

Abstract
220-kDa ankyrin-B is required for coordinated assembly of Na/Ca exchanger, Na/K ATPase, and inositol trisphosphate (InsP3) receptor at transverse-tubule/sarcoplasmic reticulum sites in cardiomyocytes. A loss-of-function mutation of ankyrin-B identified in an extended kindred causes a dominantly inherited cardiac arrhythmia, initially described as type 4 long QT syndrome. Here we report the identification of eight unrelated probands harboring ankyrin-B loss-of-function mutations, including four previously undescribed mutations, whose clinical features distinguish the cardiac phenotype associated with loss of ankyrin-B activity from classic long QT syndromes. Humans with ankyrin-B mutations display varying degrees of cardiac dysfunction including bradycardia, sinus arrhythmia, idiopathic ventricular fibrillation, catecholaminergic polymorphic ventricular tachycardia, and risk of sudden death. However, a prolonged rate-corrected QT interval was not a consistent feature, indicating that ankyrin-B dysfunction represents a clinical entity distinct from classic long QT syndromes. The mutations are localized in the ankyrin-B regulatory domain, which distinguishes function of ankyrin-B from ankyrin-G in cardiomyocytes. All mutations abolish ability of ankyrin-B to restore abnormal Ca2+ dynamics and abnormal localization and expression of Na/Ca exchanger, Na/K ATPase, and InsP3R in ankyrin-B+/- cardiomyocytes. This study, considered together with the first description of ankyrin-B mutation associated with cardiac dysfunction, supports a previously undescribed paradigm for human disease due to abnormal coordination of multiple functionally related ion channels and transporters, in this case the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor.
Primary arrhythmogenic disorders of the heart are a major cause of sudden cardiac death in otherwise healthy, and frequently young individuals. These arrhythmias are likely to be caused by aberrant function of ion channels leading to abnormal electrical properties of the heart. Mutations in genes encoding ion channels have emerged in the last decade as the basis for a variety of inherited arrhythmias, including long QT syndrome types 1-3 and 5-7, Brugada syndrome, and catecholaminergic polymorphic ventricular tachycardia (1). However, 30-45% of cases with arrhythmia cannot be explained by mutation of currently identified genes (2-4, ∥).
Physiological function of ion channels in the context of the heart requires precise cellular targeting in addition to activity at a single-channel level. This consideration suggests the possibility of a class of arrhythmias caused by defective cellular processing of channels. In support of this idea, mutation of the gene encoding the membrane adaptor protein ankyrin-B (Ank2, E1425G) was identified as the cause of type 4 long QT syndrome (5). Type 4 long QT syndrome has been characterized in a single large French kindred (5, 6). Affected family members display many atypical features compared to other long QT syndromes, including pronounced sinus bradycardia, polyphasic T waves, and atrial fibrillation (5). Moreover, cardiac repolarization defects in this family (heart-rate-corrected QT interval, or QTc) are not as severe as in other patients with long QT syndromes (4-7). Nonetheless, humans with ankyrin-B mutations experience polymorphic ventricular arrhythmia, syncope, and sudden death in response to exercise or emotional stress (5, 6).
Mice heterozygous for a null mutation in ankyrin-B (ankyrin-B+/- mice) display a cardiac phenotype similar to humans with the ankyrin-B E1425G mutation (5). Adult cardiomyocytes from ankyrin-B+/- mice have elevated Ca2+ transients and catecholamine-induced afterdepolarizations and extrasystoles (5). Adult ankyrin-B+/- cardiomyocytes also exhibit loss of the sub-population of ankyrin-B localized at transverse (T)-tubule/sarcoplasmic reticulum (SR) membrane sites (5). Moreover, the Na/K ATPase, Na/Ca exchanger, and inositol 1,4,5 trisphosphate (InsP3) receptor, all ankyrin-binding proteins, also are coordinately reduced at T-tubule/SR membrane sites (5). Elevated calcium transients in ankyrin-B+/- cardiomyocytes can be rationalized by reduction of the Na/K ATPase, which would mimic the action of cardiac glycosides in elevating intracellular Na+, and thus inhibit exchange of extracellular Na+ for intracellular Ca2+ by the Na/Ca exchanger.
Ankyrin family polypeptides (ankyrin-R, -B, and -G) are closely related and often are coexpressed in the same cell types (reviewed in ref. 8). However, unlike many protein families, ankyrin gene products have distinct functions (5, 9-12). Specific function of ankyrin-B in cardiomyocytes is determined by its divergent regulatory domain (13).
We report here four previously undescribed ankyrin-B mutations, all located in the regulatory domain, in humans with cardiac dysfunction including sudden death. We identified eight unrelated probands harboring ankyrin-B mutations whose clinical features distinguish the cardiac phenotype associated with loss of ankyrin-B activity from classic long QT syndromes. All four of these mutations cause loss of function of ankyrin-B in restoring normal Ca2+ dynamics and normal expression of Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in neonatal cardiomyocytes.
