

Abstract
Hulless barley is an important cereal crop worldwide, especially in Tibet of China. However, this crop is usually susceptible to powdery mildew caused by Blumeria graminis f. sp. hordei. In this study, we aimed to understand the functions and pathways of genes involved in the disease resistance by transcriptome sequencing of a Tibetan barley landrace with high resistance to powdery mildew. A total of 831 significant differentially expressed genes were found in the infected seedlings, covering 19 functions. Either “cell,” “cell part,” and “extracellular region” in the cellular component category or “binding” and “catalytic” in the category of molecular function as well as “metabolic process” and “cellular process” in the biological process category together demonstrated that these functions may be involved in the resistance to powdery mildew of the hulless barley. In addition, 330 KEGG pathways were found using BLASTx with an E-value cut-off of <10−5. Among them, three pathways, namely, “photosynthesis,” “plant-pathogen interaction,” and “photosynthesis-antenna proteins” had significant matches in the database. Significant expressions of the three pathways were detected at 24 h, 48 h, and 96 h after infection, respectively. These results indicated a complex process of barley response to powdery mildew infection.
1. Introduction
Hulless barley (Hordeum vulgare L. var. nudum) is a diploid (2n = 7x = 14) monocot and belongs to the family of Poaceae. Hulless barley is also a form of domesticated barley with an easier-to-remove hull. Hulless barley is an important cereal crop worldwide, especially for beer brewing and poultry feed [1]. This crop is often attacked by barley powdery mildew fungus (Blumeria graminis f. sp. hordei), which is one of the most destructive pathogens of barley. Powdery mildew causes considerable damage and severe loss of grain yield [2]. It is crucial to collect genetic resources resistant to this disease and further identify underlying genes of resistance to powdery resources. In barley, a great number of landraces have been cultivated across the world and present large genetic variation in many desirable traits, including disease resistance. Indeed, most of the genes for resistance to powdery mildew in currently used cultivars were found in barley landraces [3–7]. A few resistance genes against powdery mildew have been studied in barley, such as mla [8], mlo [9], mlg [10], mlhb [11], and mlf [12]. Nevertheless, many resistance genes have lost their effectiveness as new races of the pathogen have evolved. Hence, discovering new candidate genes in barley genome sequence is of particular importance. To date, the resistance mechanisms for powdery mildew at physiological and gene levels remain unknown, so comprehensive transcriptomic sequencing of barley varieties with the disease resistance may improve our understanding of plant reaction to pathogen infection.
In the past few years, it has been widely demonstrated that high-throughput next generation sequencing technology makes it possible to carry out genome-wide studies of transcriptomes in a cost-efficient way to explore genes and expression profiling of model and nonmodel organisms [13–15]. Transcriptome sequencing and characterization using Illumina II sequencing technology have been successfully used to interrogate transcriptomes of many organisms such as yeast [16, 17], sweet potato [18], rice [19], taxus [20], migratory locust [21], and giant panda [22]. Despite its obvious potential, Illumina second generation sequencing has not been applied to barley variety for powdery mildew resistance analysis.
The present study was undertaken to provide a broad survey of genes associated with barley resistance to powdery mildew, by transcriptome analysis using Illumina technology. The main goals of this work were as follows: (1) to discover new genes related to powdery mildew resistance; (2) to characterize the gene expression profiles during pathogen infection processes; and (3) to reveal the functions and pathways of the genes involved in the disease resistance mechanism.
2. Materials and Methods
2.1. Plant Materials, Pathogen Infection, and RNA Extraction
Hulless barley cultivar “Gan Nong Da 7,” displaying high resistance to powdery mildew (unpublished work), was used in this study. Barley seeds were sown in the greenhouse. Three weeks later, the seedlings were infected by powdery mildew (isolated from the field infected barley) and then kept in dark at 18°C for 24 hours and finally kept in light for 14 hours every day. Barley leaves were harvested at six growth stages after infection: 0 h, 24 h, 48 h, 72 h, 96 h, and 120 h, respectively (see Supplementary Figure 1 in Supplementary Material available online at http://dx.doi.org/10.1155/2014/594579). More information about the samples can be found out in Supplementary Table 1. For Illumina sequencing, the total RNA of each of the samples was isolated using an RNAiso Plus (TaKaRa, Japan) protocol and then further purified with RNase-free DNase I (TaKaRa, Japan).
