

Abstract
Transthyretin (TTR) amyloidosis causes heart failure from cardiac deposition of TTR amyloid fibrils, the by-product of TTR homotetramer disassembly. Wild-type (WT) TTR deposition leads to senile amyloidosis, predominantly manifesting with cardiomyopathy. Missense mutations in the TTR gene result in familial TTR amyloidosis. Certain mutations are more likely to affect the heart, while others cause more neurologic involvement. Extracellular fibril deposition triggers intracellular stress response, upregulation of the inflammatory cascades, apoptosis, and organ dysfunction. Recent studies suggest that TTR cardiac amyloid may be a significant contributor to the pathogenesis of heart failure with preserved ejection fraction (HFpEF). Summarized in this review are the molecular pathways underlying the cellular toxicity of TTR amyloid fibrils and the emerging therapies aimed at TTR tetramer stabilization, abrogation of TTR synthesis in the liver, or inhibition of amyloidogenesis.
Keywords: cardiac amyloidosis, transthyretin (TTR), heart failure with preserved ejection fraction (HFpEF)
Introduction
Transthyretin (TTR) amyloidosis is a disease caused by systemic deposition of wild-type (WT) or mutant TTR fibrils, resulting in heart failure when deposition occurs in the heart. Mutant TTR deposition leads to familial TTR amyloid. Accumulation of the normal TTR protein causes WT cardiac amyloidosis (also known as senile amyloidosis). Predominantly synthesized in the liver, TTR protein forms a homotetramer complex that acts as a transporter for thyroxine and retinol-binding protein. Mutation nomenclature typically uses the mature protein of 127 amino acid residues after cleavage of a 20-amino acid signal peptide. Over 100 different amyloidogenic mutations in the TTR gene have been described, including a point mutation in codon 122 that causes an amino acid change from valine to isoleucine (p. Val122Ile).1–3 While other TTR mutations result in familial amyloid polyneuropathy (FAP), Val122Ile is associated with predominantly cardiac involvement.
Interestingly, the Val122Ile allele is fairly common among African Americans, with 3–4% of self-identified African Americans carrying this mutation, typically in heterozygosity.4–6 It is extremely rare in other ethnic groups.4 The frequency of Val122Ile in populations with heart failure remains largely unknown. In the Beta-Blocker Evaluation of Survival Trial (BEST) study, where cardiac amyloidosis was an exclusion criterion, the prevalence of Val122Ile was as high as 10% among African Americans older than 60 years of age with NYHA classes III–IV heart failure and an ejection fraction (EF) of 35% or less.7 In another case control study of asymptomatic Val122Ile carriers, this allele appears to confer a significant risk for the development of heart failure.8 Once symptomatic, Val122Ile carriers with heart failure have a worse prognosis than those with WT cardiac amyloid, with a median survival of just over two years.9 Taken together, among African Americans who present with heart failure and preserved ejection fraction, the TTR Val122Ile mutation might be a severely underdiagnosed cause.
In recent years, heart failure with preserved ejection fraction (HFpEF) has become increasingly prevalent among individuals hospitalized for acute decompensated heart failure.10 However, no therapies with survival benefits have been identified in randomized clinical trials, owing in part to a heterogeneous patient population with different responses to treatments. It has been postulated that survival of patients with HFpEF can only be improved if they are phenotyped more selectively and their treatments tailored accordingly.11
A recent autopsy series provided pivotal evidence that TTR amyloidosis is more prevalent among HFpEF population.12 Of the 109 Caucasian patients seen at Mayo Clinic hospitals between 1986 and 2001 with subsequent autopsy, 5% were found to have moderate or severe WT TTR deposits in the left ventricle, consistent with WT systemic amyloidosis as the primary etiology of heart failure. In addition, mild interstitial and/or variable severity of intramural coronary vascular WT TTR deposition occurred in 12% of this cohort. None of these patients carried an antemortem diagnosis of cardiac amyloid.
How TTR amyloidosis contributes to the development of HFpEF is not known. We can only hypothesize that the accumulation of dense TTR amyloid likely worsens diastolic function. Slow accumulation of pathologic TTR amyloid deposits in the heart may initially cause asymptomatic left ventricular (LV) hypertrophy, with relatively late diagnosis because of its gradual progression. However, these individuals may have other comorbidities (diabetes, hypertension, chronic kidney disease, atrial fibrillation) that contribute independently to impaired ventricular relaxation, and also promote a pro-oxidative state, and therefore may accelerate myocardial TTR deposition as a secondary phenomenon. This latter hypothesis might explain why a significant number of mutant TTR carriers remain asymptomatic without evidence of cardiac involvement until older age, when co-morbidities such as hypertension and diabetes have taken their toll on the heart. Regardless, it should be emphasized that based on the Mayo clinic autopsy series, TTR amyloidosis might represent a significant portion of the population labeled as HFpEF, when, in fact, such patients have an entirely different disease, namely, cardiac amyloidosis.
