

Abstract
Inhalation of ambient and workplace particulate air pollution is associated with increased risk of cardiovascular disease. One proposed mechanism for this association is that pulmonary inflammation induces a hepatic acute phase response, which increases risk of cardiovascular disease. Induction of the acute phase response is intimately linked to risk of cardiovascular disease as shown in both epidemiological and animal studies. Indeed, blood levels of acute phase proteins, such as C-reactive protein and serum amyloid A, are independent predictors of risk of cardiovascular disease in prospective epidemiological studies. In this review, we present and review emerging evidence that inhalation of particles (e.g., air diesel exhaust particles and nanoparticles) induces a pulmonary acute phase response, and propose that this induction constitutes the causal link between particle inhalation and risk of cardiovascular disease. Increased levels of acute phase mRNA and proteins in lung tissues, bronchoalveolar lavage fluid and plasma clearly indicate pulmonary acute phase response following pulmonary deposition of different kinds of particles including diesel exhaust particles, nanoparticles, and carbon nanotubes. The pulmonary acute phase response is dose-dependent and long lasting. Conversely, the hepatic acute phase response is reduced relative to lung or entirely absent. We also provide evidence that pulmonary inflammation, as measured by neutrophil influx, is a predictor of the acute phase response and that the total surface area of deposited particles correlates with the pulmonary acute phase response. We discuss the implications of these findings in relation to occupational exposure to nanoparticles.
How to cite this article: WIREs Nanomed Nanobiotechnol 2014, 6:517–531. doi: 10.1002/wnan.1279
INTRODUCTION
Epidemiological studies provide strong evidence to support a link between exposure to particulate air pollution and risk of cardiovascular disease.1–3 For example, in a natural intervention study in Ireland, a ban on coal sales for heating in private households led to a reduction in the level of black smoke from 50.2 to 14.6 µg/m3.3 The reduction in black smoke was accompanied by a 10.3% (95% CI 8–13) reduction in deaths associated with cardiovascular events. An association has also been established between air pollution and blood levels of C-reactive protein (CRP, a marker of acute phase response, inflammation, and cardiovascular effects) in a cross-sectional study.4 In APOE −/− mice fed a Western diet, a model for atherosclerosis, pulmonary exposure to ambient air particles, welding fumes, and nanoparticles increased plaque progression in aorta.5–8 Thus, evidence indicates that pulmonary exposure to particles in air pollution increases cardiovascular risk. Increased industrial use of nanotechnology, in particular nanoparticles, has raised the specific concern that (occupational) exposure to nanoparticles may increase risk cardiovascular disease.9 Knowledge of the mechanism of action driving the hazardous effects associated with nanoparticle exposure would enable assessment of the magnitude of this risk and identify possible preventive strategies.
The acute phase response is the primary alarm response induced in the body by a variety of factors including bacterial infections, trauma, and surgery.10 The acute phase response and accompanying increased levels of acute phase proteins in blood are risk factors for cardiovascular disease.11–13 Conditions that lead to increased acute phase response are associated with increased risk of cardiovascular disease in epidemiological studies.14–16 For example, periodontal pathogen and viral infections are associated with risk of cardiovascular disease17,18; periodontal infection has been shown to induce acute phase response and to accelerate plaque progression in APOE −/− mice.19 Overall, it is established that blood levels of the acute phase proteins CRP and serum amyloid A (SAA) are predictive of future cardiovascular disease in prospective studies.20–22
Various investigators have proposed that the mechanism underlying the association between acute phase response and cardiovascular effects involves the inhalation of particles leading to pulmonary inflammation and subsequent systemic circulation of cytokines, which in turn induce a hepatic acute phase response.13,23–25 We have studied the possible adverse effects resulting from inhalation or pulmonary deposition of diesel exhaust particles and nanomaterials in mice for several years.26–42 Over the course of this research we have been unable to detect the induction of any hepatic acute phase response by analysis of liver mRNA or protein levels of acute phase genes in mice exposed by inhalation to diesel exhaust particles or carbon black particles in our own experiments.43 Instead, our results demonstrates that inhalation of particles induces a strong pulmonary acute phase response. In the present review article we discuss our alternative proposed mechanism of action governing an association between pulmonary acute phase response and cardiovascular disease, and the resulting implications.
ACUTE PHASE RESPONSE
The acute phase response is characterized by altered blood levels of acute phase proteins including CRP, SAA, and fibrinogen10 (Figure 1) and (Box 1). Although this pathway is generally conserved across mammals, there are some notable differences; in particular, noteworthy differences exist between humans and mice. In mice, Saa3 is expressed in various tissues including the lung, whereas Saa1 and Saa2 are considered liver specific.44 In contrast, in humans SAA3 is a pseudogene,45 and SAA1 and SAA2 are expressed hepatically and extra-hepatically.46 Moreover, Crp is only moderately induced by inflammatory stimuli in mice.47,48 Thus, these differences must be taken into consideration when extrapolating results derived from mouse models to humans.
Fig 1.
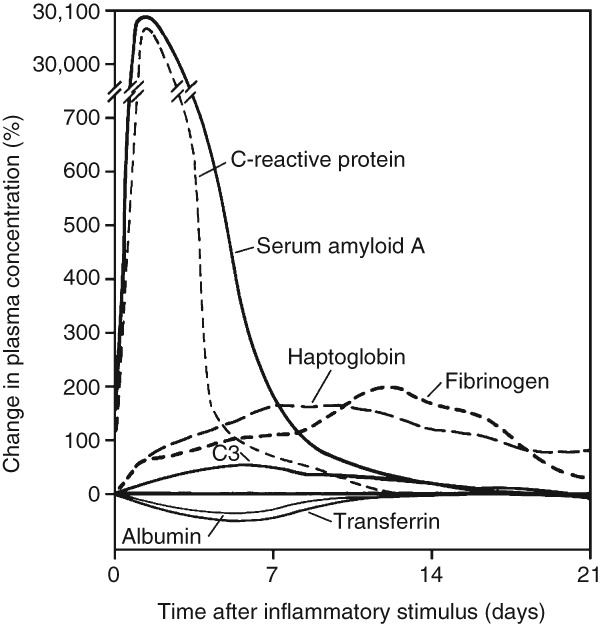
Characteristic patterns of change in plasma concentrations of some acute-phase proteins after a moderate inflammatory stimulus. (Reprinted with permission from Ref 10. Copyright 1999, Massachusetts Medical Society)
BOX 1 PHYSIOLOGICAL FUNCTIONS OF THE ACUTE PHASE RESPONSE.
The acute phase response is defined as the systemic changes induced by cytokines released from inflamed areas. It is termed the acute phase response even though it accompanies both acute and chronic inflammation. The acute phase response is characterized by changes in the concentration of plasma proteins, the so-called acute phase proteins (e.g., SAA and CRP). In addition, the acute phase response is accompanied by other acute phase phenomena. These include changes at the neuroendocrine, hematopoietic, metabolic, and hepatic level, among others. Examples of neuroendocrine changes are fever, somnolence, and anorexia. Hematopoietic changes include anemia and leuco- and thrombocytosis, while examples of metabolic changes are osteoporosis and decreased gluconeogenesis.
The acute phase response is believed to be beneficial. For example, the opsonization of microorganisms by CRP makes it easier for macrophages to recognize and engulf invading microorganisms. Another example is somnolence, which is a characteristic in patients with inflammatory conditions. The reduced energy demand associated with somnolence is beneficial in disease states. However, when the inflammatory condition and the accompanying acute phase response become chronic, it may cause adverse effects. Secondary amyloidosis is the result of an accumulation of SAA in individuals with chronic inflammatory disorders. Another example of a non-beneficial effect of a chronic acute phase response is anemia.
Since plasma concentrations of acute phase proteins reflect the level of inflammation these are used clinically to evaluate the progression of disease and to predict risk of future disease. Increasing evidence suggest that some of these changes are causal and not just passive bystanders of disease.