Materials and Methods
Genotyping and DNA Analysis. Genomic DNA was prepared from peripheral blood lymphocytes by using standard procedures. All exons of ankyrin-B (Ank2), with exception of brain-specific exon 38, were screened by single-strand conformation polymorphism (SSCP) or by denaturing HPLC (see Supporting Methods, which is published as supporting information on the PNAS web site). Of the 664 probands tested, 370 displayed long QT, 226 displayed idiopathic ventricular fibrillation, 21 displayed Andersen syndrome, 12 displayed LQT plus syndactyly, 27 displayed sick sinus syndrome, three displayed atrial fibrillation, and five displayed unexpected sudden death. Phenotypic criteria for long QT syndrome were as described (ref. 14; also see Supporting Methods).
Isolated Cardiomyocytes. Animal care was in accordance with Duke University Medical Center institutional guidelines. Neonatal cardiomyocytes were isolated from postnatal day 1 (P1) or P2 mice and transfected as described (5, 13). After transfection, isolated cardiomyocyte spontaneous contraction rates and/or Ca2+ dynamics (Fluo-3 AM) were monitored as described (5, 13). Immunolocalization experiments were performed as described (5, 13). Expression of GFP-220-kDa ankyrin-B constructs could not be visualized in transfected cardiomyocytes without the use of GFP antibody due to low expression levels. We observed no difference in the localization between endogenous ankyrin-B and any of the GFP-220-kDa ankyrin-B constructs. Nav1.5 polyclonal antibody was a generous gift of W. Catterall (University of Washington, Seattle).
Ankyrin-B Mutagenesis. Single amino acid mutations were introduced into human GFP-220-kDa ankyrin-B by using QuikChange mutagenesis (Stratagene). Mutated regions were sequenced and then subcloned into original vectors to ensure that no additional mutations were introduced into the vector.
Results
Cardiac Dysfunction in Humans Associated with Ank2 Mutations. We screened a large group of patients (664) for mutations to ankyrin-B (Ank2). These patients were selected based on presence of cardiac arrhythmias (long QT syndrome, catecholaminergic polymorphic ventricular tachycardia, idiopathic ventricular fibrillation, Andersen's syndrome, sick sinus syndrome, and unexplained sudden death) and lack of mutation in KCNQ1, HERG, SCN5A, KCNE1, KCNE2, and KCNJ2. None of the patients had structural heart disease as assessed by MRI and echocardiography. All exons of ankyrin-B (Ank2), with the exception of neuron-specific exon 38, were screened by SSCP in 344 probands. Denaturing HPLC was used to screen an additional 325 probands for mutations in Ank2 exons 36 and 42-45. No abnormal variants in the probands were detected within exons encoding the ankyrin-B membrane-binding domain (exons 1-23; Fig. 1A).
Fig. 1.

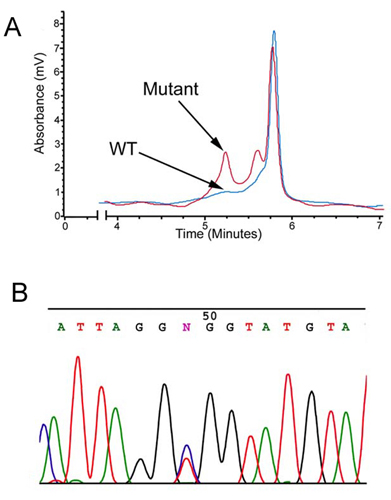
Identification of ankyrin-B mutations in human probands with cardiac arrhythmia. (A) Domain organization of 220-kDa ankyrin-B with previously identified E1425G mutation and L1622I, T1626N, R1788W, and E1813K mutations denoted. Yellow box denotes death domain, red box denotes C-terminal domain. (B) ECG recordings from European proband with ankyrin-B R1788W mutation. ECG recordings show normal resting ECG and polymorphic ventricular tachycardia induced by exercise.
Only one SSCP variant was identified within the ankyrin-B spectrin-binding domain (exons 24-36; Fig. 1A), and this proband was heterozygous for the previously identified A4274G (E1425G) mutation. This Caucasian female is clinically unaffected, and, in contrast to previously identified E1425G patients (5), had a normal QTc of 410 msec with a heart rate of 60 beats per min. The proband's 67-year-old mother is a carrier of the E1425G variant with slightly elevated QTc (430-450 msec) and moderately low heart rate (63 beats per min; see Fig. 2 and Table 1, Kindred A1). Three siblings of the proband died young of sudden death at 25, 17, and 15 years of age. The 25-year-old died while winning a prize. The 17-year-old died in the shower and had previously experienced syncopal events associated with athletics. The 15-year-old died getting out of a pool after swimming. DNA and QTc data were unattainable from the siblings. The E1425G mutation was not observed in 550 control individuals.
Fig. 2.

Pedigrees of probands with Ank2 mutations. Females are denoted by circles, and males are denoted by squares. Hatched symbols represent humans with uncertain or unknown LQT phenotype. Open symbols represent individuals without LQT phenotype at rest. Filled symbols represent individuals diagnosed with LQT phenotype. Mutation carriers are denoted by an asterisk. Probands are marked by a star. Sudden death is denoted by SD, and death is indicated by a diagonal slash. Roman numerals indicate generations in the respective families.
