

Abstract
Calcium homeostasis is central to all cellular functions and has been studied for decades. Calcium acts as a critical second messenger for both extracellular and intracellular signaling and is fundamental in cell life and death decisions [1]. The calcium gradient in the cell is coupled with an inherent ability of the divalent cation to reversibly bind multiple target biological molecules to generate an extremely versatile signaling system [2]. Calcium signals are used by the cell to control diverse processes as development, neurotransmitter release, muscle contraction, metabolism, autophagy and cell death. “Cellular calcium overload” is detrimental to cellular health, resulting in massive activation of proteases and phospholipases leading to cell death [3]. Historically, cell death associated with calcium ion perturbations has been primarily recognized as necrosis. Recent evidence clearly associate changes in calcium ion concentrations with more sophisticated forms of cellular demise, including apoptosis [4] [5] [6] [7]. Although the endoplasmic reticulum (ER) serves as the primary calcium store in the metazoan cell, dynamic calcium release to the cytosol, mitochondria, nuclei and other organelles orchestrate diverse coordinated responses. Most evidence supports that calcium transport from the ER to mitochondria plays a significant role in regulating cellular bioenergetics, production of reactive oxygen species, induction of autophagy and apoptosis. Recently, molecular identities that mediate calcium traffic between the ER and mitochondria have been discovered [8] [9] [10]. The next questions are how they are regulated for exquisite tight control of ER – mitochondrial calcium dynamics. This review attempts to summarize recent advances in the role of calcium in regulation of ER and mitochondrial function.
Introduction
In 1883, Ringer recognized that addition of calcium (Ca2+) to heart cultures caused their contraction [11] which spawned a new field regarding how Ca2+ controls cellular function. Now it is recognized that the ubiquitous second messenger Ca2+ is intricately involved in a wide spectrum of physiological functions, including signal transduction, muscle contraction, secretion of proteins and hormones and gene expression. About 50 years ago it was recognized that energized mitochondria rapidly uptake Ca2+ in response to an acute increase in the cytosolic [Ca2+]c [12, 13]. The discovery of Ca2+ probes that measure local Ca2+ concentrations within single cells provided new tools to study Ca2+ signaling, including the Ca2+ sensitive jellyfish aequorin which are engineered to target subcellular organelles, in response to a variety of physiological stimuli [14] [15] [16]. We now know that cytosolic Ca2+ concentrations [Ca2+]c can vary by several orders of magnitude and trigger cascades of cellular events including contraction of myofilaments, secretion of hormones and neurotransmitters, induction of various forms of cell death (necrosis, apoptosis and autophagy) and, more recently neurodegenerative pathways. Under resting conditions cytosolic [Ca2+]c is finely tuned at ∼100nM by the coordinated activity of Ca2+ pumping mechanisms that include plasma membrane Ca2+ ATPases and the Na+/Ca2+ exchanger that actively mobilize Ca2+ from internal to external stores [1]. Within the cell, Ca2+ is stored in specialized compartments mainly in the endoplasmic reticulum (ER) and sarcoplasmic reticulum (SR, a specialized ER counterpart in muscle cells) as well as in other membrane-bound compartments, including the Golgi apparatus, lysosomes and endosomes [3] [17]. The fine-tuning of [Ca2+]c is accomplished through pumps, channels and buffering proteins that are located within the cytosol and in the ER/SR that coordinately regulate cellular Ca2+ homeostasis and signaling. Exquisite regulation of the Ca2+ concentration in different subcompartments of the cell is essential for cell function considering the fact that the extracellular medium has an unlimited Ca2+ reservoir, ∼1mM, and intracellular subcompartments (also known as Ca2+ stores) may have [Ca2+] of ∼100μM that facilitate rapid release through channels and reuptake through Ca2+ pumps. With the observation of the close juxtaposition of ER and mitochondria [18], interest grew in the mechanisms that drive local Ca2+ uptake from subdomains of the ER/SR to the mitochondrial matrix. The activities of pumps and channels that regulate the luminal ER [Ca2+]ER are also regulated by the [Ca2+]ER. Here, we discuss the precise role of the ER and mitochondria in Ca2+ homeostasis and allude to the significance of ER-mitochondria crosstalk in further facilitating Ca2+ trafficking to regulate bioenergetics, production of reactive oxygen species (ROS), ER protein folding and induction of apoptosis and autophagy.
ER Ca2+ homeostasis
The ER is now recognized as the major Ca2+ storage organelle of the metazoan cell (Figure 1). The ER regulates Ca2+ homeostasis through the presence of many Ca2+ binding proteins that function as buffers by having a low-affinity and large capacity for Ca2+ binding. These proteins, of which the most abundant are the protein chaperones calreticulin (CRT), calnexin (CNX), BiP/GRP78, GRP94 and protein disulfide isomerase (PDI), are responsible for maintaining ER Ca2+ concentration within physiological range of ∼100-200 μM. Ca2+ binding to molecular chaperones BiP, GRP94, PDI and ERP57 also regulates their chaperone activities [19] [20]. As a consequence, alterations in [Ca2+]ER can disrupt protein folding, cause accumulation of misfolded proteins and initiate signaling of the unfolded protein response [21] [19] [22]. BIP functions in the ER as a peptide-dependent ATPase and utilizes ATP to prevent protein aggregation [23, 24]. BIP hydrolysis of ATP may deplete luminal ATP and initiate a signal to release Ca2+ to stimulate oxidative phosphorylation to maintain the ATP/ADP ratio. CRT and CNX are molecular chaperones that interact with specific glycoforms on asparagine-linked glycans to promote proper disulfide bond formation through interaction with the thiol-disulfide isomerase ERP57 [25] and direct protein trafficking and ER-associated protein degradation [26] [27]. Finally, PDI and ERO1 provide an electron transport pathway from thiol residues to molecular oxygen during disulfide bond formation [28]. In addition to molecular chaperones, calsequestrins and chromogranins also buffer [Ca2+]ER.
Figure. 1. Schematic representing how protein folding in the ER modulates mitochondrial ATP and ROS production.
