

Abstract
Background
Primary hypophysitis causes varying degrees of endocrine dysfunction and mass effect. The natural course and best treatment have not been well established.
Methods
Medical records of 22 patients who had been diagnosed with primary hypophysitis between January 2001 and March 2013 were retrospectively reviewed. Based on the anatomical location, we classified the cases as adenohypophysitis (AH), infundibuloneurohypophysitis (INH), and panhypophysitis (PH). Clinical presentation, endocrine function, pathologic findings, magnetic resonance imaging findings, and treatment courses were reviewed.
Results
Among 22 patients with primary hypophysitis, 81.8% (18/22) had involvement of the posterior pituitary lobe. Two patients of the AH (2/3, 66.6%) and three patients of the PH (3/10, 30%) groups initially underwent surgical mass reduction. Five patients, including three of the PH (3/10, 33.3%) group and one from each of the AH (1/3, 33.3%) and INH (1/9, 11.1%) groups, initially received high-dose glucocorticoid treatment. Nearly all of the patients treated with surgery or high-dose steroid treatment (9/11, 82%) required continuous hormone replacement during the follow-up period. Twelve patients received no treatment for mass reduction due to the absence of acute symptoms and signs related to a compressive mass effect. Most of them (11/12, 92%) did not show disease progression, and three patients recovered partially from hormone deficiency.
Conclusion
Deficits of the posterior pituitary were the most common features in our cases of primary hypophysitis. Pituitary endocrine defects responded less favorably to glucocorticoid treatment and surgery. In the absence of symptoms related to mass effect and with the mild defect of endocrine function, it may not require treatment to reduce mass except hormone replacement.
Keywords: Pituitary, Hypophysitis, Diabetes insipidus, Hypopituitarism, Steroids
INTRODUCTION
Primary hypophysitis is a chronic inflammatory disease of the pituitary gland that is not secondary to infection, systemic inflammatory disorders, or tumorous conditions. Primary hypophysitis is usually classified as lymphocytic, granulomatous, or xanthomatous hypophysitis based on the histologic findings. Based on the anatomical location, it is also classified into adenohypophysitis (AH), infundibuloneurohypophysitis (INH), or panhypophysitis (PH) [1]. Diffuse infiltration of the pituitary gland by inflammatory cells results in varying degrees of endocrine dysfunction due to partial or complete deficiency of pituitary hormones. Particularly, involvement of the neurohypophysis and infundibular stem can cause diabetes insipidus [2,3]. Although the recent increasing reported cases have improved our understanding of primary hypophysitis, the natural history and best treatment have not been well established [4,5]. The authors describe a single-center experience of clinical features, endocrinological findings, and treatment courses of 22 primary hypophysitis patients.
METHODS
The study subjects were 22 patients who had been diagnosed with primary hypophysitis at Samsung Medical Center, Korea between January 2001 and March 2013. They were composed of 17 women and 5 men with a mean age of 48 years (range, 21 to 67) at the time of diagnosis. Among these patients, 11 were histologically proven cases, and the others were suspected cases diagnosed by magnetic resonance imaging (MRI) findings with clinical features. Diagnosis based on MRI findings included known features suggestive of hypophysitis, including symmetric enlargement of the pituitary gland, a thickened but rarely displaced stalk, a usually intact sellar floor, homogeneity of the pituitary mass, and its intense enhancement after gadolinium. In patients with clinical symptoms and signs of diabetes insipidus, MRI findings suggestive of INH, including the loss of precontrast T1 hyperintensity in the neurohypophysis, swelling of the posterior pituitary, and thickening of the pituitary stalk were also considered sufficient for diagnosis of hypophysitis. Combined anterior pituitary stimulation testing and MRI scans of the sella area were performed in all patients at the time of diagnosis.
Based on the anatomical location, we classified these cases into AH, INH, or PH (Table 1) [1]. Clinical presentation, endocrine function, pathologic findings, MRI findings, and treatment courses were reviewed retrospectively from electronic medical records. This study was approved by the Institutional Review Board of Samsung Medical Center.
Table 1.
Classification of 22 Patients with Primary Hypophysitis Based on the Anatomical location

A, anterior hypophysis; N, infundibuloneurohypophysis.