2.2. cDNA Library Construction and Sequencing
Briefly, Sera-Mag Magnetic Oligo (dT) Beads (Illumina, San Diego, CA) were used to isolate poly (A) mRNA after the total RNA was collected from the leaves. Fragmentation buffer was added for interrupting mRNA into short fragments. Then, by using these short fragments as templates, random hexamer (N6) primers (Illumina, San Diego, CA) were used to synthesize the first-strand cDNA. The second-strand cDNA was synthesized in the buffer containing dNTPs, RNase H, and DNA polymerase I. Paired end library was constructed by the Genomic Sample Prep kit (Illumina, San Diego, CA) according to manufacturer's instructions. Short fragments were purified with QiaQuick PCR extraction kit (Qiagen, Valencia, CA) and resolved with EB buffer for end repair and adding poly (A). After that, the short fragments were connected with the sequencing adapters. For amplification with PCR, we selected suitable fragments (200 ± 25 bp) as templates based on the result of agarose gel electrophoresis. At last, the library was sequenced using Illumina Genome Analyzer IIX (Illumina, San Diego, CA).
2.3. Mapping Reads to Reference Genome
The reference genome was downloaded from the Barley Genome Database (http://150.46.168.145/gbrowse_new/). Sequencing-received raw image data was transformed by Base Calling into raw data or raw reads. Raw sequences were transformed into clean tags by removing reads with adaptor contamination, reads of low quality (reads containing Ns > 10), and the reads with more than 50% Q ≤ 5 bases. Then, the saturation analysis was performed to check whether the number of detected genes increased along with sequencing amount (total tag number). The distribution of clean tag expressions was used to evaluate the normality of the whole data. After that, the remaining reads were aligned to the reference genome using software program TopHat 2.0.9 (Johns Hopkins University; see http://ccb.jhu.edu/software/tophat/index.shtml), following the procedure: tophat-p4-library-type, fr-unstranded-G gff.
2.4. Normalized Gene Expression Level by RNA-Seq
The expression levels of genes based on RNA-Seq was normalized by the number of reads per kilo base of exon region in a gene per million mapped reads (RPKM) [23]:
| (1) |
where RPKM is the reads per kilo base transcript per million reads, R is the number of mappable reads to a gene, N is the total mapped reads in the experiment, and L is the sum of the exons in base pairs.
RPKM is able to avoid the difference from gene length and total sequence data effect on gene expression. The cut-off value for determining the background expression level was at 95% confidence interval for all RPKM values of each gene. The results from this formula were directly used to compare the difference in the gene expressions among the samples at different time sequences.
2.5. Evaluation of DGE (Differentially Expressed Genes) Libraries
For screening of DGEs between different samples, a rigorous algorithm was developed based on the previous method [24]. P value corresponds to differential gene expression test. The threshold of P value in multiple tests was determined through manipulating the false discovery rate (FDR) value. We use FDR ≤ 0.05 as the threshold to judge the significance of DGEs.
Gene ontology (GO) terms were analyzed by the software Blast2GO v 2.3.4 [25] using the default parameters. This program was used to obtain the number of each gene term (GO annotation), and then hypergeometric tests were applied to detect GO enrichment analysis of functional significance in DEGs. The calculating formula is
| (2) |
where N is number of genes with annotation, n is the number of differently expressed genes in N, M is the number of genes that are annotated to the certain GO term, and m is the number of DEGs in M.
The Kyoto Encyclopedia of Genes and Genomes (KEGG), the major public pathway-related database, was used in the pathway enrichment analysis to identify significantly enriched metabolic pathways or signal transduction pathways in DEGs compared with the whole transcriptome background. The calculating formula is the same as that in the GO analysis: N is the number of all genes with KEGG annotation, n is the number of DEGs in N, M is the number of all genes annotated to the specific pathways, and m is the number of DEGs in M. The Q value of a test measures the proportion of false positives incurred (i.e., false discovery rate) when that particular test is called significant (http://genomics.princeton.edu/storeylab/qvalue/). Pathways with Q value ≤0.05 are significantly enriched in DEGs.
3. Results
3.1. Summary of RNA-Sequencing Data Sets
To obtain a dynamic view of the gene expression profiles of barley powdery mildew resistance at different infection progress stages, six cDNA samples were prepared from barley leaves at 0 h, 24 h, 48 h, 72 h, 96 h, and 120 h after infection. And then these samples were subjected to the Illumina sequencing platform. In total, we acquired more than 42.90 G raw reads over six time points (Table 1). After cleaning the reads with the proportion of N over 10%, over half of proportion of base quality Q less than 5 bases, and the adapter polluted reads, approximately 39.80 G clean reads were collected, with 96.25% of the Q 30 bases (base quality over 30). The average data of each sample was approximately 6.64 G in size. The following data analysis procedures were based on the modified reads.
Table 1.