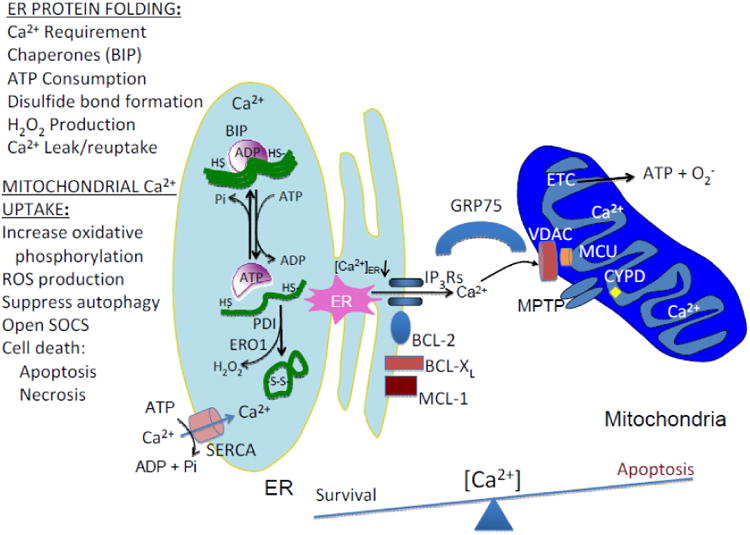
Mitochondria and ER are tethered by the actions of the MFNs, of which MFN2 is localized to the mitochondrial-associated membrane (MAM), that promote efficient Ca2+ transfer from the ER to the mitochondria. Ca2+ loading in the ER is mediated by the abundance of Ca2+-binding proteins, including CNX, CRT, as well as the protein chaperones BIP and PDI. Protein folding in the ER requires Ca2+ and ATP for chaperone function, proper glycosylation, and correct disulfide bond formation. Misfolded proteins may sequester protein chaperones that facilitates opening of Ca2+ channels to initiate Ca2+ transfer to mitochondria to stimulate oxidative phosphorylation. Ca2+ transfer occurs through the activity of several Ca2+ channels that include the ER localized inositol-1,4,5-triphosphate receptors (IP3Rs), as well as the ryanodine receptors (RyRs) and the mitochondrial-localized voltage-dependent anion channel (VDAC) and the mitochondrial Ca2+ uniporter complex MCU (MCU, including MICU1, MICU2, MCUR1 and EMRE). The IP3Rs enriched at the MAMs are linked to VDAC on the OMM by the protein chaperone GRP75. VDAC tightly controls Ca2+ permeation into mitochondria by IP3Rs-mediated Ca2+ signals. Once Ca2+ transverses the OMM it can subsequently cause depolarization of the inner mitochondrial permeability transition pore (MPTP) and induction of apoptotic stimuli. Conditions that prevent Ca2+ transfer from the ER to mitochondria include overexpression of anti-apoptotic proteins such as BCL-2 and BCL-XL and constitute survival signaling. A number of mechanisms have been proposed to cause Ca2+ leak from the ER and are depicted as red identities on the ER membrane (SEC61, SERCA1T, BCL-2, BCL-XL, MCL-1, BI-1 and IP3Rs). As Ca2+ accumulates in mitochondria, cells are predisposed to disruption of the electron transport chain (ETC) to produce ROS, MPTP, mitochondrial swelling, disruption of the OMM, release of cytochrome C and apoptosome components leading to caspase activation and apoptosis. Mechanisms the limit mitochondrial loading of Ca2+ include MPTP itself, and the mitochondrial Ca2+ exchangers NCLX and HCX. In addition to protein synthesis, ATP-utilizing processes include chaperone (BIP)-assisted protein folding in the ER lumen, SERCA-mediated Ca2+ reuptake into the ER and possibly hydrolysis of ATP by the F1/F0 ATP synthase upon collapse of the IMM electrochemical potential. Finally, in addition to superoxide production from the ETC, disulfide bond formation mediated by the protein thiol-disulfide isomerases (PDI, ERP57) and ER oxidase 1 (ERO1) generates hydrogen peroxide upon electron transport to molecular O2 as the acceptor. The balance between the amount of Ca2+ stored in the ER lumen and the amount loaded into the mitochondrial matrix may be a determinant in the decision between survival and death.
Ca2+ accumulation in the ER lumen is mediated by the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA). The SERCAs are encoded by three genes (SERCA1, SERCA2, and SERCA3), but their variety and activity is diversified by the existence of splice variants [29]. The SERCAs have four domains: a nucleotide binding domain, a phosphorylation domain, an actuator domain, and transmembrane domains that contain bindings sites for Ca2+ which are joined by short ER luminal loops [30, 31] [32]. SERCA2b is most widely expressed, exhibits the highest Ca2+ affinity and is primarily responsible for maintaining the ER luminal [Ca2+]ER. SERCAs pump two Ca2+ ions for each molecule of ATP hydrolyzed. An increase in [Ca2+]c stimulates SERCA activity. SERCA-mediated Ca2+ uptake occurs exclusively at the ER. ER resident proteins including CNX and CRT inhibit ER Ca2+ uptake by reducing SERCA activity [33] [19] [34] [35]. Due to the activity of SERCA, [Ca2+]ER is maintained at ∼100 μM, thus allowing rapid release of Ca2+ upon opening of Ca2+ channels residing in the ER membrane, including inositol 1, 4, 5-triphsphate (IP3) receptors. Under physiological and or pathological conditions where ER Ca2+ depletion occurs, a phenomenon known as capacitive Ca2+ entry (CCE) is triggered through opening of store-operated Ca2+ channels (SOCs) on the plasma membrane [36]. A protein identified as Stromal interaction molecule 1 (STIM1) is an intraluminal ER Ca2+ sensor that plays an essential role in activation of CCE by communicating [Ca2+]ER to SOCs [37].
An important addition to the enigmatic Ca2+ influx into the cell was the identification of mammalian Transient Receptor Potential (TRP) channels which were first discovered by investigating visual mutants in Drosophila [38]. The protein encoded by the trp gene is a Ca2+ permeable cation channel activated downstream of the phospholipase C (PLC) pathway. Subsequently, cloning of seven mammalian TRPCs revealed that these channels are activated by cell surface receptors that couple to PLC and opening of these channles increases Ca2+ influx and depolarization [39].
IP3Rs are encoded by three genes (IP3R1, IP3R2 and IP3R3), each having splicing variants that each display varying degrees of IP3 binding affinity and Ca2+ oscillations [40]. They form tetrameric channels and are not distributed evenly throughout the ER, but rather form clusters. Knockdown studies in CHO cells showed type 1 knockdown and type 3 knockdown reduce mitochondrial Ca2+ uptake. Type 1 IP3Rs localize to the bulk ER to mediate Ca2+ efflux into the cytosol, whereas type 3 IP3Rs reside at the direct ER-mitochondrial contacts termed MAMs (mitochondria-associated ER membranes) and facilitate flux of Ca2+ into mitochondria [41]. Another class of Ca2+ release channels is composed of the ryanodine receptors (RyR), which are encoded by three genes (RyR1, RyR2, and RyR3). RyR1 and RyR2 are expressed at high levels in the SR of skeletal and cardiac myocytes, respectively. RyRs are also expressed in numerous other cell types, including neurons, hepatocytes, pancreatic acinar cells and smooth muscle cells, although their expression is generally much lower than IP3Rs. However, they may still play a significant role in the Ca2+ signal because at each opening, they release ∼20 times more Ca2+ than IP3Rs [42]. Finally, in addition to the regulated Ca2+ release mediated by RyRs and IP3Rs, there are a number of proposed Ca2+ leak mechanisms including the translocon/BIP [43, 44], Bcl-2 family members [45], Bax inhibitor 1 (BI-1) [46], and C-terminal truncated SERCA1T variants [47], which were recently reviewed [42].
