

Abstract

Carbamoyl anions were found to smoothly react with chiral N-phosphonyl imines in toluene at −78 °C to r.t. using LiHMDS as the base. Group-assisted purification (GAP) has been utilized to give the pure amides without using column chromatography or recrystallization. The asymmetric reaction resulted in chiral N-phosphonyl amino amides with good to excellent yields (71–99%) and good crude diastereoselectivities (dr 84:16–95:5). In this GAP procedure, the crude solids are washed with diethyl ether to afford the pure products, as revealed by 1H NMR analysis; GAP washing consistently increases the diastereopurity of the products, resulting in excellent diastereoselectivities, often with final dr > 99:1. Interestingly, the diastereoenriched products can be obtained either in the ether solution or as the suspended solid, depending on the substrate.
Introduction
Chiral amines are ubiquitous throughout natural products and pharmaceuticals, and their efficient and economical synthesis is still a highly active research area.1−6 The most important subclass of chiral amines are chiral α-amino amides, which form the basic chemical unit of amino acids and peptides.7 The synthesis of unnatural α-amino amides is often carried out via a Strecker-type process.8−11 This process frequently employs the cyanide ion, which requires two additional steps using harsh conditions to achieve the desired amides. Strong acids and heat are usually needed to convert the cyanide group into the carboxylic acid, followed by amide formation.12−14 Phenylglycine has been shown to racemize under these conditions,15 and therefore, the synthesis of phenylglycine amides would be better accomplished through other routes. One alternative for the synthesis of phenylglycine amides is through the direct addition of a carbamoyl anion to the corresponding imine. Carbamoyl anions can be easily generated from the appropriate formamide through simple deprotonation with amide bases, such as lithium diisopropyl amide.16 Senanayake and co-workers recently reported the asymmetric carbamoyl anion addition to imines as a direct synthetic route to chiral α-amino amides, through the use of chiral N-sulfinyl auxiliary chemistry.17 In their report, the carbamoyl anion can directly add to the imine of choice, affording the α-amino amide in one step under mild conditions.
Over the past few years, our research group has focused on the synthesis of various chiral N-phosphonyl and N-phosphinyl auxiliaries, for use in asymmetric syntheses of chiral amines from the chiral auxiliary-protected imines.18−21 This new class of chiral auxiliaries has been applied to various asymmetric reactions, such as borylation, the Umpolung reaction, the aza-Henry reaction, and several others.22−25 In addition to their ability to effectively induce asymmetry in a variety of ways, these new auxiliaries also have the added bonus of simplifying the purification process by eliminating the need for silica column chromatography. While silica column chromatography is a standard purification method used in organic synthesis, it is often unsuitable for large-scale purification due to the high cost of silica. Also, the large amount of solvent waste generated can prove harmful to the environment. In contrast, compounds containing our phosphonyl auxiliaries can be purified by washing the crude solid products with simple solvents or solvent mixtures.22−25 This cuts costs by reducing labor and by eliminating the expensive silica gel associated with column chromatography; it also greatly reduces solvent waste, creating a more environmentally friendly procedure. In some cases, this washing procedure can even increase the diastereomeric and/or enantiomeric purity of the final product.24−26 This simplified and more economical purification method has been termed GAP chemistry: A chemistry for organic synthesis that avoids traditional purification methods such as chromatography and/or recrystallization. GAP chemistry requires considerations in regard to (1) adequate stability and chemical reactivity of functional group-attached substrates; (2) solid products that are soluble in some solvents (e.g., THF and DCM), but not well soluble in some other solvents (e.g., petroleum ethers, hexane, and their cosolvents with EtOAc, etc.); (3) stereoselectivity control in asymmetric reactions (for chiral GAP reagents); (4) substrate scope; (5) cleavage and recyclability for reuse, etc. In this paper, we report the asymmetric addition of carbamoyl anions to chiral N-phosphonyl imines for the synthesis of various derivatized phenylglycine amides, followed by GAP (Scheme 1).
Scheme 1.

Results and Discussion
Our studies began by optimizing the reaction conditions (Table 1). We started by testing the reaction in toluene at −78 °C using LDA, one of the most common amide bases and a relatively standard condition for carbamoyl anion additions.16,17,27 Our starting substrate was 1-protected benzylidine imine; to our surprise, no desired product was observed. 31P and 1H NMR analysis of the reaction mixture revealed that the imine was suffering from decomposition, affording benzaldehyde and 1-NH2 as the reaction products. In an attempt to stabilize the imine, various solvents were tested, including THF, DCM, acetonitrile, and DMF, but none were successful as all gave the same, undesired decomposition product (entries 2–5). In an effort to stabilize the imine by weakening the electron-withdrawing nature of the auxiliary, we changed from N-phosphinyl 1 to N-phosphonyl 2. The change proved effective, and we obtained the desired product in 70% conversion with dr = 75:25 (entry 6). Changing the base to LiHMDS further improved the reaction, increasing the conversion to 87% (entry 7). Changing the solvent to THF reduced the conversion to 61%, affording a greater portion of the decomposition byproducts (entry 8). In an effort to increase the dr, we changed the auxiliary to 3. Using toluene as the solvent, with LiHMDS as the base, we obtained our best result with 91% conversion to product and dr = 85:15 (entry 9).
Table 1. Optimization of Reaction Conditions and Auxiliary.
| entry | aux* | solvent | base | yield (%)a | drb |
|---|---|---|---|---|---|
| 1 | 1 | PhCH3 | LDA | 0c | N/A |
| 2 | 1 | THF | LDA | 0c | N/A |
| 3 | 1 | DCM | LDA | 0c | N/A |
| 4 | 1 | MeCNd | LDA | 0c | N/A |
| 5 | 1 | DMFd | LDA | 0c | N/A |
| 6 | 2 | PhCH3 | LDA | 70 | 75:25 |
| 7 | 2 | PhCH3 | LiHMDS | 87 | 75:25 |
| 8 | 2 | THF | LiHMDS | 61 | 75:25 |
| 9 | 3 | PhCH3 | LiHMDS | 91 | 85:15 |
% conversion based on 31P NMR of reaction mixture.