When to Suspect Cardiac Amyloidosis
The diagnosis of TTR cardiac amyloidosis is often missed until very late in the disease course, as it is an indolent illness affecting the same elderly population with HFpEF. Unlike light chain (AL) amyloidosis, there is no readily available blood test for misfolded TTR protein. Diagnostic algorithms, including non-invasive imaging modalities and endomyocardial biopsy, have been published elsewhere.13,14 Yet these algorithms can only be applied if cardiac amyloid is suspected. Table 1 summarizes our proposal of criteria that, when present, should raise a clinician’s suspicion for cardiac amyloidosis. We have tried to make these criteria as specific to TTR amyloidosis as possible, though many of them, such as low-voltage electrocardiogram (ECG) with echocardiographic evidence of LV hypertrophy, are found in other forms of infiltrative cardiomyopathy.
Table 1.
When to suspect cardiac amyloidosis in a patient with heart failure.
| CATEGORY | CRITERIA |
|---|---|
| History | • New-onset HFpEF at age >60 years • Family history of unexplained HF at age >60 years • Peripheral neuropathy • Carpal tunnel syndrome, particularly with recurrence, or with lack of improvement after surgery • Easy bruising without anticoagulation medications |
| Physical exam | • Orthostatic hypotension • Macroglossia • Thenar/hypothenar muscle wasting • Unexplained dermal bruising |
| Medications | • Beta-blocker intolerance • Vasodilator intolerance |
| ECG | • Low voltage (limb QRS <5 mm, precordial QRS <10 mm) • Discordance between ECG and echocardiogram regarding LV hypertrophy • Atrial fibrillation/flutter • AV conduction delay • Unexplained bundle branch block • Pseudo-infarction pattern |
| Echocardiogram | • Unexplained LV hypertrophy • Unexplained RV hypertrophy • Increased atrial septal thickness • Biatrial enlargement • Restrictive filling (high E/A and E/e’ ratios) • Pericardial effusion |
Given the increasing awareness of TTR cardiac amyloidosis, this review summarizes the molecular mechanisms and proposes a unifying model to explain the disease phenotype. Knowledge of the underlying molecular pathways is vital in the search for novel therapies that can alter the course of this progressive and fatal disease.
Cytotoxicity of TTR Amyloid Fibrils
TTR is a tetramer of four identical β-pleated sheet-rich subunits, each composed of 127 amino acids. The crystal structures of WT and mutant TTR in complex with its natural ligands, thyroxine and retinol-binding protein, have all been solved.15,16 TTR is predominantly synthesized in the liver, but a small amount is also made in the retinal pigment epithelium of the eye and choroid plexus of the brain. Much research effort has been devoted to the identification of disease-causing mutations, now numbered at 120 to date17 (http://amyloidosismutations.com/mut-attr.php). Certain mutations such as Val30Met are linked to devastating peripheral neuropathy, while others such as Val122Ile predominantly cause heart failure. Symptomatic patients as well as their asymptomatic mutation-carrying family members can be registered in the Transthyretin Amyloidosis Outcomes Survey (THAOS), an international, longitudinal, observational study designed to investigate the disease course and its management.18
How mutations in the TTR gene trigger amyloid fibril formation is well known19 (Fig. 1). Certain mutations destabilize the tetramer structure, either thermodynamically or kinetically, causing protein misfolding, aggregation, and fibril formation. The natural progression and phenotypic manifestations of WT and mutant TTR amyloidoses have been described and reviewed extensively elsewhere.1,13,20 However, there remain numerous unanswered questions regarding the underlying cellular signaling pathways responsible for disease severity, organ tropism, male predominance, and the age-dependent onset of TTR cardiac amyloidosis.
Figure 1.