Mitochondrial Ca2+ homeostasis
Ca2+ mobilization was first associated with mitochondrial function by the observation of rapid accumulation of a positively charged ion in the mitochondrial matrix [12] [13] [48] [49]. This finding was a predecessor to Mitchell's chemiosomotic hypothesis [50]. Mitochondria act as a Ca2+ buffer to prevent cytosolic overload upon release from the ER. Accumulation of Ca2+ in the mitochondrial matrix requires the crossing of two membranes, the outer and inner mitochondrial membranes (OMM and IMM, respectively). OMM permeability is primarily attributed to the abundant expression of voltage-dependent anion channels (VDACs). VDAC represents the major OMM protein that forms a voltage-dependent anion-selective channel (VDAC), acting as a general diffusion pore for small hydrophilic molecules, including ATP, ADP, cytochrome C, pyruvate, malate and other metabolites. Although the precise role of VDACs in regulating mitochondrial Ca2+ is debated, VDAC forms Ca2+ tunnels with IP3R3 at the MAM via linkage with GRP75 to tightly control ER Ca2+ signals into the mitochondria [3, 51].
In contrast to the OMM, which is permeable to ions and small molecules, the IMM is very impermeable and requires specific transporters for traffic between the inner mitochondrial matrix and cytosol. Ca2+ uptake into the mitochondrial matrix occurs predominantly through the IMM via the ruthenium red-sensitive mitochondrial Ca2+ uniporter (MCU) that rapidly imports Ca2+ against a steep electrochemical gradient. However, the molecular identity of the channel-forming subunit of the MCU complex identified as CCDC109A, or now called MCU, was only recently discovered using elegant bioinformatic approaches [52] [53]. Since this discovery, there has been an explosion of information regarding the macromolecular identity of the MCU, which is now regarded as a molecular complex [54]. MCU encodes a 40 kDa protein with a 5 kDa mitochondrial targeting signal that is cleaved upon import into the IMM. Both the N- and C- termini of MCU extend into the mitochondrial matrix [55]. There are two putative transmembrane domains, suggesting the functional Ca2+ channel exists as an oligomer. MCU acts as a highly-selective low conductance Ca2+ channel. PAGE on blue native gels suggested that the MCU complex migrates with an apparent molecular weight of ∼480 kDa [52], indicating the potential for numerous different regulatory subunits. One regulatory element, MICU1 (mitochondrial Ca2+ uptake 1 protein) was identified, actually before the discovery of MCU [56], and originally proposed to be required for agonist-mediated rapid Ca2+ uptake into mitochondria. MCU and MICU1 exhibit the same evolutionary pattern of expression and tissue specific expression, and physically interact [57]. MICU1 is a single transmembrane domain present on the IMM that contains two EF hand Ca2+ binding motifs. However, knockdown of MICU1 caused mitochondria to be loaded with Ca2+, the opposite of what would be expected as a component necessary for MCU activity [9]. It is now recognized that MICU1 acts as a brake on MCU-mediated Ca2+ uptake [9]. MICU2 and MICU3 are two paralogs of MICU1. Although MICU3 does not exhibit a tight localization with mitochondria, MICU2 is a mitochondrial-localized protein. Although knockdown of MICU2 did not alter the mitochondrial membrane potential or oxidative phosphorylation, it did reduce mitochondrial clearance of Ca2+. Knockdown and overexpression studies suggest that MICU1 and MICU2 display overlapping functions and they both exist in a complex with MCU [58]. Recently, it was demonstrated that deletion of MCU in cells and tissues of mice prevented Ca2+ uptake into the mitochondrial matrix, thus confirming the requirement for MCU in Ca2+ uptake. However, surprisingly, although there was a defect in mitochondrial Ca2+ uptake, there was not a significant effect on opening of the mitochondrial inner membrane permeability transition pore (MPTP) on the inner mitochondrial membrane or apoptosis [59]. Thus, Ca2+ influx into the mitochondrial matrix may play an indirect role in MPTP opening and cell death.
Additional components have been identified to associate with the MCU complex. The Mitochondrial Ca2+ Uniporter Regulator 1 (MCUR1) was identified in a siRNA screen as an essential regulator of Ca2+ uptake [8]. MCUR1 interacts with MCU, but not MICU1, and it was suggested that these proteins do not exist in the same complex. MCUR1 overexpression increased [Ca2+]m in an MCU-dependent manner. In addition, MCU overexpression did not restore [Ca2+]m in MCUR1-depleted cells, suggesting both are required for Ca2+ uptake. Finally, an Essential MCU Regulator (EMRE) was identified to interact with MICU1 and MCU in the IMM [10]. It was proposed the EMRE may act as a link to couple Ca2+ sensing between MICU1/MICU2 and the channel MCU.
The mechanism of Ca2+ release from the mitochondrion remains an enigmatic problem. Recently, NCLX was identified that has a molecular identity similar to plasma membrane NCX. NCLX localizes to the mitochondria and mediates a low affinity Ca2+ exchange with Na+ [60]. Mitochondrial H+/Ca2+ exchangers (HCX) also limit Ca2+ mitochondrial matrix accumulation caused by MCU. Importantly, Ca2+ can also escape the mitochondrial matrix through the opening of the MPTP. Although the molecular identity of the MPTP has been disputed for years, the only constituent demonstrated to be necessary for its formation is cyclophilin D (CYPD), a mitochondrial matrix protein encoded by the peptidyl-prolyl cis-trans isomerase F gene (PPIF). Other proteins associated with MPTP formation include proteins identified to interact with CYPD; the adenine nucleotide transporter (ANT), VDAC and the F0/F1 ATP synthase. For recent reviews see [61] [62] [63].
The MAMs
The ER and mitochondria interact to form specialized contacts, the MAMs, a location where membrane and luminal contents can interact and intermix. MAMs were originally identified as the site for lipid synthesis and transfer between ER and mitochondria [64]. The composition of the MAM responds rapidly in response to external and internal stimuli. Many of the MAM proteins are associated with ER tubule formation, mitochondrial fission and fusion events and cellular organelle distribution. The composition of the MAM is under intense scrutiny and different reports describe different results based on isolation and methods of characterization [65]. The MAM architecture involves proteins with varying functions including the Ca2+ transfer channels IP3R and VDAC with the mitochondrial chaperone GRP75 [66] [67] [68].
GRP75, also known as mortalin or HSPA9, is a member of the heat shock 70 protein family that displays peptide-dependent ATPase activity, although it is not induced by heat shock. It couples the IP3R to the VDAC to facilitate Ca2+ transfer from the ER lumen to the mitochondrial matrix, without affecting the degree of ER and mitochondrial contact [69]. Mutations in HSPA9 have been observed in patients with Parkinson's disease and its loss is associated with immortality in embryonic fibroblasts.
Some MAM proteins are involved in mitochondrial dynamics of fusion and fission including the mitofusin MFN2 [70]. The mitofusins MFN1 and MFN2 are dynamin-related GTPases that act on the mitochondria. MFN2 is enriched at the MAM and its absence affects ER and mitochondrial morphology, and reduces the number of ER-mitochondrial contacts [70]. MFN2 on the ER is required for connection with mitochondria by interacting directly with MFN1 or MFN2 on the OMM. Where a decrease in MFN2 decreased Ca2+ traffic to mitochondria, overexpression of MFN2 caused apoptosis [71]. ER stress induces expression of MFN2, and in the absence of MFN2, ER-stressed cells are more prone to apoptosis [72]. MFN2-dependent ER-mitochondrial tethering is increased by a ubiquitin ligase (MITOL), where ubiquitination increases MFN2 affinity for GTP causing oligomerization of MFN2 and stimulating MFN2 activity [73]
The Sigma-1 receptor is an ER chaperone enriched at the MAM. Sigma-1 receptor interacts with the chaperone BIP in a Ca2+ dependent manner [74]. A decrease in ER Ca2+ causes their dissociation where both proteins become functional chaperones. In addition, extranuclear promyelocytic leukemia protein (PML) was recently shown to be associated with MAMs where it promotes Ca2+ release from the ER by recruiting PP2A that dephosphorylates PKB/AKT to reduce its kinase activity toward the IP3R. PKB/AKT-mediated phosphorylation of IP3R reduces Ca2+ release from the ER [75]. Therefore, it is proposed that PML at the MAM increases Ca2+ release through reducing phosphorylation of IP3R to promote MPTP.