Based on Gabay and Kushner.10
PULMONARY ACUTE PHASE RESPONSE
Although the acute phase response is defined by changes in blood concentrations of specific proteins, the cellular origin of these proteins is not well characterized. The underlying assumption has been that they are of hepatic origin.12,22–24 We assessed transcriptional changes in acute phase response genes to determine the origin of the acute phase response in mice. Inhalation and instillation of various particles, including carbon black and TiO2 nanoparticles, diesel exhaust particles, dust samples and both single and multiwalled carbon nanotubes, induced a pulmonary acute phase response, whereas the hepatic response was barely detectable or entirely absent.11,28,35,39,40,43,49 Studies supporting this finding are shown in Table1. For example, in one study mice were exposed daily for 1 h by inhalation to 40 mg/m3 nano TiO2 over 11 consecutive days. This daily dose corresponds to half the dose of an 8-h working day at the Danish Occupational Exposure limit of 10 mg/m3. Mice were sampled 5 days after the final dose. Global transcriptional profiling in these mice revealed that the acute phase genes Saa3, Saa1, and fibrinogen were the most differentially expressed genes in lung.39 The acute phase response was dose-dependent; higher doses led to statistically significantly greater gene expression of more acute phase genes and to a dose-dependent increase in the expression of the acute phase genes at the mRNA level. In another study mice were sampled 24 h after intratracheal instillation of nano-TiO2; we observed statistically significant increases in 8, 21, and 44 acute phase response genes in the low (18 µg/mouse), medium (54 µg/mouse), and high (162 µg/mouse) dose groups in lung tissue using a 1.3 fold change as a cut-off.28 The same pattern was observed applying a 1.5-fold cut-off (Table1). In all of the studies, Saa3 was the most upregulated acute phase gene and in many cases was also the most differentially expressed gene. Consequently, we have used Saa3 mRNA levels as a biomarker of pulmonary acute phase response.11,27,28,39,40,42 This analysis showed that a clear dose-dependency is observed in the expression levels of Saa3 mRNA. For example, 24 h after intratracheal instillation of nano-TiO2, Saa3 mRNA levels increased by 1.8-fold for the low, 87-fold for the medium and 368-fold for the high dose groups11 (Table1). The duration of the acute phase responses was dose- and particle-dependent. The largest response was observed 1 day post exposure for most particles, and the response declined over time. However, mice exposed to carbon black at the medium and high dose (54 µg and 162 µg/mouse) exhibited 5-fold and 22-fold increases in pulmonary Saa3 mRNA levels compared to vehicle-exposed controls, respectively, 28 days after the exposure.11 These doses correspond to 3 or 9 days of exposure to Printex 90 carbon black at the current Danish occupational exposure limit to carbon black (3.5 mg/m3), respectively. For nano-TiO2, Saa3 mRNA was only statistically significantly up-regulated after 28 days for the highest dose (162 µg/mouse). The studies described above clearly demonstrate that pulmonary transcriptional changes in the acute phase response are induced following particle inhalation and remain elevated for a prolonged period of time.
Differential Expression of Murine Acute Phase Genes and Saa3 Expression Levels after Exposure to Different Nanomaterials and at Different Time Points
| Post Exposure Day | 1 | 3 | 28 | Ref | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Dose/Animal | 18 µg | 54 µg | 162 µg | 18 µg | 54 µg | 162 µg | 18 µg | 54 µg | 162 µg | |
| TiO2 nanoparticles | ||||||||||
| N acute phase genes1 | 0 | 5 | 10 | 3 | 1 | 3 | 1 | 2 | 3 | 28 |
| Fold increase of Saa3 mRNA2 | 1.8 | 87 | 368 | 1.1 | 2.6 | 19 | 1 | 1.8 | 5.5 | 11 |
| Carbon Black nanoparticles | ||||||||||
| N acute phase genes1 | 0 | 7 | 10 | 0 | 0 | 4 | 0 | 0 | 2 | 42 |
| Fold increase of Saa3 mRNA2 | 63 | 237 | 294 | 8.3 | 24 | 51 | 1.1 | 5 | 22 | 11 |
| Multiwalled Carbon nanotubes | ||||||||||
| N acute phase genes1 | 5 | 5 | 10 | ND | ND | ND | ND | 1 | ND | 35 |
| Fold increase of Saa3 mRNA2 | 52 | 151 | 95 | 39 | 152 | 612 | 7.9 | 29 | 88 | 11 |
ND, not determined.
Table 1The number of differentially expressed acute phase genes as observed in DNA microarray. Murine acute phase genes were identified through the Gene category on NCBI. The following genes were included: ACADM, AHSG, ANGPTL4, APEX1, CD163, CREB3L3, CRP, CSF3, EPO, ESRRA, F2, F8, FGF21, FN2, HAMP, HNF4A, HP, IL1RN, IL22, IL6, IL6ST, INS2, ITIH4, LBP, LCN2, MRGPRA3, NR1H4, NR5A2, ORM1, ORM2, PRLR, REG3A, REG3B, REG3G, RELA, SEPP1, SERPINA1B, SERPINA3N, SERPINF2, SIGIRR, SIM2, SMAD3, STAT3, STAT5B, SULT2A1, SAA1, SAA2, SAA3, SAA4, SAAL3, TSC2, and VIMP. Genes with a fold change ± 1.5 and FDR P-values ≤ 0.05 were considered as differentially expressed.
Table 2Fold increase of mRNA Saa3 was determined using qRT-PCR. Saa3 levels were normalized to 18S levels.
Induction of pulmonary acute phase response has also been demonstrated at the protein level in mice. For example, SAA3 protein levels measured by Western blotting were increased 2.2 fold in lung tissue 5 days after inhalation exposure to nano-TiO2.39 SAA levels were also elevated in bronchoalveolar fluid and plasma after intratracheal instillation of multiwalled carbon nanotubes.11 Protein levels of two other acute phase proteins, granulocyte colony-stimulating factor (GCSF), and granulocyte-macrophage colony-stimulating-factor (GM-CSF), were increased 6-fold and 2.5-fold, respectively, in lung tissue 24 h after instillation of nanosized TiO228 and 3-fold and 1.3-fold increased, respectively, 26–27 days after instillation of nanosized carbon black.29 In both cases, GCSF protein levels increased in a dose-dependent manner. Moreover, dose-dependent increases in the protein levels of IL-1β, IL-6, and CXCL1 were also found in lung tissues 26–27 days after instillation of nanosized carbon black.29 Teeguarden et al. reported increased protein levels of the acute phase proteins C3, Fn1, S100A8 and S100A9 in the lung tissues of mice exposed to single-walled carbon nanotubes by aspiration.50 Moreover, the level of the acute phase protein haptoglobin was increased in BALF 24 h after a single intratracheal instillation of 200 µg nanosized carbon black/mouse.51 These observed protein level changes are consistent with, and support, the large changes observed in the transcriptional network of acute phase response genes in lung tissues. Moreover, it is worth noting that changes in pulmonary expression of acute phase response genes are correlated with increased concentrations of acute phase proteins in systemic circulation. Elevated SAA levels were found in plasma both 1 and 28 days after intratracheal instillation of nanosized carbon black42 and 3 days following pulmonary exposure to multiwalled carbon nanotubes.11
Overall, the results described above indicate that expression changes in pulmonary acute phase response genes are the strongest measured response following pulmonary exposure to nanoparticles. The acute phase response has been demonstrated at both the transcript and protein level in lung tissue, bronchoalveolar lavage fluid and in plasma. Finally, the response is dose-dependent and long lasting at doses that are comparable to relatively short exposures at the current Danish Occupational Exposure Limits for carbon black and TiO2.
PULMONARY ACUTE PHASE RESPONSE CORRELATES WITH NEUTROPHIL INFLUX AND SURFACE AREA OF DEPOSITED PARTICLES
We recently reported a close correlation between Saa3 mRNA levels in lung tissue and the number of neutrophil cells in bronchoalveolar lavage fluid (Figure 2).11 This is in agreement with the observation that SAA is a strong neutrophil chemoattractant.52,53 Pulmonary Saa3 mRNA is a biomarker of a pulmonary acute phase response. Therefore, this correlation has important implications. Neutrophil influx has been shown to correlate with the total surface area of deposited nanoparticles in the lung.54,55 By extension, this necessitates that the pulmonary acute phase response correlates with the total surface area of the deposited particles in the lung; this is shown for pulmonary instillation of TiO2 and carbon black nanoparticles 1 and 28 days after instillation (Figure 3(a) and (b)). Thus, inhalation of smaller particles will cause a larger acute phase response per mass unit of deposited particles than larger particles. The total surface area increases proportionally to the decrease in diameter, such that a 10-times reduction in particle size results in 10-times larger total surface area per mass unit. In addition, nanoparticles deposit deeper in the respiratory tract than larger particles and therefore have longer retention time. Therefore, smaller particles produce prolonged inflammation in terms of neutrophil influx and prolonged acute phase response compared to larger particles.11,23,34 In accordance with this, pulmonary acute phase response was evident 28 days after exposure to nanosized TiO2, nanosized carbon black, SWCNT and MWCNT and about four weeks after inhalation exposure to nanosized TiO2 and carbon black.11 Thus, the total surface area of all deposited particles correlates with neutrophil influx and with pulmonary acute phase response. Because acute phase response predicts future cardiovascular risk, these findings establish a direct link between the total surface area of pulmonary deposited particles and cardiovascular risk.
Fig 2.
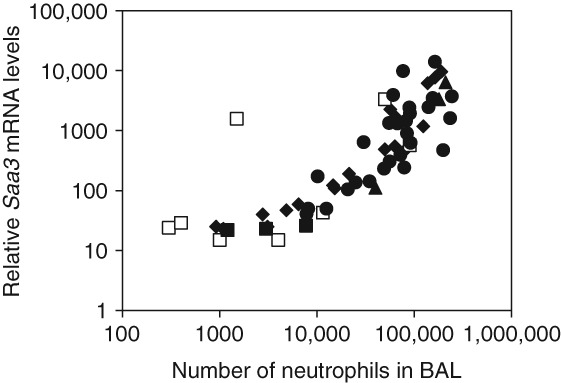
Correlation between neutrophil influx and normalized Saa3 mRNA levels in lung tissue 1–28 days following instillation or inhalation of various nanomaterials or dust collected at a biofuel plant. Saa3 mRNA levels were quantified by real-time PCR and normalized to 18S. Exposures include inhalation of carbon black and TiO2 nanoparticles (open squares), instillation of carbon black and TiO2 nanoparticles (filled diamonds), instillation of carbon nanotubes (filled circles), instillation of biofuel dust (filled triangles), instillation of vehicle (negative control) (filled squares). (Reprinted with permission from Ref 11. Copyright 2013 under Creative Commons Attribution License)
Fig 3.