RNA-sequencing data filtering analysis.
| Library | A* | B | C | D | E | F | Average | Total |
|---|---|---|---|---|---|---|---|---|
| Original reads number (G) | 7.17 | 6.40 | 6.88 | 7.79 | 7.14 | 7.51 | 7.15 | 42.90 |
| Modified reads number (G) | 6.66 | 5.92 | 6.38 | 7.24 | 6.61 | 7.00 | 6.64 | 39.83 |
| Modified Q30 bases rate (%) | 96.22 | 96.21 | 96.27 | 96.19 | 96.2 | 96.41 | 96.25 | — |
| Mapped rate (%) | 86.34 | 86.21 | 86.99 | 86.82 | 86.89 | 86.55 | 86.63 | — |
| Multimap rate (%) | 12.82 | 9.82 | 14.03 | 12.59 | 12.81 | 14.13 | 12.7 | — |
*A–F: the samples collected at 0 h, 24 h, 48 h, 72 h, 96 h, and 120 h after infection.
3.2. Evaluation of the Sequencing Data Quality
To assess the quality and coverage of the sequencing data, mean quality distribution and base distribution were analyzed. Sequencing error rate is not only related to base quality but also influenced by sequencer, reagent, sample, and so forth. Each base sequencing error can be judged by Q phred (Phred score), which is given by a model of prediction base judging error probability during Base Calling. The sequencing base mean quality distributions of six samples were similar to each other. For example, the mean quality distribution of Sample A (the sample at 0 h after infection, namely, TR130348) was illustrated in Supplementary Figure 2. The base position in reads is aligned as the x-axis and the mean Q phred as the y-axis. High proportion of Q 30 reads indicted high-quality sequence.
Base distributions of all samples were also similar and that in Sample A, as an example, was illustrated in Supplementary Figure 3. The base position in reads is aligned as the x-axis and the percentage of ATGC base as the y-axis. In general, the equal proportions of bases between T and A and between G and C were found, indicating no preference during sequencing. The GC percentage of each sample accounted for approximately 54% of the total.
3.3. Mapping Reads Coverage
The mapping results were listed in Table 1. Each of the samples had the mapped read rates greater than 86%, which indicates that most sequencing data are consistent with the reference genome of barley.
Based on the mapped reads, the proportion of exon mapping, intron mapping, and intergene mapping of sample A at 0 h since infection 5 were illustrated in Figure 1. The highest exon mapping (60.3%) was found in Sample A, while the lowest (52.1%) was found in Sample F (C120, TR130353). The intron mapping coverage ranged from 8.3% (A) to 10.8% (B, C24, TR130349). The average of intergene mapping was 35.2%. There was no affinity with reference genome annotation.
Figure 1.
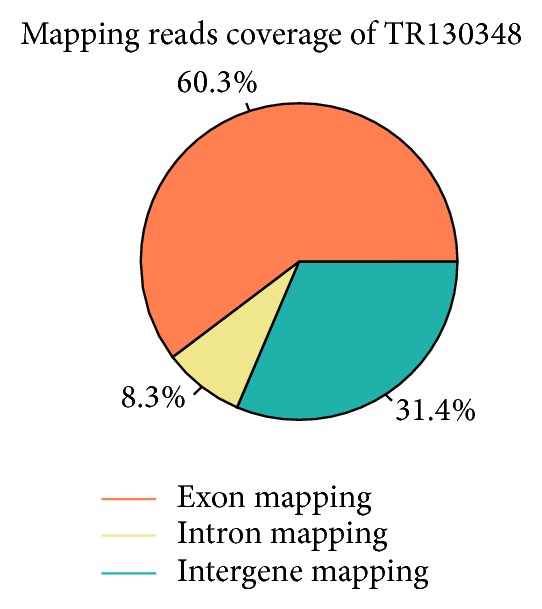
Mapping reads (exon, intron, and intergene) coverage of A (C0, TR130348).
In order to detect the depth of bases, exon gene was divided into 100 parts. The relative positions of genes are aligned as x-axis, and the number of reads is aligned as y-axis. The line charts of all samples were similar to each other, and Sample A was illustrated in Supplementary Figure 4. There was a little preference of gene exon to the base depth.
3.4. Gene Expressions
Gene saturation of each sample was also similar, and Sample A was illustrated in Supplementary Figure 5. Comparisons of gene expressions in each subsample to the whole sample showed less than 15% of the difference between them, indicating fine expression genes in the current size of sequencing data. The results represented an accurate dataset to detect highly expressed genes. The distribution density of gene global expressions of each sample was illustrated in Figure 2, which exhibits similar expressed gene distributions among these samples. Cuffdiff software (http://cufflinks.cbcb.umd.edu/index.html) was used to compare the gene expressions between sample pairs. Differently expressed genes were identified based on genes with q < 0.05 (q is the corrected P value). The results were listed in Supplementary Table 2. Total DEGs from each sample were clustered in Supplementary Figure 6. The first subgroup contained only A, the second one included B and C (C48, TR130350), and the last one covered the rest samples of D (C72, TR130351), E (C96, TR130352), and F. Euclidean distance was used to estimate the distance of gene expression between sample pairs. The clusters of A and B were illustrated in Figure 3. In the histogram, the red color indicates upregulation and the green color downregulation. Compared with Sample A, gene expressions of B and C were significantly different. There were similar distributions between A and D, between A and E, and between A and F.
