

Abstract
Background
Oxidative mitochondrial damage is closely linked to inflammation and to cell death, but low levels of reactive oxygen and nitrogen species serve as signals that involve mitochondrial repair and resolution of inflammation. More specifically, cytoprotection relies on the elimination of damaged mitochondria by selective autophagy (mitophagy) during mitochondrial quality control.
Objective
To identify and localize mitophagy in mouse lung as a potentially up-regulatable redox response to S. aureus sepsis.
Methods
Anesthetized C57BL/6 and B6.129X1-Nfe2l2tm1Ywk/J (Nrf2−/−) mice had fibrin clots loaded with S. aureus (1×107 CFU) implanted abdominally. At the time of implantation, mice were given Vancomycin (6mg/kg) and fluid resuscitation. Mouse lungs were harvested at 0, 6, 24, and 48 hours for bronchoalveolar lavage (BAL), Western blot analysis and qRT-PCR. To localize mitochondria with autophagy protein LC3, we used lung immunofluorescence staining in LC3-GFP transgenic mice.
Results
In C57BL/6 mice, sepsis-induced pulmonary inflammation was detected by significant increases in mRNA for the inflammatory markers IL-1β and TNF-α at 6h and 24h respectively hours. BAL cell count and protein increased. Sepsis suppressed lung Beclin-1 protein, but not mRNA, suggesting activation of canonical autophagy. Notably sepsis also increased the LC3-II autophagosome marker, as well as the lung’s non-canonical autophagy pathway as evidenced by loss of p62, a redox-regulated scaffolding protein of the autophagosome. In LC3-GFP mice lungs, immunofluorescence staining showed co-localization of LC3-II to mitochondria, mainly in Type 2 epithelium and alveolar macrophages. In contrast, marked accumulation of p62, as well as attenuation of LC3-II in Nrf2 KO mice supported an overall decrease in autophagic turnover.
Conclusions
The down-regulation of canonical autophagy during sepsis may contribute to lung inflammation while the switch to non-canonical autophagy selectively removes damaged mitochondria and accompanies tissue repair and cell survival. Furthermore, mitophagy in the alveolar region appears to depend on activation of Nrf2. Thus, efforts to promote mitophagy may be a useful therapeutic adjunct for acute lung injury in sepsis.
Keywords: Acute lung injury, sepsis, autophagy, mitochondria, oxidative stress, Nrf2, inflammation, p62, LC3, mitophagy
INTRODUCTION
Sepsis is now the leading cause of death in critically ill patients [1] and 40% of these patients develop acute lung injury (ALI), which is characterized by inflammatory cell infiltration and loss of type I alveolar epithelium. This leads to pulmonary capillary leak and acute respiratory failure [2]. ALI is also accompanied by severe inflammation and associated with oxidative stress caused by excessive reactive oxygen and nitrogen species (ROS/RNS) production [3]. ROS/RNS at low levels serve as signaling molecules to activate redox-sensitive genes [4], but uncontrolled ROS generation eventually overwhelms cells, causing structural damage, particularly to mitochondria. More specifically, this excessive ROS production directly inhibits oxidative phosphorylation[5]. Furthermore, loss of mitochondrial function results in epithelial apoptosis causing epithelial barrier breakdown and ALI [6]. The resolution of mitochondrial oxidative stress is linked directly to survival in severe sepsis [7].
Mitochondrial-derived reactive oxygen species (mROS) are produced normally as byproducts of oxidative phosphorylation; however, inflammation leads to excessive mROS efflux that damages mitochondria [8],[9]. Damaged mitochondria are a primary source of cellular oxidative stress. In response, nuclear-encoded mitochondrial gene expression is induced in order to match mitochondrial function with cellular energy demands [10]. Moreover, oxidative mitochondrial damage results in activation of genes that contain antioxidant response elements (ARE) that contribute to both mitochondrial antioxidant defense and mitochondrial quality control [11].
Mitochondrial quality control during oxidative stress requires a key transcription factor, Nrf2 (nuclear factor E2-related factor 2), which is responsible for the expression of SOD2 and more than 100 other cytoprotective genes [12]. Nfe2l2/Nrf2 is part of the major cellular defenses against oxidative stress after nuclear localization and transcription of ARE-containing genes by small Maf proteins [13,14]. Nrf2 is normally sequestered in the cytosol by Keap1 (Kelch-like ECH-associating protein 1), which results in its ubiquination and degradation by the 26S proteasome [15]. During oxidant stress, cysteine residues on Keap1 are oxidized, and allows Nrf2 to translocate to the nucleus and to transcribe ARE-containing genes including selected mitochondrial quality control genes [16].
Mitochondrial quality control also involves the clearance of irreparably damaged mitochondria via the process of selective macroautophagy or mitophagy [17]. These damaged mitochondria are segregated from the functional mitochondrial network, engulfed in autophagosomes, and then degraded by lysosomal proteases [18].
Current literature on mitophagy has suggested that autophagy may select mitochondria for removal through either Atg-dependent or -independent pathways [19,20]. Mitophagy is a normal physiological processes [21], but also found in aging [22], as well as in pathophysiological states such as sepsis [23]. Mitophagy in sepsis has been studied previously in the liver [24,25], and autophagy occurs in the lung in ALI [26], but mitophagy has not been characterized in the lung in sepsis.
The purpose of this investigation was to test the hypothesis that S. aureus sepsis in murine peritonitis induces sufficient oxidative stress to damage parenchymal lung mitochondria and that these mitochondria are removed by mitophagy, which in conjunction with mitochondrial biogenesis, allows restoration of the lung’s oxidative balance during sepsis resolution.
METHODS
Materials
Antibodies were purchased as follows: HO-1 (Assay Designs), Beclin-1 (Cell Signaling), Nrf2 (Santa Cruz), p62 (Abcam), LC3 (Sigma-Aldrich), SOD-2 (Abcam), 8-OhdG (Genetex). Primers used for quantitative real-time reverse transcriptase polymerase chain reaction (qRT-PCR) were all obtained from Life Technologies (Atg5, Atg12, Beclin-1, IL-1β, TNF-α, SOD-2, HO-1). Bafilomycin A1 (BFA) was purchased from LC Laboratories.
ssDNA probe
The ssDNA probe used was obtained from Millipore (MAB3299) and has been used in many previous experiments in multiple tissue types to detect apoptotic cells by immunohistochemistry [27–30].
Mice
The mouse studies were preapproved by the Duke IAUCUC. C57Bl6/J (WT) as well as B6.129X1-Nfe2l2tm1Ywk/J (Nrf2−/−) mice were obtained from Jackson Laboratory (Bar Harbor, ME). S. aureus (clinical isolate; ATCC#25923) was prepared, counted, and embedded in fibrin clots before being implanted surgically in the abdomen [7]. Wild-type C57BL/6 mice received 1×107 CFU while Nrf2−/− mice received 5×106 CFU due to their increased sensitivity to sepsis. Mouse lungs were harvested at indicated times for protein and RNA and stored at −80°C.
LC3-GFP reporter mice
The mice were purchased from the RIKEN bio-resource center in Japan (RBRC00806). A transgenic vector containing an enhanced GFP-LC3 was inserted between a CAG promoter and the SV40 late polyadenylation signal. The GFP is fused to the N-terminus to not affect PE conjugation [31]. These mice were also implanted with infected abdominal clots and the lungs were harvested and inflation-fixed in 10% formalin for immunofluorescence microscopy.
Real-time Polymerase Chain Reaction
qRT-PCR was performed with TaqMan primers on StepOnePlus (Applied Biosystems). 18s rRNA was used as endogenous control. Gene expression was determined by relative quantification.