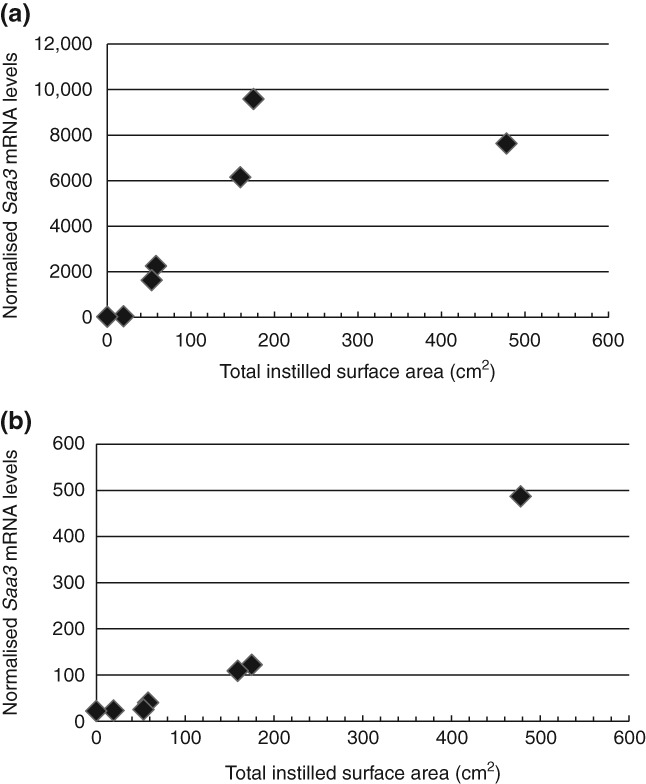
Correlation between neutrophil influx and total surface area of deposited nanoparticles. Doses of 0, 18, 54, or 162 µg/mouse carbon black or TiO2 nanoparticles were deposited in lungs of mice by intratracheal instillation and the number of neutrophils in bronchial lavage fluid was determined as described previously.74 N = 6 per group, except for vehicle controls where n = 22: (a) 24 h after instillation and (b) 28 days post exposure.
LITTLE OR NO HEPATIC ACUTE PHASE RESPONSE
The hepatic acute phase response following inhalation or pulmonary instillation of nanoparticles is much smaller than the observed pulmonary acute phase response, or may even be entirely absent. Indeed, no hepatic acute phase response was detected by global gene expression analysis following inhalation exposure to 20 mg/m3 carbon black or diesel exhaust particles 90 min daily for 4 consecutive days.11,43 In contrast, Saa3 mRNA levels were increased 4.4- and 17.4-fold in lung tissue following inhalation of nanosized carbon black and diesel exhaust particles, respectively.11 Similarly, little or no hepatic acute phase response was found following instillation of nanosized carbon black as determined by Saa3 mRNA levels,11,41,42 and no hepatic acute phase response was found after instillation of multiwalled carbon nanotubes, even when the Saa3 levels in lung tissue were induced 600-fold.11
Erdely et al. exposed mice to different kinds welding fume particles by aspiration at a dose of 340 µg/mouse.56 They found up to 15-fold increases in the levels of Saa1 in liver 24 h post exposure. They did not assess Saa1 or Saa3 expression in lung tissue. The used dose was almost twice the dose in our studies (162 µg/mouse). The welding fume particles were larger than the TiO2 nanoparticles and since specific surface areas were not specified, dose comparison in terms of surface area is not possible. We observed a 268-fold increased Saa3 expression in lung tissue 24 h after exposure to the 162 µg TiO2 nanoparticles and a 10-fold lower 30-fold increase in expression of the chemokine Ccl2 which was also assessed by Erdely.28 Erdely et al. found up to 58-fold increased Ccl2 expression.56 Provided that gene expression patterns in response to welding fumes are somewhat similar to that observed for TiO2 nanoparticles, we would predict that pulmonary Saa3 expression could be up to 600-fold increased following exposure to welding fumes. Thus, the hepatic acute phase response would be small compared to the pulmonary acute phase response.
Overall, the studies described above indicate that pulmonary exposure to particles leads to a large and robust pulmonary acute phase response and, to a much lesser degree, to a hepatic acute phase response.
MICRORNA EXPRESSION IN LUNG TISSUE
It was recently suggested that microRNAs (miRNAs) may be involved in controlling acute phase response following influenza infection in pigs.57 MiRNAs were differentially expressed in lung tissue 1, 3, or 14 day after aerosol exposure to H1N2 influenza virus. Among the perturbed miRNAs, miR-21 levels were increased 2-fold 3 days after the exposure. MiR-21 was predicted to target CXCL10, IL12A, IL10, IL1B, and SAA. Interestingly, miR-21 expression levels were also increased 1.5-fold in lung tissue in mice 5 days after inhalation exposure to TiO239 and 2-fold 26–28 days after instillation or inhalation of carbon black nanoparticles.41 Among the proposed targets for miR-21, increased protein levels of IL12, IL10, IL1B, and SAA have been found in lung tissue after pulmonary exposure to TiO2 and carbon black nanoparticles.11,28,49 Among the other differentially expressed miRNAs observed, only increased expression of miR-223 was found both after inhalation of TiO2 in mice and inhalation of influenza virus in pigs.39,57 The miRNA showing the clearest dose-response relationship following TiO2 and carbon black nanoparticle exposure is miRNA-135b.39,41 MiR-135b was recently shown to be regulated by IL-1-receptor activation.58 More studies are needed to understand the role of miRNAs in nanotoxicology.
CHOLESTEROL HOMEOSTASIS AFTER PULMONARY PARTICLE DEPOSITION
Induction of acute phase response affects cholesterol homeostasis. Cholesterol synthesis mainly takes place in the liver and cholesterol circulates in the blood as part of high density (HDL) and low density (LDL) lipoprotein molecules. In keeping with this, induction of the acute phase response in mice leads to profound changes in cholesterol biosynthesis and in blood levels of lipids.42,59 In a study where the acute phase was induced by subcutaneous injection of silver nitrate, blood levels of HDL cholesterol and LDL/VLDL cholesterol increased and followed the same time course trajectory as the acute phase response (Figure 4).59 During particle-induced pulmonary acute phase by carbon black, plasma levels of HDL decreased 3 and 28 days post exposure and plasma levels of LDL increased 28 days post exposure compared to vehicle-exposed control animals.42 Furthermore, cholesterol biosynthesis was upregulated in both liver and lung at the level of gene expression.42 Hepatic cholesterol levels were 12–22% increased, but the effect was not statistically significant (P = 0.06).42 Therefore, it seems that in mice the increased blood levels of cholesterol have at least two sources. During a hepatic acute phase response, cholesterol biosynthesis is upregulated in liver. In addition, a lowered reverse cholesterol transport will also decrease the excretion of cholesterol through the bile and thus contribute to increases in blood cholesterol levels.59
Fig 4.
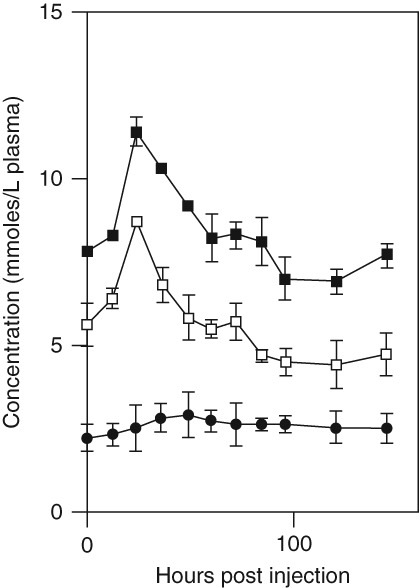
Change in plasma total cholesterol (solid squares), free cholesterol (open squares), and cholesterol esters (solid circles) concentrations following the induction of an acute subcutaneous inflammatory reaction with 2% silver nitrate. Values are the mean ± SEM, n = 12. (Reprinted with permission from Ref 59. Copyright 1997, Elsevier)
Mice and humans are expected to differ in blood lipid response because mice lack the cholesterol ester transfer protein that transfers cholesterol esters from HDL to LDL.59 When humans undergo acute phase response, LDL synthesis is increased, but LDL levels in blood decrease due to upregulation of LDL receptor activity.60 HDL blood levels decrease, and blood levels of triglycerides increase.60 Patients with acute myocardial infarction undergo acute phase response. Cholesterol biosynthesis was assessed in 34 patients hospitalized with acute myocardial infarction and cholesterol biosynthesis was found to be 23 and 29% increased 1 and 2 days after hospitalization, respectively.11,61
Thus, cholesterol biosynthesis is consistently increased during an acute phase response, and increased cholesterol biosynthesis may take place in lung and liver.