Figure 2.
Distributed density of gene global expression of each sample.
Figure 3.

Euclidean distance was used to establish the distance of expression between A (C0, TR130348) and B (C24, TR130349).
3.5. Functional Classification by GO
GO is an international standardized gene functional classification system which offers a dynamic-updated controlled vocabulary and a strictly defined concept to comprehensively describe the properties of genes and their products in any organism. GO has three ontologies: molecular function, cellular component, and biological process. In total, 39,197 reads with BLASTx matches to known proteins were assigned to gene ontology classes, with 2,654 functional terms. Of them, assignments to the biological process made up the majority (1344, 50.64%), followed by molecular function (1060, 39.94%) and cellular component (250, 9.42%). These functional classifications by GO were summarized in Table 2. Comparison of GO classification between A and B was presented in Figure 4.
Table 2.
Functional classification by GO and KEGG.
| Category | Q-value | Function |
|---|---|---|
| A_B cellular component | ||
| GO:0044464 | 0.03867023 | Cell part |
| GO:0005576 | 3.05E − 06 | Extracellular region |
| GO:0044436 | 0.02673204 | Thylakoid part |
| GO:0009521 | 0.02559572 | Photosystem |
| GO:0009523 | 0.01277032 | Photosystem II |
| GO:0009654 | 0.00933201 | Oxygen evolving complex |
| GO:0019898 | 0.00908508 | Extrinsic to membrane |
| B_E cellular component | ||
| GO:0005576 | 0.04652511 | Extracellular region |
| A_C molecular function | ||
| GO:0004519 | 0.01846397 | Endonuclease activity |
| GO:0004540 | 0.00563478 | Ribonuclease activity |
| GO:0004521 | 0.00325892 | Endoribonuclease activity |
| GO:0016894 | 2.10E − 05 | Endonuclease activity, active with either ribo- or deoxyribonucleic acids and producing 3′-phosphomonoesters |
| GO:0016892 | 2.10E − 05 | Endoribonuclease activity, producing 3′-phosphomonoesters |
| GO:0033897 | 2.10E − 05 | Ribonuclease T2 activity |
| A_E molecular function | ||
| GO:0016894 | 0.04079433 | Endonuclease activity, active with either ribo- or deoxyribonucleic acids and producing 3′-phosphomonoesters |
| GO:0016892 | 0.04079433 | Endoribonuclease activity, producing 3′-phosphomonoesters |
| GO:0033897 | 0.04079433 | Ribonuclease T2 activity |
| C_E molecular function | ||
| GO:0043531 | 0.01381725 | ADP binding |
| B_E biological process | ||
| GO:0006091 | 0.01070069 | Generation of precursor metabolites and energy |
| GO:0009765 | 4.88E − 05 | Photosynthesis, light harvesting |
| B_F biological process | ||
| GO:0009765 | 0.00971227 | Photosynthesis, light harvesting |
| C_E biological process | ||
| GO:0006952 | 0.00970112 | Defense response |
| GO:0006950 | 0.00970112 | Response to stress |
| A_B kegg | ||
| map00195 | 0.0422721 | Photosynthesis |
| A_C kegg | ||
| map04626 | 0.00085699 | Plant-pathogen interaction |
| B_E kegg | ||
| map00196 | 6.56E − 05 | Photosynthesis-antenna proteins |
| B_F kegg | ||
| map00196 | 0.00126333 | Photosynthesis-antenna proteins |
| C_E kegg | ||
| map04626 | 0.00751478 | Plant-pathogen interaction |
| map00196 | 0.04943251 | Photosynthesis-antenna proteins |
Figure 4.

Histogram presentation of gene ontology classification between A (C0, TR130348) and B (C24, TR130349). The results are summarized in three main categories: biological process, cellular component, and molecular function. The right y-axis indicates the number of genes in a category. The left y-axis indicates the percentage of a specific category of genes in that main category.
