

SUMMARY
This protocol describes a method for the laboratory synthesis of chiral tetrahydroisoquinolines, bicyclic organic framework present in a wide assortment of natural and synthetic biologically important compounds. The methodology involves the use of a two-catalyst system: an achiral strong Brønsted acid, together with a chiral urea derivative. The anion-binding properties of the urea lead to association of the ion pair that results from protonation of the imine substrate. Cycloaddition with electron-rich olefins in a [4+2] pathway, followed by spontaneous proton loss and rearomatization leads to the tetrahydroisoquinoline products in highly enantioenriched form.
INTRODUCTION
Strong Brønsted acids (HX) accelerate a wide variety of important reactions by protonating neutral substrates and thereby enhancing their electrophilicity and therefore their reactivity toward nucleophiles (Nu–H).1 While the simple proton (H+) itself is not chiral, asymmetric induction in Brønsted acid-catalyzed reactions can be achieved through the design of strong acids with chiral conjugate bases (X*−). Chiral phosphoric acids,2,3 N-triflyl phosphoramides,4 aryl sulfonic acids,5 and Lewis acid-assisted strong Brønsted acids6 are representative examples of chiral strong Brønsted acids7 that have been developed successfully (Figure 1A).
Figure 1.

Asymmetric catalysis by strong Brønsted acids. A) Use of chiral acids (H-X*). B) Use of achiral Brønsted acids (HX) together with a chiral urea co-catalyst.
The anion-binding properties of neutral, chiral H-bond donors8–10 introduce an alternate strategy for asymmetric induction in strong Brønsted acid-catalyzed reactions (Figure 1B). In this scenario, a chiral H-bond donor such as a urea can associate with a protonated substrate through the negatively charged conjugate base, and it may control the facial selectivity of subsequent nucleophilic addition reactions through appropriate non-covalent interactions in the resulting ion pair. This strategy was exploited successfully in the context of formal [4+2] cycloadditions of N-aryl imines with electron-rich olefins, also known as the Povarov11 reaction (Figure 2).12,13 This reaction is co-catalyzed by (R,R,R)-sulfinamide urea 117 and o-nitrobenzenesulfonic acid (NBSA), and affords tetrahydroquinolines with up to three contiguous stereogenic centers directly from simple achiral precursors. Alternative and complementary enantioselective variants of the Povarov reaction have also been uncovered recently using chiral Lewis acid14 and phosphoric acid catalysts.15,16
Figure 2.
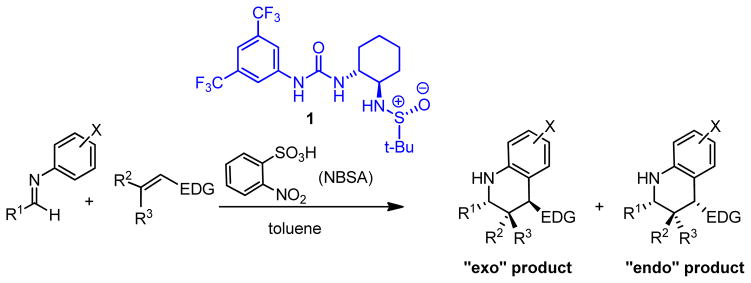
Generalized Povarov reaction co-catalyzed by o-nitrobenzenesulfonic acid and chiral urea 1 (R1, R2, R3, and X are organic substuents; EDG = electron-donating group).