Protein Methods
Fresh lung tissue was homogenized in RIPA buffer and then sonicated on ice. The samples were then placed in a Laemelli buffer. Protein content was measured with bicinchoninic acid using BSA standards. For Western blotting, protein was resolved by gradient and/or non-gradient sodium dodecyl sulfate-polyacrylamide gel electrophoresis (4–20% or 8%), transferred to Immobilion P, blocked with 5% nonfat milk and incubated at 4°C overnight with polyclonal anti-HO-1 (Enzo, ADI-SPA-895-D), Beclin-1 (Cell Signaling 3738S), Nrf2, p62 (Abcam, ab91526), or LC3α/β (all 1:1000). The membranes were washed in TBST and incubated with HRP-conjugated secondary antibodies (Santa Cruz at 1:5000). The membranes were rewashed and developed with chemiluminescence reagents. Digitized images were quantified in the mid-dynamic range using ImageJ software. Quantified data was analyzed, and graphed and presented in GraphPad Prism 6.
Immunofluorescence Microscopy
Lung samples from WT and LC3-GFP were inflation-fixed in formalin and embedded in paraffin, cut into 5-μm, mounted on slides, and probed with anti-citrate synthase (GeneTex GTX110624 1:100) and anti-SOD2 (Abcam 13533 1:100). The slides were incubated in secondary goat anti-rabbit conjugated to Alexa 594 (Invitrogen) or Alexa 488 (Invitrogen). For single-stranded DNA (ssDNA), sections were deparaffinized and incubated in 50% formamide at 60°C for 30 min. Nonspecific binding to primary antibodies was blocked by incubation in 0.1% bovine serum albumin (BSA) for 15 min. Then the sections were incubated with anti-ssDNA polyclonal antibody (Dako, diluted at 1:100) overnight. An additional washing in 0.05 M Tris-buffered saline (TBS) followed, and sections were incubated with Alexa 594 –immunoglobulin M (IgM) diluted at 1:500 for 30 min. Sections were rinsed 3 times with 0.05 M TBS for 5 min. The nuclei were counterstained with DAPI purchased from Molecular Probes. The slides were observed and images captured under a Nikon Eclipse 50i fluorescence microscope.
For in situ oxidative damage studies, the anti-8Oh-dG antibody (Genetex GTX10802) was used. Alexa Fluor 488-labeled secondary antibodies were used to develop the signal for oxidized necleic acids. Photomicrographs were taken on a Nikon E100 (Nikon Instruments, Inc., Melville, NY)microscope at 400ope at 400s, Inc.of nuclei (DAPI blue) and or cytoplasmic stained (green) cells were counted per four field (n = 3 sections per mouse strain and treatment group).
TUNEL assay
Using the TUNEL (terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick-end labeling) method, lung sections were stained for the detection of fragmented DNA, which is indicative of apoptotic cells. Sections were incubated with TdT-reaction solution and nuclei were visualized using TUNEL reagents in a kit (Promega, Madison, WI) and DAPI nuclear stain. Fluorescence images were obtained on a Nikon E100 microscope. Quantification of TUNEL-positive cells was by determining the percentage of TUNEL-positive (red) cells in multiple high power fields (n = 3 sections per mouse strain and treatment group). Data are reported as mean percentage of TUNEL-positive cells per lung section ± SEM. Data are reported as mean percentage of positive cells per lung section ± SEM.
Statistical Analysis
Data for quantified protein densitometry and qRT-PCR were expressed as means ± SEM for 5 to 6 samples (n = 3 for Nrf2−/− mice). Significance was tested by two-way ANOVA and the Student’s t test. P<0.05 was accepted as significant.
Results
Pulmonary inflammation and oxidative stress during sepsis
To assess the pulmonary inflammatory responses to S. aureus peritonitis, we measured lung mRNA levels for the early phase pro-inflammatory cytokines IL-1β and TNF-α at 0, 6, 24 and 48h. The IL-1β and TNF-α mRNA levels increased sharply at 24h to 48h after clot implantation (Figure 1A&B). Cellular inflammation was also greatly increased as shown by significantly elevated BAL white blood cell count and BAL protein at 24h (Figure 1C&D). To indirectly measure levels of oxidative stress, we stained WT mouse lungs for 8-OhdG which is a well known measure of oxidative damage [32,33]. By 24h after clot implantation, there were significantly more cells that were 8-OhdG positive compared to 0h control mice lungs (Figure 2G). By immunofluorescence, ssDNA (red staining) colocalized with SOD2 (green staining) was visualized in WT mice and used to assess the amount of oxidative damage at 24h after clot implantation. Figure 2A&B indicates that there was significant cytosolic ssDNA staining after 24h in lungs of sepsis versus control mice, especially in mitochondria. We also assessed cell death as a measure of severity of the inflammatory insult. A TUNEL assay was performed and panels in Figure 2E&F were quantified. At 24h after clot implantation, there is significantly increased DNA fragmentation compared to 0h WT controls consistent with greater apoptosis.
Figure 1.
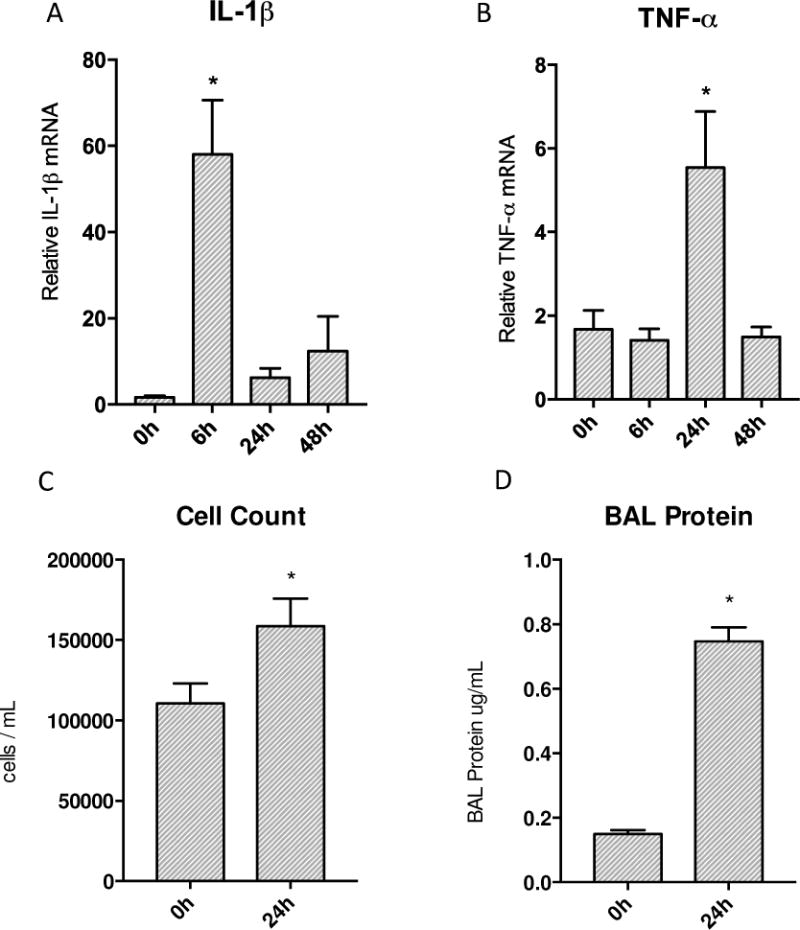
Transcription of inflammatory cytokines and end organ inflammation produced by S. aureus peritonitis. A&B) Transcription of IL-1β and TNF-α increased significantly post inoculation in the lung. IL-1β showed a robust increase at 6h then returned to baseline values while TNF-α increased significantly by 24h and then returned to base line afterwards. C&D) Significant increases seen in both cell counts and protein in bronchial alveolar lavage. (* P < 0.05 vs. time 0h)
Figure 2.