Ca2+ Flux and Mitochondrial Oxidative Phosphorylation
Protein folding in the ER is a very energy-requiring process as many of the molecular chaperones (BIP and GRP94) hydrolyze ATP during their binding and release cycles (Figure 1). In addition, Ca2+ re-uptake into the ER requires ATP hydrolysis by SERCA. Therefore, depletion of intraluminal ER ATP may be an energy deprivation signal to stimulate Ca2+ release for uptake into mitochondria. In response, Ca2+ loading of the mitochondrial matrix stimulates mitochondrial respiration and ATP production. Ca2+ stimulates the activities of TCA cycle enzymes either directly (α-ketoglutarate and isocitrate dehydrogenases) or indirectly (pyruvate dehydrogenase) [76] [77]. Basal Ca2+ release through the IP3R is essential for ATP production and prevents autophagy [78, 79].
ER-Mitochondrial Flux and Apoptosis
The role of Ca2+ signals in apoptosis is a widely investigated topic. The initiation steps of the intrinsic apoptotic cascade involve release of apoptosome components, such as cytochrome C from the mitochondria [80] [81]. This process is usually accompanied by MPTP opening and organelle fragmentation and numerous studies have revealed that the most important trigger for MPTP opening is Ca2+ that acts in concert with a variety of apoptotic signals. Studies that support a role for Ca2+ homeostasis in apoptosis involve the analysis of the anti-apoptotic proteins of Bcl-2 (B cell lymphoma 2) family members that are localized to organelles that are involved in Ca2+ handling. Bcl-2 is the prototype of a large family of proteins that exhibit either anti-apoptotic or proapoptotic functions [82]. The anti-apoptotic family members, including BCL-2 and BCL-XL, contain 4 BCL-2 homology (BH) domains. The proapoptotic members have either 3 BH domains (BH1, BH2, and BH3), as in BAX and BAK, or only a single BH3 domain, as in BIM, BAD, and BID [83].
Both BCL-2 and BCL-XL are tail-anchored proteins consisting of hydrophobic a-helix which function as a membrane insertion device. The TM domain of BCL-XL in particular possesses an X-TMB sequence that is flanked by two basic amino acids and specifically targets it to the outer mitochondrial membrane. BCL-2 on the other hand contains an X/2-TMB sequence within its TM domain that is far less basic and has no sequence homology when compared with X-TMB sequence BCL-XL [84]. BCL-2 therefore cannot be targeted to mitochondria and is observed largely at the ER. Thus, BCL-2 relies on the mitochondrial chaperone protein FKBP38, an atypical member of the FK506-binding immunophilin protein family, to shuttle to the mitochondrial membrane [85]. Interestingly, BCL-2 is enriched AT the MAMs [86]. A small fraction of BCL-XL was detected on the ER membrane due to interactions with reticulon (RTN) family members [87]. MCL-1 is detected at the OMM but curiously lacks a mitochondrial targeting sequence in its TM domain [88]. Mitochondrial targeting is achieved by the first 79 amino acids on the NH2 terminus of MCL-1, which contains a PEST (Pro-Glu-Ser-Thr rich) domain and several phosphorylation sites that promote its association with mitochondria. Deletion of the amino terminus diminishes mitochondrial targeting and anti-apoptotic function of the protein [89]. Although anti-apoptotic proteins reside mainly at the OMM and/or ER membranes, they have also been localized to other cellular locations as well [90]. On the other hand, proapoptotic BCL-2 family proteins, such as BAK mainly localize to the OMM and integrate via C-terminal TM domains [91]. BAK contains a C-terminal TM domain that targets to the ER membrane [92] [93]. The hydrophobic C-terminal TM domain of PUMA predominantly targets the mitochondria but is expressed at very low levels in cells, unless there is an increase in cytosolic Ca2+ or inactivation of P53 [94] [95]. Most other forms of BH3 only proteins, such as BID, BAD and BIM, are found in the cytosol and they serve to detect apoptotic stimuli in cells and are characterized as activators or sensitizers.
Although the anti-apoptotic BCL-2 family members (BCL-2, BCL-XL and MCL-1) bind to the IP3R, the exact mechanism by which these family members regulate ER Ca2+ levels is unclear. It was demonstrated that cells deleted in BAX and BAK, which are resistant to MPTP, have decreased [Ca2+]ER that is accompanied by: 1) an increased amount of BCL-2 bound to IP3R, 2) increased PKA-dependent phosphorylation of IP3R, and 3) increased Ca2+ leak from the ER [96]. Thus, in the absence of BAX and BAK there is hyperphosphorylation and hyperactivation of the IP3R, leading to a decrease in the releasable ER Ca2+ store. In addition, BCL-2 inhibits the IP3-induced Ca2+ release from the ER [97]. Finally, BCL-Xl can bind to all IP3R isoforms to sensitize them to IP3 and increase Ca2+ leak from the ER [98]. Although overexpression of BCL-XL provides resistance to apoptotic stimuli, this effect was not observed in cells with all 3 IP3Rs deleted [99].
BH3 only proapototic proteins also regulate luminal ER Ca2+. Studies using BAX-/BAK- double knockout cells (DKO) murine fibroblasts showed a decrease in ER luminal Ca2+ stores, which resulted in reduced flux of Ca2+ from ER into the cytosol and mitochondria compared to wild-type cells under thapsigargin (Tg) stimulation. Expression of recombinant BAX in DKO cells restored ER Ca2+ to nearly wild type levels; however, expression of mitochondria-targeted BAX in DKO cells had no effect on ER Ca2+ stores. Thus, the expression of ER targeted BAX/BAK may function to increase the ER luminal Ca2+ concentration [100] [96]. Following ER Ca2+ depletion by thapsigargin, transcriptional upregulation of PUMA, a proapoptotic protein was observed with activation of caspase 3, 8 and 9 and BID, as well as release of cytochrome C into the cytosol [101].
The relative amounts of anti and pro-apoptotic proteins at the ER membrane determines whether a cell remains viable or enters apoptosis [102]. The balance between the levels of these proteins determines the steady state ER-Ca2+ content, possibly by modulating Ca2+ leak [103]. In normal cells anti-apoptotic BCL-2 proteins dominate and function at the ER, mitochondria, nuclear envelope and plasma membrane to mediate Ca2+ homeostasis, IP3 mediated Ca2+ signaling and mitochondrial Ca2+ uptake maintain physiological Ca2+ homeostasis in the cell. Sustained release of Ca2+ into mitochondria can switch from physiological functioning to apoptosis initiation [3], leading to translocation of BCL-2 family proteins to the mitochondrial membrane. If the death signal prevails the MPTP switches from a low conductive state to a high conductive state [104] [105]. Due to their important role in regulating apoptosis, today there is much effort going into developing BH3-mimetics as potential anti-cancer drugs [106, 107] [108].