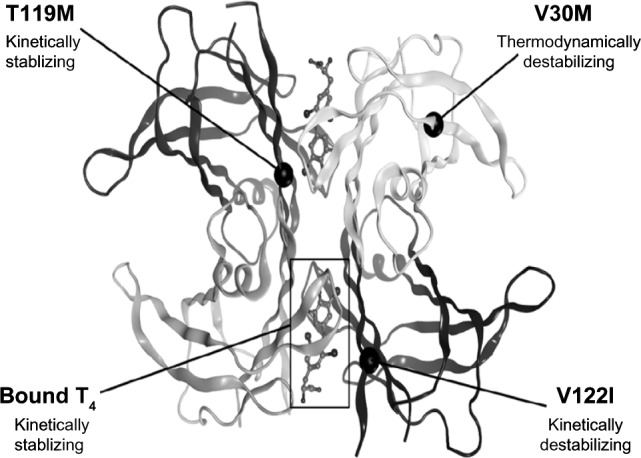
A crystal structure of TTR homotetramer (reproduced with permission from Dr. Isabella Graef).19 The natural ligand T4 provides kinetic stability to the TTR homotetramer, as does T119M if found in the presence of a thermodynamically destabilizing mutation such as Val30Met. V122I is one of the most common TTR mutations found worldwide.
Disease severity
Though TTR misfolding can cause amyloidosis in organs where it is made (as in vitreous amyloid), the burden of disease predominantly occurs as systemic TTR deposition, most importantly involving the heart and peripheral nerves. Research in the field of FAP provides much insight into the pathogenesis of TTR misfolding, which is inferred to be similar in the heart. Owing to the lack of readily available cardiac tissues or a transgenic animal model that can accurately recapitulate disease phenotype, in vivo studies examining the cardiotoxicity of TTR misfolding are limited. Certain mutations such as Leu55Pro are associated with early onset, and devastating and widespread amyloidosis, while others cause more indolent disease.21 The answer to this mystery might be the efficiency of protein secretion in the liver. Despite structural instability, mutant TTR tetramers are secreted with the same efficiency as wild type if they possess a thermodynamic and kinetic profile favorable enough to escape the machinery that controls protein folding, ie, the endoplasmic reticulum (ER)-associated degradation (ERAD) pathway. Alternatively, mutant tetramers can be stabilized by a small molecule chaperone such as thyroxine and, thus, efficiently secreted.22,23 In short, if a mutation is particularly amyloidogenic (ie, causing highly unstable TTR tetramers) but can be secreted normally either because of its ligand binding or favorable thermodynamic profile, the disease manifestations are likely more widespread and severe.
As in the case of other amyloidoses such as Alzheimer’s disease, extracellular deposition of TTR (mutant or WT) triggers intracellular stress response, suggesting the presence of a plasma membrane receptor for TTR, or a signaling cascade triggered by an indirect interaction between TTR and a transmembrane molecule. In lung and nerve tissues isolated from patients with FAP (Val30Met), both soluble, ligand-less TTR tetramer and unfolded TTR fibrils have been shown to bind RAGE (receptor for advanced glycation end-product), a scavenger receptor of the immunoglobulin superfamily capable of binding β-pleated sheet-rich proteins.24 Signal transduction via the interaction between RAGE and hyperglycemia-induced advanced glycation end products (AGEs) has been implicated in diabetic cardiovascular complications.25 Binding of RAGE triggers NF-κB activation, nuclear translocation, and upregulation of downstream proinflammatory cascades that result in oxidative damage and cell death.24 Extracellular TTR fibrils have also been shown to trigger the intracellular unfolded-protein response involving endoplasmic reticular stress pathways and cytosolic Ca2+ overload,26 as well as downregulation of proteasomal activity, further compromising the cell’s protection against oxidative stress.27 Once bound to retinol-binding protein (its natural ligand), the TTR tetramer is stabilized and loses its ability to interact with RAGE, thus no longer able to induce cytotoxic stress response.24 Strategies to stabilize the tetramer, either by TTR’s natural ligands or by a small molecule such as tafamidis, are one key to novel therapeutic developments as described below.
Organ tropism
Why the heart is preferentially affected in WT cardiac amyloidosis or Val122Ile TTR is not known. In contrast, Val30Met TTR amyloidosis typically causes earlier and more severe involvement of the peripheral nerves, with later cardiac manifestations. Cardiac tissues isolated from mutation carriers have been shown to harbor both WT and mutant TTR fibrils.28 Curiously, in an autopsy series of Val30Met FAP patients, older men had more WT than mutant TTR in their hearts, whereas their kidney, peripheral nerve, and gastrointestinal tissues exhibited no such correlation between age and the amount of TTR deposition.29 A cardio-specific receptor for TTR has not been found, but aging is a significant factor in the pathogenesis of TTR cardiac amyloidosis.