RESULTS
Clinical presentation
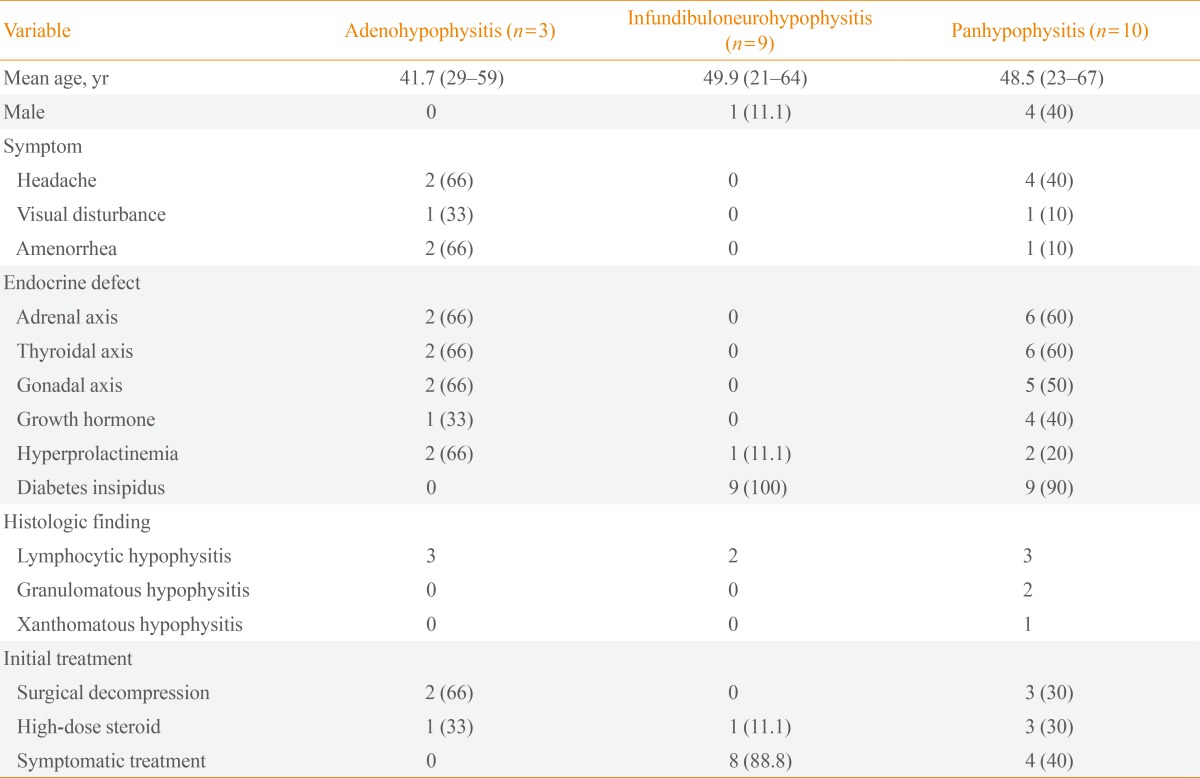
The most common clinical symptoms of this study group were polyuria and polydipsia (18/22, 81.8%), followed by headache (6/22, 27.2%), general weakness (5/22, 22%), and amenorrhea (3/22, 13%). Visual disturbance, such as diplopia and visual field defect, was noted in two patients (9%). Based on the anatomical location classification, the most common symptoms in AH were headache and amenorrhea (2/3, 66%) (Table 2). In INH, general weakness was the only symptom except for polyuria and polydipsia. Only one case was associated with a recent pregnancy, and her initial symptoms of headache and visual disturbance appeared during the third trimester. Two patients had coexisting autoimmune disease, one with Hashimoto's thyroiditis and another with limited Wegener's granulomatosis.
Table 2.
Clinical Features, Pathologic Findings, and Treatments of 22 Patients with Primary Hypophysitis Based on the Anatomical Location
Values are expressed as mean (range) or number (%).
Endocrinological assessment
Among 22 patients with primary hypophysitis, 81.8% (18/22) had involvement of the posterior pituitary lobe resulting in diabetes insipidus. Exclusive involvement of the infundibular stem or the posterior lobe was found in 40.9% (9/22) of cases (Table 2). The most frequently observed defect in the anterior pituitary was corticotrophs (8/22, 36.3%) and thyrotrophs (8/22, 36.3%), followed by gonadotrophs (7/22, 31.8%). Impaired function of somatotrophs was seen in five of 22 cases (22.7%). Prolactin levels were increased in 22.7% (5/22) of cases. In patients with AH, deficiency in adrenocorticotropic hormone (ACTH), thyroid stimulating hormone (TSH), and luteinizing hormone (LH)/follicle stimulating hormone (FSH) was seen in 66.6% (2/3), and prolactin levels were also increased in 66.6% (2/3) of cases. All patients with INH and PH had dysfunction in secretion of antidiuretic hormone. In patients with PH, deficiency of ACTH and TSH was noted equal in 60% (6/10), followed by LH/FSH deficiency (5/10, 50%) and growth hormone (GH) deficiency (4/10, 40%) (Table 2).