Role of Ca2+ in autophagy
Numerous studies suggest that intracellular Ca2+ significantly regulates autophagy, however the specific mechanism(s) is unknown. AMP-activated protein kinase (AMPK) may play a pivotal role in this regulation. Constitutive Ca2+ leak through the IP3R to the mitochondrial matrix stimulates enzymes of the TCA cycle to increase ATP production, thereby inhibiting AMPK. However, massive Ca2+ release, via thapsigargin, although not physiological, increases cytosolic Ca2+ to activate the Ca2+/calmodulin-dependent kinase β(CaMKKβ) leading to activation of AMPK [109], which subsequently activates mammalian TOR (mTOR)-dependent autophagy [79] [110]. Inhibition of mTOR, such as by rapamycin, recruits Beclin to IP3Rs to stimulate Ca2+ release and activate autophagy. It was also suggested that a novel-type protein kinase C family member (PKCθ) is required for ER stress-induced autophagy, via Ca2+ release [111]. Ca2+ induces PKCθ phosphorylation within the activation loop that promotes localization of LC3-II in punctate cytoplasmic structures. Reduction of PKCθ prevented the ER stress-induced autophagic response. Interestingly, PKCθ activation was not required for autophagy induced by amino acid starvation, and PKCθ activation in response to ER stress did not require either mTOR kinase or the UPR pathways. However, although UPR signaling may not be essential for ER stress-induced autophagy it may potentiate other pathways to generate a strong autophagic response. For example, PERK mediated phosphorylation of eIF2α promotes autophagy [112] [113], possibly through increased expression of the transcription factors ATF4 and CHOP which activate transcription of numerous autophagy genes [114].
Ca2+ loading of mitochondria can also activate mitophagy. One current model posits that Ca2+ loading causes depolarization of the IMM to cause PINK1 translocation to the OMM leading to recruitment of the E3 ubiquitin ligase PARKIN that activates mitophagy through ubiquitination [115]. PINK1-mediated phosphorylation of MFN2 may directly recruit PARKIN to the mitochondria [116]. Obviously, more studies are required to explore roles of physiologically relevant Ca2+ signals in both normal, as well as stressed cells, and how these signals impact the autophagic response.
Conclusions
The communication between mitochondria and ER to coordinate cellular Ca2+ homeostasis is critical to numerous cell functions that extend beyond bioenergetics, metabolism and protein folding and secretion. Although much evidence supports the notion that protein misfolding in the ER causes Ca2+ release and uptake into mitochondria to activate oxidative phosphorylation, this notion needs to be experimentally tested. Altered protein folding in the ER may provide an intricate sensing mechanism to control cellular ATP levels to ensure an adequate supply for the cell as it is challenged by insults that disrupt the protein-folding environment of the ER. What is less clear is whether disturbances in mitochondrial function can disrupt protein folding in the ER. Recent studies suggest that mitochondrial stress stimulates gluconeogenic enzymes in the liver leading to insulin resistance and ER stress [117]. Further studies are required to dissect the role of Ca2+ signaling in the interplay between ER and mitochondrial functions in cell biology.
Highlights.
Protein misfolding in the ER regulates mitochondrial function through Ca2+ traffic.
Mitochondrial Ca2+ uptake controls bioenergetics, autophagy and apoptosis.
ER and mitochondrial function are coordinated by Ca2+ traffic.
Many molecular identities that regulate ER to mitochondrial Ca2+ traffic are identified.
Understanding Ca2+ traffic will lead to therapeutics for many degenerative diseases.
Acknowledgments
We thank Drs. Jing Yong and Nina Grankvist for review of this manuscript. RJK is supported by NIH grants DK042394, DK088227 and HL052173 and the Crohn's and Colitis Foundation of America.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 2.Case RM, Eisner D, Gurney A, Jones O, Muallem S, Verkhratsky A. Evolution of calcium homeostasis: from birth of the first cell to an omnipresent signalling system. Cell Calcium. 2007;42:345–350. doi: 10.1016/j.ceca.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008;27:6407–6418. doi: 10.1038/onc.2008.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kruman I, Guo Q, Mattson MP. Calcium and reactive oxygen species mediate staurosporine-induced mitochondrial dysfunction and apoptosis in PC12 cells. J Neurosci Res. 1998;51:293–308. doi: 10.1002/(SICI)1097-4547(19980201)51:3<293::AID-JNR3>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 5.Tombal B, Denmeade SR, Isaacs JT. Assessment and validation of a microinjection method for kinetic analysis of [Ca2+]i in individual cells undergoing apoptosis. Cell Calcium. 1999;25:19–28. doi: 10.1054/ceca.1998.0005. [DOI] [PubMed] [Google Scholar]
- 6.Lynch K, Fernandez G, Pappalardo A, Peluso JJ. Basic fibroblast growth factor inhibits apoptosis of spontaneously immortalized granulosa cells by regulating intracellular free calcium levels through a protein kinase Cdelta-dependent pathway. Endocrinology. 2000;141:4209–4217. doi: 10.1210/endo.141.11.7742. [DOI] [PubMed] [Google Scholar]
- 7.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 8.Mallilankaraman K, Cardenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenar T, Csordas G, Madireddi P, Yang J, Muller M, Miller R, Kolesar JE, Molgo J, Kaufman B, Hajnoczky G, Foskett JK, Madesh M. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol. 2012;14:1336–1343. doi: 10.1038/ncb2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, Rothberg BS, Mak DO, Foskett JK, Madesh M. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell. 2012;151:630–644. doi: 10.1016/j.cell.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sancak Y, Markhard AL, Kitami T, Kovacs-Bogdan E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O, Mootha VK. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science. 2013;342:1379–1382. doi: 10.1126/science.1242993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ringer S. A third contribution regarding the Influence of the Inorganic Constituents of the Blood on the Ventricular Contraction. J Physiol. 1883;4:222–225. doi: 10.1113/jphysiol.1883.sp000127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deluca HF, Engstrom GW. Calcium uptake by rat kidney mitochondria. Proc Natl Acad Sci U S A. 1961;47:1744–1750. doi: 10.1073/pnas.47.11.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vasington FD, Murphy JV. Ca ion uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. The Journal of biological chemistry. 1962;237:2670–2677. [PubMed] [Google Scholar]
- 14.Miyawaki A, Griesbeck O, Heim R, Tsien RY. Dynamic and quantitative Ca2+ measurements using improved cameleons. Proc Natl Acad Sci U S A. 1999;96:2135–2140. doi: 10.1073/pnas.96.5.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rudolf R, Mongillo M, Rizzuto R, Pozzan T. Looking forward to seeing calcium. Nat Rev Mol Cell Biol. 2003;4:579–586. doi: 10.1038/nrm1153. [DOI] [PubMed] [Google Scholar]