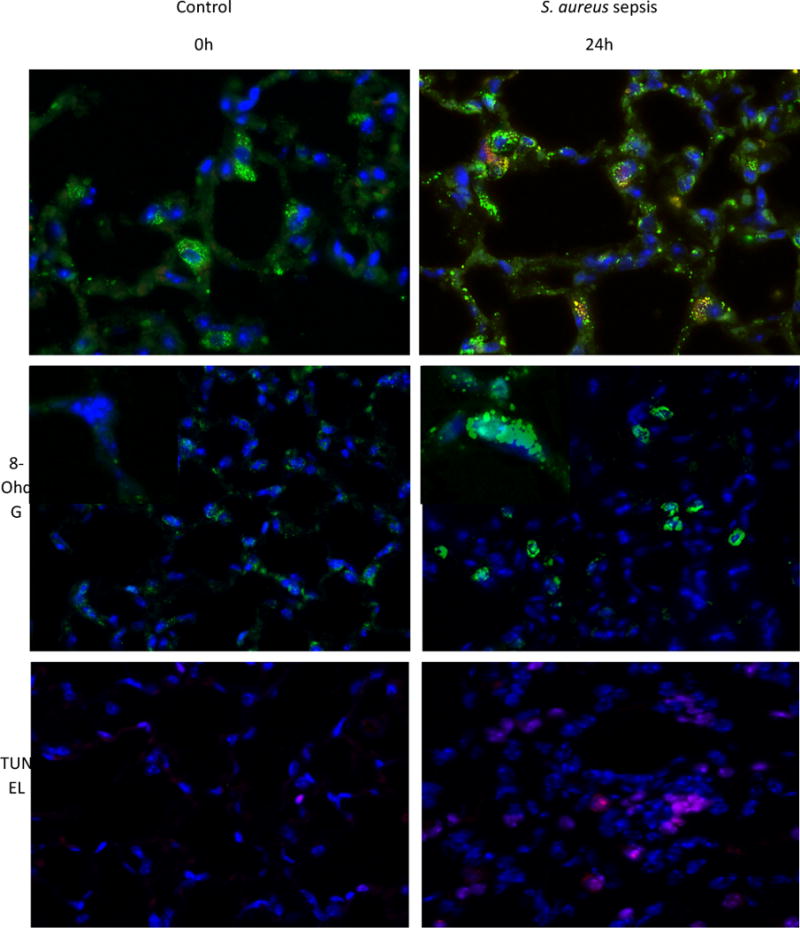
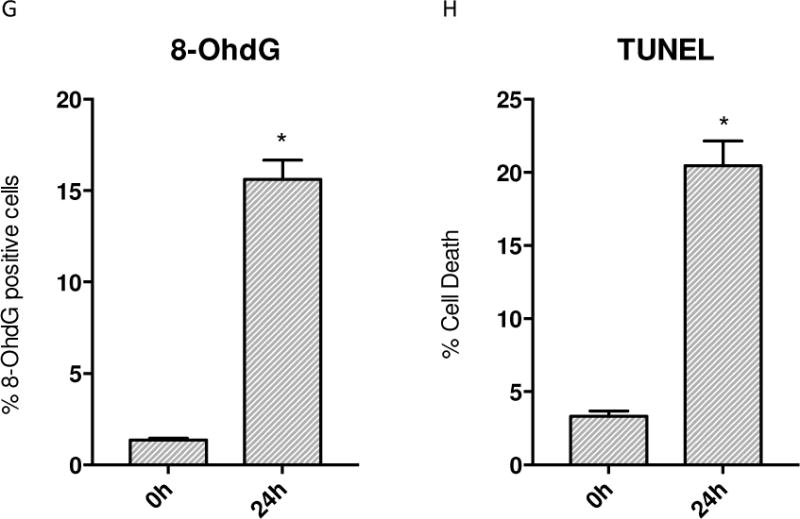
Immunofluorescence of WT mice given S. aureus 1×107 CFU at 24h stained for ssDNA. A) The control mouse shows low fluorescence of ssDNA (stained red) that is colocalized with SOD2 (green) B) Sepsis increases the amount of ssDNA present. C) Control WT mouse lung stained for 8-OhdG (green) D) WT mouse lung stained for 8-OhdG at 24h after clot implantation. E) Control WT mouse lung with TUNEL staining at 0h. F) WT mouse lung with TUNEL staining after 24h after clot implantation. G) Quantified panels C&D for 8-OhdG positive cells. H) Quantified data from panels E&F for presence of cell death. (* P < 0.05 vs. time 0h)
Lung inflammation induces a strong antioxidant response
To gauge the level of antioxidant response in the mouse lung during sepsis we measured SOD2 expression. SOD2 mRNA levels were heavily increased at 24h and showed a parallel significant elevation in protein levels by 48h (Figure 3A&B). The oxidative burden in the lung induced by S. aureus was determined by measuring Nfe2l2/Nrf2 and HO-1 proteins. Nrf2 was significantly elevated early at the protein level, rising almost 3 fold at 6h after clot implantation compared to 0h and then decreased to baseline levels by 24h onwards in Figure 3F. HO-1 protein showed significant up-regulation by 24h compared to 0h control levels, then returned to baseline by 48h as shown in Figure 3D.
Figure 3.
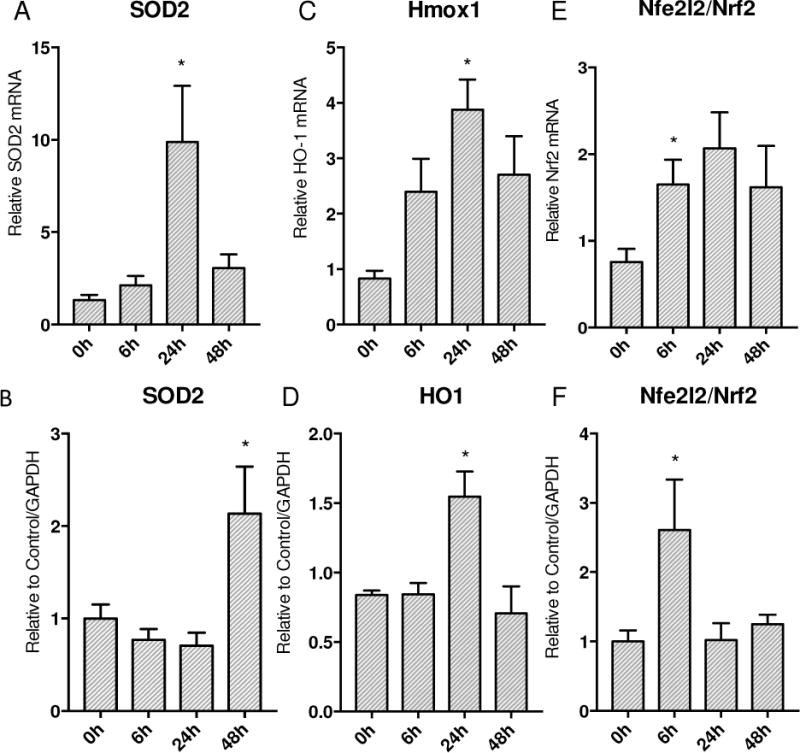
Response of ARE regulated gene expression to S. aureus 1×107 CFU murine sepsis in the lung. A&B) SOD2 mRNA transcription expression increase by 24h with protein levels following by 48h. C&D) HO-1 mRNA transcription increases significantly by 24h with subsequent protein increase at 24h. E) mRNA transcription of Nrf2 increases significantly at 24h. F) Nrf2 protein shows early up-regulation by 6h. (* P < 0.05 vs. time 0h)
Mitochondrial biogenesis is induced in the lung during murine sepsis
Mitochondrial biogenesis involves the up-regulation of several hundred genes; therefore we assessed carefully selected biomarkers including the PGC1-α co-regulator and the mitochondrial transcription factor (Tfam) necessary for mitochondrial DNA transcription and replication as well as the mitochondrial matrix TCA cycle protein, citrate synthase. PGC1-α increased more than 6 fold early on at 6h and remained elevated (Figure 4B). The Tfam mRNA increased 4 fold by 24h then decreased by 48h (Figure 4A). The mitochondrial citrate synthase protein showed a significant 3 fold increase at 24h that was sustained through 48h (Figure 4C).
Figure 4.