SAA EXPRESSION
SAA may play a central role in promoting atherosclerosis and thus risk of cardiovascular disease. As described above increased expression of three isogenes, Saa3, Saa1, and Saa2, was observed following pulmonary exposure to nanoparticles in mice.39,42 The same response was observed when mice inhaled lipopolysaccharide (LPS) over 20 min,62 which mimics a lung bacterial infection. Gene expression changes quantified 1, 2, 4, 8, 12, and 24 h post exposure to LPS showed evidence of an early inflammatory response, measured as increased expression of macrophage inflammatory genes, which peaked 1 h post exposure. This was followed by induction of proinflammatory interleukin and chemokine genes, which peaked 2 or 4 h post exposure. Among these, IL1-β, Il-6 and TNF are strongly induced and peak 2–4 h after LPS exposure.62 IL-1β, IL-6, and TNF all induce Saa transcription.52 The expression of Saa1 and Saa3 was already increased just 1 h after exposure, but the increased expression peaked 12 h post exposure.62 Thus, pulmonary Saa expression is likely induced by proinflammatory cytokines and Saa expression peaks later than proinflammatory cytokine expression, ie after 12 h following LPS challenge and somewhat later following particle exposure.
A number of cell types present in the lung are capable of expressing SAA including macrophages,63 Clara cells,64 and alveolar type II cells.64 Macrophages are capable of expressing Saa1, Saa2, and Saa363 and all three SAA proteins are incorporated into HDL after induction of acute phase response.63 The exact cellular origin of the pulmonary acute phase response is not at present clear, although several cell types that are present in the lungs are capable of expressing SAA.
SAA PROTEIN FUNCTION IN HDL
SAA likely promotes cardiovascular disease directly. When SAA expression is induced during the acute phase response, SAA replaces ApolipoproteinA-1 in HDL, resulting in partial depletion of ApolipoproteinA-1 from HDL.65,66 ApolipoproteinA-1 is a cofactor for lecithin-cholesterol acyltransferase (LCAT), which esterifies cholesterol for transport. Thus, SAA indirectly influences cholesterol efflux through its effects on ApolipoproteinA-1.59 In addition, SAA has been shown to directly inhibit LCAT activity.67 In a study of patients with acute coronary syndrome, stable coronary artery disease and healthy controls, serum levels of ApolipoproteinA-1 were the best predictor of cholesterol efflux compared to total cholesterol, triglycerides, HDL, LDL, ApoB, CRP, and SAA.68 Thus, incorporation of SAA into HDL leads to accumulation of cholesterol in the peripheral tissues at least partly caused by reduced cholesterol efflux.
When macrophages are exposed to SAA-containing HDL, they turn into foam cells in a dose-dependent manner69 as a consequence of increased uptake of LDL69 and increased uptake and hydrolysis of cholesteryl esters from SAA-containing HDL.70 Foam cells are cholesterol-loaded macrophages that are a principal component of plaques. It has been demonstrated that virus-mediated overexpression of Saa1 was sufficient to promote plaque progression in APOE −/− mice.71
SAA seems to signal by Toll-like receptor 4 to activate NF-kβ-dependent transcription72 and induces IL-1 and IL-8 in neutrophil cells (which accumulate during pulmonary inflammation). It promotes monocyte chemotaxis and adhesion, and induces matrix-metalloproteinases.73 In addition, SAA potentiates prothrombotic and proinflammatory events by induction of tissue factors.73
Therefore, formation of acute phase HDL lipoproteins that contain more SAA and less ApolipoproteinA-1 may constitute an important link between acute phase response and cardiovascular disease. The acute phase HDL is less proficient in reverse cholesterol transport, thereby leading to perturbed cholesterol homeostasis and increased accumulation of cholesterol. In addition, macrophages exposed to SAA or acute phase HDL change behavior and turn into foam cells, thus further promoting plaque progression and atherosclerosis.
A MECHANISM FOR PARTICLE-INDUCED PULMONARY ACUTE PHASE RESPONSE AND ATHEROSCLEROSIS
On the basis of the data presented above, we propose a mechanism of action that may contribute to particle-induced acute phase response and increased risk of cardiovascular disease (Figure 5).
Fig 5.
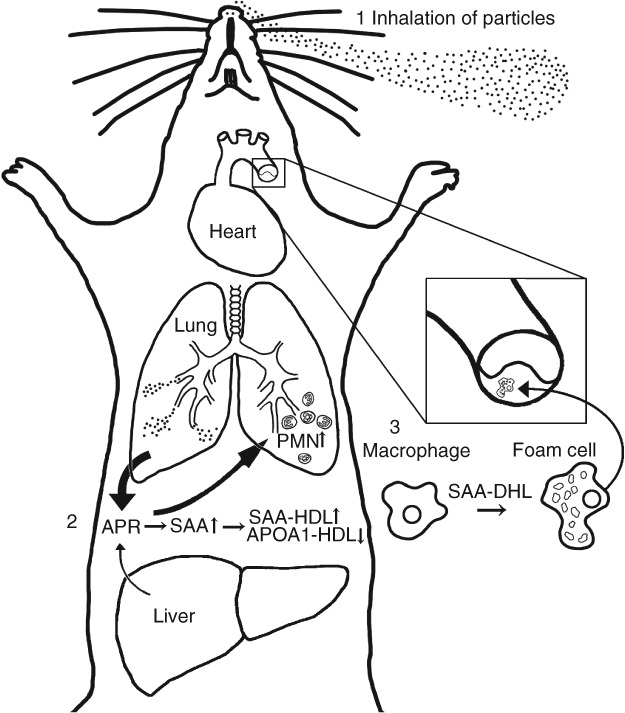
The proposed mechanism of action of particle-induced cardiovascular disease. Inhalation of particles (indicated by 1 in the figure) leads to a pulmonary acute phase response (2). The accumulation of SAA in lungs leads to attraction of neutrophils. SAA is incorporated into HDL (3), replacing APO-A1 and goes into systemic circulation. SAA-HDL is deficient in reverse cholesterol transport and promotes foam cell formation from macrophages, leading to plaque progression.
Inhalation of particles induces a pulmonary acute phase response, which depends on the total surface area of the deposited particles and the clearance of the particles from the alveoli. Nanoparticles have a large surface to mass ratio and prolonged retention in lung; therefore, inhalation of nanoparticles induces a strong and prolonged acute phase response. The cholesterol biosynthetic pathway is upregulated at the level of transcription in the lung and/or liver. In the alveoli, macrophages engulf particles and produce SAA, once induced by proinflammatory cytokines. The excreted SAA functions as a neutrophil attractant in the lung, resulting in neutrophil influx into the lung. SAA is incorporated into HDL where it replaces ApolipoproteinA-1. SAA probably goes into systemic circulation as SAA-HDL. There are no indications that the macrophages residing in the lung enter the systemic circulation, since macrophages in blood smears from mice following pulmonary exposure to carbon black do not contain carbon black nanoparticles in contrast to macrophages isolated from bronchiolar lavage fluid (LM Pedersen, P Jackson, H Wallin, and U Vogel, unpublished observations). Lee et al. recently proposed a role for SAA-HDL in the pathogenesis of atherosclerosis.69 They note that normal HDL inhibits foam cell formation by facilitating cholesterol efflux from macrophages and participates in reverse cholesterol transport, enabling cholesterol excretion.69 In contrast, the combined absence of Apo-A1 and presence of SAA leads to lowered reverse cholesterol transport from peripheral tissues by HDL and furthermore SAA-HDL stimulates foam cell formation of peripheral macrophages.69 In addition, SAA stimulates MMP9 expression in monocytes. MMP9 triggers atherosclerotic rupture possibly leading to myocardial ischemia.69 There is no direct evidence that pulmonary acute phase response and pulmonary Saa expression cause plaque progression; however, we have shown that pulmonary exposure to TiO2 nanoparticles will promote plaque progression7 and in parallel that both inhalation39 and pulmonary exposure by instillation11,28,74 of the very same TiO2 nanoparticles induces pulmonary acute phase response while causing very little or no hepatic acute phase response.11 Thus, the downstream consequences of induced acute phase response and increased SAA expression may be promotion of foam cell formation and plaque progression.
LIFESTYLE FACTORS THAT AFFECT ACUTE PHASE RESPONSE IN HUMANS
Some lifestyle factors influence SAA levels. Body mass index (BMI) is highly associated with plasma SAA levels21 because white adipocytes express SAA.75 SAA transcription is induced by the proinflammatory cytokines IL6, IL1β, and TNF. Alcohol suppresses LPS-induced SAA production in mice.76 A possible mechanism may be an ethanol-mediated inhibition of NLRP3 inflammasome activation in macrophages,77 thus inhibiting IL-1β mediated activation of Saa transcription. If this is an important mechanism for the protective effect of alcohol in relation to acute coronary heart disease,78 then alcohol intake should have the largest effect among smokers and others exposed to acute phase response inducers. In a study of 2989 men, the effect of smoking and alcohol intake on coronary artery occlusion was assessed.79 The observed protective effect of alcohol intake on artery occlusion was largest among heavy smokers and least among non-smokers. However, there was no statistically significant interaction. Non-steroidal anti-inflammatory drug (NSAID) use also inhibits induction of proinflammatory cytokines, and thus likely inhibits the acute phase response. In line with this view, aspirin has a protective effect against future myocardial infarction especially among those with high baseline CRP levels.80 Furthermore, it has been shown that the NSAID ibuprofen inhibits IL1B synthesis and amyloidosis in a mouse model of Alzheimer's disease.81 Taken together, this indicates that controlled human exposures ideally should take alcohol consumption, BMI and NSAID use into account. Thus, it may prove more difficult to detect increases in acute phase protein levels in people with suppressed acute phase response caused by low-level aspirin, pain-killers, or high alcohol consumption.