Under optimized conditions, the asymmetric catalytic Povarov reaction was found to proceed effectively in cycloadditions of enamide 3 or ene-carbamate 4 with a wide variety of N-aryl imines (Figure 3). High levels of enantioselectivity were observed in reactions that were performed under cryogenic conditions with a 2:1 ratio of catalyst 1 to NBSA. Reactions of benzaldimines 2 with vinylpyrrolidinone 3 afford pyrrolidinone-substituted tetrahydroquinolines 5exo with high enantio- and diastereoselectivities (Figure 4). Tricyclic hexahydropyrrolo-[3,2-c]quinolines 6exo were obtained under the same conditions through the cyclization of N-Cbz–protected 2,3-dihydropyrrole 4 with 2 (Figure 3). Although lower diastereoselectivities favoring the exo isomer were obtained in this reaction (6exo/6endo = 1.4 to 4.2:1), the exo product was generated in high ee (90 to 98% ee) and could be isolated in diastereomerically pure form in useful yields (45 to 73%, Figure 5).
Figure 3.
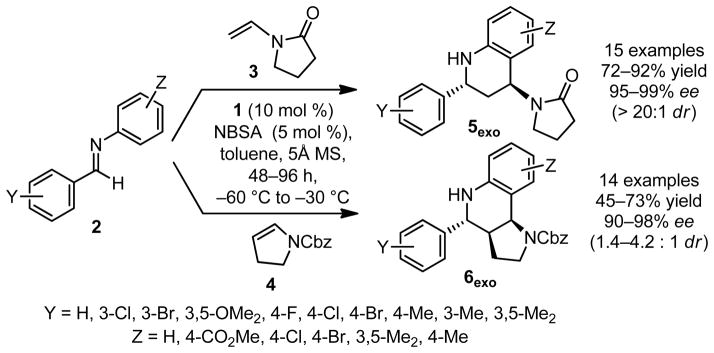
Asymmetric Povarov reactions catalyzed by 1/NBSA with enamide 3 or enecarbamate 4 as the nucleophilic reacting partners
Figure 4.
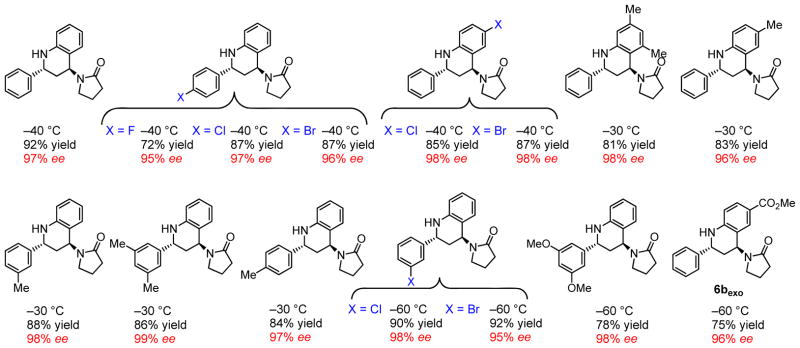
Scope of the catalytic asymmetric Povarov reaction of N-aryl imines and enamide 3.
Figure 5.

Scope of the catalytic asymmetric Povarov reaction of N-aryl imines and enecarbamate 4.
A very similar reaction protocol could be applied to highly enantioselective Povarov reactions between glyoxylate imines 7a or 7b with 2,3-dihydropyrrole 4 (Figure 6A). In this case, the catalytic reaction selectively affords the endo products (dr > 20:1, 95–97% ee). This transformation provides a direct route to the core tetrahydroquinoline structure of a variety of biologically active compounds, including martinelline (9, Figure 6B), a naturally occurring nonpeptide natural product that has been identified as a bradykinin B1 and B2 receptor antagonist.18,19,20 An analogous reaction has also been applied successfully to the preparation of a 2328-membered library of 2,3,4-trisubstituted tetrahydroquinolines (Figure 6C).13
Figure 6.

A) Catalytic asymmetric Povarov reaction of ethyl 2-(arylimino)acetates and enecarbamate 4; B) The enantioselective synthesis of 8b constitutes a formal enantioselective synthesis of martinelline (9). C) Application to a 2328-membered library of 2,3,4-trisubstituted tetrahydroquinolines (R1, R2, R3 = organic diversity elements.