- 16.Bonora M, Giorgi C, Bononi A, Marchi S, Patergnani S, Rimessi A, Rizzuto R, Pinton P. Subcellular calcium measurements in mammalian cells using jellyfish photoprotein aequorin-based probes. Nat Protoc. 2013;8:2105–2118. doi: 10.1038/nprot.2013.127. [DOI] [PubMed] [Google Scholar]
- 17.Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. 2006;86:369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- 18.Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- 19.Gorlach A, Klappa P, Kietzmann T. The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid Redox Signal. 2006;8:1391–1418. doi: 10.1089/ars.2006.8.1391. [DOI] [PubMed] [Google Scholar]
- 20.Prins D, Michalak M. Organellar calcium buffers. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a004069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annual review of biochemistry. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 22.Michalak M, Groenendyk J, Szabo E, Gold LI, Opas M. Calreticulin, a multiprocess calcium-buffering chaperone of the endoplasmic reticulum. The Biochemical journal. 2009;417:651–666. doi: 10.1042/BJ20081847. [DOI] [PubMed] [Google Scholar]
- 23.Dorner AJ, Wasley LC, Kaufman RJ. Protein dissociation from GRP78 and secretion are blocked by depletion of cellular ATP levels. Proc Natl Acad Sci U S A. 1990;87:7429–7432. doi: 10.1073/pnas.87.19.7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hendershot LM. The ER function BiP is a master regulator of ER function. Mt Sinai J Med. 2004;71:289–297. [PubMed] [Google Scholar]
- 25.Zapun A, Darby NJ, Tessier DC, Michalak M, Bergeron JJ, Thomas DY. Enhanced catalysis of ribonuclease B folding by the interaction of calnexin or calreticulin with ERp57. The Journal of biological chemistry. 1998;273:6009–6012. doi: 10.1074/jbc.273.11.6009. [DOI] [PubMed] [Google Scholar]
- 26.Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annual review of biochemistry. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 27.Hebert DN, Molinari M. Flagging and docking: dual roles for N-glycans in protein quality control and cellular proteostasis. Trends in biochemical sciences. 2012;37:404–410. doi: 10.1016/j.tibs.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tu BP, Weissman JS. Oxidative protein folding in eukaryotes: mechanisms and consequences. The Journal of cell biology. 2004;164:341–346. doi: 10.1083/jcb.200311055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vandecaetsbeek I, Vangheluwe P, Raeymaekers L, Wuytack F, Vanoevelen J. The Ca2+ pumps of the endoplasmic reticulum and Golgi apparatus. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a004184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olesen C, Picard M, Winther AM, Gyrup C, Morth JP, Oxvig C, Moller JV, Nissen P. The structural basis of calcium transport by the calcium pump. Nature. 2007;450:1036–1042. doi: 10.1038/nature06418. [DOI] [PubMed] [Google Scholar]
- 31.Toyoshima C. How Ca2+-ATPase pumps ions across the sarcoplasmic reticulum membrane. Biochim Biophys Acta. 2009;1793:941–946. doi: 10.1016/j.bbamcr.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 32.Moeller CW, Welch KC. Prevention and management of complications in sphenoidotomy. Otolaryngol Clin North Am. 2010;43:839–854. doi: 10.1016/j.otc.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 33.John LM, Lechleiter JD, Camacho P. Differential modulation of SERCA2 isoforms by calreticulin. The Journal of cell biology. 1998;142:963–973. doi: 10.1083/jcb.142.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roderick HL, Lechleiter JD, Camacho P. Cytosolic phosphorylation of calnexin controls intracellular Ca(2+) oscillations via an interaction with SERCA2b. The Journal of cell biology. 2000;149:1235–1248. doi: 10.1083/jcb.149.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arnaudeau S, Frieden M, Nakamura K, Castelbou C, Michalak M, Demaurex N. Calreticulin differentially modulates calcium uptake and release in the endoplasmic reticulum and mitochondria. The Journal of biological chemistry. 2002;277:46696–46705. doi: 10.1074/jbc.M202395200. [DOI] [PubMed] [Google Scholar]
- 36.Smyth JT, Hwang SY, Tomita T, DeHaven WI, Mercer JC, Putney JW. Activation and regulation of store-operated calcium entry. J Cell Mol Med. 2010;14:2337–2349. doi: 10.1111/j.1582-4934.2010.01168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carrasco S, Meyer T. STIM proteins and the endoplasmic reticulum-plasma membrane junctions. Annual review of biochemistry. 2011;80:973–1000. doi: 10.1146/annurev-biochem-061609-165311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rohacs T. Regulation of transient receptor potential channels by the phospholipase C pathway. Adv Biol Regul. 2013;53:341–355. doi: 10.1016/j.jbior.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vazquez G, Wedel BJ, Aziz O, Trebak M, Putney JW., Jr The mammalian TRPC cation channels. Biochim Biophys Acta. 2004;1742:21–36. doi: 10.1016/j.bbamcr.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 40.Parys JB, Decuypere JP, Bultynck G. Role of the inositol 1,4,5-trisphosphate receptor/Ca2+-release channel in autophagy. Cell communication and signaling : CCS. 2012;10:17. doi: 10.1186/1478-811X-10-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mendes CC, Gomes DA, Thompson M, Souto NC, Goes TS, Goes AM, Rodrigues MA, Gomez MV, Nathanson MH, Leite MF. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. The Journal of biological chemistry. 2005;280:40892–40900. doi: 10.1074/jbc.M506623200. [DOI] [PubMed] [Google Scholar]
- 42.Kiviluoto S, Vervliet T, Ivanova H, Decuypere JP, De Smedt H, Missiaen L, Bultynck G, Parys JB. Regulation of inositol 1,4,5-trisphosphate receptors during endoplasmic reticulum stress. Biochim Biophys Acta. 2013;1833:1612–1624. doi: 10.1016/j.bbamcr.2013.01.026. [DOI] [PubMed] [Google Scholar]
- 43.Van Coppenolle F, Vanden Abeele F, Slomianny C, Flourakis M, Hesketh J, Dewailly E, Prevarskaya N. Ribosome-translocon complex mediates calcium leakage from endoplasmic reticulum stores. J Cell Sci. 2004;117:4135–4142. doi: 10.1242/jcs.01274. [DOI] [PubMed] [Google Scholar]
- 44.Alder NN, Shen Y, Brodsky JL, Hendershot LM, Johnson AE. The molecular mechanisms underlying BiP-mediated gating of the Sec61 translocon of the endoplasmic reticulum. The Journal of cell biology. 2005;168:389–399. doi: 10.1083/jcb.200409174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pinton P, Ferrari D, Magalhaes P, Schulze-Osthoff K, Di Virgilio F, Pozzan T, Rizzuto R. Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. The Journal of cell biology. 2000;148:857–862. doi: 10.1083/jcb.148.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bultynck G, Kiviluoto S, Henke N, Ivanova H, Schneider L, Rybalchenko V, Luyten T, Nuyts K, De Borggraeve W, Bezprozvanny I, Parys JB, De Smedt H, Missiaen L, Methner A. The C terminus of Bax inhibitor-1 forms a Ca2+-permeable channel pore. The Journal of biological chemistry. 2012;287:2544–2557. doi: 10.1074/jbc.M111.275354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chami M, Oules B, Szabadkai G, Tacine R, Rizzuto R, Paterlini-Brechot P. Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol Cell. 2008;32:641–651. doi: 10.1016/j.molcel.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lehninger AL, Rossi CS, Greenawalt JW. Respiration-dependent accumulation of inorganic phosphate and Ca ions by rat liver mitochondria. Biochem Biophys Res Commun. 1963;10:444–448. doi: 10.1016/0006-291x(63)90377-2. [DOI] [PubMed] [Google Scholar]