Findings on MRI
In the AH and PH (n=13) cases, symmetric enlargement of the entire pituitary gland with homogeneous enhancement after gadolinium was seen in seven patients. Among these 13 patients, thickening of the pituitary stalk was seen in eight patients (61.5%), and suprasellar extension was seen in six patients (46.1%). In one patient, the finding was seen as prominent cystic area on MRI. In the INH (n=9) cases, all patients showed thickening of the pituitary stalk and loss of T1 hyperintensity in the neurohypophysis on MRI. One of these patients also demonstrated suprasellar extension. One case showed swelling of the posterior pituitary gland. Among all patients (n=22), an abnormally thickened infundibular stalk was found in 17 patients, and loss of T1 hyperintensity in the posterior lobe was seen in 18 patients.
Pathological examination
A histological diagnosis was available in 11 patients (50%), including six patients who underwent surgical treatment for mass reduction and five patients who underwent biopsy for diagnosis through a transsphenoidal approach. Among these patients, hypophysitis was diagnosed as lymphocytic in eight patients, granulomatous in two patients, and xanthomatous in one patient (Table 2). The histologic diagnosis in the patient with a prominent cystic lesion on MRI was xanthomatous hypophysitis. Patients with lymphocytic hypophysitis showed varied anatomical involvement: three patients had deficits in the anterior pituitary, two patients in the posterior pituitary, and three patients in both pituitary lobes. Patients with xanthomathous hypophysitis and granulomatous hypophysitis had deficits in both the anterior and posterior pituitary (Table 2).
Treatment and patient outcomes
Two patients of the AH and three patients of the PH groups initially underwent surgical mass reduction for decompression through a transsphenoidal approach (Table 2). Five patients, including three from the PH and one from each of the AH and INH groups, initially received high-dose glucocorticoid treatment. Eight patients with INH and four patients with PH received no treatment for mass reduction due to the absence of acute symptoms and signs related to mass effect such as headache, visual field defect, or optic chiasm compression.
Fourteen patients (63.6%) underwent further anterior pituitary stimulation testing at least once during the follow-up period. Eight other patients with INH followed only basal pituitary hormone. In cases of surgical or high-dose steroid treatment, MRI scans were repeated within 3 months of treatment. Follow-up MRI scans were then performed regularly every 1 or 2 years in all patients (except those lost to follow-up) during the remaining follow-up period. Mean duration of follow-up was 57 months (range, 7 to 138; median, 48).
All of the patients who initially underwent surgical treatment and four of five patients who initially received high-dose glucocorticoids showed mass reduction on follow-up MRI images (Table 3). One patient in the high-dose glucocorticoid treatment group demonstrated no response to treatment; however, this patient has not experienced disease progression during 19 months of follow-up without alternative treatment, as seen in most patients (11 of 12) in the non-treatment group. During the follow-up period, two (one in each treatment group) of 10 patients who responded to initial surgical or high-dose glucocorticoid treatment presented with a recurring pituitary mass, and one patient in the non-treatment group had an increase in pituitary mass size. These three patients required a second alternative treatment to reduce the pituitary mass (Tables 3, 4). Among the patients who underwent surgical resection, recurring pituitary mass was seen in one patient with xanthomatous hypophysitis, developed visual disturbance due to suprasellar extension 10 months after surgery. This patient underwent adjunctive high-dose glucocorticoid treatment (methylprednisolone 500 mg intravenous for 3 days) and has shown good response in mass reduction without disease progression during 15 months of follow-up (Fig. 1) [6]. One patient treated with high-dose glucocorticoids (1 mg/kg dose of prednisolone) showed good response in headache and mass reduction; however, she was unable to reduce her prednisolone below 15 mg due to headache. Unfortunately, during long-term treatment with low-dose prednisolone, she gained 30 kg, developed diabetes, and experienced an increase in the size of the pituitary mass. Twelve months after initial steroid treatment, she underwent hypophysectomy, and lymphocytic hypophysitis was histologically confirmed. Her headaches resolved without disease progression during 19 months after surgery. She then died from cerebral infarction. The panhypopituitarism in these two patients did not improve at all despite treatment. Among patients without surgical or high-dose steroid treatment, one patient with INH received high-dose glucocorticoids (1 mg/kg dose of prednisolone) due to progression in mass size during 19 months of follow-up (Fig. 1). Hormone deficiency remained unchanged and required desmopressin despite the high-dose steroid treatment. A follow-up MRI could not be performed, as the patient was lost to follow-up.