Experimental design
The catalytic asymmetric synthesis of a pyrrolidinone-substituted tetrahydroisoquinoline 5b and a tricyclic hexahydropyrrolo-[3,2-c]quinoline 8b are described in this protocol as two representative examples (Figure 7).
Figure 7.

Protocols for the asymmetric synthesis of 5b and 8b.
MATERIALS
REAGENTS
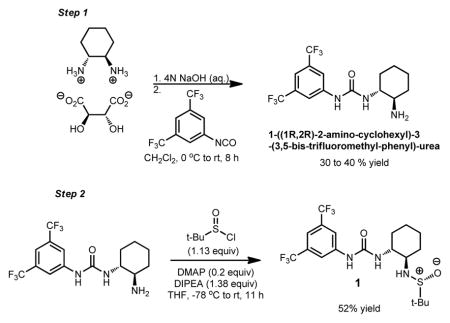
Catalyst 1 was prepared by the procedure in Box 1, as adapted from the literature procedure.17
Compounds 2b, 4 and 7b were prepared in accordance with literature procedures.21,22,23
2-Nitrobenzenesulfonic acid (Aldrich, cat. no. 127698)
2, 3-Dihydrofuran (Aldrich, cat. no. 200018)
1-Vinylpyrrolidin-2-one (3) (1-Vinyl-2-pyrrolidinone, Aldrich, cat. no. V3409)
Pyrrolidine (Aldrich, cat. no. W352316)
Benzyl chloroformate (Aldrich, cat. no. 119938)
Ethyl glyoxalate (Aldrich, cat. no. 50705)
4-Chloroaniline (Aldrich, cat. no. 477222)
Benzaldehyde (Aldrich, cat. No. B1334)
Methyl 4-aminobenzoate (Aldrich, cat. No. 274186)
Toluene, anhydrous
5 Å powdered molecular sieves
Et3N (Aldrich)
Dichloromethane
Methanol
Hexanes
Ethyl acetate
Sodium sulfate, anhydrous
Brine (saturated aqueous NaCl solution)
Saturated aqueous NaHCO3
Thin-layer chromatography (TLC) (Silica gel 60F254, layer thickness 250 μm, EMD Chemicals Inc.)
Silica gel (Silica Gel for Flash Chromatography, 60Å, 40–63 μm, Sorbent Technologies, cat. no. 40930-25)
Box 1. Synthesis of Povarov catalyst 1.
REAGENTS
3,5-Bis(trifluoromethyl)phenyl isocyanate (Aldrich, cat. No. 374857)
(R,R)-1,2-Diammoniumcyclohexane mono-(+)-tartrate24
N,N-Diisopropylethylamine (Aldrich, cat. No. 387649)
4-(Dimethylamino)pyridine (Aldrich, cat. No. 107700)
tert-butylsulfinyl chloride (Aldrich, cat. No. 569437)
1N HCl (EMD Chemicals Inc., cat. No. HX0603-4) solution
Saturated NaHCO3 (Mallinckrodt Chemicals, cat. No. 7412-06) aqueous solution
4N Sodium hydroxide (Macron Fine Chemicals, cat. No. 7708-06) aqueous solution
THF, anhydrous (VWR)
Dichloromethane (VWR)
Methanol (VWR)
Aqueous ammonia, 28~30% (wt/vol) (BDH, cat. No. BDH3014)
Anhydrous sodium sulfate (VWR)
Thin-layer chromatography (TLC) (Silica gel 60F254, layer thickness 250 μm, EMD Chemicals Inc.)
Silica gel (Silica Gel for Flash Chromatography, 60Å, 40–63 μm, Sorbent Technologies, cat. no. 40930-25)
EQUIPMENT
Syringe pump
Rotary evaporator
Chromatographic columns
Step 1 Synthesis of 1-((1R,2R)-2-amino-cyclohexyl)-3-(3,5-bis-trifluoromethyl-phenyl)-urea
● TIMING 10 h
PROCEDURE
Weigh out (R,R)-1,2-Diammoniumcyclohexane mono-(+)-tartrate (7.77 g, 29.4 mmol) in a 125 mL Erlenmeyer flask, and dissolve in 20 mL dichloromethane.