Table 1. Cardiac phenotypes of humans with Ank2 mutations.
| Individual | Age | HR | QTc | Mutation | Phenotype |
|---|---|---|---|---|---|
| A1-II4 | 67 | 63 | 430 | E1425G | Asymptomatic |
| A1-III4 | 28 | 60 | 410 | E1425G | Asymptomatic |
| A1-III5 | 25 | NA | NA | Untested | Sudden death after stress |
| A1-III6 | 17 | NA | NA | Untested | History of syncope with athletics, sudden death |
| A1-III7 | 15 | NA | NA | Untested | Sudden death after exercise |
| A2-I2 | 70 | 74 | 430 | T1626N | Asymptomatic |
| A2-II2 | 48 | 67 | 430 | T1626N | Asymptomatic |
| A2-II4 | 44 | 86 | 440 | T1626N | Asymptomatic |
| A2-II5 | 46 | 72 | 450 | T1626N | Long QT, syncope |
| A2-III2 | 19 | NA | NA | T1626N | Sudden death, previously asymptomatic |
| A2-III3 | 24 | 83 | 430 | T1626N | Asymptomatic |
| A2-III4 | 18 | 66 | 430 | T1626N | Asymptomatic |
| A3-II2 | 58 | 63 | 420 | T1626N | Asymptomatic |
| A3-II3 | 53 | 65 | 440 | T1626N | Asymptomatic |
| A3-II5 | 51 | 50-110 | 450 | T1626N | Sinus arrhythmia |
| A4-I2 | 43 | 71 | 440 | L16221 | Asymptomatic |
| A4-II1 | 23 | 63 | 445 | L16221 | Long QT |
| A4-II2 | 20 | 71 | 435 | L16221 | Ventricular tachycardia, ventricular fibrillation |
| A4-II3 | 17 | 66 | 430 | L16221 | Asymptomatic |
| A5-I1 | 37 | 60 | 530 | R1788W | Syncope during sleep Long QT, bradycardia |
| A5-II1 | NA | NA | NA | Untested | Long QT |
| A6-I2 | 52 | 67 | 425 | R1788W | Asymptomatic |
| A6-II1 | 35 | 62 | 430 | R1788W | Syncope with exercise; supraventricular and ventricular arrhythmia |
| Proband 7 | 61 | 64 | 395 | E1813K | Recurring arrhythmia; ventricular tachycardia |
| Proband 8 | 24 | 62 | 490 | E1813K | Idiopathic ventricular fibrillation |
HR, heart rate in beats per min; NA, not available.
SSCP/denaturing HPLC and DNA sequence analyses identified four potential ankyrin-B loss-of-function mutations in probands with varied arrhythmia phenotypes. All four mutations are localized to the ankyrin-B C-terminal regulatory domain previously demonstrated to confer ankyrin-B-specific activity in cardiomyocytes (Fig. 1A; ref. 13). We identified two unrelated probands with identical C-to-A mutations in exon 42 (C4877A) leading to a threonine-to-asparagine substitution at amino acid 1626 (T1626N, Fig. 1A). Both Caucasian probands were from the United States and had marginally elevated QTc (450 and 440-450 msec). One 46-year-old female proband (Fig. 2 and Table 1, Kindred A2) with a QTc of 450 msec had experienced syncope, but had a normal resting heart rate of 72 beats per min. The daughter of this proband was also heterozygous for the T1626N mutation and died of sudden death at age 19 with no previous cardiac symptoms (QTc and heart rate data are unavailable). Two siblings, two sons, and the mother of the same proband are also carriers of the mutation and have resting QTc in the normal range (430, 440, 440, 430, and 440 msec, respectively). To date, these carriers are asymptomatic. The second T1626N proband is a 51-year-old Caucasian male from the United States. This patient displays mildly elevated QTc (450 msec) with sinus arrhythmia (heart rate 50-110 beats per min). Two of the proband's siblings with normal QTc (420 and 400 msec) are heterozygous for the ankyrin-B T1626N mutation (Fig. 2 and Table 1, Kindred A3) but are asymptomatic to date. This mutation was not observed in 550 control individuals.
We identified a C-to-A mutation (C4864A) in exon 42 resulting in a leucine-to-isoleucine substitution at amino acid 1622 (L1622I) in one European female proband with a normal QTc (445 msec) (Fig. 1A). This Caucasian proband displayed polymorphic ventricular tachycardia, ventricular fibrillation, and a resting heart rate of 63 beats per min. The proband's mother, sister, and brother are carriers of the mutation (Fig. 2 and Table 1, Kindred A4), but to date all are asymptomatic with normal resting QTc (440 msec, 435 msec, and 430 msec, respectively) and normal heart rates (71, 71, and 66 beats per min, respectively). The L1622I mutation was not observed in control individuals.