CONTROLLED HUMAN EXPOSURES
A number of controlled human exposures to various kinds of particles have been performed where blood markers of inflammation and acute phase response have been assessed. Only a few of these studies have assessed SAA levels, whereas more have examined CRP. In cross-sectional studies, CRP and SAA levels are highly correlated.20,21 However, since there are differences in the kinetics of CRP and SAA synthesis and degradation, there may be differences in controlled human studies. None of the controlled exposures assessed exposure to engineered nanoparticles. However, we propose that the total surface area of pulmonary deposited particles will predict the pulmonary acute phase response. Therefore, we hypothesize that inhalation of all kinds of particles will induce a pulmonary acute phase response in a dose-dependent manner.
Blood levels of CRP are associated with risk of coronary heart disease in prospective studies, but genetically determined differences in CRP levels are not associated with risk.82 This indicates that CRP is not causally related to risk of coronary heart disease, but rather co-varies with the causal factor, which could be SAA.82,83
The influence of particle exposure on serum levels of acute phase proteins has been addressed in a number of controlled human exposure studies. Here, we summarize some of these.
Both SAA and CRP were assessed in a controlled human exposure study using a 4-h exposure to 0.24–0.28 mg/m3 wood smoke where about half the particles were less than 100 nm.84 The 13 test subjects were healthy non-smokers who did not consume excessive amounts of alcohol, exhibited no allergic symptoms and were free of infections over the week prior to the experiment.84 Blood levels of SAA were 2-fold increased 3 and 24 h after the exposure, whereas no changes were detected for CRP. Exposure to clean air had no effect on either acute phase protein at any time point.84
The effects of wood smoke exposure were assessed in a random cross over study.82 Portable air filters were used to clean the air in households with wood-stoves during two 7-day periods, one with HEPA filtration and one without. HEPA filtration of the air reduced particulate matter 2.5 microns in diameter and less (PM2.5) from 11.3 to 4.6 µg/m3. In this study the participants were non-smokers. CRP levels were assessed after 7-day exposures and a 32.6% reduction in CRP levels was observed after HEPA filtration of the air.85 SAA levels were not assessed in this study.
In a second random cross-over study on 42 non-smoking elderly volunteers, indoor air was either HEPA filtrated or unfiltered for 48 h.86 HEPA filtration reduced PM2.5 from 9.4 to 4.6 µg/m3. SAA and CRP were assessed in blood sampled in the morning following each 48-h exposure scenario. No changes were observed for either SAA or CRP.
In a study of occupational exposure to welding, cutting, grinding, and foundries, the mean level of total dust was 0.93 mg/m3 with PM2.5 0.31 mg/m3.87 CRP levels (but not SAA levels) were assessed at four time-points: pre-shift, and post-shift day 1, day 2, and day 4. Pre-shift was on the first day after summer vacation. There were 73 subjects and smoking was the only factor taken into account. Twelve of seventy-three were current smokers. CRP levels were 19% increased post-shift day 2 but not altered on post-shift days 1 or 4.
Fifty-two elderly persons with ischemic heart disease were followed for six months and CRP levels were correlated with ambient air pollution.88 The average ambient PM2.5 was 8.7 µg/m3. SAA was not assessed. CRP levels were statistically significantly associated with PM2.5, with the strongest associations observed with 1 and 2 lag days.
Twelve male patients with stable coronary heart disease and 12 age-matched volunteers were exposed to concentrated ambient fine and ultrafine particles for 2 h in a randomized double-blinded cross-over design.89 Participants were non-smokers and were asked to abstain from alcohol intake 24 h before exposure. All twelve coronary heart disease patients took aspirin. The PM2.5 level was 190 µg/m3. Individuals exposed to clean air had PM2.5 levels of 0.5 µg/m3. CRP levels were unaffected by exposure after 6 and 24 h. SAA was not assessed.
Taken together, some but not all controlled exposure studies find increased levels of SAA and/or CRP after inhalation exposure to particles. In general, increased levels of acute phase proteins were found in studies with either larger particle doses, longer durations or which included test subjects who did not take medication that could interfere with acute phase response. Thus, evidence from available controlled human exposures partly supports the notion of acute phase response as a possible mediator of systemic effects following particle inhalation. However, the human exposures provide no information regarding the tissue of origin of the acute phase proteins.
ENDOTHELIAL DYSFUNCTION
Inhalation of particles promotes cardiovascular disease by other means in addition to the acute phase response, and pulmonary acute phase response likely also promotes cardiovascular risk by other means in addition to the proposed acute phase HDL model.
Endothelial dysfunction has been proposed as a pro-atherosclerotic mechanism of particle inhalation.90 In very elegantly designed experiments, it was shown that serum from volunteers exposed to diesel exhaust particles was able to increase expression of vascular cell adhesion molecule (VCAM-1) in primary human coronary artery endothelial cells.90 This identified ‘a circulating factor’ as the mediator of the particle-induced endothelial dysfunction. In a subsequent and equally elegant study,91 CD36 +/+ and −/− mice were exposed to ozone, and vasorelaxation was assessed in aorta from unexposed mice exposed to serum ex vivo as a measure of endothelial dysfunction. Neutrophil influx was observed in CD36+/− mice but not in Cd36−/− mice. It was shown that while the CD36 receptor was not required for production of the circulating factor, CD36 was required for impairment of vasorelaxation responses. CD36 is a SAA receptor92 and it is expressed on macrophages and on endothelial cells.91 SAA has been shown to induce endothelial dysfunction.93 Thus, the study by Robertson91 suggests that ozone inhalation induces a pulmonary acute phase response independently of CD36 status, and that SAA-containing acute phase HDL or SAA alone are possible soluble factors mediating the CD36-dependent endothelial dysfunction (Box 1).
CONCLUSION
In this review we summarize evidence that pulmonary exposure to particles and nanoparticles induces a pulmonary acute phase response in mice, whereas acute phase response is reduced or absent in the liver following particle inhalation. We suggest that the acute phase response, and in particular SAA, may contribute significantly to the pathogenesis of cardiovascular disease. We suggest that SAA is produced in the lungs and enters systemic circulation as part of acute phase HDL. Acute phase HDL is deficient in reverse cholesterol transport, and stimulates foam cell formation from macrophages in peripheral tissues leading to plaque formation.
There is overwhelming evidence that the acute phase response is causally related to risk of cardiovascular disease from both epidemiological studies and mechanistic studies in vivo and in vitro. Thus, the fact that pulmonary exposure to particles induces pulmonary acute phase response in mice and accompanying systemic circulation of acute phase protein provides evidence of a causal link between particle inhalation and risk of cardiovascular disease. The association is further supported by controlled human exposure studies demonstrating increased blood levels of acute phase proteins following exposure to different kinds of particles. Acute phase proteins in systemic circulation can originate from many different tissues and therefore only increased tissue transcription can be used as evidence of origin. In addition, we suggest a mechanism involving SAA related foam cell formation, which would also explain the lack of association between genetically determined differences in CRP levels and risk of coronary heart disease.
Our finding of an association between neutrophil influx and pulmonary acute phase response has important implications. Neutrophil influx has also been shown to correlate closely with the total surface area of deposited particles. This implies that inhalation of nanosized particles would induce a much stronger acute phase response and that the accompanying risk of cardiovascular disease should be proportional to the increase in total surface area compared to the same mass of larger particles. This is especially relevant in relation to the consideration of nanoparticle-specific occupational exposure levels, and underscores cardiovascular disease as an occupational disease. In epidemiological studies, even modest differences in blood levels of CRP and SAA are associated with risk of future cardiovascular disease11–13 and ambient air pollution was found to correlate with CRP levels.88 Taken together, this could indicate that even ambient levels of particles such as those in air pollution may evoke a pulmonary acute phase response and modify risk of cardiovascular disease correspondingly.
Further work is required to establish the slope of the correlation between deposited surface area and acute phase proteins levels in blood, to more definitively establish the correlation between acute phase protein levels in blood and future risk of cardiovascular risk. In addition, the findings and proposed models provide the basis for the development of in vitro testing protocols if macrophage-SAA synthesis can be used to group and rank particles (including nanoparticles) for their potential to cause cardiovascular risk.
Acknowledgments
This work was supported by the grant Danish Centre for Nanosafety (grant 20110092173/3) from the Danish Working Environment Research Fund.