Introduce 4N sodium hydroxide solution (40 mL) into the flask and stir the biphasic mixture with a magnetic stirring bar for 10 minutes.
Separate the two phases in a 250 mL separatory funnel, and wash the aqueous phase with dichloromethane (3×20 mL)
Dry the combined organic phases with anhydrous sodium sulfate (10 g), and filter the mixture through a funnel lined with filter paper.
Concentrate the filtrate in a 250 mL round-bottom flask by rotary evaporation to obtain crude (R,R)-1,2-Diammoniumcyclohexane.
Dissolve the crude (R,R)-1,2-Diammoniumcyclohexane in 40 mL dichloromethane and cool the solution to 0 °C.
Add a solution of 3,5-bis(trifluoromethyl)phenyl isocyanate (2.5 g, 9.8 mmol) in dichloromethane (10 mL) by syringe pump (2 mL/h).
After the completion of addition, stir the solution at room temperature for 3 hours, and then concentrate the solution by rotary evaporation.
Pack a chromatography column (4.0 cm i.d. x 16 cm length) with silica gel (100 g) using a mixture of dichloromethane/methanol/aqueous ammonia (95:5:1, vol/vol/vol) as eluent.
Dissolve the crude product in eluent (4 mL) and load it to the column.
Elute the column using the mixture of DCM/methanol/aqueous ammonia (gradient from 95:5:1 to 90:10:1, vol/vol/vol)
Analyze the contents of the collected fractions by thin-layer chromatography (DCM/methanol/aqueous ammonia, 90:10:1, vol/vol/vol); the Rf of the product is at approximately 0.1.
Combine fractions containing the product, dry with anhydrous sodium sulfate (50 g), filter, and concentrate by rotary evaporation.
Dry the product under high vacuum.
Step 2. Synthesis of Povarov catalyst 1
● TIMING 12 h
PROCEDURE
Add THF (50 mL) into a 250 mL round-bottom flask, and cool the flask to −78 °C.
Add tert-butylsulfinyl chloride (0.23 mL, 1.84 mmol), N,N-Diisopropylethylamine (0.39 mL, 2.25 mmol), and 4-(Dimethylamino)pyridine (40 mg, 0.33 mmol) subsequently into the flask.
After stirring for 5 minutes, add 1-((1R,2R)-2-amino-cyclohexyl)-3-(3,5-bis-trifluoromethyl-phenyl)-urea (0.60 g, 1.63 mmol) in single portion into the flask.
Stir the reaction mixture at −78 °C for 4 hours and then warm up to room temperature slowly in 7 hours.
Quench the reaction with MeOH (5 mL), and remove the solvent using the rotary evaporator.
Dissolve the residue in ethyl acetate (20 mL) and wash the organic layer with 1N HCl (20 mL) and Saturated NaHCO3 aqueous solution (20 mL)
Dry the organic layer with anhydrous sodium sulfate (10 g) and filter by filter funnel.
Remove the solvent in the filtrate using the rotary evaporator.
Pack a chromatography column (4.0 cm i.d. x 16 cm length) with silica gel (100 g) using a mixture of DCM/methanol (99.25:0.75, vol/vol) as eluent.
Dissolve the crude product in eluent (2 mL) and load it to the column.
Elute the column using the mixture of DCM/methanol (gradient from 99.25:0.75 to 97:3, vol/vol)
Analyze the contents of the collected fractions by thin-layer chromatography (DCM/methanol, 95:5, vol/vol); Rf of the product is found at 0.2.
Combine fractions containing the product and evaporate the solvent using rotary evaporator.
Dry the product under high vacuum.