We observed two unrelated probands with identical C-to-T mutations (C5336T) in exon 45 of Ank2 leading to arginine-to-tryptophan substitution at amino acid 1788 (R1788W; Fig. 1A and Fig. 7, which is published as supporting information on the PNAS web site). One proband was a 37-year old-Caucasian female from the United States (Fig. 2 and Table 1, Kindred A5). She presented with syncope (originally treated as a seizure) at age 12. This woman subsequently had multiple episodes of syncope associated with sleep, and torsades de pointes ventricular tachycardia was documented. β-Blocker therapy failed to eliminate symptoms, and she was treated with an implantable cardiac defibrillator. ECGs revealed a heart rate of 60 beats per min, prominent T-U waves, and prolongation of the QT interval with a QTc of 530 msec. The proband's son has also been diagnosed with long QT syndrome, although DNA from this subject is unattainable.
The second proband, heterozygous for a R1788W mutation, is a Caucasian female from Europe with multiple episodes of exercise-associated syncope. She presented with supraventricular and ventricular tachycardias that were reproducibly elicited by exercise tests. The patient has normal QTc at rest (430 msec), but prolongation of 470 msec was observed after a syncopal event. This woman was successfully treated with β-blockers. However, exercise-induced nonsustained supraventricular and ventricular arrhythmias persisted and were recorded in a clinical setting (see ECGs in Fig. 1B). The father of the proband is a carrier of the mutation (Fig. 2 and Table 1, Kindred A6) with QTc of 430 msec and a heart rate of 67 beats per min. The R1788W mutation was not identified in 280 DNA samples obtained from individuals with normal ECG.
Finally, we identified two unrelated probands with a fourth ankyrin-B mutation, a G-to-A mutation (G5437A) in exon 45 leading to a glutamate-to-lysine substitution at amino acid 1813 (E1813K, Fig. 1A). The first proband was a 24-year-old Caucasian female from Europe diagnosed with recurring arrhythmia and documented torsades de pointes ventricular tachycardia (Fig. 2 and Table 1, Proband 7). The proband presented with an elevated resting QTc of 490 msec and mild bradycardia (62 beats per min). We were unable to obtain DNA samples from family members. The second proband is a 60-year-old Caucasian male from the United States (Fig. 2 and Table 1, Proband 8). This proband displayed idiopathic ventricular fibrillation (15) with a normal QTc (395 msec), a normal resting ECG, and a heart rate of 64 beats per min. No DNA was available from close relatives (Fig. 2; Kindred G). The E1813K mutation was found in five DNA samples obtained from individuals with normal ECGs.
In summary, we have identified four ankyrin-B mutations (T1626N, L1622I, R1788W, and E1813K) in humans with cardiac dysfunction. We have also identified an additional proband with an ankyrin-B E1425G mutation. Probands with ankyrin-B mutations display varying degrees of cardiac dysfunction including bradycardia, sinus arrhythmia, idiopathic ventricular fibrillation, catecholaminergic polymorphic ventricular tachycardia, and sudden death. In contrast to the original E1425G kindred, ankyrin-B mutation carriers do not necessarily display a prolonged QTc or significant bradycardia at rest.
Human Mutations Abolish Function of Ankyrin-B in Cardiomyocytes. Functional consequences of the four human ankyrin-B mutations were evaluated by using a neonatal cardiomyocyte rescue assay (13). Compared with neonatal cardiomyocytes derived from wild-type mice, ankyrin-B+/- neonatal cardiomyocytes display reduced frequency of spontaneous contractions and abnormal Ca2+ dynamics (Fig. 3 and ref. 13). Additionally, ankyrin-B+/- cardiomyocytes display abnormal localization and expression of InsP3 receptor, Na/K ATPase, and Na/Ca exchanger (Figs. 4 and 5, and Figs. 8-10, which are published as supporting information on the PNAS web site). In contrast, ankyrin-B+/- cardiomyocytes display normal localization and expression of endoplasmic and sarcoplasmic reticulum proteins (SERCA2, calreticulin, calsequestrin, ryanodine receptor 2), plasma membrane Ca2+ channels (DHPR, PMCA2), ion channels and associated subunits (Nav1.5, Nav1.6, KCNQ1, MinK, Kir1.2, Kir2.3, ERG1), structural proteins (α-actinin, dystrophin), and connexin 43 (5). Abnormal ankyrin-B+/- phenotypes can be restored to normal by transfection of GFP-220-kDa ankyrin-B human cDNA into ankyrin-B+/- cardiomyocytes (Figs. 3, 4, 5, refs. 5 and 13). In contrast, GFP-220-kDa ankyrin-B containing the human E1425G mutation is unable to rescue spontaneous contractions, Ca2+ dynamics, or abnormal ion channel and transporter localization when expressed in ankyrin-B+/- cardiomyocytes (ref. 5, see Figs. 8 and 9). Therefore, this cardiomyocyte rescue assay can be used to test potential functional consequences of ankyrin-B mutations.
Fig. 3.