Based on analysis of 1H and 31P NMR data of reaction mixture.
Imine decomposed into phosphinamide and benzaldehyde.
Reaction mixture solidified at −78 °C, then liquefied with slow warming to r.t.
We next examined the GAP capabilities of this product using compound 5a. Washing the crude (after extraction) with diethyl ether showed promise by providing a white solid after washing. To our delight, NMR analysis of the solid after washing showed slight diastereoenrichment. We later found that suspension of the crude solid in diethyl ether for 24 h, followed by filtration, could afford a highly diastereoenriched product as the suspended solid. During our examination of the substrate scope, we also discovered that some substrates were diastereoenriched in the ether solution, and not in the suspended solid. This is in direct contrast to our previous observations of GAP chemistry, where washing has consistently enriched the solid products by removing the minor isomer in the wash solution. We have termed this observation as a reverse-GAP stereoselectivity enrichment, due to its reverse nature from the previously observed GAP phenomenon. As can be seen in Table 2, both GAP and reverse-GAP enhancements are highly effective at diastereoenrichment, with seven examples showing dr > 99:1. As an added bonus, the phase not containing the enriched product generally has a diastereomeric ratio near 50:50. This phenomenon is equally true for both the GAP and the reverse-GAP enhancements, and results in high yields for the isolated, enriched products. This can be easily seen through 31P NMR analysis of two substrates: 5c for the GAP process, and 5d for the reverse-GAP stereoselectivity enhancement (Figure 1). For the reverse-GAP enhancement, pure enriched products can be precipitated from the diethyl ether solution via addition of petroleum ether, to remove any nondiastereomeric impurities, which tend to stay in the ether solution during GAP ether washing. In order to isolate the α-amino amides, the auxiliary can be cleaved from the products under mild conditions, using a 5:1 mixture of methanol:1.0 N HCl (aq) at room temperature over 1 h, followed by Boc protection, affording 6c, which is a known compound28 (Scheme 2).
Table 2. Substrate Scope with GAP and Reverse-GAP Purification.
| imine | product | R | NR′2 | yield (%)a | drb | GAP yield (%)c | GAP dr (g/rg)d |
|---|---|---|---|---|---|---|---|
| 4a | 5a | Ph | NMe2 | 74 | 85:15 | 45 | 93:7 (g) |
| 4b | 5b | Ph | pyrrolidine | 99 | 84:16 | 55 | 93:7 (g) |
| 4c | 5c | Ph | morpholine | 97 | 87:13 | 63 | >99:1 (g) |
| 4d | 5d | 1-Nap | morpholine | 90 | 85:15 | 54 | >99:1 (rg) |
| 4e | 5e | p-FPh | morpholine | 73 | 86:14 | 60 | 92:8 (g) |
| 4f | 5f | p-ClPh | morpholine | 86 | 86:14 | 50 | >99:1 (rg) |
| 4g | 5g | m-BrPh | morpholine | 81 | 86:14 | 52 | 95:5 (g) |
| 4h | 5h | p-MePh | morpholine | 84 | 86:14 | 41 | >99:1 (rg) |
| 4i | 5i | o-MePh | morpholine | 71 | 84:16 | 36 | >99:1 (g) |
| 4j | 5j | p-BnOPh | morpholine | 99 | 85:15 | 62 | >99:1 (rg) |
| 4k | 5k | tBu | morpholine | 75 | 95:5 | 59 | >99:1 (rg) |
Isolated crude yields after workup and extraction.
Crude dr as determined by 1H and 31P NMR analysis of the crude solids.
Isolated total yields of purified compounds after GAP ether washing.
Products purified by either the GAP (g) or reverse-GAP (rg) enrichment; dr values after GAP ether washing as determined by 1H and 31P NMR analysis.
Figure 1.

31P NMR spectra of solid and solution phase after GAP ether washing for substrates 5c and 5d.
Scheme 2.
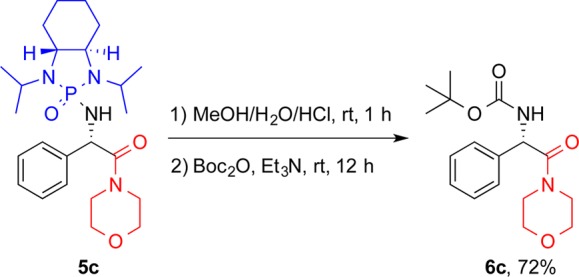
Absolute configuration was determined from X-ray analysis of 5c, revealing the newly formed stereocenter to be (S). The crystal structure is shown in Figure 2. From this information, we can propose a possible transition state for this transformation. As seen in Figure 3, we propose a six-membered transition state, with the lithium coordinating both to the phosphonyl and the carbamoyl oxygens, as well as the carbamoyl carbon. Studies by Senanayake and co-workers have shown through 13C NMR data that, after carbamoyl anion formation, the lithium is coordinated to both the carbamoyl carbon and oxygen, and so this has been included in our transition state.17 The position of the isopropyl group partially below the ring creates a strong 1,3-diaxial steric interaction, causing the imine to orient itself such that the R group is in the equatorial position. This facilitates Re face attack by the carbamoyl carbon, selectively forming the (S) isomer.
Figure 2.