EQUIPMENT
Round-bottomed flasks
Dual argon vacuum manifold with vacuum line
Rubber septa
Disposable syringes and injection needles
Rotary evaporator
Chromatographic columns
Immersion cooler
1H NMR and 13C NMR spectrometers
High Performance Liquid Chromatography (HPLC) or Supercritical Fluid Chromatography (SFC) and chiral analytical stationary phase
Electrospray (ESI) mass spectrometer
Infrared spectrometer
Polarimeter
REAGENT SETUP
All commercially available reagents were used as received unless noted otherwise. 2,3-Dihydrofuran and 1-vinylpyrrolidin-2-one were distilled prior to use. 5 Å molecular sieves were activated by flame-drying in a flask under vacuum and then storing in a vacuum oven at 120 °C. 2-Nitrobenzenesulfonic acid (NBSA) is obtained commercially as a hydrate. It can be used as a solid hydrate as described in the Procedure for the preparation of 5b, or 0.1 M stock solutions can be prepared by dissolving NBSA·xH2O in anhydrous diethyl ether with 5A activated sieves (20 mg sieves/1 mL solvent) and stored under argon up to 24 h.
PROCEDURE
Synthesis of 5b. Total time required: 80 h
In a 10 mL oven-dried round-bottom flask, charge N-benzylidene-4-chloroaniline 2b (86 mg, 0.4 mmol), 2-nitro-benzenesulfonic acid (4.0 mg, 0.02 mmol), catalyst 1 (18.8 mg, 0.04 mmol), activated 5 Å molecular sieves (40 mg) at room temperature.
Introduce anhydrous toluene (5 mL) and a teflon-coated magnetic stir bar to the flask under a nitrogen atmosphere.
Cool the reaction mixture to −40 °C with an immersion cooler and stir for 10 min.
Add 1-vinylpyrrolidin-2-one 3 (0.30 mL of a 2.0 M toluene solution, 0.60 mmol) dropwise and allow the resulting solution to stir vigorously for 72 h.
Quench the reaction with pre-cooled Et3N (−40 °C, 0.28 mL, 2.0 mmol).
Pour the resulting mixture into a separatory funnel filled with 5 mL of saturated NaHCO3. Extract the organic material with ethyl acetate (10 mL x 2) and combine the organic phases.
Wash the organic phase with brine (10 mL).
Dry the organic phase over anhydrous sodium sulfate (ca. 10 g) and filter.
Transfer the organic solution into a 50-mL round-bottomed flask, and concentrate by rotary evaporation at 35 °C.
Purify the desired product by a flash chromatography column packed with silica gel (1.5 cm i.d. x 20 cm length) with hexanes/EtOAc as a eluent (gradient from 10:1 to 1:1) to afford 5b as a colorless oil.
Synthesis of 8b Timing 4 h
Charge a 10-mL oven-dried round-bottom flask with freshly made (E)-methyl 4-((2-ethoxy-2-oxoethylidene)amino)benzoate 7b (94 mg, 0.4 mmol), catalyst 1(7.6 mg, 0.016 mmol), benzyl 2,3-dihydropyrrole-1-carboxylate 4 (0.22 mL of a 2.0 M toluene stock solution, 0.44 mmol), and activated 5 Å molecular sieves (40 mg) at room temperature.
Introduce anhydrous toluene (5 mL) and a teflon-coated magnetic stir bar to the flask under a nitrogen atmosphere.
Cool the reaction mixture to −60 °C with an immersion cooler and stir for 10 min.
Add 2-nitrobenzenesulfonic acid (1.6 mg, 0.008 mmol, as 0.1M stock solution in Et2O), dropwise and allow the resulting solution to stir vigorously for 1.5 h at −60 °C.
Quench the reaction with pre-cooled Et3N (−60 °C, 0.28 mL, 2.0 mmol).