A total of 831 significant DEGs were found by Cufflink. The assigned functions of these genes covered a broad range of GO categories. Under the cellular component category, cell, cell part, and extracellular region, including “thylakoid part,” “photosystem,” and “photosystem II,” were prominently represented, indicating that some powdery mildew-related metabolic activities of photosynthesis occurred in the leaf of the Tibetan barley landrace. Interestingly, many genes were assigned to “oxygen evolving complex.” It was also noteworthy that a large number of genes were involved in “extrinsic to membrane.” Under the category of molecular function, binding and catalysis, including “ADP binding” and “auxiliary transport protein,” represented the majorities of the category. Among the genes assigned to auxiliary transport protein, “endonuclease activity” represented the most abundant classification, followed by “ribonuclease activity,” “endoribonuclease activity,” and “ribonuclease T2 activity.” For the biological process category, many genes were classified into the metabolic process and cellular process, including “photosynthesis, light harvesting,” whereas only a few genes were assigned to “defense response,” “response to stress,” and “generation of precursor metabolites and energy.”
3.6. Functional Classification by KEGG
KEGG is a public database recording the networks of molecular interactions in the cells and variants of them specific to particular organisms. Pathway-based analysis helps to further understand the biological functions and interactions of genes. First, based on comparison with the KEGG database using BLASTx with an E-value cut-off of <10−5, 330 KEGG pathways were detected. Among them, three pathways, that is, “photosynthesis,” “plant-pathogen interaction,” and “photosynthesis-antenna proteins,” had significant matches in the database. As shown in Table 2, the “photosynthesis” pathway became distinct at the stage of 24 h after infection, the “plant-pathogen interaction” pathway also differed significantly at the time, and the “photosynthesis-antenna proteins” and “photosynthesis” pathway was remarkable 96 h after infection. These results indicated a dynamic and complex process of barley response to powdery mildew.
4. Discussion
4.1. Illumina Paired End Sequencing and Assembly
In this study, the mRNA of the barley plants infected with powdery mildew pathogen was sequenced using Illumina Genome Analyzer, with Sera-Mag Magnetic Oligo (dT) Beads. A clear bioinformatic map of mRNA involved in multiple biological processes was produced. As a result, 42.9 G data was collected from six samples over infection time. After filtering, the average data size of each sample was 6.64 G, and the reads number was 66.38 M, which met the requirements for further analysis. Saturability analysis indicated a qualified coverage of most genes based on our data size. In addition, the clean reads of Q 30 occupied over 95% of the total, suggesting high-quality sequencing.
TopHat package was used to blast the transcriptome data to the reference genome. It has been found that 86% of the reads were mapped to the reference genome. Multiblasted reads were greater than 10%, which might suggest that they were repeatable in this species. Further analysis of the mapping reads showed that the average of intergene mapping reads was more than 30%, which might be due to inadequate annotation of the genome, as reported by Luo et al. [26].
4.2. Functional Annotation of DEGs
On the basis of extensive examination of the DEGs between samples, 831 significant DEGs were found across nineteen functions. These functions were related to cell, cell part, and extracellular region in the cellular component category, binding and catalytic in the category of molecular function, and metabolic process and cellular process in the biological process category. This indicated that these functions were likely involved in powdery mildew-resistant hulless barley. Hulbert et al. [27] summarised that the powdery mildew resistance genes carry motifs found in other receptor and signal transduction proteins, such as nucleotide-binding site domains and kinase domains. Active oxygen in some species has been found to play a number of critical roles in defence responses during plant-pathogen interactions [28–30]. Warren et al. [31] reported that it functioned in defense response signaling of an Arabidopsis mutation since it interfered with resistance conferred by several other nucleotide-binding site genes. The cellular components and processes reflect where resistance genes interact with their corresponding elicitors. The cell membrane and extracellular leucine-rich repeats indicated the association between transmembrane domain and the corresponding kinase [32–34]. The observed interaction with intracellular resistance genes products should stimulate researches into how these diverse organisms deliver elicitors into plant cells.
Furthermore, KEGG was used to annotate the DEGs by enrichment analysis and revealed the significant pathways involved in the disease resistance. Three pathways occurred in different stages: the infection firstly acted on “photosynthesis” of leaves and then caused “pathogen recognition interaction” and defense response signaling and finally affected “photosynthesis-antenna proteins.” This event sequence exhibited a dynamic process of barley responding to powdery mildew. Once the recognition of pathogen occurred, the defense responses were triggered. These are often characterized as a hypersensitive response, which involves the death of the first cell or cells infected and the local accumulation of antimicrobial compounds [35].