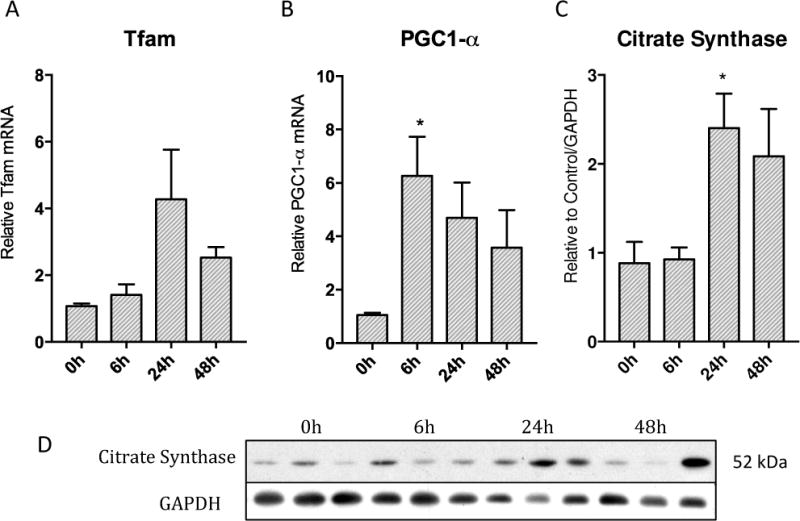
Activation of PGC1-α co-activator, nuclear encoded Tfam, and resulting increase in mitochondrial protein citrate synthase over 48h in lung parenchyma after S. aureus sepsis. A) Tfam mRNA increases in transcription robustly at 24h then starts to decrease by 48h. B) PGC1-α mRNA transcription increases significantly early on at 6h then remains elevated throughout 48h C) Mitochondrial protein citrate synthase is significantly increased by 24h and remains elevated through 48h. D) Western blots of above proteins (* p < 0.05)
Autophagy induction in the lung during sepsis
To assess autophagy, the canonical autophagy proteins Beclin and LC3-II, were assayed by western blot analysis. The ARE-regulated gene for the scaffolding protein p62/SQSTM1 was also examined. To validate our findings, we also surveyed these proteins after administering BFA, a V-ATPase autophagy inhibitor, to evaluate changes in autophagic flux [34].
LC3-II, an integral protein of autophagosome biogenesis, showed a 2.5 fold increase at the protein level at 24h compared to 0h and then returned to baseline by 48h (Figure 5E). When mice were treated with BFA, a robust elevation of LC3-II of almost 3 fold was observed in treated mice at 24h compared to untreated mice at 24h (Figure 5F).
Figure 5.
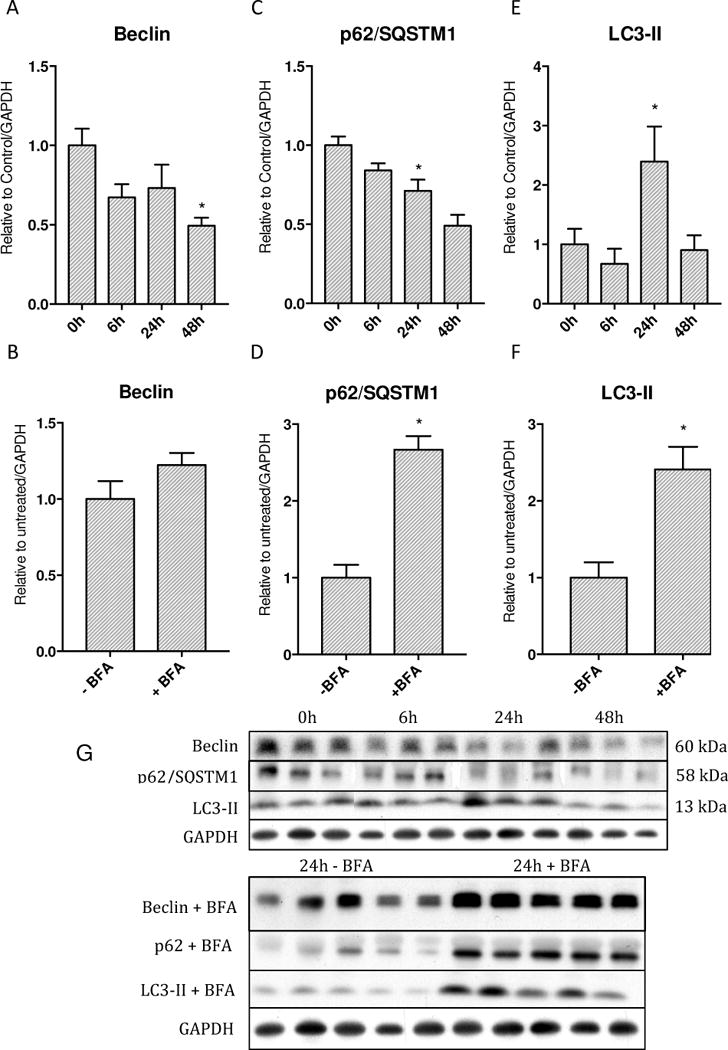
Levels of autophagy proteins and assessment of autophagic flux with Bafilomycin A. A&B) Beclin protein levels decrease starting at 6h and continue to decrease through 48h, however Bafilomycin produces nonsignificant increase. C&D) p62 protein levels decrease starting at 6h and reach significance vs. 0h at 48h while inhibition of autophagy with Bafilomycin shows significant accumulation E&F) LC3-II protein levels increase significantly at 24h then return to baseline by 48h. Bafilomycin produces significant accumulation G) Western blots of above proteins. (* p < 0.05).
Beclin 1, an autophagy initiation protein, displayed a steady decrease in protein from 0h by almost half by 48h (Figure 5A). However, after the mice were treated with BFA, Beclin 1 showed a small increase compared to untreated mice at 24h (Figure 5B).
The protein levels for SQSTM1/p62, a critical redox-sensitive scaffolding protein that contains a LC3 interacting domain as well as an Nrf2 interaction domain, mirrored Beclin 1 with a significant drop in the protein levels by 48h to almost half of 0h levels (Figure 5C). When septic mice were treated with BFA, there was an almost 3 fold increase in protein levels at 24h (Figure 5D).
Mitophagy occurs in key lung cells
To localize the major site of autophagy, we performed immunofluorescence studies in the sepsis model in LC3-GFP reporter mice. We probed these lungs at 24h for LC3 and for mitochondria by co-staining for citrate synthase. We observed LC3 sites (puncta) largely in alveolar type II cells, but also in macrophages, along with significant co-localization with citrate synthase staining of the mitochondria (Figure 6C). The number of LC3 puncta was higher in S. aureus sepsis compared to the control setting (Figures 6A&B). There was also a noticeable increase in citrate synthase in sepsis compared to control consistent with mitochondrial biogenesis (Figures 6C&D). When the LC3 signal and citrate synthase signal were overlaid, there was strong localization of LC3 puncta to mitochondria consistent with increased mitophagy.
Figure 6.