Age-dependent onset
The typical age of onset for WT TTR cardiac amyloidosis is in the seventh or eighth decade, and Val122Ile mutation carriers characteristically develop symptomatic heart failure after 60 years of age.8,30 It is unclear why WT TTR tetramers disassemble and deposit in the heart with increasing age, and why life-long mutation carriers may remain asymptomatic until the seventh or eighth decade. Age-dependent protein oxidation has been implicated in many disease models. As shown in an in vitro study, oxidized WT and Val122Ile TTR isoforms exert a more cytotoxic effect than their unoxidized counterparts.31 Another possibility is that the decline of thyroid function or retinol-binding protein in octogenarians32,33 accounts for TTR’s increased susceptibility to misfolding, once the serum levels of its natural ligands are lower.
Male sex prevalence
Clinically apparent WT TTR cardiac amyloidosis is mainly a disease of older men.34,35 In the autopsy series of Val30Met FAP patients described above, there appears to be a negative correlation between female sex and the amount of WT TTR deposits in the heart.29 This finding suggests that sex hormones might play a role in mediating the male prevalence of TTR amyloidosis or perhaps even later onset of WT TTR amyloidosis among women. TTR production by the liver of castrated mice increases several folds in response to exogenous 17 β-estradiol (E2) or 5α-dihydrotestosterone (DHT), though the latter seems to elicit more robust TTR production.36 While women’s ovarian estradiol levels drop sharply after menopause, men’s testosterone production, in contrast, is a steady decline, which may explain the prevalence of WT cardiac amyloidosis in male patients.
In short, Figure 2 summarizes the molecular mechanisms of TTR cytotoxicity as inferred from many studies to date. These mechanisms lend credence to the search for TTR amyloidosis therapies.
Figure 2.

A proposed mechanism for the pathogenesis of TTR amyloidosis. Stable WT or mutant TTR tetramers are synthesized in the liver and must pass through the protein folding quality control machinery in the ER (ERAD) prior to being secreted. Highly unstable mutant tetramers such as Asp18Gly (D18G) are degraded via the ubiquitin–proteasome system, while WT TTR and stabilized mutants such as Val122Ile (V122I) are efficiently secreted into the serum. With aging-related oxidation, coupled with thermodynamic instability caused by certain mutations, TTR tetramers disassociate, forming amyloid fibrils that deposit extracellularly in target organs, such as the heart. Amyloid fibrils bind RAGE, triggering intracellular inflammatory cascades that result in apoptosis, organ dysfunction, and death. Novel treatments include antisense oligonucleotides and siRNA aimed at inhibiting TTR synthesis. Tafamidis and diflunisal are small molecule stabilizers. The lysine-rich molecular tweezers inhibit amyloidogenesis, while anakinra suppresses the downstream inflammatory cascade.
Emerging Therapies for the Treatment of TTR Cardiac Amyloidosis
Small molecule stabilizers
Stabilization of the TTR homotetramer proves to be a viable approach in the search for novel therapies. This approach was initially considered after recognition that a naturally occurring mutation in TTR, Thr119Met, effectively stabilizes the TTR tetramer in the context of a trans-destabilizing mutation37 (Fig. 1). Though thyroxine is a natural tetramer stabilizer, it is not a viable therapy because of its hormonal property. A search for small molecule stabilizers yielded two promising drugs, diflunisal, a non-steroidal anti-inflammatory drug (NSAID), and tafamidis meglumine (Vyndaqel™, Pfizer), an NSAID analog without NSAID activity. In a multinational, randomized, placebo-controlled trial, diflunisal was shown to reduce the progression of neurological damage in patients with FAP.38 Diflunisal appears to be safe if given at low doses to compensated patients with TTR cardiac amyloidosis.39
Tafamidis meglumine is a benzoxazole lacking NSAID activity, which makes it ideal for heart failure patients for whom the fluid retention and renal dysfunction caused by NSAIDs may worsen disease manifestations. Tafamidis kinetically stabilizes TTR and inhibits amyloidogenesis.40 In a double-blind, placebo-controlled, randomized trial of patients with FAP, tafamidis appears to reduce peripheral neurologic impairment, though the co-primary endpoints, based on the intention to treat two different neuropathy symptom-scoring systems, were not significantly different between treatment and placebo arms.41 In a smaller cohort of individuals with non-Val30Met TTR amyloid and systemic TTR amyloidosis (including cardiomyopathy), tafamidis similarly stabilized the TTR tetramer with good tolerability.42 Approved in Europe and other locations for early stage FAP, tafamidis is still under review by the US Food and Drug Administration (FDA), and more clinical trials are underway to evaluate its efficacy in TTR cardiac amyloid patients. Other high-throughput drug screens continue to identify small molecule stabilizers that bind and stabilize mutant and WT TTR tetramers with high affinity.19