KEGG analysis revealed that resistance gene action was coupled to a complex series of biochemical defense pathways. It is therefore more likely that resistance genes may function together in recognizing pathogen elicitors, possibly as coreceptors. The similarity in structure of the tomato Cf proteins [36] to the rice Xa21 protein [37] implies that the transmembrane domain genes may also include a kinase in their defense-signaling pathway. Mla resistance protein, containing recognition complexes, may be activated by RAR1/SGT1 (two conserved-interacting proteins in mutants of barley Rar1) [38]. Mlo resistance genes were triggered by a rapid formation of enlarged cell wall appositions below the fungus's encounter sites and of a physical and chemical barrier that the infection peg can rarely penetrate [9]. Mlo allele encodes a putative membrane protein, which may be a negative regulator of certain defense responses [39], whereas Ror (required for mlo-specified resistance) genes act as positive regulators of a non-race-specific resistance response [40]. Barley lines that are homozygous for the nonfunctional alleles show spontaneous defense responses like cell wall appositions in the epidermal cells and even some cell death [41]. Piffanelli et al. [42] inferred that the CIS- (cytokine-induced SH2-containing protein-) dependent perturbation of transcription machinery assembly by transcriptional interference in Mlo-11 plants is a likely mechanism leading to disease resistance.
5. Conclusions
This work presents a first report of the transcriptome sequencing of the Tibetan barley landrace with powdery mildew resistance and brings a major genomic resource for barley resistance to this disease. A large number of genes in the hulless barley were characterized by DEG analysis using Illumina sequencing technology. The transcriptome and DEG analyses also provided us with a genome-wide view of the transcriptional mechanisms to improve genome annotation and enabled us to understand some related biological progress of hulless barley disease resistance. The data in this study is consistent with those using multiple approaches including QTL mapping and FISH, indicating the reliability of the results from the mRNA-Seq and DEG analysis. Therefore, further work is needed to find additional linked DNA markers for these DEGs. It is necessary to develop new, reliable, PCR-based markers tightly linked to the resistance genes and this will greatly facilitate gene transfer into currently used varieties.
Supplementary Material
Two supplementary tables and six figures are available online at http://dx.doi.org/10.1155/2014/594579. Sample information is illustrated in table 1 and figure 1. The evaluation of sequence data is illustrated in figures 2-5. Different gene expression (DGE) is illustrated in table 2 and figure 6.
Acknowledgments
This work was supported by Special Funds for Preliminary Research of 973 Plan (2012CB723006), China National Scientific and Technological Support Plan (2012BAD03B01), and Specific Financial Funds in Tibet Autonomous Region (2011XZCZZX001).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors’ Contribution
Xing-Quan Zeng and Xiao-Mei Luo contributed equally to this work.
References
- 1.Bhatty R. S. β-glucan and flour yield of hull-less barley. Cereal Chemistry. 1999;76(2):314–315. doi: 10.1094/cchem.1999.76.2.314. [DOI] [Google Scholar]
- 2.McDonald B. A., Linde C. The population genetics of plant pathogens and breeding strategies for durable resistance. Euphytica. 2002;124(2):163–180. doi: 10.1023/a:1015678432355. [DOI] [Google Scholar]
- 3.Fischbeck G., Jahoor A. The transfer of genes for mildew resistance from Hordeum spontaneum . In: Jørgensen J. H., editor. Integrated Control of Cereal Mildews: Virulence Patterns and Their Change. Roskilde, Denmark: Risø National Laboratory; 1991. pp. 247–255. http://agris.fao.org/agris-search/search.do?recordID=DK9420942. [Google Scholar]
- 4.Jørgensen J. H., Wolfe P. M. Genetics of powdery mildew resistance in barley. Critical Reviews in Plant Sciences. 1994;13(1):97–119. doi: 10.1080/07352689409701910. [DOI] [Google Scholar]
- 5.Helms Jørgensen J., Jensen H. P. Powdery mildew resistance in barley landrace material. I. Screening for resistance. Euphytica. 1997;97(2):227–233. doi: 10.1023/A:1003032424968. [DOI] [Google Scholar]
- 6.Silvar C., Dhif H., Igartua E., Kopahnke D., Gracia M. P., Lasa J. M., Ordon F., Casas A. M. Identification of quantitative trait loci for resistance to powdery mildew in a Spanish barley landrace. Molecular Breeding. 2010;25(4):581–592. doi: 10.1007/s11032-009-9354-z. [DOI] [Google Scholar]