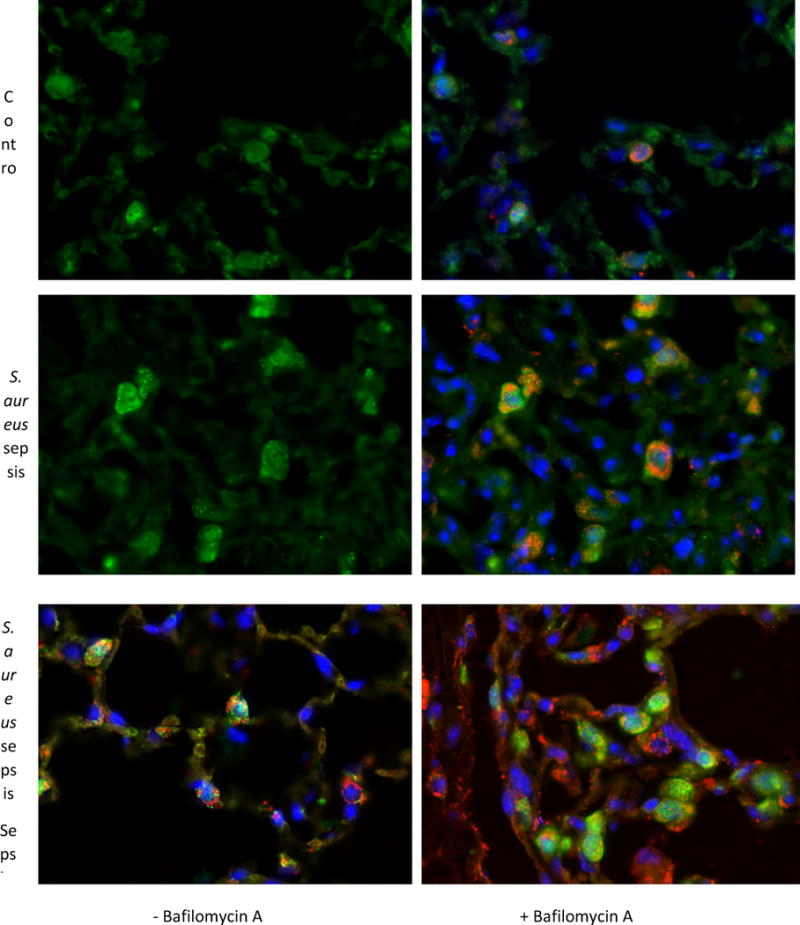
Immunofluorescence of LC3-GFP reporter mice lungs after S. aureus 1×107 CFU at 24 hours as well as Bafilomycin A. A) The control mouse shows a low number of LC3 puncta B) Citrate synthase is stained red and nuclei are stained blue. C) Sepsis induces more LC3 to form puncta at 24 hours namely in alveolar type 2 as well as macrophages. D) Sepsis induces increased citrate synthase as well as a LC3 puncta expression in alveolar type 2 cells and macrophages. There is more co-localization of LC3 to citrate synthase than in panels A and B. E) There are definite LC3 puncta in alveolar type 2 cells as well as a macrophage that are co-localized with mitochondria as well. F) With Bafilomycin A, there is an increase in LC3 puncta as well as citrate synthase.
To characterize autophagic flux, we also performed immunofluorescence on LC3-GFP mice that had been given S. aureus and either a saline control injection or 1mg/kg BFA. There was a significantly increased number of LC3 puncta, representing autophagosomes, present in septic mice compared to control mice (Figure 7). Likewise, increased LC3 puncta co-localizing with citrate synthase was observed in alveolar type II and macrophages at 24h supporting a robust activation of mitophagy induced by sepsis (Figure 7).
Figure 7.
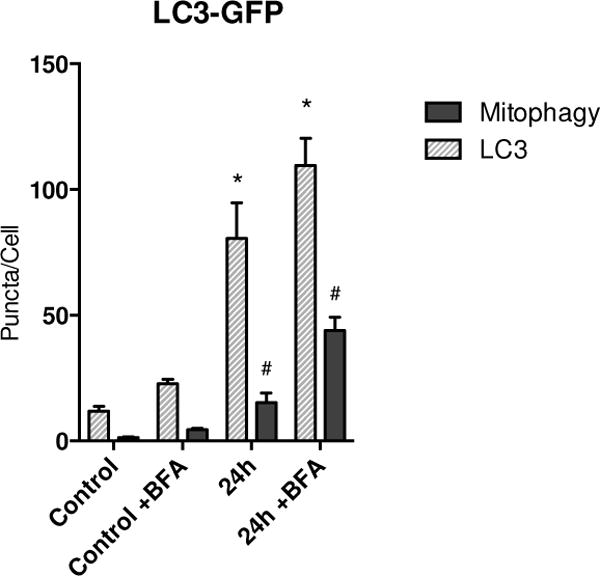
Number of autophagic and mitophagic sites (puncta) in LC3-GFP mice during sepsis. Sepsis causes a significant increase in GFP puncta per cell. Colocalization of GFP with mitochondria shows an increase in mitophagic puncta per cell in the setting of sepsis. ( * = p < 0.05 compared to LC3 puncta in WT control; # = p < 0.05 compared to colocalization in WT control)
Autophagy regulation by the Nrf2 antioxidant response
To ascertain whether redox stress induced mitophagy during sepsis, changes in LC3-II and p62 protein expression was measured in Nrf2−/− mice after S. aureus clot implantation. LC3-II protein levels were significantly decreased in these mice at 24h compared to wild-type mice at 24h (Figure 8B). With BFA, a large accumulation of LC3-II suggests an overall decrease in autophagic flux in comparison to WT mice. The scaffolding protein p62 also showed significant up-regulation at 24h in Nrf2−/− mice compared to 0h and 24h WT septic mice (Figure 8C). The transcription of the p62 gene assessed by mRNA levels shows a significant increase at 24h in Nrf2−/− mice compared to Nrf2−/− mice at 0h as well as to WT mice at 0h (Figure 8D). However, there was a much smaller increase in mRNA levels compared to 24h WT septic mice indicating that the increase in p62 protein is due mainly to its accumulation and not to increased.
Figure 8.
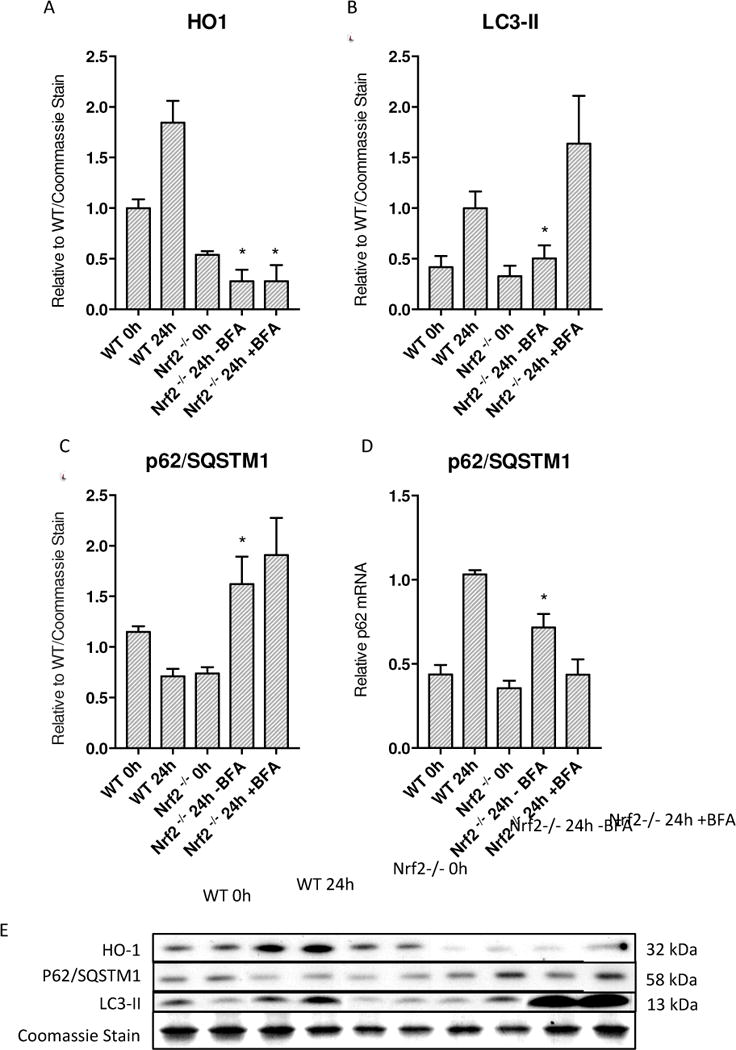
Decreased autophagy in Nrf2 deficient mice during S. aureus 5×106 peritonitis. A) HO-1 in Nrf2 KO mice show an overall diminished response to sepsis. At 24h Nrf2 KO mice had significantly lower HO-1 levels than WT mice at 24h in the same sepsis model. B) LC3-II is significantly decreased in Nrf2 deficient mice at 24h compared to wild-type C57BL/6 mice. C) p62 levels show accumulation at 24h at the protein level significantly compared to wild-type mice at 24h. D) p62 mRNA shows increased transcription compared to 0h wild-type and Nrf2 deficient mice although decreased copy number compared to wild-type mice at 24h. E) Western blots of above proteins. (* = p < 0.05 compared to WT 24h)
Discussion
Our data demonstrate for the first time that S. aureus sepsis induces mitophagy in the distal lung by a redox sensitive pathway. Moreover, this redox pathway plays a key role in intracellular mitochondria quality control, specifically in the turnover of damaged mitochondria as well as in mitochondrial biogenesis. In our mouse model, we found increased early-phase inflammatory cytokine elaboration in the lung, i.e. IL-1β and TNF-α, as well as evidence of end organ inflammation and ALI represented by increased cell count and protein in BAL fluid at 24h. In response to increased inflammation, the up-regulation of SOD2 protein indicated a mitochondrial adaptive response to the intracellular oxidative stress. The early response to oxidative stress as measured by SOD2 protein levels closely mirrors the ROS-sensitive up-regulation of Nrf2 and the subsequent downstream transcription of HO-1, further highlighting the lung’s anti-oxidant response to oxidative stress.