Table 3.
Treatment Courses with Follow-Up Endocrine and Image Outcomes in 22 Patients
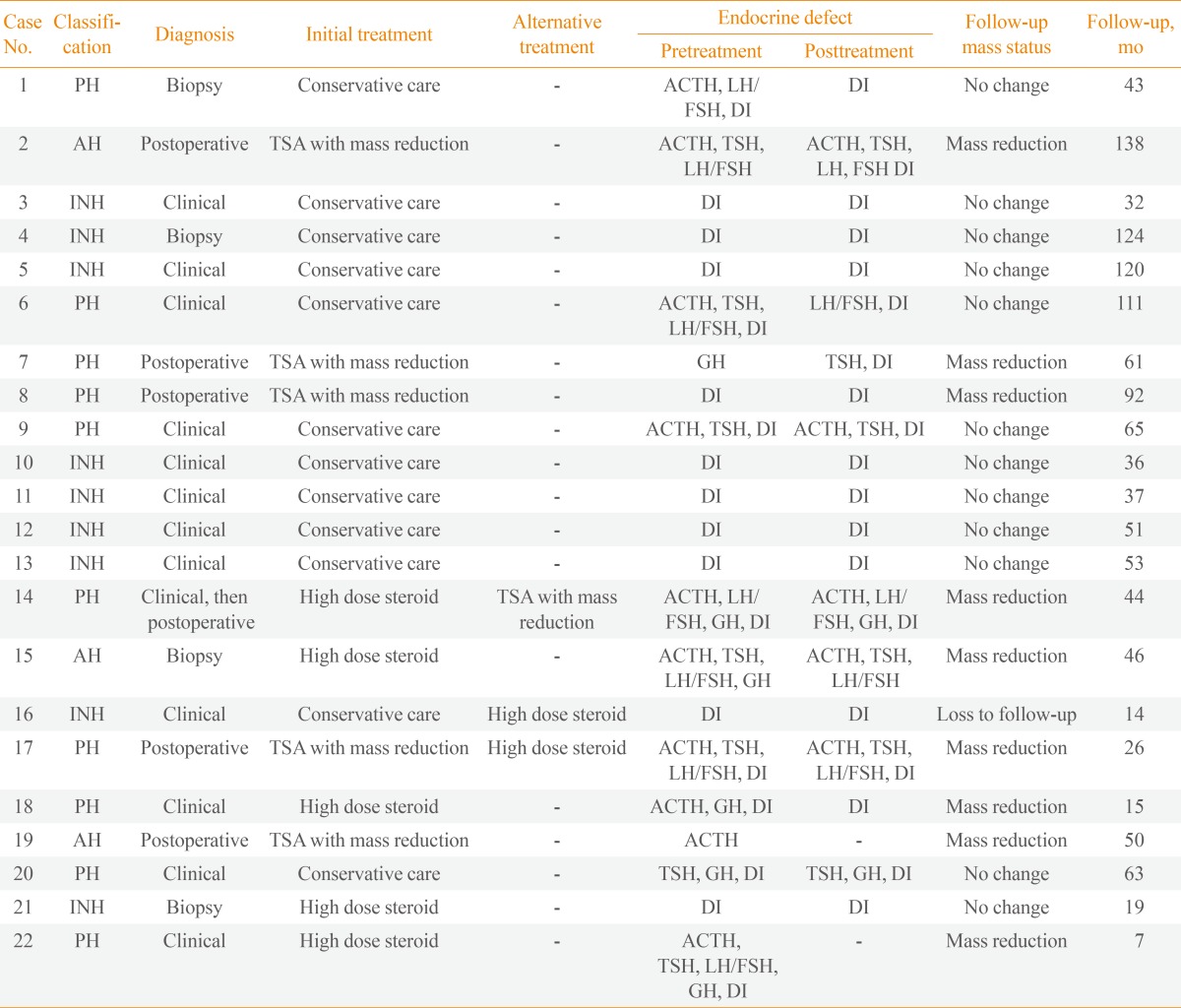
PH, panhypophysitis; ACTH, adrenocorticotropic hormone; LH, luteinizing hormone; FSH, follicle stimulating hormone; DI, diabetes insipidus; AH, adenohypophysitis; TSA, transsphenoidal approach; TSH, thyroid stimulating hormone; INH, infundibuloneurohypophysitis; GH, growth hormone.