- 7.Silvar C., Casas A. M., Kopahnke D., Habekuß A., Schweizer G., Gracia M. P., Lasa J. M., Ciudad F. J., Molina-Cano J. L., Igartua E., Ordon F. Screening the Spanish barley core collection for disease resistance. Plant Breeding. 2010;129(1):45–52. doi: 10.1111/j.1439-0523.2009.01700.x. [DOI] [Google Scholar]
- 8.Wei F., Gobelman-Werner K., Morroll S. M., et al. The Mla (powdery mildew) resistance cluster is associated with three NBS-LRR gene families and suppressed recombination within a 240-kb DNA interval on chromosome 5S (1HS) of barley. Genetics. 1999;153(4):1929–1948. doi: 10.1093/genetics/153.4.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jørgensen I. H. Discovery, characterization and exploitation of Mlo powdery mildew resistance in barley. Euphytica. 1992;63(1-2):141–152. doi: 10.1007/bf00023919. [DOI] [Google Scholar]
- 10.Falak I., Falk D. E. Doubled haploids as a tool for studying resistance to powdery mildew (Erysiphe graminis f. sp. hordei) Barley Newsletter. 1993;36, article 208 [Google Scholar]
- 11.Pickering R. A., Hill A. M., Michel M., Timmerman-Vaughan G. M. The transfer of a powdery mildew resistance gene from Hordeum bulbosum L to barley (H. vulgare L.) chromosome 2 (2I) Theoretical and Applied Genetics. 1995;91(8):1288–1292. doi: 10.1007/bf00220943. [DOI] [PubMed] [Google Scholar]
- 12.Schönfeld M., Ragni A., Fischbeck G., Jahoor A. RFLP mapping of three new loci for resistance genes to powdery mildew (Erysiphe graminis f. sp. hordei) in barley. Theoretical and Applied Genetics. 1996;93(1-2):48–56. doi: 10.1007/s001220050246. [DOI] [PubMed] [Google Scholar]
- 13.Barakat A., Diloreto D. S., Zhang Y., Smith C., Baier K., Powell W. A., Wheeler N., Sederoff R. Comparison of the transcriptomes of American chestnut (Castanea dentata) and Chinese chestnut (Castanea mollissima) in response to the chestnut blight infection. BMC Plant Biology. 2009;9, article 51 doi: 10.1186/1471-2229-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J., Wang W., Li R., et al. The diploid genome sequence of an Asian individual. Nature. 2008;456(7218):60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parchman T. L., Geist K. S., Grahnen J. A., Benkman C. W., Buerkle C. A. Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery. BioMed Central Genomics. 2010;11(1, article 180) doi: 10.1186/1471-2164-11-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagalakshmi U., Wang Z., Waern K., Shou C., Raha D., Gerstein M., Snyder M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320(5881):1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilhelm B. T., Marguerat S., Watt S., Schubert F., Wood V., Goodhead I., Penkett C. J., Rogers J., Bähler J. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 2008;453(7199):1239–1243. doi: 10.1038/nature07002. [DOI] [PubMed] [Google Scholar]
- 18.Wang Z., Fang B., Chen J., Zhang X., Luo Z., Huang L., Chen X., Li Y. De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweet potato (Ipomoea batatas) BMC Genomics. 2010;11(1, article 726) doi: 10.1186/1471-2164-11-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang G., Guo G., Hu X., Zhang Y., Li Q., Li R., Zhuang R., Lu Z., He Z., Fang X., Chen L., Tian W., Tao Y., Kristiansen K., Zhang X., Li S., Yang H., Wang J., Wang J. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Research. 2010;20(5):646–654. doi: 10.1101/gr.100677.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hao D. C., Ge G., Xiao P., Zhang Y., Yang L. The first insight into the tissue specific taxus transcriptome via illumina second generation sequencing. PLoS ONE. 2011;6(6) doi: 10.1371/journal.pone.0021220.e21220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen S., Yang P., Jiang F., Wei Y., Ma Z., Kang L. De Novo analysis of transcriptome dynamics in the migratory locust during the development of phase traits. PLoS ONE. 2010;5(12) doi: 10.1371/journal.pone.0015633.e15633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins L. J., Biggs P. J., Voelckel C., Joly S. An approach to transcriptome analysis of non-model organisms using short-read sequences. Genome Informatics. 2008;21:3–14. [PubMed] [Google Scholar]
- 23.Mortazavi A., Williams B. A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 24.Audic S., Claverie J.-M. The significance of digital gene expression profiles. Genome Research. 1997;7(10):986–995. doi: 10.1101/gr.7.10.986. [DOI] [PubMed] [Google Scholar]
- 25.Conesa A., Götz S., García-Gómez J. M., Terol J., Talón M., Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 26.Luo X., Wight C. P., Zhou Y., Tinker N. A. Characterization of chromosome-specific genomic DNA from hexaploid oat. Genome. 2012;55(4):265–268. doi: 10.1139/g2012-011. [DOI] [PubMed] [Google Scholar]