The cell relies on mitophagy to remove damaged mitochondria, which are the major source of intracellular ROS [35]. This prevents further deleterious ROS production and subsequent damage to DNA, lipids, and other proteins [36]. Here we found that systemic S. aureus sepsis produces lung mitochondrial damage as visualized by cytoplasmic ssDNA [37]. In addition to the removal of these damaged mitochondria, the cell must also replace these organelles in order to restore cellular bioenergetics. This requires mitochondrial biogenesis.
Earlier studies in this S. aureus model also suggest that sepsis-induced inflammation is limited by activation of mitochondrial biogenesis in part through up-regulation of the anti-inflammatory cytokine, IL-10 [13,38,39]. The present work demonstrated significant increases in a critical co-activator of mitochondrial biogenesis, PGC1-α protein [40], as well as Tfam, which is necessary for mtDNA replication and transcription. An increase in mitochondrial density in the distal lung was documented by increases in citrate synthase protein levels as well as with visual confirmation by in situ staining. Adaptive mitochondrial biogenesis during sepsis has been reported as an inducible process that rescues mice from lethal sepsis [11,41]. Recent studies have noted the protective role for mitochondrial biogenesis and autophagy in murine sepsis [42–44] as well as for mitochondrial biogenesis in human sepsis [45]. Our data suggest that mitochondrial biogenesis and autophagy are very closely linked to bioenergetic homeostasis in the distal lung.
Loss of autophagic regulation has been documented to occur in the settings of nutrient deprivation, hypoxia, and oxidative stress [46,47]. Recent evidence has highlighted the importance of p62/sequestosome 1/SQSTM1 protein in the selective degradation of ubiquinated proteins [48–50]. This protein binds directly to LC3-II through its LC3-interacting region (LIR) which ultimately results in lysosomal degradation of proteins [51]. Earlier observations of mitophagy have emphasized that during collapse of membrane potential and generation of excess ROS, mitochondria are disposed of by p62-dependent pathways [52,53]. In the setting of S. aureus sepsis, it has been noted that damage to mitochondria results in the deleterious generation of ROS leading to activation of mitophagy [25,26]. Previous studies on autophagy have described elevations of LC3-II protein as a marker for increased autophagosome formation and decreases in p62 protein as a marker for increases in autophagic turnover [54–56]. The p62 protein has been demonstrated to be an integral part of intracellular polyubiquinated protein inclusions and as a marker of the autophagic machinery [57]. Similarly, inhibition of lysosomal degradation of autophagosomes has been correlated with increased levels of p62 [58]. We demonstrated a significant increase in LC3-II protein associated with a significant decrease in p62 protein at 24h of sepsis, which we interpret as enhanced mitochondrial autophagy in the lung.
The proof of principle of redox-regulated mitophagy in sepsis was its significant attenuation in Nrf2−/− mice. This observation was confirmed by administering BFA, which nullified this decrease in autophagic induction. BFA inhibits the vacuolar ATPase that governs the fusion of autophagosomes to lysosomes during autophagy as well as prevents acidification of lysosomes, which ultimately halts the autophagosome at its degradation state [59]. This results in intracellular accumulation of proteins involved with autophagic turnover, including LC3-II and p62 [60]. BFA is widely used to evaluate autophagic flux since inhibition of lysosome degradation will allow assessment of autophagosomes either at a protein level or in situ immunofluorescence [34]. Our data from wild type and Nrf2−/− mice with and without BFA during S. aureus sepsis demonstrate that loss of Nrf2 decreases LC3-II production and also leads to a buildup of p62 and Beclin-1 suggesting an overall decrease in autophagic flux. This effect was reversed with BFA. Although we did not measure the effect of BFA on survival in the mice here, the literature indicates that this agent has a negative effect on survival in a CLP sepsis model in mice [26]. Our data showing that Nrf2 knockout produces decreases in LC3-II as well as in the selective autophagy receptor protein p62, suggest that Nrf2 plays a key role in the redox-sensitive induction of mitochondrial biogenesis as well as redox-sensitive mitophagy; however, our findings are associative and not sufficient for causation.
Nrf2 is responsible for the transcription of many genes with ARE in their promoter regions. For instance, Komatsu et al. mapped a motif on p62 that allows interaction with Keap1. Keap1 regulates levels of Nrf2 by binding directly to it and causing it to be degraded by the 26s proteasome. The competitive interaction with Keap1 increases Nrf2 regulated genes. In vitro, through a luciferase assay they show decreased transactivation of Nrf2 activity with increased expression of Keap1 while overexpression of p62 nullified this effect. Komatsu et al. then demonstrated p62 dependent Nrf2 stabilization by using p62−/− hepatocytes transfected with p62 and p62 mutants that show increased downstream transcription of Nrf2 target genes with p62 and not the p62 mutants. The authors concluded that p62 activates Nrf2 through inactivation of Keap1 [61].
Jain et al. furtherposited that p62-Nrf2 interactions through Keap1 create a feed forward loop where increased Nrf2 levels will further increase p62 in a p62 dependent fashion. Using p62−/− MEFs, Jain et al. transfected with a p62-Luc reporter plasmid as well as an expression vector for Myc-p62. Increased activation of luciferase displayed evidence that p62 was able to induce its own promoter. Using a mutant that did not interact with Keap1 showed this process occurred in a Keap1-dependent manner [62].
Our data supports this idea of a p62 driven, Nrf2 dependent, p62 expression when examining our experiments with Nrf2−/− mice. In figure 8C, at 0h there is decreased overall p62 expression in the Nrf2−/− compared with WT mice. Interestingly, at 24h despite increased p62 protein (Figure 8C), there is decreased mRNA transcription of p62 (Figure 8D). We interpret this data to show that p62 expression is dependent on Nrf2.
An earlier report from our laboratory suggests a protective role for Nrf2 in the lung during S. aureus- induced acute lung injury by restoring mitochondrial biogenesis and activation of key antioxidant response genes [63]. Likewise, it now appears that Nrf2 may also play a protective role through regulation of lung mitophagy, which allows for the control of excess ROS as well as activation of mitochondrial biogenesis through the HO-1 system.
In conclusion, this work supports that cellular redox homeostasis involves an equilibrium for mitophagic flux and mitochondrial biogenesis in S. aureus sepsis-induced lung injury. The dysregulation of this equilibrium can lead to degradation of mitochondrial quality control, ultimately resulting in bioenergetic collapse and subsequent cell death.