Table 4.
Pretreatment and Posttreatment Endocrine Assessment of the Patients Based on the Treatment
Values are expressed as mean (range).
Fig. 1.

Summary of treatment courses of 22 patients with primary hypophysitis. TSA, transsphenoidal approach.
aDue to regrowth of mass; bDue to steroid related side effect and progress with steroid tapering; cDue to progression in mass size.
Seven patients received high-dose glucocorticoids initially or alternatively, and five patients showed mass reduction. Among these five patients, four patients had symptoms for less than 3 months before steroid treatment. One patient who didn't respond to high-dose steroids had symptoms for 5 months.
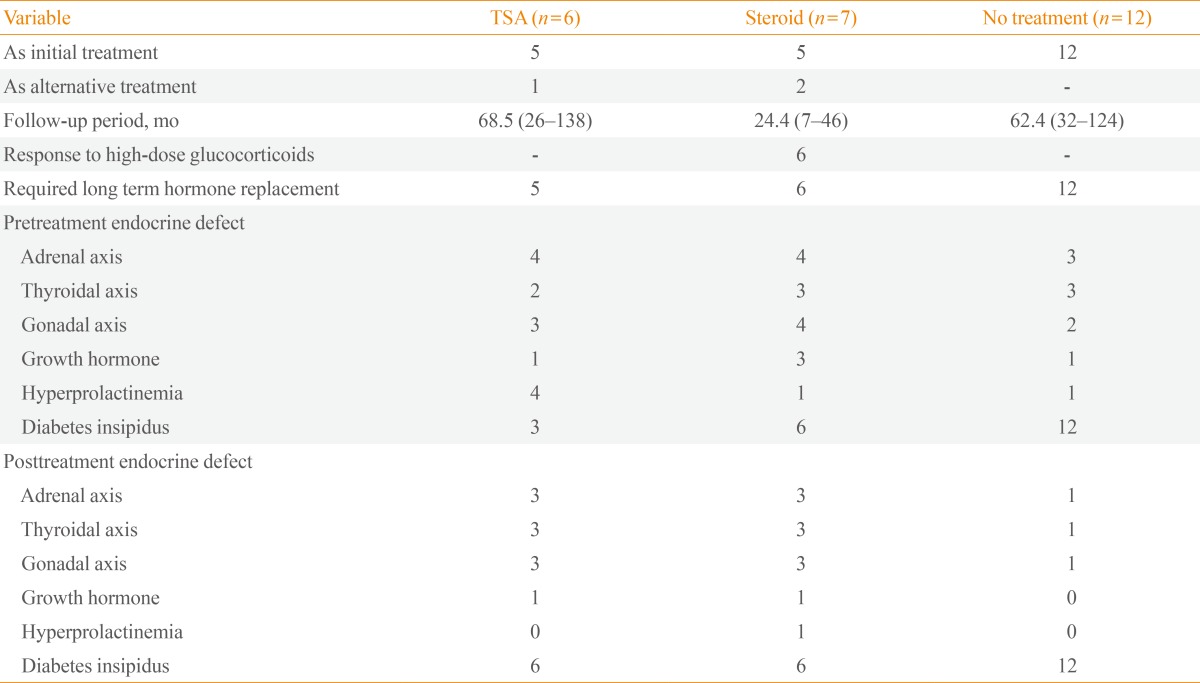
Nearly all of the patients treated with surgery or high-dose steroids (9/11, 82%) required continuous hormone replacement during the follow-up period. Only two patients have shown complete improvement after mass reduction without need for hormone replacement (Table 4). One patient had PH due to granulomatous hypophysitis associated with Wegener's granulomatosis, and another patient had AH due to lymphocytic hypophysitis. Pituitary mass reduction was achieved by high-dose steroid treatment and surgery, respectively. Among patients without surgical or high-dose steroid treatment, two patients showed spontaneous partial recovery in hormone deficiency (Table 3). Also, two patients treated with high-dose steroids recovered partially from hormone deficiency. All patients treated with high-dose steroids had weight gain, and one patient developed avascular necrosis of the femoral head. Three of six patients who received tumor removal surgery developed additional hypopituitarism, such as diabetes insipidus or hypothyroidism, after surgery (Table 4).