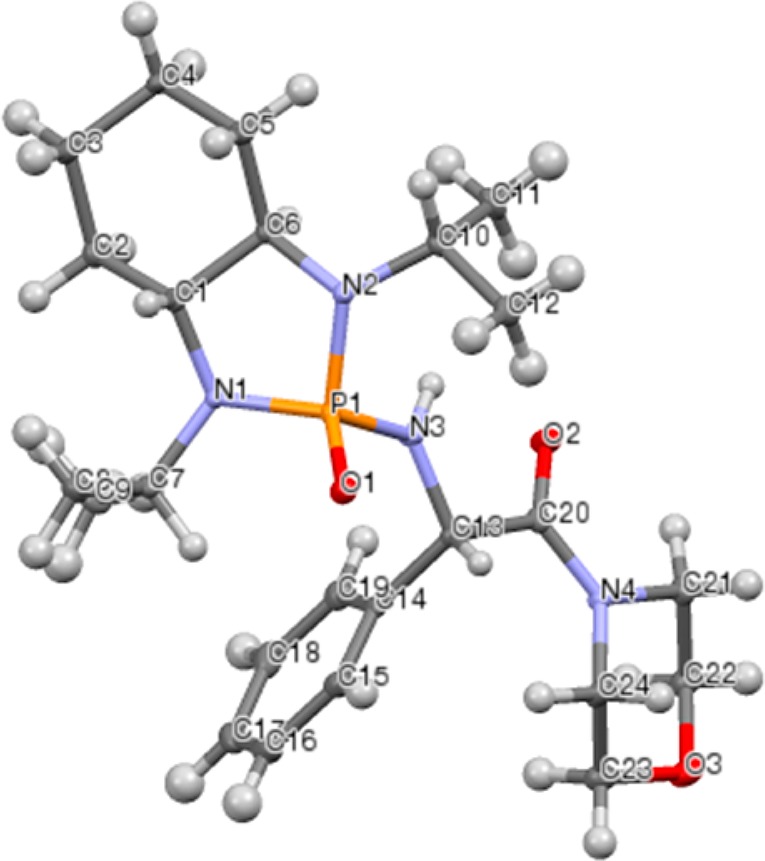
X-ray structure of 5c.
Figure 3.
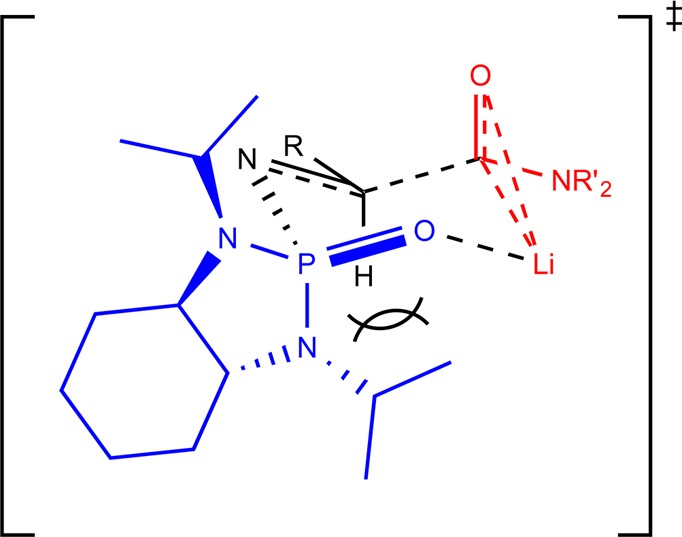
Proposed six-membered transition state.
Conclusion
In summary, we have been able to successfully apply the carbamoyl anion addition to chiral N-phosphonyl imines for the direct synthesis of phenylglycine amide derivatives, without using column chromatography or recrystallization. The amides can be synthesized in good yields and diastereoselectivities, with excellent diastereoselectivities after purification. Both GAP and reverse-GAP enrichments were developed for the purification of these isomeric products. Simply washing the crudes via suspension in ether can afford the diastereoenriched products in either the suspended solid (GAP enrichment) or the ether solution (reverse-GAP enrichment). Selection for either process is determined by the chemical nature of the substrate.
Experimental Section
General Methods
ACS grade toluene was distilled over CaH2 under argon prior to use. LiHMDS in THF/ethylbenzene was purchased and used within 1 month after arrival. HRMS analysis of new compounds was performed using an Orbitrap mass analyzer.
Synthesis of Compounds 4a–j
These compounds were synthesized according to the procedures found in the literature.19,21,221H and 31P NMR data for these compounds match the literature data.
Synthesis of Compound 4k
3-NH2 (736 mg, 2.84 mmol), pivalaldehyde (316 μL, 2.84 mmol), DCM (12.0 mL), and triethylamine (1.20 mL, 8.52 mmol) were added, respectively, in a 20 mL scintillation vial and stirred at rt until dissolved. TiCl4 (2.41 mL of 1.0 M solution in DCM) was added dropwise, the vial was capped, and the solution was stirred at rt overnight. After completion of the reaction, the reaction mixture was filtered through a short pad of silica. The silica pad was washed with DCM to remove any unreacted aldehyde, and then washed with ethyl acetate to recover the product. Evacuation of the solvent afforded 4k (563 mg, 61%) as a yellow oil. The compound was not pure enough for analysis and was carried on to the next step as an intermediate without further purification. This course of action was taken due to the instability of the compound on silica, preventing further purification.