Supplementary Material
Acknowledgments
The authors would like to thank the Duke Center for Hyperbaric Medicine and Environmental Physiology as well as the Eugene E. Stead Foundation for their generous scholarships and support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Annane D, Aegerter P, Jars-Guincestre MC, Guidet B. Current Epidemiology of Septic Shock. American Thoracic Society; 2012. http://dx.doi.org/10.1164/rccm.2201087. [DOI] [PubMed] [Google Scholar]
- 2.Hotchkiss RS, Karl IE. The Pathophysiology and Treatment of Sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- 3.Galley HF. Oxidative stress and mitochondrial dysfunction in sepsis. British Journal of Anaesthesia. 2011;107:57–64. doi: 10.1093/bja/aer093. [DOI] [PubMed] [Google Scholar]
- 4.Cash TP, Pan Y, Simon MC. Reactive oxygen species and cellular oxygen sensing. Free Radical Biology and Medicine. 2007;43:1219–1225. doi: 10.1016/j.freeradbiomed.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singer M. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence Landes Bioscience. 2013;5:0–1. doi: 10.4161/viru.26907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun S, Sursal T, Adibnia Y, Zhao C, Zheng Y, Li H, Otterbein LE, Hauser CJ, Itagaki K. Mitochondrial DAMPs Increase Endothelial Permeability through Neutrophil Dependent and Independent Pathways. PLoS ONE Public Library of Science. 2013;8:e59989. doi: 10.1371/journal.pone.0059989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haden D, Suliman H, Carraway M, Welty-Wolf K, Ali A, Shitara H, Yonekawa H, Piantadosi C. Mitochondrial biogenesis restores oxidative metabolism during Staphylococcus aureus sepsis. Am J Respir Crit Care Med. 2007;176:768–777. doi: 10.1164/rccm.200701-161OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009;16:939–946. doi: 10.1038/cdd.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giuseppe Filomeni EDSCGR, Ciriolo MR. Under the ROS: Thiol network is the principal suspect for autophagy commitment. Autophagy Landes Bioscience. 2010;6:999–1005. doi: 10.4161/auto.6.7.12754. [DOI] [PubMed] [Google Scholar]
- 10.Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem. 2010;47:69–84. doi: 10.1042/bse0470069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.MacGarvey NC, Suliman HB, Bartz RR, Fu P, Withers CM, Welty-Wolf KE, Piantadosi CA. Activation of Mitochondrial Biogenesis by Heme Oxygenase-1–mediated NF-E2–related Factor-2 Induction Rescues Mice from Lethal StaphylococcusaureusSepsis. Am J Respir Crit Care Med. 2012;185:851–861. doi: 10.1164/rccm.201106-1152OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stępkowski TM, Kruszewski MK. Molecular cross-talk between the NRF2/KEAP1 signaling pathway, autophagy, and apoptosis. Free Radical Biology and Medicine. 2011;50:1186–1195. doi: 10.1016/j.freeradbiomed.2011.01.033. [DOI] [PubMed] [Google Scholar]
- 13.Nguyen T, Nioi P, Pickett CB. The Nrf2-Antioxidant Response Element Signaling Pathway and Its Activation by Oxidative Stress. Journal of Biological Chemistry. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. American Society for Biochemistry and Molecular Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Venugopal R, Jaiswal AK. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant respons…: EBSCOhost. Oncogene. 1998 doi: 10.1038/sj.onc.1202237. [DOI] [PubMed] [Google Scholar]
- 15.Itoh K, Wakabayashi N, Katoh Y, Ishii T, O’Connor T, Yamamoto M. Keap1 regulates both cytoplasmic – nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes to Cells. 2003;8:379–391. doi: 10.1046/j.1365-2443.2003.00640.x. Blackwell Science Ltd. [DOI] [PubMed] [Google Scholar]
- 16.Itoh K, Tong KI, Yamamoto M. Molecular mechanism activating nrf2–keap1 pathway in regulation of adaptive response to electrophiles. Free Radical Biology and Medicine. 2004;36:1208–1213. doi: 10.1016/j.freeradbiomed.2004.02.075. [DOI] [PubMed] [Google Scholar]
- 17.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. Nature Publishing Group. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanki T, Klionsky DJ. The molecular mechanism of mitochondria autophagy in yeast. Molecular Microbiology. 2010;75:795–800. doi: 10.1111/j.1365-2958.2009.07035.x. Blackwell Publishing Ltd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012;393:547–564. doi: 10.1515/hsz-2012-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Charleen T, Chu JZ, Dagda RK. Beclin 1-Independent Pathway of Damage-Induced Mitophagy and Autophagic Stress: Implications for Neurodegeneration and Cell Death. Autophagy. 2007;3:663–666. doi: 10.4161/auto.4625. Landes Bioscience. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, Ney PA. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. PNAS. 2007;104:19500–19505. doi: 10.1073/pnas.0708818104. National Acad Sciences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mammucari C, Rizzuto R. Signaling pathways in mitochondrial dysfunction and aging. Mechanisms of Ageing and Development. 2010;131:536–543. doi: 10.1016/j.mad.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watanabe E, Muenzer JT, Hawkins WG, Davis CG, Dixon DJ, McDunn JE, Brackett DJ, Lerner MR, Swanson PE, Hotchkiss RS. Sepsis induces extensive autophagic vacuolization in hepatocytes a clinical and laboratory-based study. Laboratory Investigation. 2009;89:549–561. doi: 10.1038/labinvest.2009.8. Nature Publishing Group. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carchman EH, Rao J, Loughran PA, Rosengart MR, Zuckerbraun BS. Heme oxygenase-1–mediated autophagy protects against hepatocyte cell death and hepatic injury from infection/sepsis in mice. Hepatology. 2011;53:2053–2062. doi: 10.1002/hep.24324. Wiley Subscription Services, Inc., A Wiley Company. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carchman EH, Whelan S, Loughran P, Mollen K, Stratamirovic S, Shiva S, Rosengart MR, Zuckerbraun BS. Experimental sepsis-induced mitochondrial biogenesis is dependent on autophagy, TLR4, and TLR9 signaling in liver. The FASEB Journal. 2013:fj.13–fj.229476. doi: 10.1096/fj.13-229476. Federation of American Societies for Experimental Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lo S, Yuan SSF, Hsu C, Cheng YJ, Chang YF, Hsueh HW, Lee PH, Hsieh YC. Lc3 Over-Expression Improves Survival and Attenuates Lung Injury Through Increasing Autophagosomal Clearance in Septic Mice. Annals of Surgery. 2013;257:352–363. doi: 10.1097/SLA.0b013e318269d0e2. [DOI] [PubMed] [Google Scholar]
- 27.Rocchi P, Jugpal P, So A, Sinneman S, Ettinger S, Fazli L, Nelson C, Gleave M. Small interference RNA targeting heat-shock protein 27 inhibits the growth of prostatic cell lines and induces apoptosis via caspase-3 activation in vitro. BJU International. 2006;98:1082–1089. doi: 10.1111/j.1464-410X.2006.06425.x. Blackwell Publishing Ltd. [DOI] [PubMed] [Google Scholar]
- 28.Fung SJ, Xi M, Zhang J, Sampogna S, Chase MH. Apnea produces excitotoxic hippocampal synapses and neuronal apoptosis. Experimental Neurology. 2012;238:107–113. doi: 10.1016/j.expneurol.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 29.Frankfurt OS, Robb JA, Sugarbaker EV, Villa L. Apoptosis in breast carcinomas detected with monoclonal antibody to single-stranded DNA: relation to bcl-2 expression, hormone receptors, and lymph node metastases. Clin Cancer Res. 1997;3:465–471. American Association for Cancer Research. [PubMed] [Google Scholar]
- 30.Suliman HB, Carraway MS, Tatro LG, Piantadosi CA. A new activating role for CO in cardiac mitochondrial biogenesis. J Cell Sci. 2007;120:299–308. doi: 10.1242/jcs.03318. [DOI] [PubMed] [Google Scholar]