- 49.Carafoli E. Historical review: mitochondria and calcium: ups and downs of an unusual relationship. Trends in biochemical sciences. 2003;28:175–181. doi: 10.1016/S0968-0004(03)00053-7. [DOI] [PubMed] [Google Scholar]
- 50.Mitchell P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature. 1961;191:144–148. doi: 10.1038/191144a0. [DOI] [PubMed] [Google Scholar]
- 51.Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N, Hall MN. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci U S A. 2013;110:12526–12534. doi: 10.1073/pnas.1302455110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patron M, Raffaello A, Granatiero V, Tosatto A, Merli G, De Stefani D, Wright L, Pallafacchina G, Terrin A, Mammucari C, Rizzuto R. The mitochondrial calcium uniporter (MCU): molecular identity and physiological roles. The Journal of biological chemistry. 2013;288:10750–10758. doi: 10.1074/jbc.R112.420752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martell JD, Deerinck TJ, Sancak Y, Poulos TL, Mootha VK, Sosinsky GE, Ellisman MH, Ting AY. Engineered ascorbate peroxidase as a genetically encodedreporter for electron microscopy. Nat Biotechnol. 2012;30:1143–1148. doi: 10.1038/nbt.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature. 2010;467:291–296. doi: 10.1038/nature09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bick AG, Calvo SE, Mootha VK. Evolutionary diversity of the mitochondrial calcium uniporter. Science. 2012;336:886. doi: 10.1126/science.1214977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, Taneja N, Oshea J, Koteliansky V, Mootha VK. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One. 2013;8:e55785. doi: 10.1371/journal.pone.0055785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–1472. doi: 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takeuchi A, Kim B, Matsuoka S. The mitochondrial Na+-Ca2+ exchanger, NCLX, regulates automaticity of HL-1 cardiomyocytes. Sci Rep. 2013;3:2766. doi: 10.1038/srep02766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elrod JW, Molkentin JD. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ J. 2013;77:1111–1122. doi: 10.1253/circj.cj-13-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D, Sekler I. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci U S A. 2010;107:436–441. doi: 10.1073/pnas.0908099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim B, Takeuchi A, Koga O, Hikida M, Matsuoka S. Pivotal role of mitochondrial Na(+)(-)Ca(2)(+) exchange in antigen receptor mediated Ca(2)(+) signalling in DT40 and A20 B lymphocytes. J Physiol. 2012;590:459–474. doi: 10.1113/jphysiol.2011.222927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rusinol AE, Cui Z, Chen MH, Vance JE. A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. The Journal of biological chemistry. 1994;269:27494–27502. [PubMed] [Google Scholar]
- 65.Marchi S, Patergnani S, Pinton P. The endoplasmic reticulum-mitochondria connection: One touch, multiple functions. Biochim Biophys Acta. 2013 doi: 10.1016/j.bbabio.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 66.Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. The Journal of cell biology. 2006;175:901–911. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Poston CN, Duong E, Cao Y, Bazemore-Walker CR. Proteomic analysis of lipid raft-enriched membranes isolated from internal organelles. Biochem Biophys Res Commun. 2011;415:355–360. doi: 10.1016/j.bbrc.2011.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Poston CN, Krishnan SC, Bazemore-Walker CR. In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM) J Proteomics. 2013;79:219–230. doi: 10.1016/j.jprot.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 69.Flachbartova Z, Kovacech B. Mortalin - a multipotent chaperone regulating cellular processes ranging from viral infection to neurodegeneration. Acta Virol. 2013;57:3–15. doi: 10.4149/av_2013_01_3. [DOI] [PubMed] [Google Scholar]
- 70.de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 71.Guo X, Chen KH, Guo Y, Liao H, Tang J, Xiao RP. Mitofusin 2 triggers vascular smooth muscle cell apoptosis via mitochondrial death pathway. Circ Res. 2007;101:1113–1122. doi: 10.1161/CIRCRESAHA.107.157644. [DOI] [PubMed] [Google Scholar]
- 72.Ngoh GA, Papanicolaou KN, Walsh K. Loss of mitofusin 2 promotes endoplasmic reticulum stress. The Journal of biological chemistry. 2012;287:20321–20332. doi: 10.1074/jbc.M112.359174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sugiura A, Nagashima S, Tokuyama T, Amo T, Matsuki Y, Ishido S, Kudo Y, McBride HM, Fukuda T, Matsushita N, Inatome R, Yanagi S. MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Mol Cell. 2013;51:20–34. doi: 10.1016/j.molcel.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 74.Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007;131:596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 75.Giorgi C, Ito K, Lin HK, Santangelo C, Wieckowski MR, Lebiedzinska M, Bononi A, Bonora M, Duszynski J, Bernardi R, Rizzuto R, Tacchetti C, Pinton P, Pandolfi PP. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science. 2010;330:1247–1251. doi: 10.1126/science.1189157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Decuypere JP, Monaco G, Bultynck G, Missiaen L, De Smedt H, Parys JB. The IP(3) receptor-mitochondria connection in apoptosis and autophagy. Biochim Biophys Acta. 2011;1813:1003–1013. doi: 10.1016/j.bbamcr.2010.11.023. [DOI] [PubMed] [Google Scholar]
- 77.Gunter TE, Yule DI, Gunter KK, Eliseev RA, Salter JD. Calcium and mitochondria. FEBS Lett. 2004;567:96–102. doi: 10.1016/j.febslet.2004.03.071. [DOI] [PubMed] [Google Scholar]
- 78.Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, Thompson CB, Birnbaum MJ, Hallows KR, Foskett JK. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142:270–283. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cardenas C, Foskett JK. Mitochondrial Ca(2+) signals in autophagy. Cell Calcium. 2012;52:44–51. doi: 10.1016/j.ceca.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell. 2002;9:423–432. doi: 10.1016/s1097-2765(02)00442-2. [DOI] [PubMed] [Google Scholar]
- 81.Yuan S, Akey CW. Apoptosome structure, assembly, and procaspase activation. Structure. 2013;21:501–515. doi: 10.1016/j.str.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cory S, Huang DC, Adams JM. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene. 2003;22:8590–8607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- 83.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 84.Kaufmann T, Schlipf S, Sanz J, Neubert K, Stein R, Borner C. Characterization of the signal that directs Bcl-x(L), but not Bcl-2, to the mitochondrial outer membrane. The Journal of cell biology. 2003;160:53–64. doi: 10.1083/jcb.200210084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Portier BP, Taglialatela G. Bcl-2 localized at the nuclear compartment induces apoptosis after transient overexpression. The Journal of biological chemistry. 2006;281:40493–40502. doi: 10.1074/jbc.M606181200. [DOI] [PubMed] [Google Scholar]