General Procedure for the Synthesis of Compounds 5a–k
A 10 mL Schlenk tube was equipped with a stir bar, flame-dried under vacuum, and backfilled with argon under positive pressure. Imine (1.0 mmol), toluene (3.0 mL), and then formamide (3.0 mmol) were added, respectively, with stirring. The reaction was cooled to −78 °C with CO2/acetone, followed by the dropwise addition of LiHMDS (3.0 mL of a 1.0 M solution in THF/ethylbenzene). After stirring overnight with slow warming to room temperature, the reaction was quenched with 0.3 M citric acid. The bilayer was separated, the organic layer washed ×2 with citric acid, and the aqueous layer washed ×1 with toluene. The combined organic layers were washed ×2 with 30% methanol/water to remove the formamide, and the aqueous layer was washed again ×1 with toluene. Finally, the combined organic layers were washed with brine, dried over MgSO4, filtered, and evacuated to afford the crude product.
General Procedure for GAP Purification (Solid Enrichment)
At room temperature, the crude solid was suspended in an appropriate amount of diethyl ether, such that there was 30 μL ether/mg solid. The suspension was stirred overnight, after which the suspended solid was removed via filtration. The filtered solid was washed once with diethyl ether, then dried under vacuum to afford the diastereoenriched and purified product.
General Procedure for Reverse-GAP Purification (Liquid Enrichment)
At room temperature, the crude solid was suspended in an appropriate amount of diethyl ether, such that there was 10 μL ether/mg solid. The suspension was stirred overnight, after which the suspended solid was removed via filtration. The filtered solution was evacuated, and the resulting solid was redissolved in a minimal amount of diethyl ether. Addition of petroleum ether caused a white solid precipitate, which solid was recovered via a second filtration as the diastereoenriched and purified product.
Data for Pure Compounds 5a–k
Compound 5a
White solid; yield 454 mg, 74%; mp 162–164 °C; [α]D10 = +14.82° (c 0.162, CHCl3); 1H NMR (400 MHz, CDCl3): δ = 7.42–7.39 (m, 2H), 7.32–7.28 (m, 2H), 7.25–7.21 (m, 1H), 5.35–5.31 (t, J = 9.2 Hz, 1H), 3.75–3.70 (t, J = 10.4 Hz, 1H), 3.40–3.28 (m, 1H), 3.12–2.97 (m, 1H), 2.95 (s, 6H), 2.93–2.85 (m, 1H), 2.79–2.72 (m, 1H), 2.03–1.94 (m, 2H), 1.77–1.68 (m, 2H), 1.35–1.09 (m, 4H), 1.19–1.17 (d, J = 6.8 Hz, 3H), 1.12–1.10 (d, J = 6.8 Hz, 3H), 1.02–1.01 (d, J = 6.8 Hz, 3H), 0.88–0.87 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ = 171.86–171.79 (d, J = 6.6 Hz), 140.85, 128.80, 127.68, 127.62, 60.04–59.93 (d, J = 10.5 Hz), 59.01–58.90 (d, J = 10.5 Hz), 56.38–56.35 (d, J = 2.9 Hz), 53.56, 44.18–44.14 (d, J = 3.8 Hz), 44.03–44.00 (d, J = 2.7 Hz), 37.24, 36.17, 31.73, 31.24–31.13 (d, J = 11.4 Hz), 30.36–30.26 (d, J = 9.6 Hz), 24.49, 23.05–22.99 (d, J = 5.7 Hz), 22.79, 22.20–22.17 (d, J = 2.9 Hz), 20.09, 19.82, 14.26; 31P NMR (162 MHz, CDCl3): δ = 21.91; HRMS (ESI): m/z calcd for [C22H37N4O2P + H]+: 421.2732, found: 421.2736.
Compound 5b
White solid; yield 446 mg, 99%; mp 163–165 °C; [α]D10 = +3.22° (c 0.250, CHCl3); 1H NMR (400 MHz, CDCl3): δ = 7.42–7.39 (m, 2H), 7.31–7.27 (m, 2H), 7.25–7.20 (m, 1H), 5.14–5.10 (t, J = 9.0 Hz, 1H), 3.75–3.70 (t, J = 10.4 Hz, 1H), 3.65–3.59 (m, 1H), 3.55–3.47 (m, 1H), 3.43–3.27 (m, 2H), 3.20–3.00 (m, 2H), 2.92–2.84 (m, 1H), 2.79–2.71 (m, 1H), 2.04–1.94 (m, 2H), 1.94–1.76 (m, 4H), 1.76–1.67 (m, 2H), 1.38–1.18 (m, 4H), 1.12–1.15 (d, J = 6.8 Hz, 3H), 1.10–1.08 (d, J = 6.4 Hz, 3H), 1.04–1.02 (d, J = 6.8 Hz, 3H), 0.90–0.89 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ = 170.21–170.14 (d, J = 6.7 Hz), 140.66, 128.70, 127.75, 127.66, 60.03–59.92 (d, J = 10.5 Hz), 59.01–58.91 (d, J = 9.5 Hz), 57.81, 46.43, 46.24, 44.12–44.08 (d, J = 3.8 Hz), 31.20–31.09 (d, J = 11.4 Hz), 30.37–30.28 (d, J = 9.6 Hz), 26.08, 24.47, 24.18, 22.99–22.94 (d, J = 5.7 Hz), 22.25, 20.03, 19.83; 31P NMR (162 MHz, CDCl3): δ = 21.98; HRMS (ESI): m/z calcd for [C24H39N4O2P + H]+: 447.2889, found: 447.2893.
