Saeki A.; Tanimura N.; Tsutsui H.; Miyake K.; Suda Y.; Fujimoto Y.; Fukase K. Chem.—Eur. J. 2011, 17, 14464–14474. [DOI] [PubMed] [Google Scholar]
- For examples, see:; a Kosma P.; Schulz G.; Brade H. Carbohydr. Res. 1988, 183, 183–199. [DOI] [PubMed] [Google Scholar]; b Solomon D.; Fridman M.; Zhang J.; Baasov T. Org. Lett. 2001, 3, 4311–4314. [DOI] [PubMed] [Google Scholar]; c Tanaka H.; Takahashi D.; Takahashi T. Angew. Chem., Int. Ed. 2006, 45, 770–773. [DOI] [PubMed] [Google Scholar]; d Ichiyanagi T.; Fukunaga M.; Tagashira R.; Hayashi S.; Nanjo M.; Yamasaki R. Tetrahedron 2011, 67, 5964–5971. [Google Scholar]; e Młynarski J.; Banaszek A. Tetrahedron: Asymmetry 2000, 11, 3737–3746. [Google Scholar]
- Hofinger A.; Kosma P.; Christian R.; Bock K.; Brade H. Carbohydr. Res. 1993, 243, 273–291. [DOI] [PubMed] [Google Scholar]
- a Oscarson S.; Hansson J. Curr. Org. Chem. 2000, 4, 535–564. [Google Scholar]; b Kosma P. In Microbial Glycobiology; Moran A. P., Holst O., Brennan P. J., von Itzstein M., Eds.; Elsevier: Amsterdam, 2009; p 429. [Google Scholar]; c Li L.-S.; Wu Y.-L. Curr. Org. Chem. 2003, 7, 447–475. [Google Scholar]
- For selected examples see:; a Kusumoto S.; Kusunose N.; Kamikawa T.; Shiba T. Tetrahedron Lett. 1988, 29, 6325–6326. [Google Scholar]; b Kiso M.; Fujita M.; Ogawa Y.; Ishida H.; Hasegawa A. Carbohydr. Res. 1990, 196, 59–73. [DOI] [PubMed] [Google Scholar]; c Kosma P.; Bahnmüller R.; Schulz G.; Brade H. Carbohydr. Res. 1990, 208, 37–50. [DOI] [PubMed] [Google Scholar]; d Paulsen H.; Krogmann C. Liebigs Ann. Chem. 1989, 1203–1213. [Google Scholar]
- Wimmer N. PhD thesis, University of Natural Resources and Life Sciences-Vienna, 2001. [Google Scholar]
- For selected examples, see:; a Paulsen H.; Heitmann A. C. Liebigs Ann. Chem. 1989, 655–663. [Google Scholar]; b Paulsen H.; Brenken M. Liebigs Ann. Chem. 1991, 1113–1126. [Google Scholar]; c Auzanneau F. I.; Charon D.; Szilágy L.; Szabó L. J. Chem. Soc., Perkin Trans. 1 1991, 803–809. [Google Scholar]; d Boons G. J. P. H.; van Delft F. L.; van der Klein P. A. M.; van der Marel G. A.; van Boom J. H. Tetrahedron 1992, 48, 885–904. [Google Scholar]; e Paulsen H.; Höffgen E. Liebigs Ann. Chem. 1993, 543–550. [Google Scholar]; f Paulsen H.; Höffgen E. Liebigs Ann. Chem. 1993, 531–541. [Google Scholar]; g Ekelöf K.; Oscarson S. Carbohydr. Res. 1995, 278, 289–300. [DOI] [PubMed] [Google Scholar]; h Ekelöf K.; Oscarson S. J. Org. Chem. 1996, 61, 7711–7718. [DOI] [PubMed] [Google Scholar]; i Bernlind C.; Oscarson S. J. Org. Chem. 1998, 63, 7780–7788. [Google Scholar]; j Yang Y.; Martin C. E.; Seeberger P. H. Chem. Sci. 2011, 3, 896–899. [Google Scholar]; k Boltje T. J.; Zhong W.; Park J.; Wolfert M. A.; Chen W.; Boons G.-J. J. Am. Chem. Soc. 2012, 134, 14255–14262. [DOI] [PMC free article] [PubMed] [Google Scholar]; l Yi R.; Ogaki A.; Fukunaga M.; Nakajima H.; Ichiyanagi T. Tetrahedron 2014, 70, 3675–3682. [Google Scholar]
- Pokorny B.; Kosma P.. Chem.—Eur. J. 2014, in press (doi: 10.1002/chem.201405424). [DOI] [Google Scholar]
- Previously obtained via selective cleavage from the 4:5;7:8-di-O-isopropylidene derivative in a reported yield of 42%:; a Charon D.; Auzanneau F.-I.; Mérienne C.; Szabó L. Tetrahedron Lett. 1987, 28, 1393–1396. [Google Scholar]; b Auzanneau F.-I.; Charon D.; Szabó L. Carbohydr. Res. 1988, 179, 125–136. [DOI] [PubMed] [Google Scholar]
- Claesson A.; Luthman K. Acta Chem. Scand. B 1982, 36, 719–720. [Google Scholar]
- Kosma P.; Sekljic H.; Balint G. J. Carbohydr. Chem. 1996, 15, 701–714. [Google Scholar]
- Disaccharide 21 was stable during high-temperature measurements in d8-tol for several hours.
- Unger F. M.; Stix D.; Schulz G. Carbohydr. Res. 1980, 80, 191–195. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.