Human ankyrin-B mutations do not restore abnormal spontaneous contractions or abnormal Ca2+ dynamics in ankyrin-B+/- cardiomyocytes. Compared to wild-type cardiomyocytes, ankyrin-B+/- cardiomyocytes display decreased spontaneous contraction rates (A) and abnormal calcium dynamics (B). Abnormal ankyrin-B+/- cardiomyocyte contraction rates and calcium dynamics are rescued by transfection of GFP-220-kDa ankyrin-B, but not GFP-220-kDa ankyrin-B mutants E1425G, L1622I, T1626N, R1788W, or E1813K (n > 200 cells; P < 0.05). In B, calcium dynamics (expressed in Hz) were measured by using Fluo-3 AM. (C) Raw data of Ca2+ levels in cardiomyocytes (F/Fo). Graphs represent untransfected wild-type and ankyrin-B+/- neonatal cardiomyocytes and GFP-220-kDa ankyrin-B and GFP-220-kDa ankyrin-B R1788W transfected ankyrin-B+/- myocytes.
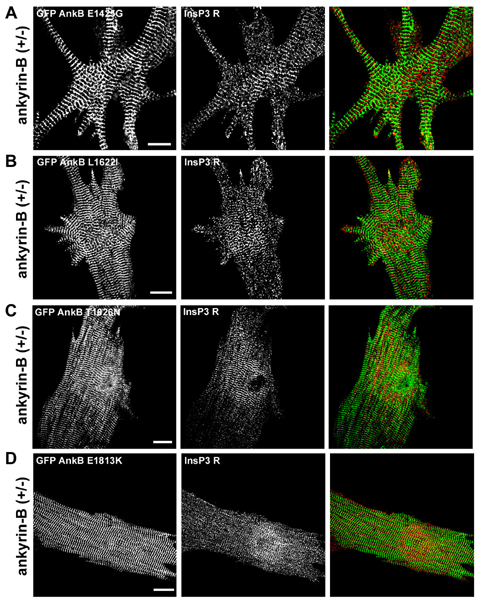
Fig. 4.

Human mutations are ankyrin-B loss-of-function mutations in mouse cardiomyocytes. (A) Immunolocalization of endogenous ankyrin-B (Left) and InsP3 receptor (Right) in wild-type neonatal cardiomyocytes. (Scale bar, 10 μm.) (B) Immunolocalization of ankyrin-B (Left) and InsP3 receptor (Right) in ankyrin-B+/- neonatal mouse cardiomyocytes. Reduction in ankyrin-B levels leads to loss of InsP3 receptor expression and organization (compare InsP3 receptor staining in A and B). (Scale bar, 10 μm.) (C) Abnormal localization of InsP3 receptor in ankyrin-B+/- cardiomyocytes is rescued by transfection of GFP-220-kDa ankyrin-B. GFP-220-kDa ankyrin-B was immunolocalized by using affinity-purified GFP antibody. (Scale bar, 10 μm.) (D) R1788W is an ankyrin-B loss-of-function mutation. Expression of GFP-220-kDa ankyrin-B R1788W does not rescue abnormal expression or localization of InsP3 receptor in ankyrin-B+/- neonatal cardiomyocytes. GFP-220-kDa ankyrin-B was immunolocalized by using affinity-purified GFP antibody. (Scale bar, 10 μm.) GFP-220-kDa ankyrin-B, GFP-220-kDa ankyrin-B R1788W, and endogenous ankyrin-B (in +/+ cell) are localized in the same striated pattern. The perimeter of the cell has been outlined in B and D.
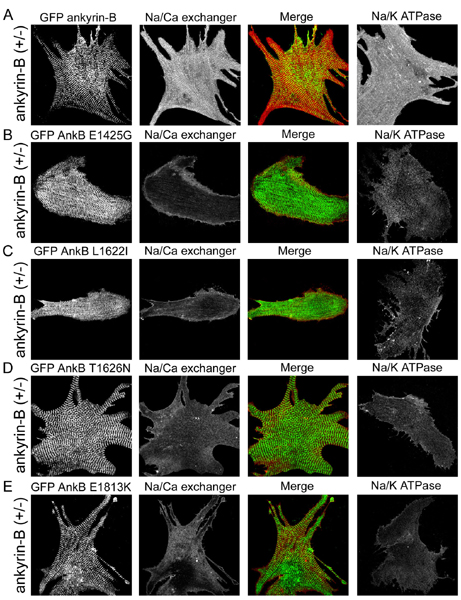
Fig. 5.