- 31.Mizushima N. Chapter 2 Methods for Monitoring Autophagy Using GFP-LC3 Transgenic Mice. Autophagy in Mammalian Systems, Part B. 2009:13–23. doi: 10.1016/S0076-6879(08)03602-1. Elsevier. [DOI] [PubMed] [Google Scholar]
- 32.Chiou CC, Chang PY, Chan EC, Wu TL, Tsao KC, Wu JT. Urinary 8-hydroxydeoxyguanosine and its analogs as DNA marker of oxidative stress: development of an ELISA and measurement in both bladder and prostate cancers. Clinica Chimica Acta. 2003;334:87–94. doi: 10.1016/s0009-8981(03)00191-8. [DOI] [PubMed] [Google Scholar]
- 33.Wu LL, Chiou CC, Chang PY, Wu JT. Urinary 8-OHdG: a marker of oxidative stress to DNA and a risk factor for cancer, atherosclerosis and diabetics. Clinica Chimica Acta. 2004;339:1–9. doi: 10.1016/j.cccn.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 34.Klionsky D, Abdalla F, Abeliovich H. Guidelines for the use and interpretation of assays for monitoring autophagy. 2012:1–100. [Google Scholar]
- 35.Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free Radical Biology and Medicine. 2009;47:333–343. doi: 10.1016/j.freeradbiomed.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 36.Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013;20:31–42. doi: 10.1038/cdd.2012.81. Nature Publishing Group. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Degtyareva NP, Heyburn L, Sterling J, Resnick MA, Gordenin DA, Doetsch PW. Oxidative stress-induced mutagenesis in single-strand DNA occurs primarily at cytosines and is DNA polymerase zeta-dependent only for adenines and guanines. Nucleic Acids Res. 2013;41:8995–9005. doi: 10.1093/nar/gkt671. Oxford University Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Element AR. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J Biochem Mol Biol. 2004 doi: 10.5483/bmbrep.2004.37.2.139. [DOI] [PubMed] [Google Scholar]
- 39.Piantadosi CA, Withers CM, Bartz RR, MacGarvey NC, Fu P, Sweeney TE, Welty-Wolf KE, Suliman HB. Heme oxygenase-1 couples activation of mitochondrial biogenesis to anti-inflammatory cytokine expression. Journal of Biological Chemistry. 2011;286:16374–16385. doi: 10.1074/jbc.M110.207738. American Society for Biochemistry and Molecular Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 41.Lancel S, Hassoun SM, Favory R, Decoster B, Motterlini R, Neviere R. Carbon Monoxide Rescues Mice from Lethal Sepsis by Supporting Mitochondrial Energetic Metabolism and Activating Mitochondrial Biogenesis. Journal of Pharmacology and Experimental Therapeutics. 2009;329:641–648. doi: 10.1124/jpet.108.148049. American Society for Pharmacology and Experimental Therapeutics. [DOI] [PubMed] [Google Scholar]
- 42.Takahashi W, Hatano H, Hirasawa H, Oda S. Critical Care | Abstract | Protective role of autophagy in mouse cecal ligation and puncture-induced sepsis model. Critical Care. 2013 doi: 10.1186/cc12839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsieh CH, Pai PY, Hsueh HW, Yuan SS, Hsieh YC. Complete induction of autophagy is essential for cardioprotection in sepsis. Annals of Surgery. 2011;253:1190–1200. doi: 10.1097/SLA.0b013e318214b67e. [DOI] [PubMed] [Google Scholar]
- 44.Seonmin L, Seon-Jin L, A CA, E FL, Wol CS, A PM, Kiichi N, W RS, K CAM. Carbon Monoxide Confers Protection in Sepsis by Enhancing Beclin 1-Dependent Autophagy and Phagocytosis. Mary Ann Liebert, Inc; 140 Huguenot Street, 3rd Floor New Rochelle, NY 10801 USA: 2014. http://dx.doi.org/10.1089/ars.2013.5368. [Google Scholar]
- 45.Carre JE, Orban JC, Re L, Felsmann K, Iffert W, Bauer M, Suliman HB, Piantadosi CA, Mayhew TM, Breen P, Stotz M, Singer M. Survival in Critical Illness Is Associated with Early Activation of Mitochondrial Biogenesis. Am J Respir Crit Care Med. 2010;182:745–751. doi: 10.1164/rccm.201003-0326OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature. 2011;13:132–141. doi: 10.1038/ncb2152. Nature Publishing Group. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, Huang L, Xue P, Li B, Wang X, Jin H, Wang J, Yang F, Liu P, Zhu Y, Sui S, Chen Q. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nature. 2012;14:177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 48.Moscat J, Diaz-Meco MT. p62 at the Crossroads of Autophagy, Apoptosis, and Cancer. Cell. 2009;137:1001–1004. doi: 10.1016/j.cell.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mathew R, Karp C, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, Dipaola R, Karantza-Wadsworth V, White E. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nezis IP, Stenmark H. p62 at the interface of autophagy, oxidative stress signaling, and cancer. Antioxid Redox Signal. 2012;17:786–793. doi: 10.1089/ars.2011.4394. [DOI] [PubMed] [Google Scholar]
- 51.Lin X, Li S, Zhao Y, Ma X, Zhang K, He X, Wang Z. Interaction Domains of p62: A Bridge Between p62 and Selective Autophagy. DNA Cell Biol. 2013;32:220–227. doi: 10.1089/dna.2012.1915. [DOI] [PubMed] [Google Scholar]
- 52.Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 53.Narendra DP, Youle RJ. Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid Redox Signal. 2011;14:1929–1938. doi: 10.1089/ars.2010.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang QJ, Ding Y, Kohtz S, Mizushima N, Cristea IM, Rout MP, Chait BT, Zhong Y, Heintz N, Yue Z. Induction of Autophagy in Axonal Dystrophy and Degeneration. J Neurosci. 2006;26:8057–8068. doi: 10.1523/JNEUROSCI.2261-06.2006. Society for Neuroscience. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. Journal of Biological Chemistry. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. American Society for Biochemistry and Molecular Biology. [DOI] [PubMed] [Google Scholar]
- 56.Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Øvervatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. Rockefeller Univ Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata JI, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura SI, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, Yamamoto M, Yue Z, Uchiyama Y, Kominami E, Tanaka K. Homeostatic Levels of p62 Control Cytoplasmic Inclusion Body Formation in Autophagy-Deficient Mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 58.Guo X, Dong Y, Yin S, Zhao C, Huo Y, Fan L, Hu H. Cell Death and Disease – Abstract of article: Patulin induces pro-survival functions via autophagy inhibition and p62 accumulation. Cell Death & Disease. 2013 doi: 10.1038/cddis.2013.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shacka J, Klocke B, Roth K. Autophagy, bafilomycin and cell death: the “a-B-cs” of plecomacrolide-induced neuroprotection. Autophagy. 2006;2:228–230. doi: 10.4161/auto.2703. [DOI] [PubMed] [Google Scholar]
- 60.Zhu J, Dagda RK, Chu CT. Monitoring Mitophagy in Neuronal Cell Cultures. Neurodegeneration. 2011:325–339. doi: 10.1007/978-1-61779-328-8_21. Humana Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong K, Kim M, Nishito Y, Iemura SI, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nature cell biology. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 62.Jain A, Lamark T, Sjøttem E, Larsen KB, Awuh JA, Øvervatn A, McMahon M, Hayes JD, Johansen T. p62/SQSTM1 Is a Target Gene for Transcription Factor NRF2 and Creates a Positive Feedback Loop by Inducing Antioxidant Response Element-driven Gene Transcription. Journal of Biological Chemistry. 2010;285:22576–22591. doi: 10.1074/jbc.M110.118976. American Society for Biochemistry and Molecular Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Athale J, Ulrich A, Chou Macgarvey N, Bartz RR, Welty-Wolf KE, Suliman HB, Piantadosi CA. Nrf2 promotes alveolar mitochondrial biogenesis and resolution of lung injury in Staphylococcus aureus pneumonia in mice. Free Radical Biology and Medicine. 2012;53:1584–1594. doi: 10.1016/j.freeradbiomed.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.