Compound 5c
White solid; yield 446 mg, 97%; mp 176–178 °C; [α]D10 = +5.91° (c 0.238, CHCl3); 1H NMR (400 MHz, CDCl3): δ = 7.39–7.37 (m, 2H), 7.32–7.28 (m, 2H), 7.26–7.22 (m, 1H), 5.36–5.32 (t, J = 8.8 Hz, 1H), 3.68–3.46 (m, 7H), 3.40–3.30 (m, 2H), 3.17–3.07 (m, 1H), 3.07–2.95 (m, 1H), 2.91–2.85 (m, 1H), 2.76–2.72 (m, 1H), 2.03–1.92 (m, 2H), 1.78–1.67 (m, 2H), 1.37–1.14 (m, 4H), 1.21–1.19 (d, J = 7.2 Hz, 3H), 1.13–1.11 (d, J = 7.2 Hz, 3H), 1.01–0.99 (d, J = 6.8 Hz, 3H), 0.86–0.84 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ = 170.46–170.38 (d, J = 7.6 Hz), 140.88–140.86 (d, J = 1.9 Hz), 128.96, 127.85, 127.42, 66.80, 66.34, 59.99–59.87 (d, J = 12.4), 58.96–58.86 (d, J = 9.5 Hz), 56.33–56.30 (d, J = 2.9 Hz), 46.12, 44.17–44.13 (d, J = 3.8 Hz), 43.99–43.96 (d, J = 2.8 Hz), 42.86, 31.18–31.07 (d, J = 11.4 Hz), 30.29–30.19 (d, J = 9.5 Hz), 24.45, 23.04–22.97 (d, J = 6.7 Hz), 22.13–22.11 (d, J = 2.9 Hz), 20.17, 19.84; 31P NMR (162 MHz, CDCl3): δ = 21.79; HRMS (ESI): m/z calcd for [C24H39N4O3P + H]+: 463.2838, found: 463.2839.
Compound 5d
White solid; yield 461 mg, 90%; mp 105–107 °C; [α]D10 = +15.11° (c 0.225, CHCl3); 1H NMR (400 MHz, CDCl3): δ = 8.71–8.69 (d, J = 8.0 Hz, 1H), 7.85–7.83 (d, J = 8.0 Hz, 1H), 7.79–7.77 (d, J = 8.1 Hz, 1H), 7.61–7.56 (m, 1H), 7.52–7.48 (m, 2H), 7.42–7.38 (t, J = 7.6 Hz, 1H), 6.11–6.06 (t, J = 9.6 Hz, 1H), 3.79–3.29 (m, 8H), 3.07–2.75 (m, 5H), 2.01–1.89 (m, 2H), 1.74–1.65 (m, 2H), 1.35–1.07 (m, 4H), 1.24–1.23 (d, J = 6.8 Hz, 3H), 1.16–1.15 (d, J = 6.8 Hz, 3H), 0.86–0.84 (d, J = 6.8 Hz, 3H), 0.54–0.52 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ = 171.25–171.19 (d, J = 5.7 Hz), 137.01–136.98 (d, J = 3.0 Hz), 134.29, 130.61, 128.88–128.79 (d, J = 9.5 Hz), 126.82, 126.12, 125.55, 124.32, 66.79, 66.12, 59.90–59.79 (d, J = 11.4 Hz), 59.06–58.96 (d, J = 10.5 Hz), 53.96, 45.99, 44.16–44.12 (d, J = 3.8 Hz), 43.78–43.74 (d, J = 3.8 Hz), 43.06, 31.26-31.15 (d, J = 11.5 Hz), 30.54–30.44 (d, J = 9.5 Hz), 24.48, 22.92–22.85 (d, J = 6.7 Hz), 22.56–22.53 (d, J = 3.8 Hz), 20.08, 19.42; 31P NMR (162 MHz, CDCl3): δ = 21.67; HRMS (ESI): m/z calcd for [C28H41N4O3P + H]+: 513.2995, found: 513.3005.
Compound 5e
White solid; yield 340 mg, 73%; mp 215–217 °C; [α]D10 = +7.76° (c 0.438, CHCl3); 1H NMR (400 MHz, CDCl3): δ = 7.39–7.35 (m, 2H), 7.02–6.96 (t, J = 8.8 Hz, 2H), 5.37–5.33 (t, J = 8.8 Hz, 1H), 3.67–3.48 (m, 7H), 3.39–3.26 (m, 2H), 3.20–3.13 (m, 1H), 3.04–2.94 (m, 1H), 2.90–2.84 (m, 1H), 2.75–2.69 (m, 1H), 1.35–1.09 (m, 4H), 1.22–1.20 (t, J = 7.2 Hz, 3H), 1.13–1.12 (t, J = 6.8 Hz, 3H), 1.00–0.98 (t, J = 6.8 Hz, 3H), 0.88–0.86 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ = 170.29–170.23 (d, J = 6.7 Hz), 163.54, 161.09, 136.92, 129.23–129.14 (d, J = 8.5 Hz), 115.91, 155.67, 66.81, 66.37, 59.95–59.84 (d, J = 10.5 Hz), 58.97–58.86 (d, J = 10.5 Hz), 55.49–55.45 (d, J = 3.8 Hz), 46.11, 44.18–44.14 (d, J = 3.8 Hz), 44.04–44.01 (d, J = 2.9 Hz), 42.84, 31.09–30.97 (d, J = 11.5 Hz), 30.23–30.14 (d, J = 9.6 Hz), 24.43, 22.98–22.91 (d, J = 6.7 Hz), 22.08–22.04 (d, J = 3.8 Hz), 20.22, 19.88; 31P NMR (162 MHz, CDCl3): δ = 21.72; HRMS (ESI): m/z calcd for [C24H38FN4O3P + H]+: 481.2744, found: 481.2760.