TTR silencers
To date the only approved method for curing familial TTR amyloidosis is orthotopic liver transplantation. However, this approach is problematic in many ways. TTR patients are generally older, and many do not qualify for organ transplantation. In some cases, cardiac and peripheral nerve deposition of amyloid continues to progress despite liver transplant.43,44 Degradation of TTR mRNAs either by short interfering RNA (siRNA) or antisense oligonucleotides has been shown to be an effective method in lowering TTR serum level and halting the progression of amyloid formation.45,46 Isis Pharmaceuticals and Alnylam Pharmaceuticals are conducting Phase II/III clinical trials at sites in the US and other countries to evaluate the efficacy of antisense oligonucleotides and siRNA, respectively.47
Novel therapies
As described earlier, binding of TTR fibrils to RAGE leads to NF-κB activation and upregulation of downstream proinflammatory cascades involving TNF-α, macrophage colony-stimulating factor, and IL-1β. In a transgenic FAP mouse model of human Val30Met TTR, administration of anakinra, an IL-1 type 1 receptor inhibitor, prevents extracellular deposition of TTR fibrils as well as neuronal apoptosis.48 Another avenue of novel therapeutics involves lysine-specific molecular tweezers, small molecules that specifically bind lysine residues and interfere with hydrophobic and electrostatic interactions that are vital for the oligomerization process.49 In Val30Met FAP mice, CLR01, a molecular tweezer, inhibits TTR deposition in the peripheral nervous system and gastrointestinal tract, with a concomitant decrease in cellular stress response, protein oxidation, and apoptosis.50
Other small molecules that hold therapeutic promises for TTR are resveratrol, doxycycline, and epigallocatechin-3--gallate (EGCG). Resveratrol is a phenolic compound found in grape skin. It kinetically stabilizes the tetramer in the same way as tafamidis and diflunisal by occupying the thyroxine-binding pocket, and has been shown to accelerate the formation of non-toxic native TTR tetramers in vitro.51 Doxycycline, on the other hand, prevents TTR deposits in genetically predisposed mice, especially when given together with a biliary acid, tauroursodeoxycholic acid (TUDCA).52,53 The efficacy of doxycycline and TUDCA combination is being tested in a phase II clinical trial.17
EGCG is a major polyphenolic component of green tea. Unlike tafamidis and diflunisal, EGCG stabilizes the homotetramer by binding to a site at the interface of two dimers. This molecule also has the ability to disrupt TTR fibrils. EGCG-treated old FAP mice have less organ TTR deposits, and more importantly, show signs of amyloid deposit disaggregation, the latter an extremely promising outcome as most patients have extensive amyloid deposition at the time of diagnosis.54
Conclusion
TTR cardiac amyloidosis is a progressive, fatal disease that affects older, predominantly male patients. The disease is caused by extracellular deposition of amyloid fibrils, formed from the misfolding of WT or mutant TTR tetramers. Extracellular fibril deposition leads to upregulation of the intracellular stress response, inflammation, and ultimately, programmed cell death. Whether TTR is a major cause of HFpEF, or HFpEF triggers TTR misfolding and amyloidogenesis, remains unknown. Regardless, as diagnostic technologies advance and TTR amyloid is more readily detected, it is increasingly important to understand the molecular pathogenesis of the disease in order to create new treatments aimed at improving the quality of life and survival of individuals with this condition.
Footnotes
Author Contributions
Wrote the first draft of the manuscript: VKT. Contributed to the writing of the manuscript: VKT, MM, DPJ. Agree with manuscript results and conclusions: VKT, MM, DPJ. Jointly developed the structure and arguments for the paper: VKT, MM, DPJ. Made critical revisions and approved final version: VKT, MM, DPJ. All authors reviewed and approved of the final manuscript.
ACADEMIC EDITOR: Thomas Vanhecke, Editor in Chief
FUNDING: Authors disclose no funding sources.
COMPETING INTERESTS: DPJ discloses grants and consulting fees from Pfizer Inc and consulting fees from Alnylam Pharmaceuticals Inc, outside the work presented here. Other authors disclose no competing interests.
Paper subject to independent expert blind peer review by minimum of two reviewers. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE). Provenance: the authors were invited to submit this paper.