- 27.Hulbert S. H., Webb C. A., Smith S. M., Sun Q. Resistance gene complexes: evolution and utilization. Annual Review of Phytopathology. 2001;39:285–312. doi: 10.1146/annurev.phyto.39.1.285. [DOI] [PubMed] [Google Scholar]
- 28.Sutherland M. W. The generation of oxygen radicals during host plant responses to infection. Physiological and Molecular Plant Pathology. 1991;39(2):79–93. doi: 10.1016/0885-5765(91)90020-i. [DOI] [Google Scholar]
- 29.Tzeng D. D., DeVay J. E. Role of oxygen radicals in plant disease development. In: Andrews J. H., Tommerup I. C., editors. Advance in Plant Pathology. Vol. 10. New York, NY, USA: Academic Press; 1993. pp. 1–34. [Google Scholar]
- 30.Baker C. J., Orlandi E. W. Active oxygen in plant pathogenesis. Annual Review of Phytopathology. 1995;33:299–321. doi: 10.1146/annurev.py.33.090195.001503. [DOI] [PubMed] [Google Scholar]
- 31.Warren S. G., Brandt R. E., Hinton P. O. Effect of surface roughness on bidirectional reflectance of Antarctic snow. Journal of Geophysical Research E: Planets. 1998;103(11):25789–25807. doi: 10.1029/98je01898. [DOI] [Google Scholar]
- 32.van den Ackerveken G. F., van Kan J. A. L., de Wit P. J. G. M. Molecular analysis of the avirulence gene avr9 of the fungal tomato pathogen Cladosporium fulvum fully supports the gene-for-gene hypothesis. Plant Journal. 1992;2(3):359–366. doi: 10.1111/j.1365-313x.1992.00359.x. [DOI] [PubMed] [Google Scholar]
- 33.Joosten M. H. A., Cozijnsen T. J., de Wit P. J. Host resistance to a fungal tomato pathogen lost by a single base-pair change in an avirulence gene. Nature. 1994;367(6461):384–386. doi: 10.1038/367384a0. [DOI] [PubMed] [Google Scholar]
- 34.Botella M. A., Parker J. E., Frost L. N., Bittner-Eddy P. D., Beynon J. L., Daniels M. J., Holub E. B., Jones J. D. G. Three genes of the Arabidopsis RPP1 complex resistance locus recognize distinct Peronospora parasitica avirulence determinants. Plant Cell. 1998;10(11):1847–1860. doi: 10.1105/tpc.10.11.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones J. D. G., Dangl J. L. The plant immune system. Nature. 2006;444(7117):323–329. doi: 10.1038/nature05286. [DOI] [PubMed] [Google Scholar]
- 36.Dixon M. S., Jones D. A., Keddie J. S., Thomas C. M., Harrison K., Jones J. D. G. The tomato Cf-2 disease resistance locus comprises two functional genes encoding leucine-rich repeat proteins. Cell. 1996;84(3):451–459. doi: 10.1016/s0092-8674(00)81290-8. [DOI] [PubMed] [Google Scholar]
- 37.Song W.-Y., Pi L.-Y., Wang G.-L., Gardner J., Holsten T., Ronald P. C. Evolution of the rice Xa21 disease resistance gene family. The Plant Cell. 1997;9(8):1279–1287. doi: 10.1105/tpc.9.8.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shen Q.-H., Zhou F., Bieri S., Haizel T., Shirasu K., Schulze-Lefert P. Recognition specificity and RAR1/SGT1 dependence in barley Mla disease resistance genes to the powdery mildew fungus. The Plant Cell. 2003;15(3):732–744. doi: 10.1105/tpc.009258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Büschges R., Hollricher K., Panstruga R., Simons G., Wolter M., Frijters A., Van Daelen R., Van der Lee T., Diergaarde P., Groenendijk J., Töpsch S., Vos P., Salamini F., Schulze-Lefert P. The barley Mlo gene: a novel control element of plant pathogen resistance. Cell. 1997;88(5):695–705. doi: 10.1016/s0092-8674(00)81912-1. [DOI] [PubMed] [Google Scholar]
- 40.Freialdenhoven A., Peterhänsel C., Kurth J., Kreuzaler F., Schulze-Lefert P. Identification of genes required for the function of non-race-specific mlo resistance to powdery mildew in barley. Plant Cell. 1996;8(1):5–14. doi: 10.1105/tpc.8.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wolter M., Hollricher K., Salamini F., Schulze-Lefert P. The mlo resistance alleles to powdery mildew infection in barley trigger a developmentally controlled defence mimic phenotype. Molecular and General Genetics. 1993;239(1-2):122–128. doi: 10.1007/BF00281610. [DOI] [PubMed] [Google Scholar]
- 42.Piffanelli P., Ramsay L., Waugh R., Benabdelmouna A., D'Hont A., Hollricher K., Jørgensen J. H., Schulze-Lefert P., Panstruga R. A barley cultivation-associated polymorphism conveys resistance to powdery mildew. Nature. 2004;430(7002):887–891. doi: 10.1038/nature02781. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Two supplementary tables and six figures are available online at http://dx.doi.org/10.1155/2014/594579. Sample information is illustrated in table 1 and figure 1. The evaluation of sequence data is illustrated in figures 2-5. Different gene expression (DGE) is illustrated in table 2 and figure 6.