Compound 5f
White solid; yield 427 mg, 86%; mp 207–209 °C; [α]D10 = +4.00° (c 0.200, CHCl3); 1H NMR (400 MHz, CDCl3): δ = 7.36–7.34 (d, J = 8.4 Hz, 2H), 7.31–7.28 (d, J = 8.4 Hz, 2H), 5.38–5.34 (t, J = 9.0 Hz, 1H), 3.68–3.45 (m, 7H), 3.40–3.20 (m, 3H), 3.07–2.96 (m, 1H), 2.92–2.86 (m, 1H), 2.77–2.72 (m, 1H), 2.04–1.96 (m, 2H), 1.79–1.71 (m, 2H), 1.37–1.10 (m, 4H), 1.23–1.22 (d, J = 6.8 Hz, 3H), 1.15–1.13 (d, J = 6.4 Hz, 3H), 1.02–1.00 (d, J = 7.2 Hz, 3H), 0.90–0.89 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ = 170.14–170.06 (d, J = 7.6 Hz), 139.52–139.50 (d, J = 1.9 Hz), 133.75, 129.14, 128.88, 66.83, 66.42, 59.98–59.88 (d, J = 10.5 Hz), 59.02–58.91 (d, J = 10.5 Hz), 55.56–55.54 (d, J = 2.8 Hz), 46.14, 44.21–44.17 (d, J = 3.8 Hz), 44.10–44.08 (d, J = 2.9 Hz), 31.12–31.00 (d, J = 11.5 Hz), 30.26–30.16 (d, J = 9.5 Hz), 24.46, 23.01–22.96 (d, J = 5.7 Hz), 22.12–22.08 (d, J = 3.8 Hz), 20.27, 19.94; 31P NMR (162 MHz, CDCl3): δ = 21.72; HRMS (ESI): m/z calcd for [C24H38ClN4O3P + H]+: 497.2448, found: 497.2460.
Compound 5g
White solid; yield 438 mg, 81%; mp 212–214 °C; [α]D10 = +1.86° (c 0.700, CHCl3); 1H NMR (400 MHz, CDCl3): δ = 7.56–7.55 (t, J = 1.8 Hz, 1H), 7.40–7.36 (m, 1H), 7.34–7.30 (m, 1H), 7.19–7.15 (t, J = 7.8 Hz, 1H), 5.37–5.32 (t, J = 8.8 Hz, 1H), 3.69–3.49 (m, 7H), 3.40–3.20 (m, 3H), 3.07–2.94 (m, 1H), 2.91–2.84 (m, 1H), 2.77–2.68 (m, 1H), 2.03–1.93 (m, 2H), 1.78–1.67 (m, 2H), 1.37–1.10 (m, 4H), 1.21–1.19 (d, J = 7.2 Hz, 3H), 1.13–1.11 (d, J = 6.8 Hz, 3H), 1.02–1.00 (d, J = 6.8 Hz, 3H), 0.90–0.89 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ = 169.87–169.80 (d, J = 7.6 Hz), 143.15, 131.01, 130.48, 130.31, 126.14, 122.96, 66.82, 66.39, 59.94–59.85 (d, J = 10.5 Hz), 58.98–58.87 (d, J = 10.5 Hz), 55.64–55.60 (d, J = 3.8 Hz), 46.15, 44.17–44.13 (d, J = 3.8 Hz), 44.08–44.05 (d, J = 2.9 Hz), 42.88, 31.11–30.99 (d, J = 11.4 Hz), 30.20–30.11 (d, J = 9.5 Hz), 24.42, 23.04–22.98 (d, J = 6.7 Hz), 22.02–21.99 (d, J = 2.9 Hz), 20.23, 19.85; 31P NMR (162 MHz, CDCl3): δ = 21.67; HRMS (ESI): m/z calcd for [C24H38BrN4O3P + H]+: 541.1943, found: 541.1960.
Compound 5h
White solid; yield 400 mg, 84%; mp 195–197 °C; [α]D10 = +4.32° (c 0.162, CHCl3); 1H NMR (400 MHz, CDCl3): δ = 7.27–7.24 (d, J = 8.4 Hz, 2H), 7.11–7.09 (d, J = 8.0 Hz, 2H), 5.33–5.29 (t, J = 9.0 Hz, 1H), 3.67–3.46 (m, 7H), 3.40–3.28 (m, 2H), 3.18–3.13 (m, 1H), 3.11–3.00 (m, 1H), 2.91–2.85 (m, 1H), 2.77–2.70 (m, 1H), 2.31 (s, 3H), 2.20–1.94 (m, 2H), 1.77–1.68 (m, 2H), 1.38–1.90 (m, 4H), 1.21–1.19 (d, J = 6.8 Hz, 3H), 1.13–1.11 (d, J = 6.8 Hz, 3H), 1.03–1.02 (d, J = 6.8 Hz, 3H), 0.88–0.86 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ = 170.67–170.60 (d, J = 6.7 Hz), 137.81, 137.53, 129.59, 127.32, 66.82, 66.40, 60.02–59.90 (d, J = 11.5 Hz), 59.01–58.90 (d, J = 10.5 Hz), 55.94, 46.13, 44.15–44.01 (d, J = 14.3 Hz), 42.84, 31.21–31.10 (d, J = 11.5 Hz), 30.34–30.24 (d, J = 9.6 Hz), 24.47, 23.08–23.02 (d, J = 5.7 Hz), 22.15, 21.25, 20.18, 19.89; 31P NMR (162 MHz, CDCl3): δ = 21.94; HRMS (ESI): m/z calcd for [C25H41N4O3P + H]+: 477.2995, found: 477.2994.