Pour the reaction mixture into a separatory funnel filled with 5 mL of saturated NaHCO3. Extract the organic material with ethyl acetate (10 mL x 2) and combine the organic phases.
Wash the organic phase with brine (10 mL).
Dry the organic phase over anhydrous sodium sulfate (ca. 10 g) and filter.
Transfer the organic solution into a 50-mL round-bottomed flask, and concentrate by rotary evaporation at 35 °C.
Purify the desired product by a flash chromatography column packed with silica gel (1.5 cm i.d. x 20 cm length) with hexanes/EtOAc as a eluent, gradient from 10:1 to 1:2) to afford 8b as a white foam.
TROUBLESHOOTING
Low yield
A low yield can result from acid-catalyzed imine hydrolysis due to adventitious water. Since the commercially available NBSA exists in its hydrated form, freshly activated 5 Å molecular sieves should be used in order achieve reproducibly high yields.
ANTICIPATED RESULTS
Analytical data
1-((2R,4S)-6-chloro-2-phenyl-1,2,3,4-tetrahydroquinolin-4-yl)pyrrolidin-2-one (5b)
Yield 82–86%
5b was determined to be 98% ee by Chiral SFC analysis (Pirkle Covalent (S, S) Whelk, 3.0 mL/min, 230 nm, 22% MeOH in supercritical CO2, tr(minor) = 7.63 min, tr(major) = 6.59 min). [α]25D= −83.1° (c = 3.1, CH2Cl2)
IR (film) νmax, 3336 (m), 1667 (s), 1605 (m), 1490 (m), 1420 (s), 1285 (m), 1269 (s) cm−1
1H NMR (500 MHz, CDCl3) δ ppm 7.25–7.37 (m, 5 H) 7.03 (dd, J = 8.58, 2.40 Hz, 1 H) 6.95 (d, J = 2.29 Hz, 1 H) 6.54 (d, J = 8.70 Hz, 1 H) 5.19 (t, J = 5.27 Hz, 1 H) 4.43 (dd, J = 9.61, 3.66 Hz, 1 H) 4.34 (br. s., 1 H) 3.35–3.42 (m, 1 H) 3.16–3.23 (m, 1 H) 2.44 (t, J = 8.13 Hz, 2 H) 2.23–2.30 (m, 1 H) 1.96–2.14 (m, 3 H)
13C NMR (125 MHz, CDCl3) δ ppm 174.8, 143.7, 142.9, 128.7, 128.5, 127.8, 126.2, 126.2, 122.0, 119.1, 115.6, 53.4, 45.7, 45.2, 35.6, 31.2, 18.3.
HRMS (ESI-TOF) for C19H19ClN2O [M + Na+] calculated 349.1078, found 349.1078.
(3aR,4R,9bR)-1-benzyl 4-ethyl 8-methyl 3,3a,4,5-tetrahydro-2H-pyrrolo[3,2-c]quinoline-1,4,8(9bH)-tricarboxylates (8b)
Yield 73–79%
8b was determined to be 95% ee by Chiral SFC analysis (Pirkle Covalent (S, S) Whelk, 3.0 mL/min, 230 nm, 20% MeOH, tr(minor) = 14.31 min, tr(major) = 13.31 min). [α]25D= 125.3° (c = 4.1, CH2Cl2)
IR (film) νmax, 3366 (m), 2951 (m), 1737 (s), 1698 (s), 1415 (s), 1281 (s), 1209 (m), 1131 (m), 1102 (m) cm−1
1H NMR (500 MHz, CDCl3) rotamers, δ ppm 8.26 (s, 0.5 H) 8.15 (s, 0.5 H) 7.74 (d, J = 7.33 Hz, 0.5 H) 7.50 (d, J = 7.33 Hz, 0.5 H) 7.28–7.45 (m, 5 H) 6.55 (d, J = 8.70 Hz, 1 H) 5.18–5.48 (m, 2.5 H) 4.78 (d, J = 12.13 Hz, 0.5 H) 4.21–4.41 (m, 4 H) 3.82 (d, J = 8.70 Hz, 3 H) 3.50–3.72 (m, 1 H) 3.32–3.46 (m, 1 H) 2.92 (d, J = 7.10 Hz, 1 H) 1.79–2.00 (m, 2 H) 1.33 (t, J = 7.21 Hz, 3 H)
13C NMR (125 MHz, CDCl3) rotamers, δ ppm 170.4, 166.8, 156.3, 155.3, 145.4, 132.3, 131.9, 130.2, 128.5, 128.4, 127.8, 120.3, 113.8, 67.6, 67.0, 61.9, 55.8, 53.3, 53.2, 51.6, 44.7, 38.8, 38.3, 25.4, 23.3, 22.3, 14.2.