REFERENCES
- 1.Dungu JN, Anderson LJ, Whelan CJ, Hawkins PN. Cardiac transthyretin amyloidosis. Heart. 2012;98(21):1546–54. doi: 10.1136/heartjnl-2012-301924. [DOI] [PubMed] [Google Scholar]
- 2.Coelho T, Maurer MS, Suhr OB. THAOS – the transthyretin amyloidosis outcomes survey: initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr Med Res Opin. 2013;29(1):63–76. doi: 10.1185/03007995.2012.754348. [DOI] [PubMed] [Google Scholar]
- 3.Jacobson DR, Gorevic PD, Buxbaum JN. A homozygous transthyretin variant associated with senile systemic amyloidosis: evidence for a late-onset disease of genetic etiology. Am J Hum Genet. 1990;47(1):127. [PMC free article] [PubMed] [Google Scholar]
- 4.Yamashita T, Asl KH, Yazaki M, Benson MD. A prospective evaluation of the transthyretin Ile122 allele frequency in an African-American population. Amyloid. 2005;12(2):127–30. doi: 10.1080/13506120500107162. [DOI] [PubMed] [Google Scholar]
- 5.Jacobson DR, Pastore RD, Yaghoubian R, et al. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997;336(7):466–73. doi: 10.1056/NEJM199702133360703. [DOI] [PubMed] [Google Scholar]
- 6.Buxbaum J, Alexander A, Koziol J, Tagoe C, Fox E, Kitzman D. Significance of the amyloidogenic transthyretin Val122Ile allele in African Americans in the arteriosclerosis risk in communities (ARIC) and cardiovascular health (CHS) studies. Am Heart J. 2010;159(5):864–70. doi: 10.1016/j.ahj.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buxbaum J, Jacobson DR, Tagoe C, et al. Transthyretin V122I in African Americans with congestive heart failure. J Am Coll Cardiol. 2006;47(8):1724–5. doi: 10.1016/j.jacc.2006.01.042. [DOI] [PubMed] [Google Scholar]
- 8.Jacobson D, Tagoe C, Schwartzbard A, Shah A, Koziol J, Buxbaum J. Relation of clinical, echocardiographic and electrocardiographic features of cardiac amyloidosis to the presence of the transthyretin V122I allele in older African American men. Am J Cardiol. 2011;108(3):440–4. doi: 10.1016/j.amjcard.2011.03.069. [DOI] [PubMed] [Google Scholar]
- 9.Ruberg FL, Maurer MS, Judge DP, et al. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the transthyretin amyloidosis cardiac study (TRACS) Am Heart J. 2012;164(2):222.e–8. doi: 10.1016/j.ahj.2012.04.015. [DOI] [PubMed] [Google Scholar]
- 10.Bishu K, Redfield MM. Acute heart failure with preserved ejection fraction: unique patient characteristics and targets for therapy. Curr Heart Fail Rep. 2013;10(3):190. doi: 10.1007/s11897-013-0149-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shah AM, Pfeffer MA. The many faces of heart failure with preserved ejection fraction. Nat Rev Cardiol. 2012;9(10):555. doi: 10.1038/nrcardio.2012.123. [DOI] [PubMed] [Google Scholar]
- 12.Mohammed SF, Mirzoyev SA, Edwards WD, et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail. 2014;2(2):113–22. doi: 10.1016/j.jchf.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruberg FL, Berk JL. Transthyretin (TTR) cardiac amyloidosis. Circulation. 2012;126(10):1286–300. doi: 10.1161/CIRCULATIONAHA.111.078915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maleszewski JJ, Murray DL, Dispenzieri A, et al. Relationship between monoclonal gammopathy and cardiac amyloid type. Cardiovasc Pathol. 2013;22(3):189–94. doi: 10.1016/j.carpath.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 15.Monaco HL, Rizzi M, Coda A. Structure of a complex of two plasma proteins: transthyretin and retinol-binding protein. Science. 1995;268(5213):1039–41. doi: 10.1126/science.7754382. [DOI] [PubMed] [Google Scholar]
- 16.Blake CC, Geisow MJ, Oatley SJ, Rérat B, Rérat C. Structure of prealbumin: secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 A. J Mol Biol. 1978;121(3):339–56. doi: 10.1016/0022-2836(78)90368-6. [DOI] [PubMed] [Google Scholar]
- 17.Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31. doi: 10.1186/1750-1172-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Planté-Bordeneuve V, Suhr OB, Maurer MS, White B, Grogan DR, Coelho T. The transthyretin amyloidosis outcomes survey (THAOS) registry: design and methodology. Curr Med Res Opin. 2013;29(1):77–84. doi: 10.1185/03007995.2012.754349. [DOI] [PubMed] [Google Scholar]