REFERENCES
- 1.Pope CA, III, Thun MJ, Namboodiri MM, Dockery DW, Evans JS, Speizer FE, Heath CW., Jr Particulate air pollution as a predictor of mortality in a prospective study of U.S. adults. Am J Respir Crit Care Med. 1995;151:669–674. doi: 10.1164/ajrccm/151.3_Pt_1.669. PM:7881654. [DOI] [PubMed] [Google Scholar]
- 2.Dockery DW, Pope CA, III, Xu X, Spengler JD, Ware JH, Fay ME, Ferris BG, Jr, Speizer FE. An association between air pollution and mortality in six U.S. cities. N Engl J Med. 1993;329:1753–1759. doi: 10.1056/NEJM199312093292401. PM:8179653. [DOI] [PubMed] [Google Scholar]
- 3.Clancy L, Goodman P, Sinclair H, Dockery DW. Effect of air-pollution control on death rates in Dublin, Ireland: an intervention study. Lancet. 2002;360:1210–1214. doi: 10.1016/S0140-6736(02)11281-5. PM:12401247. [DOI] [PubMed] [Google Scholar]
- 4.Hertel S, Viehmann A, Moebus S, Mann K, Brocker-Preuss M, Mohlenkamp S, Nonnemacher M, Erbel R, Jakobs H, Memmesheimer M, et al. Influence of short-term exposure to ultrafine and fine particles on systemic inflammation. Eur J Epidemiol. 2010;25:581–592. doi: 10.1007/s10654-010-9477-x. PM:20559688. [DOI] [PubMed] [Google Scholar]
- 5.Li Z, Hulderman T, Salmen R, Chapman R, Leonard SS, Young SH, Shvedova A, Luster MI, Simeonova PP. Cardiovascular effects of pulmonary exposure to single-wall carbon nanotubes. Environ Health Perspect. 2007;115:377–382. doi: 10.1289/ehp.9688. ISI:000244651500029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen LC, Nadziejko C. Effects of subchronic exposures to concentrated ambient particles (CAPs) in mice: V. CAPs exacerbate aortic plaque development in hyperlipidemic mice. Inhal Toxicol. 2005;17:217–224. doi: 10.1080/08958370590912815. ISI:000227779500005. [DOI] [PubMed] [Google Scholar]
- 7.Mikkelsen L, Sheykhzade M, Jensen KA, Saber AT, Jacobsen NR, Vogel U, Wallin H, Loft S, Moller P. Modest effect on plaque progression and vasodilatory function in atherosclerosis-prone mice exposed to nanosized TiO(2) Part Fibre Toxicol. 2011;8:32. doi: 10.1186/1743-8977-8-32. PM:22074227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Erdely A, Hulderman T, Salmen-Muniz R, Liston A, Zeidler-Erdely PC, Chen BT, Stone S, Frazer DG, Antonini JM, Simeonova PP. Inhalation exposure of gas-metal arc stainless steel welding fume increased atherosclerotic lesions in apolipoprotein E knockout mice. Toxicol Lett. 2011;204:12–16. doi: 10.1016/j.toxlet.2011.03.030. PM:21513782. [DOI] [PubMed] [Google Scholar]
- 9.Donaldson K, Stone V, Seaton A, MacNee W. Ambient particle inhalation and the cardiovascular system: potential mechanisms. Environ Health Perspect. 2001;109(Suppl 4):523–527. doi: 10.1289/ehp.01109s4523. PM:11544157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gabay C, Kushner I. Mechanisms of disease: acute-phase proteins and other systemic responses to inflammation. New Engl J Med. 1999;340:448–454. doi: 10.1056/NEJM199902113400607. ISI:000078571000007. [DOI] [PubMed] [Google Scholar]
- 11.Saber AT, Lamson JS, Jacobsen NR, Ravn-Haren G, Hougaard KS, Nyendi AN, Wahlberg P, Madsen AM, Jackson P, Wallin H, et al. Particle-induced pulmonary acute phase response correlates with neutrophil influx linking inhaled particles and cardiovascular risk. PLoS One. 2013;8:e69020. doi: 10.1371/journal.pone.0069020. PM:23894396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaptoge S, Di AE, Pennells L, Wood AM, White IR, Gao P, Walker M, Thompson A, Sarwar N, Caslake M, et al. C-reactive protein, fibrinogen, and cardiovascular disease prediction. N Engl J Med. 2012;367:1310–1320. doi: 10.1056/NEJMoa1107477. PM:23034020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taubes G. Cardiovascular disease. Does inflammation cut to the heart of the matter? Science. 2002;296:242–245. doi: 10.1126/science.296.5566.242. PM:11951014. [DOI] [PubMed] [Google Scholar]
- 14.Lowe GD. The relationship between infection, inflammation, and cardiovascular disease: an overview. Ann Periodontol. 2001;6:1–8. doi: 10.1902/annals.2001.6.1.1. [DOI] [PubMed] [Google Scholar]
- 15.Mezaki T, Matsubara T, Hori T, Higuchi K, Nakamura A, Nakagawa I, Imai S, Ozaki K, Tsuchida K, Nasuno A, et al. Plasma levels of soluble thrombomodulin, C-reactive protein, and serum amyloid A protein in the atherosclerotic coronary circulation. Jpn Heart J. 2003;44:601–612. doi: 10.1536/jhj.44.601. [DOI] [PubMed] [Google Scholar]
- 16.Libby P, Okamoto Y, Rocha VZ, Folco E. Inflammation in atherosclerosis: transition from theory to practice. Circ J. 2010;74:213–220. doi: 10.1253/circj.cj-09-0706. ISI:000274099300001. [DOI] [PubMed] [Google Scholar]
- 17.Pussinen PJ, Tuomisto K, Jousilahti P, Havulinna AS, Sundvall J, Salomaa V. Endotoxemia, immune response to periodontal pathogens, and systemic inflammation associate with incident cardiovascular disease events. Arterioscler Thromb Vasc Biol. 2007;27:1433–1439. doi: 10.1161/ATVBAHA.106.138743. ISI:000246714600030. [DOI] [PubMed] [Google Scholar]
- 18.Estabragh ZR, Mamas MA. The cardiovascular manifestations of influenza: a systematic review. Int J Cardiol. 2013;167:2397–2403. doi: 10.1016/j.ijcard.2013.01.274. PM:23474244. [DOI] [PubMed] [Google Scholar]
- 19.Rivera MF, Lee JY, Aneja M, Goswami V, Liu L, Velsko IM, Chukkapalli SS, Bhattacharyya I, Chen H, Lucas AR, et al. Polymicrobial infection with major periodontal pathogens induced periodontal disease and aortic atherosclerosis in hyperlipidemic ApoE(null) mice. PLoS One. 2013;8:e57178. doi: 10.1371/journal.pone.0057178. PM:23451182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000;342:836–843. doi: 10.1056/NEJM200003233421202. PM:10733371. [DOI] [PubMed] [Google Scholar]
- 21.Johnson BD, Kip KE, Marroquin OC, Ridker PM, Kelsey SF, Shaw LJ, Pepine CJ, Sharaf B, Bairey Merz CN, Sopko G, et al. Serum amyloid A as a predictor of coronary artery disease and cardiovascular outcome in women: the National Heart, Lung, and Blood Institute-Sponsored Women's Ischemia Syndrome Evaluation (WISE) Circulation. 2004;109:726–732. doi: 10.1161/01.CIR.0000115516.54550.B1. PM:14970107. [DOI] [PubMed] [Google Scholar]
- 22.Pai JK, Pischon T, Ma J, Manson JE, Hankinson SE, Joshipura K, Curhan GC, Rifai N, Cannuscio CC, Stampfer MJ, et al. Inflammatory markers and the risk of coronary heart disease in men and women. N Engl J Med. 2004;351:2599–2610. doi: 10.1056/NEJMoa040967. PM:15602020. [DOI] [PubMed] [Google Scholar]
- 23.Oberdorster G, Oberdorster E, Oberdorster J. Nanotoxicology: an emerging discipline evolving from studies of ultrafine particles. Environ Health Perspect. 2005;113:823–839. doi: 10.1289/ehp.7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fang SC, Cassidy A, Christiani DC. A systematic review of occupational exposure to particulate matter and cardiovascular disease. Int J Environ Res Public Health. 2010;7:1773–1806. doi: 10.3390/ijerph7041773. ISI:000277119600030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Packard RR, Libby P. Inflammation in atherosclerosis: from vascular biology to biomarker discovery and risk prediction. Clin Chem. 2008;54:24–38. doi: 10.1373/clinchem.2007.097360. PM:18160725. [DOI] [PubMed] [Google Scholar]
- 26.Jacobsen NR, Moller P, Jensen KA, Vogel U, Ladefoged O, Loft S, Wallin H. Lung inflammation and genotoxicity following pulmonary exposure to nanoparticles in ApoE−/− mice. Part Fibre Toxicol. 2009;6:2. doi: 10.1186/1743-8977-6-2. PM:19138394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bourdon JA, Saber AT, Jacobsen NR, Jensen KA, Madsen AM, Lamson JS, Wallin H, Moller P, Loft S, Yauk CL, et al. Carbon Black Nanoparticle Instillation Induces Sustained Inflammation and Genotoxicity in Mouse Lung and Liver. Part Fibre Toxicol. 2012;9:5. doi: 10.1186/1743-8977-9-5. PM:22300514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Husain M, Saber AT, Guo C, Jacobsen NR, Jensen KA, Yauk CL, Williams A, Vogel U, Wallin H, Halappanavar S. Pulmonary instillation of low doses of titanium dioxide nanoparticles in mice leads to particle retention and gene expression changes in the absence of inflammation. Toxicol Appl Pharmacol. 2013;269:250–262. doi: 10.1016/j.taap.2013.03.018. PM:23557971. [DOI] [PubMed] [Google Scholar]