HRMS (ESI-TOF) for C24H26N2O6 [M + Na+] calculated 438.1791, found 438.1796.
Footnotes
H.X. designed and performed the experiments, and co-wrote the paper. H.Z. performed the synthesis of catalyst 1. E.N.J. designed and supervised the experiments, analysed data, and co-wrote the paper.
The authors have no competing financial interests.
References
- 1.Eigen M. Proton transfer acid-base catalysis: enzymatic hydrolysis. Part 1 elementary processes. Angew Chem Int Ed. 1964;3:1–19. [Google Scholar]
- 2.Akiyama T, Itoh J, Yokota K, Fuchibe K. Enantioselective Mannich-type reaction catalyzed by a chiral Brønsted acid. Angew Chem Int Ed. 2004;43:1566–1568. doi: 10.1002/anie.200353240. [DOI] [PubMed] [Google Scholar]
- 3.Uraguchi D, Sorimachi K, Terada M. Organocatalytic asymmetric aza-Friedel-Crafts alkylation of furan. J Am Chem Soc. 2004;126:11804–11805. doi: 10.1021/ja046185h. [DOI] [PubMed] [Google Scholar]
- 4.Nakashima D, Yamamoto H. Design of chiral N-triflyl phosphoramide as a strong chiral Brønsted acid and its application to asymmetric Diels-Alder reaction. J Am Chem Soc. 2006;128:9626–9627. doi: 10.1021/ja062508t. [DOI] [PubMed] [Google Scholar]
- 5.Hatano M, Maki T, Moriyama K, Arinobe M, Ishihara K. Pyridinium 1,1′-binaphthyl-2,2′-disulfonates as highly effective chiral Brønsted acid-base combined salt catalysts for enantioselective Mannich-type reaction. J Am Chem Soc. 2008;130:16858–16860. doi: 10.1021/ja806875c. [DOI] [PubMed] [Google Scholar]
- 6.Ishihara K, Kaneeda M, Yamamoto H. Lewis-acid assisted chiral Brønsted acid for enantioselective protonation of silyl enol ethers and ketene bis(trialkyl silyl) acetals. J Am Chem Soc. 1994;116:11179–11180. [Google Scholar]
- 7.Yamamoto H, Futatsugi K. “Designer acids”: Combined acid catalysis for asymmetric synthesis. Angew Chem Int Ed. 2005;44:1924–1942. doi: 10.1002/anie.200460394. [DOI] [PubMed] [Google Scholar]
- 8.Raheem IT, Thiara PS, Peterson EA, Jacobsen EN. Enantioselective Pictet-Spengler-type cyclizations of hydroxylactams: H-bond donor catalysis by anion binding. J Am Chem Soc. 2007;129:13404–13405. doi: 10.1021/ja076179w. [DOI] [PubMed] [Google Scholar]
- 9.Doyle AG, Jacobsen EN. Small-molecule H-bond donors in asymmetric catalysis. Chem Rev. 2007;107:5713–5743. doi: 10.1021/cr068373r. [DOI] [PubMed] [Google Scholar]
- 10.Schreiner PR, Wittkopp A. H-bonding additives act like Lewis acid catalysts. Org Lett. 2002;4:217–220. doi: 10.1021/ol017117s. [DOI] [PubMed] [Google Scholar]
- 11.Kouznetsov VV. Recent synthetic developments in a powerful imino Diels-Alder reaction (Povarov reaction): application to the synthesis of N-polyheterocycles and related alkaloids. Tetrahedron. 2009;65:2721–2750. [Google Scholar]