- 19.Penchala SC, Connelly S, Wang Y, et al. AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc Natl Acad Sci USA. 2013;110(24):9992–7. doi: 10.1073/pnas.1300761110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rapezzi C, Quarta CC, Riva L, et al. Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol. 2010;7(7):398–408. doi: 10.1038/nrcardio.2010.67. [DOI] [PubMed] [Google Scholar]
- 21.Jacobson DR, McFarlin DE, Kane I, Buxbaum JN. Transthyretin Pro55, a variant associated with early-onset, aggressive, diffuse amyloidosis with cardiac and neurologic involvement. Hum Genet. 1992;89(3):353–6. doi: 10.1007/BF00220559. [DOI] [PubMed] [Google Scholar]
- 22.Sekijima Y, Wiseman RL, Matteson J, et al. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121(1):73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 23.Sato T, Susuki S, Suico MA, et al. Endoplasmic reticulum quality control regulates the fate of transthyretin variants in the cell. EMBO J. 2007;26(10):2501–12. doi: 10.1038/sj.emboj.7601685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sousa MM, Yan SD, Stern D, Saraiva MJ. Interaction of the receptor for advanced end-glycation products (RAGE) with transthyretin triggers nuclear transcription factor kB (NF-kB) activation. Lab Invest. 2000;80(7):1101–10. doi: 10.1038/labinvest.3780116. [DOI] [PubMed] [Google Scholar]
- 25.Yan SF, Ramasamy R, Schmidt AM. The RAGE axis: a fundamental mechanism signaling danger to the vulnerable vasculature. Circ Res. 2010;106(5):842–53. doi: 10.1161/CIRCRESAHA.109.212217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teixeira PF, Cerca F, Santos SD, Saraiva MJ. Endoplasmic reticulum stress associated with extracellular aggregates: evidence from transthyretin deposition in familial amyloid polyneuropathy. J Biol Chem. 2006;281(31):21998–2003. doi: 10.1074/jbc.M602302200. [DOI] [PubMed] [Google Scholar]
- 27.Santos S, Cardoso I, Magalhães J, Saraiva M. Impairment of the ubiquitin– proteasome system associated with extracellular transthyretin aggregates in familial amyloidotic polyneuropathy. J Pathol. 2007;213(2):200–9. doi: 10.1002/path.2224. [DOI] [PubMed] [Google Scholar]
- 28.Liepnieks JJ, Wilson DL, Benson MD. Biochemical characterization of vitreous and cardiac amyloid in Ile84Ser transthyretin amyloidosis. Amyloid. 2006;13(3):170–7. doi: 10.1080/13506120600877003. [DOI] [PubMed] [Google Scholar]
- 29.Tasaki M, Ueda M, Obayashi K, et al. Effect of age and sex differences on wild-type transthyretin amyloid formation in familial amyloidotic polyneuropathy: a proteomic approach. Int J Cardiol. 2013;170(1):69–74. doi: 10.1016/j.ijcard.2013.10.033. [DOI] [PubMed] [Google Scholar]
- 30.Cornwell GG, III, Murdoch WL, Kyle RA, Westermark P, Pitkänen P. Frequency and distribution of senile cardiovascular amyloid: a clinicopathologic correlation. Am J Med. 1983;75(4):618–23. doi: 10.1016/0002-9343(83)90443-6. [DOI] [PubMed] [Google Scholar]
- 31.Zhao L, Buxbaum JN, Reixach N. Age-related oxidative modifications of transthyretin modulate its amyloidogenicity. Biochemistry. 2013;52(11):1913–26. doi: 10.1021/bi301313b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Begin ME, Langlois MF, Lorrain D, Cunnane SC. Thyroid function and cognition during aging. Curr Gerontol Geriatr Res. 2008;2008:1–11. doi: 10.1155/2008/474868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morinobu T, Tamai H, Tanabe T, et al. Plasma alpha-tocopherol, beta-carotene, and retinol levels in the institutionalized elderly individuals and in young adults. Int J Vitam Nutr Res. 1994;64(2):104–8. [PubMed] [Google Scholar]
- 34.Kyle RA, Spittell PC, Gertz MA, et al. The premortem recognition of systemic senile amyloidosis with cardiac involvement. Am J Med. 1996;101(4):395–400. doi: 10.1016/S0002-9343(96)00229-X. [DOI] [PubMed] [Google Scholar]
- 35.Ng B, Connors LH, Davidoff R, Skinner M, Falk RH. Senile systemic amyloidosis presenting with heart failure: a comparison with light chain – associated amyloidosis. Arch Intern Med. 2005;165(12):1425–9. doi: 10.1001/archinte.165.12.1425. [DOI] [PubMed] [Google Scholar]