DISCUSSION
The clinical presentations of primary hypophysitis are variable including symptoms related to sellar compression, hypopituitarism, and diabetes insipidus. The presentation depends on whether the immune system affects the anterior lobe, posterior lobe, or both [7]. In a review analyzing 379 cases of primary lymphocytic hypophysitis based on 370 articles published as case reports or small case series [1], 89.7% (340/379) had symptoms from partial or complete deficiency of the anterior pituitary hormones, and 35.3% (134/379) developed diabetes insipidus. PH presenting with both symptoms was seen in 25.1% (95/379) and INH was seen only in 10.2% (39/379). In our study, among 22 patients with primary hypophysitis, 59.1% (13/22) had symptoms due to anterior pituitary hormone deficiency. Also, 81.8% (18/22) had diabetes insipidus, and 40.9% (9/22) had this symptom exclusively. Compared with the previous review [1,8], it is interesting that symptoms due to posterior pituitary hormone deficiency were most common in our study group. Some primary hypophysitis cases may still go undiagnosed because of their indolent, subclinical course [2,9]. In particular, acute symptoms related to mass effect were rare in INH [1,7]. Thus, cases of primary hypophysitis with only polydipsia and polyuria may not be published as case reports and might result in underestimation of INH case number [9]. The most common deficit of the anterior pituitary hormones was ACTH followed by TSH, GH, and gonadotropins, similar to previous review [1,7,8].
The natural history of primary hypophysitis is variable and unpredictable [5,10,11]. Typically, it follows a progressive course in which the pituitary initially becomes inflamed, edematous, and enlarged, and then the patient develops symptoms secondary to mass effects. With destruction of pituicytes, the parenchyma is replaced by fibrosis resulting in pituitary atrophy leading to hypopituitarism [1]. In some cases, the disease course was aggressive and rapidly progressed with neurologic deficits [1,5,7,12,13]. Meanwhile, partial or full recovery of pituitary function as well as resolution of pituitary masses in the absence of any intervention has also been well documented in several case reports and reviews [14,15,16,17,18,19,20]. In our study, 12 patients had no surgical or steroid treatment because of the absence of acute symptoms and signs related to the mass effect. In these patients, no one has shown complete spontaneous recovery of pituitary function and mass; however, two patients have shown spontaneous partial recovery of pituitary function without mass reduction, and nine patients remained unchanged in pituitary function and mass size without disease progression during follow-up. Only one patient progressed in mass size without further changes in hormone deficiency.
The treatment of primary hypophysitis was only symptomatic in considerable cases, including reducing the size of the pituitary mass or replacing the deficient endocrine function [1,8]. Surgery, in addition to providing a histological diagnosis, was very effective in achieving decompression of the sellar mass and promptly resolving symptoms related to mass effect. Glucocorticoids were also effective for reducing the size of the pituitary mass or the thickened stalk because of their well-known lymphocytolytic properties [21,22,23]. There were some reports showing recovery of anterior and posterior pituitary function [21,24,25] as well as mass reduction [21] after glucocorticoid treatment. In our study, all of the patients who underwent surgical treatment and five of seven patients who received high-dose glucocorticoids showed mass reduction. However, complete recovery of pituitary function without need for hormone replacement was seen only in two patients (18%), suggesting that pituitary endocrine defect was less likely to respond to both glucocorticoid and surgical treatment despite apparent response to treatment via mass reduction. Pituitary endocrine defects are due to diffuse lymphocytic infiltration resulting in cell destruction, rather than compression of normal parenchyma by a pituitary mass [1]. Thus, as seen in our study, patients presenting with hypopituitarism or diabetes insipidus rarely benefit from surgical decompression, and response to glucocorticoid treatment varies with disease stage. In particular, it is likely that fibrous stages of hypophysitis will be unresponsive to glucocorticoids [1].
In the present study, the majority of hypophysitis cases without acute symptoms related to mass effect did not show disease progression during follow-up in the absence of any intervention. Although surgical treatment was very effective for achieving decompression of pituitary masses, it rarely improved pituitary endocrine deficiency. Surgery also carries a risk of complications such as bleeding, cerebrospinal fluid leaks, and diabetes insipidus [4,8,18]. Glucocorticoids were effective for reducing the size of the pituitary mass. In addition, although the occurrence of spontaneous recovery can be a confounder, some cases (three of seven) have shown complete or partial improvement of hormonal status in response to glucocorticoids. However, complications related to the use of glucocorticoids, such as diabetes, weight gain, and avascular necrosis of bone, developed. Appropriate management remains still controversial due to variability in the natural history of primary hypophysitis [4,5]. However, current literature suggests that surgery should be performed only in the presence of serious and progressive deficits of visual fields, visual acuity, or ocular movements, and not responsive to medical treatment [4,26]. Thus, if a diagnosis of primary hypophysitis is suspected clinically and there are no urgent visual symptoms, the use of glucocorticoids are preferable to surgery. Additionally, considering our experiences, it seems reasonable to suggest that there is no need for other treatment except for hormone replacement in the absence of symptoms related to mass effect.