Compound 5i
White solid; yield 340 mg, 71%; mp 162–164 °C; [α]D10 = +24.06° (c 0.188, CHCl3); 1H NMR (400 MHz, CDCl3): δ = 7.33–7.28 (m, 1H), 7.16–7.11 (m, 3H), 5.51–5.46 (t, J = 9.2 Hz, 1H), 3.76–3.31 (m, 8H), 3.15–3.30 (m, 1H), 3.04–2.93 (m, 2H), 2.92–2.83 (m, 1H), 2.82–2.73 (m, 1H), 2.05–1.92 (m, 2H), 1.77–1.68 (m, 2H), 1.40–1.10 (m, 4H), 1.24–1.22 (d, J = 6.8 Hz, 3H), 1.16–1.15 (d, J = 6.4 Hz, 3H), 0.98–0.96 (d, J = 6.8 Hz, 3H), 0.84–0.82 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ = 171.06–170.99 (d, J = 6.7 Hz), 139.15, 135.44, 131.25, 127.80, 126.70, 66.82, 66.25, 59.92–59.81 (d, J = 11.4 Hz), 59.09–58.99 (d, J = 10.5 Hz), 54.10, 45.80, 44.10, 43.15, 31.05–30.95 (d, J = 10.5 Hz), 30.59–30.50 (d, J = 9.5 Hz), 24.49, 22.91–22.84 (d, J = 6.7 Hz), 22.60–22.56 (d, J = 3.8 Hz), 20.05, 19.70; 31P NMR (162 MHz, CDCl3): δ = 21.84; HRMS (ESI): m/z calcd for [C25H41N4O3P + H]+: 477.2995, found: 477.3003.
Compound 5j
White solid; yield 561 mg, 99%; mp 60–62 °C; [α]D10 = +3.73° (c 0.375, CHCl3); 1H NMR (400 MHz, CDCl3): δ = 7.42–7.28 (m, 7H), 6.92–6.88 (d, J = 8.8 Hz, 2H), 5.32–5.27 (t, J = 8.6 Hz, 1H), 5.04 (s, 2H), 3.70–3.46 (m, 7H), 3.40–3.27 (m, 2H), 3.19–3.10 (m, 1H), 3.08–2.98 (m, 1H), 2.91–2.85 (m, 1H), 2.70–2.77 (m, 1H), 2.02–1.93 (m, 2H), 1.79–1.67 (m, 2H), 1.36–1.09 (m, 4H), 1.21–1.20 (t, J = 6.8 Hz, 3H), 1.13–1.12 (t, J = 6.4 Hz, 3H), 1.02–1.00 (t, J = 6.8 Hz, 3H), 0.87–0.86 (t, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ = 170.62–170.55 (d, J = 6.7 Hz), 158.32, 136.94, 133.44, 128.67, 128.08, 127.55, 115.26, 70.08, 66.82, 66.39, 59.97–59.86 (d, J = 10.5 Hz), 58.95–58.85 (d, J = 9.5 Hz), 55.62–55.59 (d, J = 2.8 Hz), 46.11, 44.17–44.13 (d, J = 3.8 Hz), 43.99–43.96 (d, J = 2.8 Hz), 42.83, 31.18–31.06 (d, J = 11.5 Hz), 30.28–30.18 (d, J = 9.5 Hz), 24.44, 23.07–23.01 (d, J = 5.7 Hz), 22.13–22.09 (d, J = 3.8 Hz), 20.18, 19.87; 31P NMR (162 MHz, CDCl3): δ = 21.88; HRMS (ESI): m/z calcd for [C31H45N4O4P + H]+: 569.3257, found: 569.3245.
Compound 5k
White solid; yield 334 mg, 75%; mp 192–194 °C; [α]D10 = +3.05° (c 0.525, CHCl3); 1H NMR (400 MHz, CDCl3): δ = 4.23–4.18 (t, J = 10.4 Hz, 3H), 3.82–3.40 (m, 9H), 3.28–3.14 (m, 1H), 2.94–2.89 (m, 1H), 2.78–2.70 (m, 2H), 2.05–1.98 (m, 2H), 1.75–1.68 (m, 2H), 1.37–1.08 (m, 16H), 0.97 (s, 9H); 13C NMR (100 MHz, CDCl3): δ = 172.52, 66.96, 66.77, 59.50–59.40 (d, J = 10.5 Hz), 58.95–58.84 (d, J = 10.5 Hz), 56.96, 47.10, 44.08–44.05 (d, J = 2.9 Hz), 43.96–43.91 (d, J = 4.8 Hz), 41.85, 35.72–35.68 (d, J = 4.8 Hz), 32.22–32.10 (d, J = 12.4 Hz), 30.92–30.81 (d, J = 10.5 Hz), 26.66, 24.77–24.69 (d, J = 7.6 Hz), 24.42, 23.29–23.24 (d, J = 4.8 Hz), 19.68; 31P NMR (162 MHz, CDCl3): δ = 23.57; HRMS (ESI): m/z calcd for [C22H43N4O3P + H]+: 443.3151, found: 443.3144.
Compound 6c
Compound 5c (100 mg, 0.216 mmol) was dissolved in 5 mL of methanol, followed by the addition of 1 mL of 1.0 N HCl (aq). After stirring for 1 h, the reaction mixture was diluted with 1.0 N NaOH (aq) and extracted ×2 with ethyl acetate. The combined organic layers were dried over MgSO4, filtered, and evacuated to afford a crude solid. This solid was dissolved in 5 mL of a 1:1 dioxane/water mixture, after which triethylamine (45 μL, 0.324 mmol) and Boc2O (52 mg, 0.238 mmol) were added, and the reaction was stirred overnight at room temperature. The reaction was then diluted with 1.0 N HCl and extracted ×2 with ethyl acetate. The combined organic layers were dried with MgSO4, filtered, and evacuated to afford 6c. Yield 50 mg, 72%; 1H NMR (400 MHz, CDCl3): δ = 7.37–7.27 (m, 5H), 6.03–6.01 (d, J = 7.6 Hz, 1H), 5.54–5.52 (d, J = 7.6 Hz, 1H), 3.77–3.60 (m, 2H), 3.60–3.47 (m, 3H), 3.45–3.37 (m, 1H), 3.28–3.20 (m, 1H), 3.12–3.05 (m, 1H), 1.40 (s, 9H). These 1H NMR data match those found in the literature.28
Acknowledgments
We thank the NIH (R33DA031860), the Robert A. Welch Foundation (D-1361), NSFC (No. 21332005, P. R. China), and the Jiangsu Innovation Programs (P. R. China) for their generous support of this research. We also thank the NSF (CHE-1048553) and the CRIF program for support of our NMR facility. We also wish to thank our current and former co-workers, Siqi Lin, Padmanabha V. Kattamuri, Shuo Qiao, Wei Zhou, Dr. Jianbo Xie, Dr. Jianbin Wu, and Dr. Guanghui An for their valuable suggestions and assistance. Special thanks to Dr. Daniel Unruh for the X-ray analysis and to Dr. Kazimirez Surowiec and Dr. Yehia Mechref for the HR-MS analysis.