GFP-220-kDa ankyrin-B, but not GFP-220-kDa ankyrin-B R1788W, rescues abnormal Na/Ca exchanger and Na/K ATPase localization in ankyrin-B+/- cardiomyocytes. (A) Immunolocalization of endogenous ankyrin-B with Na/Ca exchanger and Na/K ATPase in wild-type neonatal cardiomyocytes. (Scale bar, 10 μm.) (B) Immunolocalization of endogenous ankyrin-B with Na/Ca exchanger and Na/K ATPase in ankyrin-B+/- neonatal mouse cardiomyocytes. Reduction in ankyrin-B levels leads to loss of Na/Ca exchanger and Na/K ATPase expression and organization (compare Na/Ca exchanger and Na/K ATPase staining in A and B). (Scale bar, 10 μm.) (C) Abnormal expression and localization of Na/Ca exchanger and Na/K ATPase in ankyrin-B+/- neonatal cardiomyocytes is rescued by transfection of GFP-220-kDa ankyrin-B. GFP-220-kDa ankyrin-B was immunolocalized by using affinity-purified GFP antibody. (Scale bar, 10 μm.) (D) R1788W is an ankyrin-B loss-of-function mutation. Expression of GFP-220-kDa ankyrin-B R1788W does not rescue abnormal expression or localization of Na/Ca exchanger or Na/K ATPase in ankyrin-B+/- neonatal cardiomyocytes. (Scale bar, 10 μm.) GFP-220-kDa ankyrin-B, GFP-220-kDa ankyrin-B R1788W, and endogenous ankyrin-B (in +/+ cell) are localized in the same striated pattern. The perimeter of the cardiomyocyte in D is outlined.
Individual GFP-220-kDa ankyrin-B mutants L1622I, T1626N, R1788W, and E1813K were created by using site-directed mutagenesis. After each plasmid was sequenced to ensure that no additional mutations were introduced, the four human mutants were transfected into ankyrin-B+/- neonatal cardiomyocytes. All of the GFP-220-kDa ankyrin-B mutants were localized in the same striated pattern as endogenous ankyrin-B and GFP-220-kDa wild-type ankyrin-B (assessed by using affinity-purified GFP antibody), and were expressed at similar levels to GFP-220-kDa ankyrin-B when transfected into ankyrin-B+/- neonatal cardiomyocytes (Figs. 4, 5, 8, and 9). However, in contrast to GFP-220-kDa ankyrin-B, GFP-220-kDa ankyrin-B L1622I, T1626N, R1788W, or E1813K mutants were unable to rescue abnormal spontaneous contraction rates (Fig. 3A) or abnormal Ca2+ dynamics (Fig. 3 B and C) in ankyrin-B+/- neonatal cardiomyocytes.
We next analyzed the localization and expression of InsP3 receptor, Na/Ca exchanger, and Na/K ATPase in ankyrin-B+/- cardiomyocytes expressing individual human ankyrin-B mutants. As noted above, all GFP-220-kDa ankyrin-B mutants were expressed at levels similar to GFP-220-kDa ankyrin-B, and were normally localized in a striated pattern (Figs. 4, 5, 8, and 9). Similar to our previous results with the E1425G mutant, human ankyrin-B mutants L1622I, T1626N, R1788W, and E1813K were unable to rescue the abnormal patterns of staining for InsP3 receptor, Na/Ca exchanger, and Na/K ATPase (Figs. 4, 5, 8, and 9). In fact, in the presence of GFP-220-kDa ankyrin-B human mutants, expression of Na/Ca exchanger and Na/K ATPase is decreased to below levels observed in nontransfected ankyrin-B+/- cardiomyocytes (Figs. 5, 9, and 10). These combined results demonstrate that the four human ankyrin-B mutations (L1622I, T1626N, R1788W, E1813K) abolish function of ankyrin-B in neonatal cardiomyocytes.
Voltage-Gated Sodium Channel (Nav1.5) Is Normally Expressed and Localized in Ankyrin-B+/- Cardiomyocytes. Ankyrin-G associates with the voltage-gated Na+ channel at axon initial segments (16, 17). Therefore, we determined whether loss of ankyrin-B expression in ankyrin-B+/- mice altered the distribution and/or expression of the cardiac voltage-gated Na+ channel (Nav1.5) in isolated neonatal or adult mouse cardiomyocytes. In contrast to the striated ankyrin-B staining pattern, Nav1.5 in wild-type neonatal cardiomyocytes is localized at the peripheral sarcolemma as assessed by using an affinity-purified polyclonal antibody (Fig. 6A). The localization of Nav1.5 in ankyrin-B+/- neonatal (Fig. 6B) and adult cardiomyocytes (data not shown) is identical by immunofluorescence. Additionally, neonatal and adult cardiomyocytes derived from wild-type or ankyrin-B+/- mice have identical levels of Nav1.5 protein expression by Western blot analysis (Fig. 6 C and D; P > 0.05).
Fig. 6.

Reduction of ankyrin-B does not affect Nav1.5 expression or localization in ankyrin-B+/- cardiomyocytes. Immunolocalization of endogenous ankyrin-B and Nav1.5 in wild-type (A) and ankyrin-B+/- (B) neonatal cardiomyocytes using affinity-purified antibodies. We observed no difference in Nav1.5 expression or localization between wild-type and ankyrin-B+/- cardiomyocytes. The perimeter of the cell is outlined in B. Distance between ankyrin-B striations equals 1.8 μm. (C and D) Quantitative Western blot analysis of Nav1.5 expression in wild-type and ankyrin-B+/- adult (C) and neonatal (D) cardiomyocytes using 125I-labeled protein A. Protein levels were quantitated by PhosphorImaging. Reduction of ankyrin-B levels in ankyrin-B+/- cardiomyocytes has no affect on Nav1.5 expression (n = 3, P > 0.05; not significant).