- 86.Meunier J, Hayashi T. Sigma-1 receptors regulate Bcl-2 expression by reactive oxygen species-dependent transcriptional regulation of nuclear factor kappaB. J Pharmacol Exp Ther. 2010;332:388–397. doi: 10.1124/jpet.109.160960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tagami S, Eguchi Y, Kinoshita M, Takeda M, Tsujimoto Y. A novel protein, RTN-XS, interacts with both Bcl-XL and Bcl-2 on endoplasmic reticulum and reduces their anti-apoptotic activity. Oncogene. 2000;19:5736–5746. doi: 10.1038/sj.onc.1203948. [DOI] [PubMed] [Google Scholar]
- 88.Chou CH, Lee RS, Yang-Yen HF. An internal EELD domain facilitates mitochondrial targeting of Mcl-1 via a Tom70-dependent pathway. Mol Biol Cell. 2006;17:3952–3963. doi: 10.1091/mbc.E06-04-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Germain M, Duronio V. The N terminus of the anti-apoptotic BCL-2 homologue MCL-1 regulates its localization and function. The Journal of biological chemistry. 2007;282:32233–32242. doi: 10.1074/jbc.M706408200. [DOI] [PubMed] [Google Scholar]
- 90.Jeong SY, Gaume B, Lee YJ, Hsu YT, Ryu SW, Yoon SH, Youle RJ. Bcl-x(L) sequesters its C-terminal membrane anchor in soluble, cytosolic homodimers. EMBO J. 2004;23:2146–2155. doi: 10.1038/sj.emboj.7600225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lindsay J, Esposti MD, Gilmore AP. Bcl-2 proteins and mitochondria-- specificity in membrane targeting for death. Biochim Biophys Acta. 2011;1813:532–539. doi: 10.1016/j.bbamcr.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 92.Mathai JP, Germain M, Marcellus RC, Shore GC. Induction and endoplasmic reticulum location of BIK/NBK in response to apoptotic signaling by E1A and p53. Oncogene. 2002;21:2534–2544. doi: 10.1038/sj.onc.1205340. [DOI] [PubMed] [Google Scholar]
- 93.Zhao X, Wang L, Sun Y, Ye L, Lu J, Yuan Y, Qian G, Ge S. The endoplasmic reticulum (ER)-target protein Bik induces Hep3B cells apoptosis by the depletion of the ER Ca2+ stores. Mol Cell Biochem. 2008;312:33–38. doi: 10.1007/s11010-008-9718-4. [DOI] [PubMed] [Google Scholar]
- 94.Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 95.Reimertz C, Kogel D, Rami A, Chittenden T, Prehn JH. Gene expression during ER stress-induced apoptosis in neurons: induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. The Journal of cell biology. 2003;162:587–597. doi: 10.1083/jcb.200305149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, Korsmeyer SJ. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2005;102:105–110. doi: 10.1073/pnas.0408352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA, Velez P, Distelhorst CW. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. The Journal of cell biology. 2004;166:193–203. doi: 10.1083/jcb.200309146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, Foskett JK. The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol. 2005;7:1021–1028. doi: 10.1038/ncb1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li C, Wang X, Vais H, Thompson CB, Foskett JK, White C. Apoptosis regulation by Bcl-x(L) modulation of mammalian inositol 1,4,5-trisphosphate receptor channel isoform gating. Proc Natl Acad Sci U S A. 2007;104:12565–12570. doi: 10.1073/pnas.0702489104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 101.Luo X, He Q, Huang Y, Sheikh MS. Transcriptional upregulation of PUMA modulates endoplasmic reticulum calcium pool depletion-induced apoptosis via Bax activation. Cell Death Differ. 2005;12:1310–1318. doi: 10.1038/sj.cdd.4401659. [DOI] [PubMed] [Google Scholar]
- 102.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 103.Giam M, Huang DC, Bouillet P. BH3-only proteins and their roles in programmed cell death. Oncogene. 2008;27(Suppl 1):S128–136. doi: 10.1038/onc.2009.50. [DOI] [PubMed] [Google Scholar]
- 104.Jouaville LS, Ichas F, Mazat JP. Modulation of cell calcium signals by mitochondria. Mol Cell Biochem. 1998;184:371–376. [PubMed] [Google Scholar]
- 105.Bianchi K, Rimessi A, Prandini A, Szabadkai G, Rizzuto R. Calcium and mitochondria: mechanisms and functions of a troubled relationship. Biochim Biophys Acta. 2004;1742:119–131. doi: 10.1016/j.bbamcr.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 106.Labi V, Erlacher M, Kiessling S, Villunger A. BH3-only proteins in cell death initiation, malignant disease and anticancer therapy. Cell Death Differ. 2006;13:1325–1338. doi: 10.1038/sj.cdd.4401940. [DOI] [PubMed] [Google Scholar]
- 107.Shamas-Din A, Brahmbhatt H, Leber B, Andrews DW. BH3-only proteins: Orchestrators of apoptosis. Biochim Biophys Acta. 2011;1813:508–520. doi: 10.1016/j.bbamcr.2010.11.024. [DOI] [PubMed] [Google Scholar]
- 108.Oakes SR, Vaillant F, Lim E, Lee L, Breslin K, Feleppa F, Deb S, Ritchie ME, Takano E, Ward T, Fox SB, Generali D, Smyth GK, Strasser A, Huang DC, Visvader JE, Lindeman GJ. Sensitization of BCL-2-expressing breast tumors to chemotherapy by the BH3 mimetic ABT-737. Proc Natl Acad Sci U S A. 2012;109:2766–2771. doi: 10.1073/pnas.1104778108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hoyer-Hansen M, Bastholm L, Szyniarowski P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N, Elling F, Rizzuto R, Mathiasen IS, Jaattela M. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25:193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 110.Decuypere JP, Kindt D, Luyten T, Welkenhuyzen K, Missiaen L, De Smedt H, Bultynck G, Parys JB. mTOR-Controlled Autophagy Requires Intracellular Ca(2+), Signaling. PLoS One. 2013;8:e61020. doi: 10.1371/journal.pone.0061020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sakaki K, Wu J, Kaufman RJ. Protein kinase Ctheta is required for autophagy in response to stress in the endoplasmic reticulum. The Journal of biological chemistry. 2008;283:15370–15380. doi: 10.1074/jbc.M710209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Talloczy Z, Jiang W, Virgin HWt, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A. 2002;99:190–195. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14:230–239. doi: 10.1038/sj.cdd.4401984. [DOI] [PubMed] [Google Scholar]
- 114.B'Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41:7683–7699. doi: 10.1093/nar/gkt563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, Youle RJ. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. The Journal of cell biology. 2002;159:931–938. doi: 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen Y, Dorn GW., 2nd PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science. 2013;340:471–475. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lee JW, Kim WH, Lim JH, Song EH, Song J, Choi KY, Jung MH. Mitochondrial dysfunction: glucokinase downregulation lowers interaction of glucokinase with mitochondria, resulting in apoptosis of pancreatic beta-cells. Cell Signal. 2009;21:69–78. doi: 10.1016/j.cellsig.2008.09.015. [DOI] [PubMed] [Google Scholar]