Supporting Information Available
1H, 13C, and 31P NMR spectra of all compounds, as well as data for the X-ray crystal structure of 5c (CCDC 1022082) and the accompanying CIF file are included. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Chang M.; Liu S.; Huang K.; Zhang X. Org. Lett. 2013, 15, 4354. [DOI] [PubMed] [Google Scholar]
- Kohls H.; Steffen-Munsberg F.; Höhne M. Curr. Opin. Chem. Biol. 2014, 19, 180. [DOI] [PubMed] [Google Scholar]
- Koszelewski D.; Tauber K.; Faber K.; Kroutil W. Trends Biotechnol. 2010, 28, 324. [DOI] [PubMed] [Google Scholar]
- Nugent T. C. In Process Chemistry in the Pharmaceutical Industry; Gadamasetti K., Braish T., Eds.; CRC Press: Boca Raton, FL, 2007; Vol. 2, p 137. [Google Scholar]
- Nugent T. C.; El-Shazly M. Adv. Synth. Catal. 2010, 352, 753. [Google Scholar]
- Verrier C.; Carret S.; Poisson J.-F. Org. Lett. 2012, 14, 5122. [DOI] [PubMed] [Google Scholar]
- Gorzynski-Smith J.Organic Chemistry, 2nd ed.; McGraw-Hill: New York, NY, 2008. [Google Scholar]
- Chen Y.-J.; Chen C. Tetrahedron: Asymmetry 2008, 19, 2201. [Google Scholar]
- Denmark S. E.; Fu J. Chem. Rev. 2003, 103, 2763. [DOI] [PubMed] [Google Scholar]
- Saravanan S.; Sadhukhan A.; Khan N.-u. H.; Kureshy R. I.; Abdi S. H. R.; Bajaj H. C. J. Org. Chem. 2012, 77, 4375. [DOI] [PubMed] [Google Scholar]
- Yashin N. V.; Averina E. B.; Sedenkova K. N.; Kuznetsova T. S.; Zefirov N. S. Russ. Chem. Bull. 2013, 62, 928. [Google Scholar]
- Gu Z.; Zakarian A. Angew. Chem., Int. Ed. 2010, 49, 9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopylova N. A.; Grygorenko O. O.; Komarov I. V.; Groth U. Tetrahedron: Asymmetry 2010, 21, 2868. [Google Scholar]
- Xu K.; Zhang S.; Hu Y.; Zha Z.; Wang Z. Chem.—Eur. J. 2013, 19, 3573. [DOI] [PubMed] [Google Scholar]
- Boesten W. H. J. U. S. Patent 4,094,904, June 13, 1978.
- Fraser R. R.; Hubert P. R. Can. J. Chem. 1974, 52, 185. [Google Scholar]
- Reeves J. T.; Tan Z.; Herbage M. A.; Han Z. S.; Marsini M. A.; Li Z.; Li G.; Xu Y.; Fandrick K. R.; Gonnella N. C.; Campbell S.; Ma S.; Grinberg N.; Lee H.; Lu B. Z.; Senanayake C. H. J. Am. Chem. Soc. 2013, 135, 5565. [DOI] [PubMed] [Google Scholar]
- Han J.; Ai T.; Nguyen T.; Li G. Chem. Biol. Drug Des. 2008, 72, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kattuboina A.; Kaur P.; Nguyen T.; Li G. Tetrahedron Lett. 2008, 49, 3722. [Google Scholar]
- Kattuboina A.; Li G. Tetrahedron Lett. 2008, 49, 1573. [Google Scholar]
- Ai T.; Li G. Bioorg. Med. Chem. Lett. 2009, 19, 3967. [DOI] [PubMed] [Google Scholar]
- Kattamuri P. V.; Ai T.; Pindi S.; Sun Y.; Gu P.; Shi M.; Li G. J. Org. Chem. 2011, 76, 2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur P.; Wever W.; Pindi S.; Milles R.; Gu P.; Shi M.; Li G. Green Chem. 2011, 13, 1288. [Google Scholar]
- Pindi S.; Kaur P.; Shakya G.; Li G. Chem. Biol. Drug. Des. 2011, 77, 20. [DOI] [PubMed] [Google Scholar]
- Xie J.-b.; Luo J.; Winn T. R.; Cordes D. B.; Li G. Beilstein J. Org. Chem. 2014, 10, 746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pindi S.; Wu J.; Li G. J. Org. Chem. 2013, 78, 4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves J. T.; Lorenc C.; Camara K.; Li Z.; Lee H.; Busacca C. A.; Senanayake C. H. J. Org. Chem. 2014, 79, 5895. [DOI] [PubMed] [Google Scholar]
- Pradhan T. K.; Krishnan K. S.; Vasse J.-L.; Szymoniak J. Org. Lett. 2011, 13, 1793. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.