In conclusion, the deficit of the posterior pituitary was the most common feature in our cases of primary hypophysitis. It seems reasonable to use glucocorticoids as the first-line treatment for primary hypophysitis; however, pituitary endocrine defects responded less favorably to glucocorticoid treatment and surgery despite mass reduction. In the absence of symptoms related to mass effect and with mild defects in endocrine function, it may not require treatment to reduce mass except hormone replacement.
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.Caturegli P, Newschaffer C, Olivi A, Pomper MG, Burger PC, Rose NR. Autoimmune hypophysitis. Endocr Rev. 2005;26:599–614. doi: 10.1210/er.2004-0011. [DOI] [PubMed] [Google Scholar]
- 2.Abe T. Lymphocytic infundibulo-neurohypophysitis and infundibulo-panhypophysitis regarded as lymphocytic hypophysitis variant. Brain Tumor Pathol. 2008;25:59–66. doi: 10.1007/s10014-008-0234-8. [DOI] [PubMed] [Google Scholar]
- 3.Kojima H, Nojima T, Nagashima K, Ono Y, Kudo M, Ishikura M. Diabetes insipidus caused by lymphocytic infundibuloneurohypophysitis. Arch Pathol Lab Med. 1989;113:1399–1401. [PubMed] [Google Scholar]
- 4.Leung GK, Lopes MB, Thorner MO, Vance ML, Laws ER., Jr Primary hypophysitis: a single-center experience in 16 cases. J Neurosurg. 2004;101:262–271. doi: 10.3171/jns.2004.101.2.0262. [DOI] [PubMed] [Google Scholar]
- 5.Cosman F, Post KD, Holub DA, Wardlaw SL. Lymphocytic hypophysitis: report of 3 new cases and review of the literature. Medicine (Baltimore) 1989;68:240–256. doi: 10.1097/00005792-198907000-00006. [DOI] [PubMed] [Google Scholar]
- 6.Joung JY, Jeong H, Cho YY, Huh K, Suh YL, Kim KW, Bae JC. Steroid responsive xanthomatous hypophysitis associated with autoimmune thyroiditis: a case report. Endocrinol Metab (Seoul) 2013;28:65–69. doi: 10.3803/EnM.2013.28.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rivera JA. Lymphocytic hypophysitis: disease spectrum and approach to diagnosis and therapy. Pituitary. 2006;9:35–45. doi: 10.1007/s11102-006-6598-z. [DOI] [PubMed] [Google Scholar]
- 8.Gutenberg A, Hans V, Puchner MJ, Kreutzer J, Brück W, Caturegli P, Buchfelder M. Primary hypophysitis: clinical-pathological correlations. Eur J Endocrinol. 2006;155:101–107. doi: 10.1530/eje.1.02183. [DOI] [PubMed] [Google Scholar]
- 9.Imura H, Nakao K, Shimatsu A, Ogawa Y, Sando T, Fujisawa I, Yamabe H. Lymphocytic infundibuloneurohypophysitis as a cause of central diabetes insipidus. N Engl J Med. 1993;329:683–689. doi: 10.1056/NEJM199309023291002. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka S, Tatsumi KI, Kimura M, Takano T, Murakami Y, Takao T, Hashimoto K, Kato Y, Amino N. Detection of autoantibodies against the pituitary-specific proteins in patients with lymphocytic hypophysitis. Eur J Endocrinol. 2002;147:767–775. doi: 10.1530/eje.0.1470767. [DOI] [PubMed] [Google Scholar]
- 11.Nishiki M, Murakami Y, Ozawa Y, Kato Y. Serum antibodies to human pituitary membrane antigens in patients with autoimmune lymphocytic hypophysitis and infundibuloneurohypophysitis. Clin Endocrinol (Oxf) 2001;54:327–333. doi: 10.1046/j.1365-2265.2001.01210.x. [DOI] [PubMed] [Google Scholar]
- 12.Lury KM. Inflammatory and infectious processes involving the pituitary gland. Top Magn Reson Imaging. 2005;16:301–306. doi: 10.1097/01.rmr.0000224686.21748.ea. [DOI] [PubMed] [Google Scholar]