- 29.Jackson P, Hougaard KS, Boisen AM, Jacobsen NR, Jensen KA, Moller P, Brunborg G, Gutzkow KB, Andersen O, Loft S, et al. Pulmonary exposure to carbon black by inhalation or instillation in pregnant mice: effects on liver DNA strand breaks in dams and offspring. Nanotoxicology. 2012;6:486–500. doi: 10.3109/17435390.2011.587902. PM:21649560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saber AT, Jacobsen NR, Bornholdt J, Kjaer SL, Dybdahl M, Risom L, Loft S, Vogel U, Wallin H. Cytokine expression in mice exposed to diesel exhaust particles by inhalation. Role of tumor necrosis factor. Part Fibre Toxicol. 2006;3:4. doi: 10.1186/1743-8977-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saber AT, Bornholdt J, Dybdahl M, Sharma AK, Loft S, Vogel U, Wallin H. Tumor necrosis factor is not required for particle-induced genotoxicity and pulmonary inflammation. Arch Toxicol. 2005;79:177–182. doi: 10.1007/s00204-004-0613-9. [DOI] [PubMed] [Google Scholar]
- 32.Boisen AM, Shipley T, Jackson P, Hougaard KS, Wallin H, Yauk CL, Vogel U. NanoTIO(2) (UV-Titan) does not induce ESTR mutations in the germline of prenatally exposed female mice. Part Fibre Toxicol. 2012;9:19. doi: 10.1186/1743-8977-9-19. PM:22656316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hougaard KS, Jensen KA, Nordly P, Taxvig C, Vogel U, Saber AT, Wallin H. Effects of prenatal exposure to diesel exhaust particles on postnatal development, behavior, genotoxicity, and inflammation in mice. Part Fibre Toxicol. 2008;5:3. doi: 10.1186/1743-8977-5-3. PM:18331653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hougaard KS, Jackson P, Jensen KA, Sloth JJ, Loschner K, Larsen EH, Birkedal RK, Vibenholt A, Boisen AM, Wallin H, et al. Effects of prenatal exposure to surface-coated nanosized titanium dioxide (UV-Titan). A study in mice. Part Fibre Toxicol. 2010;7:16. doi: 10.1186/1743-8977-7-16. PM:20546558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sos PS, Jacobsen NR, Labib S, Wu D, Husain M, Williams A, Bogelund JP, Andersen O, Kobler C, Molhave K, et al. Transcriptomic analysis reveals novel mechanistic insight into murine biological responses to multi-walled carbon nanotubes in lungs and cultured lung epithelial cells. PLoS One. 2013;8:e80452. doi: 10.1371/journal.pone.0080452. PM:24260392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bornholdt J, Dybdahl M, Vogel U, Hansen M, Loft S, Wallin H. Inhalation of ozone induces DNA strand breaks and inflammation in mice. Mutat Res. 2002;520:63. doi: 10.1016/s1383-5718(02)00176-6. [DOI] [PubMed] [Google Scholar]
- 37.Dybdahl M, Risom L, Bornholdt J, Autrup H, Loft S, Wallin H. Inflammatory and genotoxic effects of diesel particles in vitro and in vivo. Mutat Res. 2004;562:119–131. doi: 10.1016/j.mrgentox.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 38.Risom L, Dybdahl M, Bornholdt J, Vogel U, Wallin H, Moller P, Loft S. Oxidative DNA damage and defence gene expression in the mouse lung after short-term exposure to diesel exhaust particles by inhalation. Carcinogenesis. 2003;24:1847–1852. doi: 10.1093/carcin/bgg144. [DOI] [PubMed] [Google Scholar]
- 39.Halappanavar S, Jackson P, Williams A, Jensen KA, Hougaard KS, Vogel U, Yauk CL, Wallin H. Pulmonary response to surface-coated nanotitanium dioxide particles includes induction of acute phase response genes, inflammatory cascades, and changes in microRNAs: a toxicogenomic study. Environ Mol Mutagen. 2011;52:425–439. doi: 10.1002/em.20639. PM:21259345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jackson P, Halappanavar S, Hougaard KS, Williams A, Madsen AM, Lamson JS, Andersen O, Yauk C, Wallin H, Vogel U. Maternal inhalation of surface-coated nanosized titanium dioxide (UV-Titan) in C57BL/6 mice: effects in prenatally exposed offspring on hepatic DNA damage and gene expression. Nanotoxicology. 2013;7:85–96. doi: 10.3109/17435390.2011.633715. PM:22117692. [DOI] [PubMed] [Google Scholar]
- 41.Bourdon JA, Saber AT, Halappanavar S, Jackson PA, Wu D, Hougaard KS, Jacobsen NR, Williams A, Vogel U, Wallin H, et al. Carbon black nanoparticle intratracheal installation results in large and sustained changes in the expression of miR-135b in mouse lung. Environ Mol Mutagen. 2012;53:462–468. doi: 10.1002/em.21706. PM:22753103. [DOI] [PubMed] [Google Scholar]
- 42.Bourdon JA, Halappanavar S, Saber AT, Jacobsen NR, Williams A, Wallin H, Vogel U, Yauk CL. Hepatic and pulmonary toxicogenomic profiles in mice intratracheally instilled with carbon black nanoparticles reveal pulmonary inflammation, acute phase response, and alterations in lipid homeostasis. Toxicol Sci. 2012;127:474–484. doi: 10.1093/toxsci/kfs119. PM:22461453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saber AT, Halappanavar S, Folkmann JK, Bornholdt J, Boisen AM, Moller P, Williams A, Yauk C, Vogel U, Loft S, et al. Lack of acute phase response in the livers of mice exposed to diesel exhaust particles or carbon black by inhalation. Part Fibre Toxicol. 2009;6:12. doi: 10.1186/1743-8977-6-12. PM:19374780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uhlar CM, Whitehead AS. Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem. 1999;265:501–523. doi: 10.1046/j.1432-1327.1999.00657.x. ISI:000083077100001. [DOI] [PubMed] [Google Scholar]
- 45.Chiba T, Han CY, Vaisar T, Shimokado K, Kargi A, Chen MH, Wang S, McDonald TO, O'Brien KD, Heinecke JW, et al. Serum amyloid A3 does not contribute to circulating SAA levels. J Lipid Res. 2009;50:1353–1362. doi: 10.1194/jlr.M900089-JLR200. ISI:000266912200010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meek RL, Urielishoval S, Benditt EP. Expression of apolipoprotein serum amyloid-A messenger-Rna in human atherosclerotic lesions and cultured vascular cells – implications for serum amyloid-A Function. Proc Natl Acad Sci USA. 1994;91:3186–3190. doi: 10.1073/pnas.91.8.3186. ISI:A1994NF96800067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whitehead AS, Zahedi K, Rits M, Mortensen RF, Lelias JM. Mouse C-reactive protein. Generation of cDNA clones, structural analysis, and induction of mRNA during inflammation. Biochem J. 1990;266:283–290. doi: 10.1042/bj2660283. PM:2310378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pepys MB, Hirschfield GM. C-reactive protein: a critical update. J Clin Invest. 2003;111:1805–1812. doi: 10.1172/JCI18921. PM:12813013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jackson P, Hougaard KS, Vogel U, Wu D, Casavant L, Williams A, Wade M, Yauk CL, Wallin H, Halappanavar S. Exposure of pregnant mice to carbon black by intratracheal instillation: toxicogenomic effects in dams and offspring. Mutat Res. 2012;745:73–83. doi: 10.1016/j.mrgentox.2011.09.018. [DOI] [PubMed] [Google Scholar]
- 50.Teeguarden JG, Webb-Robertson BJ, Waters KM, Murray AR, Kisin ER, Varnum SM, Jacobs JM, Pounds JG, Zanger RC, Shvedova AA. Comparative proteomics and pulmonary toxicity of instilled single-walled carbon nanotubes, crocidolite asbestos, and ultrafine carbon black in mice. Toxicol Sci. 2011;120:123–135. doi: 10.1093/toxsci/kfq363. PM:21135415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang CC, Chen SH, Ho SH, Yang CY, Wang HD, Tsai ML. Proteomic analysis of proteins from bronchoalveolar lavage fluid reveals the action mechanism of ultrafine carbon black-induced lung injury in mice. Proteomics. 2007;7:4388–4397. doi: 10.1002/pmic.200700164. PM:17963277. [DOI] [PubMed] [Google Scholar]
- 52.Liang TS, Wang JM, Murphy PM, Gao JL. Serum amyloid A is a chemotactic agonist at FPR2, a low-affinity N-formylpeptide receptor on mouse neutrophils. Biochem Biophys Res Commun. 2000;270:331–335. doi: 10.1006/bbrc.2000.2416. PM:10753626. [DOI] [PubMed] [Google Scholar]