A previous study reported prolonged action potentials and minor differences in Na+ channel kinetics in primary cultures of ankyrin-B-null neonatal cardiomyocytes (18). However, the INa density was only modestly affected, implying no major difference in numbers of Na+ channels. Moreover, no significant differences in the upstroke or action potential duration (including repolarization) were detected between isolated adult ventricular cardiomyocytes of wild-type and ankyrin-B heterozygous mice when two different pacing protocols were used (5). The two studies cannot be directly compared, because one study described ankyrin-B null neonatal cardiomyocytes, and the other characterized ankyrin-B heterozygous adult cardiomyocytes. However, we can conclude that Nav1.5 expression or localization is not altered in adult ankyrin-B+/- cardiomyocytes and does not contribute to elevated Ca2+ transients noted in these cells.
Discussion
This study reports clinical features of eight unrelated probands harboring loss-of-function mutations in the Ank2 gene, including four previously undescribed mutations, that define a cardiac arrhythmia syndrome associated with loss of ankyrin-B activity. All four of the mutations are localized to the ankyrin-B regulatory domain, and cause loss-of-function of ankyrin-B in neonatal cardiomyocytes. Humans with ankyrin-B mutations display varying degrees of cardiac dysfunction including bradycardia, sinus arrhythmia, idiopathic ventricular fibrillation, catecholaminergic polymorphic ventricular tachycardia, and risk of sudden death. However, prolonged QTc intervals were not a consistent feature, indicating that ankyrin-B dysfunction represents a distinct clinical entity from classic long QT syndrome.
The finding that ankyrin-B mutations are associated with complex cardiac defects is consistent with the expression of ankyrin-B in multiple cell-types of the heart. For example, ankyrin-B is localized in both atrial and ventricular cardiomyocytes, as well as cardiac Purkinje conduction fibers (data not shown). Moreover, a 440-kDa alternatively spliced variant of ankyrin-B is present in unmyelinated axons of neurons innervating the heart (not shown). Loss-of-function mutation in ankyrin-B thus is likely to affect multiple aspects of heart function with consequences for conduction, atrial and ventricular rhythm, and function of sinus and atrioventricular nodes. In addition, ankyrin-B is expressed in multiple cell types of multiple organs, including beta cells of the pancreas, the nervous system, and the retina. Therefore, humans with ankyrin-B syndrome may display phenotypes in addition to cardiac arrhythmia.
This study provides evidence for facets of ankyrin-B function that go beyond the simple role of a scaffolding protein predicted from the role of ankyrin in the erythrocyte membrane skeleton. Four different mutations in the regulatory domain of ankyrin-B, distant from binding sites in the membrane-binding domain, cause the same level of loss of expression and mis-localization of the Na/K ATPase, Na/Ca exchanger and InsP3 receptor. Thus, ankyrin-B domains are not independent modules, but work together, either through intramolecular interactions between membrane-binding and regulatory domains, and/or through binding to other proteins. One critical role for the regulatory domain is in restricting interactions of the membrane-binding domain. For example, ankyrin-G is required for targeting of voltage-gated Na+ channels at axon initial segments, whereas ankyrin-B, which has a closely related membrane-binding domain, is not required for expression of voltage-gated Na+ channels in cardiomyocytes (Fig. 6).
All of the ankyrin-B mutations are located in or near the C-terminal regulatory domain. The regulatory domain of ankyrins can modulate the activities of the N-terminal membrane- and spectrin-binding domains (19, 20). Therefore, these mutations could affect intramolecular association of the regulatory domain with membrane- or spectrin-binding domains, or interfere with intermolecular interactions. One of the ankyrin-B mutations (R1788W) directly abolishes a recently described interaction between ankyrin-B and the molecular cochaperone Hsp40/Hdj1 (21). The R1788W mutation is located on the surface of an amphipathic alpha-helix structure required for ankyrin-B function and for association with Hsp40/Hdj1 (21). R1788W mutation blocks Hsp40/Hdj1 binding with ankyrin-B in solution (Fig. 11A, which is published as supporting information on the PNAS web site) and in yeast two-hybrid assays (data not shown). Moreover, immobilized GFP-220-kDa ankyrin-B, but not GFP-220-kDa ankyrin-B R1788W, associates with Hsp40/Hdj1 from cardiac lysates (Fig. 11B). Therefore, the R1788W mutation abolishes ankyrin-B binding to Hsp40/Hdj1 and suggests that disruption of the ankyrin-B-Hsp40/Hdj1 interaction may lead to human cardiac dysfunction.
In summary, this study, considered together with the description of ankyrin-B mutation associated with cardiac dysfunction (5), supports a previously undescribed paradigm for human disease due to abnormal coordination of multiple functionally related ion channels and transporters, in this case the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor. Identification of the structural basis for ankyrin-B interactions with Na/K ATPase, Na/Ca exchanger, and InsP3 receptor and dissection of the pathway for assembly of these proteins promises to yield common elements used more generally in targeting membrane proteins to specialized membrane domains in other physiological systems.