- 13.Bellastella A, Bizzarro A, Coronella C, Bellastella G, Sinisi AA, De Bellis A. Lymphocytic hypophysitis: a rare or underestimated disease. Eur J Endocrinol. 2003;149:363–376. doi: 10.1530/eje.0.1490363. [DOI] [PubMed] [Google Scholar]
- 14.Ishihara T, Hino M, Kurahachi H, Kobayashi H, Kajikawa M, Moridera K, Ikekubo K, Hattori N. Long-term clinical course of two cases of lymphocytic adenohypophysitis. Endocr J. 1996;43:433–440. doi: 10.1507/endocrj.43.433. [DOI] [PubMed] [Google Scholar]
- 15.Castle D, de Villiers JC, Melvill R. Lymphocytic adenohypophysitis. Report of a case with demonstration of spontaneous tumour regression and a review of the literature. Br J Neurosurg. 1988;2:401–405. doi: 10.3109/02688698809001013. [DOI] [PubMed] [Google Scholar]
- 16.Leiba S, Schindel B, Weinstein R, Lidor I, Friedman S, Matz S. Spontaneous postpartum regression of pituitary mass with return of function. JAMA. 1986;255:230–232. [PubMed] [Google Scholar]
- 17.Zeller JR, Cerletty JM, Rabinovitch RA, Daniels D. Spontaneous regression of a postpartum pituitary mass demonstrated by computed tomography. Arch Intern Med. 1982;142:373–374. [PubMed] [Google Scholar]
- 18.Hashimoto K, Takao T, Makino S. Lymphocytic adenohypophysitis and lymphocytic infundibuloneurohypophysitis. Endocr J. 1997;44:1–10. doi: 10.1507/endocrj.44.1. [DOI] [PubMed] [Google Scholar]
- 19.Bitton RN, Slavin M, Decker RE, Zito J, Schneider BS. The course of lymphocytic hypophysitis. Surg Neurol. 1991;36:40–43. doi: 10.1016/0090-3019(91)90131-r. [DOI] [PubMed] [Google Scholar]
- 20.McGrail KM, Beyerl BD, Black PM, Klibanski A, Zervas NT. Lymphocytic adenohypophysitis of pregnancy with complete recovery. Neurosurgery. 1987;20:791–793. doi: 10.1227/00006123-198705000-00021. [DOI] [PubMed] [Google Scholar]
- 21.Kristof RA, Van Roost D, Klingmuller D, Springer W, Schramm J. Lymphocytic hypophysitis: non-invasive diagnosis and treatment by high dose methylprednisolone pulse therapy. J Neurol Neurosurg Psychiatry. 1999;67:398–402. doi: 10.1136/jnnp.67.3.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nussbaum CE, Okawara SH, Jacobs LS. Lymphocytic hypophysitis with involvement of the cavernous sinus and hypothalamus. Neurosurgery. 1991;28:440–444. doi: 10.1097/00006123-199103000-00019. [DOI] [PubMed] [Google Scholar]
- 23.Feigenbaum SL, Martin MC, Wilson CB, Jaffe RB. Lymphocytic adenohypophysitis: a pituitary mass lesion occurring in pregnancy. Proposal for medical treatment. Am J Obstet Gynecol. 1991;164(6 Pt 1):1549–1555. doi: 10.1016/0002-9378(91)91435-y. [DOI] [PubMed] [Google Scholar]
- 24.Yamagami K, Yoshioka K, Sakai H, Fukumoto M, Yamakita T, Hosoi M, Ishii T, Sato T, Tanaka S, Fujii S. Treatment of lymphocytic hypophysitis by high-dose methylprednisolone pulse therapy. Intern Med. 2003;42:168–173. doi: 10.2169/internalmedicine.42.168. [DOI] [PubMed] [Google Scholar]
- 25.Beressi N, Cohen R, Beressi JP, Dumas JL, Legrand M, Iba-Zizen MT, Modigliani E. Pseudotumoral lymphocytic hypophysitis successfully treated by corticosteroid alone: first case report. Neurosurgery. 1994;35:505–508. doi: 10.1227/00006123-199409000-00020. [DOI] [PubMed] [Google Scholar]
- 26.Cheung CC, Ezzat S, Smyth HS, Asa SL. The spectrum and significance of primary hypophysitis. J Clin Endocrinol Metab. 2001;86:1048–1053. doi: 10.1210/jcem.86.3.7265. [DOI] [PubMed] [Google Scholar]