- 36.Gonçalves I, Alves CH, Quintela T, et al. Transthyretin is up-regulated by sex hormones in mice liver. Mol Cell Biochem. 2008;317(1–2):137–42. doi: 10.1007/s11010-008-9841-2. [DOI] [PubMed] [Google Scholar]
- 37.Hammarström P, Schneider F, Kelly JW. Trans-suppression of misfolding in an amyloid disease. Science. 2001;293(5539):2459–62. doi: 10.1126/science.1062245. [DOI] [PubMed] [Google Scholar]
- 38.Berk JL, Suhr OB, Obici L, et al. Diflunisal Trial Consortium. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310(24):2658. doi: 10.1001/jama.2013.283815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Castaño A, Helmke S, Alvarez J, Delisle S, Maurer MS. Diflunisal for ATTR cardiac amyloidosis. Congest Heart Fail. 2012;18(6):315–9. doi: 10.1111/j.1751-7133.2012.00303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bulawa CE, Connelly S, Devit M, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci USA. 2012;109(24):9629–34. doi: 10.1073/pnas.1121005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785–92. doi: 10.1212/WNL.0b013e3182661eb1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Merlini G, Planté-Bordeneuve V, Judge DP, et al. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J Cardiovasc Transl Res. 2013;6(6):1011–20. doi: 10.1007/s12265-013-9512-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liepnieks JJ, Benson MD. Progression of cardiac amyloid deposition in hereditary transthyretin amyloidosis patients after liver transplantation. Amyloid. 2007;14(4):277–82. doi: 10.1080/13506120701614032. [DOI] [PubMed] [Google Scholar]
- 44.Liepnieks JJ, Zhang LQ, Benson MD. Progression of transthyretin amyloid neuropathy after liver transplantation. Neurology. 2010;75(4):324–7. doi: 10.1212/WNL.0b013e3181ea15d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819–29. doi: 10.1056/NEJMoa1208760. [DOI] [PubMed] [Google Scholar]
- 46.Ackermann EJ, Guo S, Booten S, et al. Clinical development of an antisense therapy for the treatment of transthyretin-associated polyneuropathy. Amyloid. 2012;19:43–4. doi: 10.3109/13506129.2012.673140. [DOI] [PubMed] [Google Scholar]
- 47.Sekijima Y. Recent progress in the understanding and treatment of transthyretin amyloidosis. J Clin Pharm Ther. 2014;39(3):225–33. doi: 10.1111/jcpt.12145. [DOI] [PubMed] [Google Scholar]
- 48.Gonçalves NP, Vieira P, Saraiva MJ. Interleukin-1 signaling pathway as a therapeutic target in transthyretin amyloidosis. Amyloid. 2014;21:1–10. doi: 10.3109/13506129.2014.927759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sinha S, Lopes DHJ, Du Z, et al. Lysine-specific molecular tweezers are broad-spectrum inhibitors of assembly and toxicity of amyloid proteins. J Am Chem Soc. 2011;133(42):16958–69. doi: 10.1021/ja206279b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ferreira N, Pereira-Henriques A, Attar A, et al. Molecular tweezers targeting transthyretin amyloidosis. Neurotherapeutics. 2014;11(2):450–61. doi: 10.1007/s13311-013-0256-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bourgault S, Choi S, Buxbaum JN, Kelly JW, Price JL, Reixach N. Mechanisms of transthyretin cardiomyocyte toxicity inhibition by resveratrol analogs. Biochem Biophys Res Commun. 2011;410(4):707–13. doi: 10.1016/j.bbrc.2011.04.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cardoso I, Martins D, Ribeiro T, Merlini G, Saraiva M. Synergy of combined doxycycline/TUDCA treatment in lowering transthyretin deposition and associated biomarkers: studies in FAP mouse models. J Transl Med. 2010;8(1):74. doi: 10.1186/1479-5876-8-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cardoso I, Saraiva MJ. Doxycycline disrupts transthyretin amyloid: evidence from studies in a FAP transgenic mice model. FASEB J. 2006;20:234–9. doi: 10.1096/fj.05-4509com. [DOI] [PubMed] [Google Scholar]
- 54.Ferreira N, Saraiva MJ, Almeida MR. Epigallocatechin-3-gallate as a potential therapeutic drug for TTR-related amyloidosis: “in vivo” evidence from FAP mice models. PLoS One. 2012;7(1):e29933. doi: 10.1371/journal.pone.0029933. [DOI] [PMC free article] [PubMed] [Google Scholar]