- 12.Xu H, Zuend SJ, Woll MG, Tao Y, Jacobsen EN. Asymmetric Cooperative Catalysis of Strong Brønsted Acid-Promoted Reactions Using Chiral Ureas. Science. 2010;327:986–990. doi: 10.1126/science.1182826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerard B, et al. Application of a catalytic asymmetric Povarov reaction using chiral ureas to the synthesis of a tetrahydroquinoline library. ACS Combi Sci. 2012;14:621–630. doi: 10.1021/co300098v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishitani H, Kobayashi S. Catalytic asymmetric aza Diels-Alder reactions using a chiral lanthanide Lewis acid. Enantioselective synthesis of tetrahydroquinoline derivatives using a catalytic amount of a chiral source. Tetrahedron Lett. 1996;37:7357–7360. [Google Scholar]
- 15.Akiyama T, Morita H, Fuchibe K. Chiral Brønsted acid-catalyzed inverse electron-demand aza Diels-Alder reaction. J Am Chem Soc. 2006;128:13070–13071. doi: 10.1021/ja064676r. [DOI] [PubMed] [Google Scholar]
- 16.Liu H, Dagousset G, Masson G, Retailleau P, Zhu JP. Chiral Brønsted acid-catalyzed enantioselective three-component Povarov reaction. J Am Chem Soc. 2009;131:4598–4599. doi: 10.1021/ja900806q. [DOI] [PubMed] [Google Scholar]
- 17.Tan KL, Jacobsen EN. Indium-mediated asymmetric allylation of acylhydrazones using a chiral urea catalyst. Angew Chem Int Ed. 2007;46:1315–1317. doi: 10.1002/anie.200603354. [DOI] [PubMed] [Google Scholar]
- 18.Witherup KM, et al. Martinelline and martinellic acid, novel G-protein linked receptor antagonists from the tropical plant Martinella iquitosensis (Bignoniaceae) J Am Chem Soc. 1995;117:6682–6685. [Google Scholar]
- 19.Xia CF, Heng LS, Ma DW. Total synthesis of (+/−)-martinelline. Tetrahedron Lett. 2002;43:9405–9409. [Google Scholar]
- 20.Batey RA, et al. A three-component coupling protocol for the synthesis of substituted hexahydropyrrolo[3,2-c]quinolines. Chem Commun. 1999:651–652. [Google Scholar]
- 21.Keinicke L, Fristrup P, Norrby PO, Madsen R. Nonradical Zinc Barbier Reaction for Diastereoselective Synthesis of Vicinal Amino Alcohols. J Am Chem Soc. 2005;127:15756–15761. doi: 10.1021/ja054706a. [DOI] [PubMed] [Google Scholar]
- 22.Kraus GA, Neuenschwander K. Facile synthesis of N-acyl-2-pyrrolines. J Org Chem. 1981;46:4791–4792. [Google Scholar]
- 23.Trost BM, Marrs CM. A [3+2] cycloaddition and [4+3] cycloaddition approach to N-heterocycles via palladium-catalyzed TMM reactions with imines. J Am Chem Soc. 1993;115:6636–6645. [Google Scholar]
- 24.Larrow JF, Jacobsen EN. (R,R)-N,N′-Bis(3,5-Di-tert-Butylsalicylidene)-1,2-Cyclohexanediamino Manganese(III) Chloride, A Highly Enantioselective Epoxidation Catalyst. Org Syn. 1998;75:1. [Google Scholar]