Supplementary Material
Acknowledgments
We thank the patients and their families for their cooperation. This work was supported National Institutes of Health Grants P50-HL-52338 and R01HL46401 (to M.T.K.), the Leducq Foundation, Telethon Grant GP0227Y01 (to C.N., G.B., and S.G.P.), the Howard Hughes Medical Institute (V.B.), and Johnson & Johnson (V.B.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: QTc, heart-rate-corrected QT interval; InsP3, inositol 1,4,5 trisphosphate; SSCP, single-strand confirmation polymorphism.
Footnotes
Napolitano, C., Ronchetti, E., Memmi, M., Nastoli, J., Faggiano, G., Schwartz, P. J. & Priori, S. G. (2001) J. Am. Coll. Cardiol. 87A (abstr.).
References
- 1.Towbin, J. A. & Vatta, M. (2001) Am. J. Med. 110, 385-398. [DOI] [PubMed] [Google Scholar]
- 2.Splawski, I., Shen, J., Timothy, K. W., Lehmann, M. H., Priori, S., Robinson, J. L., Moss, A. J., Schwartz, P. J., Towbin, J. A., Vincent, G. M. & Keating, M. T. (2000) Circulation 102, 1178-1185. [DOI] [PubMed] [Google Scholar]
- 3.Priori, S. G., Napolitano, C. & Vicentini, A. (2003) J. Interv. Cardiol. Electrophysiol. 9, 93-101. [DOI] [PubMed] [Google Scholar]
- 4.Moss, A. J. (2003) J. Am. Med. Assoc. 289, 2041-2044. [Google Scholar]
- 5.Mohler, P. J., Schott, J. J., Gramolini, A. O., Dilly, K. W., Guatimosim, S., duBell, W. H., Song, L. S., Haurogne, K., Kyndt, F., Ali, M. E., et al. (2003) Nature 421, 634-639. [DOI] [PubMed] [Google Scholar]
- 6.Schott, J. J., Charpentier, F., Peltier, S., Foley, P., Drouin, E., Bouhour, J. B., Donnelly, P., Vergnaud, G., Bachner, L., Moisan, J. P., et al. (1995) Am. J. Hum. Genet. 57, 1114-1122. [PMC free article] [PubMed] [Google Scholar]
- 7.Chiang, C. E. & Roden, D. M. (2000) J. Am. Coll. Cardiol. 36, 1-12. [DOI] [PubMed] [Google Scholar]
- 8.Bennett, V. & Baines, A. J. (2001) Physiol. Rev. 81, 1353-1392. [DOI] [PubMed] [Google Scholar]
- 9.Zhou, D., Lambert, S., Malen, P. L., Carpenter, S., Boland, L. M. & Bennett, V. (1998) J. Cell Biol. 143, 1295-1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scotland, P., Zhou, D., Benveniste, H. & Bennett, V. (1998) J. Cell Biol. 143, 1305-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tuvia, S., Buhusi, M., Davis, L., Reedy, M. & Bennett, V. (1999) J. Cell Biol. 147, 995-1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jenkins, S. M. & Bennett, V. (2001) J. Cell Biol. 155, 739-746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mohler, P. J., Gramolini, A. O. & Bennett, V. (2002) J. Biol. Chem. 277, 10599-10607. [DOI] [PubMed] [Google Scholar]
- 14.Splawski, I., Timothy, K. W., Vincent, G. M., Atkinson, D. L. & Keating, M. T. (1997) N. Engl. J. Med. 336, 1562-1567. [DOI] [PubMed] [Google Scholar]
- 15.Priori, S. G., Napolitano, C. & Grillo, M. (2001) Cardiovasc. Res. 50, 218-223. [DOI] [PubMed] [Google Scholar]
- 16.Garrido, J. J., Giraud, P., Carlier, E., Fernandes, F., Moussif, A., Fache, M. P., Debanne, D. & Dargent, B. (2003) Science 300, 2091-2094. [DOI] [PubMed] [Google Scholar]
- 17.Lemaillet, G., Walker, B. & Lambert, S. (2003) J. Biol. Chem. 278, 27333-27339. [DOI] [PubMed] [Google Scholar]
- 18.Chauhan, V. S., Tuvia, S., Buhusi, M., Bennett, V. & Grant, A. O. (2000) Circ. Res. 86, 441-447. [DOI] [PubMed] [Google Scholar]
- 19.Hall, T. G. & Bennett, V. (1987) J. Biol. Chem. 262, 10537-10545. [PubMed] [Google Scholar]
- 20.Davis, L. H., Davis, J. Q. & Bennett, V. (1992) J. Biol. Chem. 267, 18966-18972. [PubMed] [Google Scholar]
- 21.Mohler, P. J., Hoffman, J. A., Davis, J. Q., Abdi, K. M., Kim, C. R., Jones, S. K., Davis, L. H., Roberts, K. F. & Bennett, V. (2004) J. Biol. Chem., in press.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}