- 53.Badolato R, Wang JM, Murphy WJ, Lloyd AR, Michiel DF, Bausserman LL, Kelvin DJ, Oppenheim JJ. Serum amyloid A is a chemoattractant: induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J Exp Med. 1994;180:203–209. doi: 10.1084/jem.180.1.203. PM:7516407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duffin R, Tran L, Brown D, Stone V, Donaldson K. Proinflammogenic effects of low-toxicity and metal nanoparticles in vivo and in vitro: highlighting the role of particle surface area and surface reactivity. Inhal Toxicol. 2007;19:849–856. doi: 10.1080/08958370701479323. PM:17687716. [DOI] [PubMed] [Google Scholar]
- 55.Saber AT, Jensen KA, Jacobsen NR, Birkedal R, Mikkelsen L, Moller P, Loft S, Wallin H, Vogel U. Inflammatory and genotoxic effects of nanoparticles designed for inclusion in paints and lacquers. Nanotoxicology. 2012;6:453–471. doi: 10.3109/17435390.2011.587900. PM:21649461. [DOI] [PubMed] [Google Scholar]
- 56.Simeonova PP, Erdely A. Engineered nanoparticle respiratory exposure and potential risks for cardiovascular toxicity: predictive tests and biomarkers. Inhal Toxicol. 2009;21(Suppl 1):68–73. doi: 10.1080/08958370902942566. PM:19558236. [DOI] [PubMed] [Google Scholar]
- 57.Skovgaard K, Cirera S, Vasby D, Podolska A, Breum SO, Durrwald R, Schlegel M, Heegaard PM. Expression of innate immune genes, proteins and microRNAs in lung tissue of pigs infected experimentally with influenza virus (H1N2) Innate Immun. 2013;19:531–544. doi: 10.1177/1753425912473668. PM:23405029. [DOI] [PubMed] [Google Scholar]
- 58.Halappanavar S, Nikota J, Wu D, Williams A, Yauk CL, Stampfli M. IL-1 receptor regulates microRNA-135b expression in a negative feedback mechanism during cigarette smoke-induced inflammation. J Immunol. 2013;190:3679–3686. doi: 10.4049/jimmunol.1202456. PM:23440414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lindhorst E, Young D, Bagshaw W, Hyland M, Kisilevsky R. Acute inflammation, acute phase serum amyloid A and cholesterol metabolism in the mouse. Biochim Biophys Acta. 1997;1339:143–154. doi: 10.1016/s0167-4838(96)00227-0. PM:9165109. [DOI] [PubMed] [Google Scholar]
- 60.Balci B. The modification of serum lipids after acute coronary syndrome and importance in clinical practice. Curr Cardiol Rev. 2011;7:272–276. doi: 10.2174/157340311799960690. PM:22758629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pfohl M, Schreiber I, Liebich HM, Haring HU, Hoffmeister HM. Upregulation of cholesterol synthesis after acute myocardial infarction—is cholesterol a positive acute phase reactant? Atherosclerosis. 1999;142:389–393. doi: 10.1016/s0021-9150(98)00242-1. PM:10030390. [DOI] [PubMed] [Google Scholar]
- 62.Jeyaseelan S, Chu HW, Young SK, Worthen GS. Transcriptional profiling of lipopolysaccharide-induced acute lung injury. Infect Immun. 2004;72:7247–7256. doi: 10.1128/IAI.72.12.7247-7256.2004. ISI:000225453900056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meek RL, Eriksen N, Benditt EP. Murine serum amyloid A3 is a high density apolipoprotein and is secreted by macrophages. Proc Natl Acad Sci USA. 1992;89:7949–7952. doi: 10.1073/pnas.89.17.7949. PM:1518819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tomita T, Sakurai Y, Ishibashi S, Maru Y. Imbalance of Clara cell-mediated homeostatic inflammation is involved in lung metastasis. Oncogene. 2011;30:3429–3439. doi: 10.1038/onc.2011.53. PM:21399660. [DOI] [PubMed] [Google Scholar]
- 65.Cabana VG, Reardon CA, Wei B, Lukens JR, Getz GS. SAA-only HDL formed during the acute phase response in apoA-I+/+and apoA-I−/− mice. J Lipid Res. 1999;40:1090–1103. ISI:000080853900013. [PubMed] [Google Scholar]
- 66.Cabana VG, Lukens JR, Rice KS, Hawkins TJ, Getz GS. HDL content and composition in acute phase response in three species: triglyceride enrichment of HDL a factor in its decrease. J Lipid Res. 1996;37:2662–2674. ISI:A1996WD85500017. [PubMed] [Google Scholar]
- 67.Steinmetz A, Hocke G, Saile R, Puchois P, Fruchart JC. Influence of serum amyloid A on cholesterol esterification in human plasma. Biochim Biophys Acta. 1989;1006:173–178. doi: 10.1016/0005-2760(89)90192-6. PM:2512983. [DOI] [PubMed] [Google Scholar]
- 68.Hafiane A, Jabor B, Ruel I, Ling J, Genest J. High-density lipoprotein mediated cellular cholesterol efflux in acute coronary syndromes. Am J Cardiol. 2014;113:249–255. doi: 10.1016/j.amjcard.2013.09.006. PM:24210679. [DOI] [PubMed] [Google Scholar]
- 69.Lee HY, Kim SD, Baek SH, Choi JH, Cho KH, Zabel BA, Bae YS. Serum amyloid A stimulates macrophage foam cell formation via lectin-like oxidized low-density lipoprotein receptor 1 upregulation. Biochem Biophys Res Commun. 2013;433:18–23. doi: 10.1016/j.bbrc.2013.02.077. PM:23454129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Artl A, Marsche G, Lestavel S, Sattler W, Malle E. Role of serum amyloid A during metabolism of acute-phase HDL by macrophages. Arterioscler Thromb Vasc Biol. 2000;20:763–772. doi: 10.1161/01.atv.20.3.763. ISI:000085834100023. [DOI] [PubMed] [Google Scholar]
- 71.Dong Z, Wu T, Qin W, An C, Wang Z, Zhang M, Zhang Y, Zhang C, An F. Serum amyloid A directly accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Mol Med. 2011;17:1357–1364. doi: 10.2119/molmed.2011.00186. PM:21953420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hiratsuka S, Watanabe A, Sakurai Y, Kashi-Takamura S, Ishibashi S, Miyake K, Shibuya M, Akira S, Baitarani H, Maru Y. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol. 2008;10:1349–1355. doi: 10.1038/ncb1794. ISI:000260586700018. [DOI] [PubMed] [Google Scholar]
- 73.Song C, Shen Y, Yamen E, Hsu K, Yan W, Witting PK, Geczy CL, Freedman SB. Serum amyloid A may potentiate prothrombotic and proinflammatory events in acute coronary syndromes. Atherosclerosis. 2009;202:596–604. doi: 10.1016/j.atherosclerosis.2008.04.049. PM:18571179. [DOI] [PubMed] [Google Scholar]
- 74.Saber AT, Jacobsen NR, Mortensen A, Szarek J, Jackson P, Madsen AM, Jensen KA, Koponen IK, Brunborg G, Gutzkow KB, et al. Nanotitanium dioxide toxicity in mouse lung is reduced in sanding dust from paint. Part Fibre Toxicol. 2012;9:4. doi: 10.1186/1743-8977-9-4. PM:22300483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Poitou C, Viguerie N, Cancello R, De MR, Cinti S, Stich V, Coussieu C, Gauthier E, Courtine M, Zucker JD, et al. Serum amyloid A: production by human white adipocyte and regulation by obesity and nutrition. Diabetologia. 2005;48:519–528. doi: 10.1007/s00125-004-1654-6. PM:15729583. [DOI] [PubMed] [Google Scholar]
- 76.Pruett BS, Pruett SB. An explanation for the paradoxical induction and suppression of an acute phase response by ethanol. Alcohol. 2006;39:105–110. doi: 10.1016/j.alcohol.2006.08.003. PM:17134663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nurmi K, Virkanen J, Rajamaki K, Niemi K, Kovanen PT, Eklund KK. Ethanol inhibits activation of NLRP3 and AIM2 inflammasomes in human macrophages-a novel anti-inflammatory action of alcohol. PLoS One. 2013;8:e78537. doi: 10.1371/journal.pone.0078537. PM:24244322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tolstrup J, Gronbaek M. Alcohol and atherosclerosis: recent insights. Curr Atheroscler Rep. 2007;9:116–124. doi: 10.1007/s11883-007-0007-6. PM:17877920. [DOI] [PubMed] [Google Scholar]
- 79.Barboriak JJ, Anderson AJ, Hoffmann RG. Smoking, alcohol and coronary artery occlusion. Atherosclerosis. 1982;43:277–282. doi: 10.1016/0021-9150(82)90028-4. PM:7115464. [DOI] [PubMed] [Google Scholar]
- 80.Whicher J, Biasucci L, Rifai N. Inflammation, the acute phase response and atherosclerosis. Clin Chem Lab Med. 1999;37:495–503. doi: 10.1515/CCLM.1999.080. PM:10418738. [DOI] [PubMed] [Google Scholar]
- 81.Morihara T, Teter B, Yang F, Lim GP, Boudinot S, Boudinot FD, Frautschy SA, Cole GM. Ibuprofen suppresses interleukin-1β induction of pro-amyloidogenic α1-antichymotrypsin to ameliorate β-amyloid (Abeta) pathology in Alzheimer's models. Neuropsychopharmacology. 2005;30:1111–1120. doi: 10.1038/sj.npp.1300668. PM:15688088. [DOI] [PubMed] [Google Scholar]
- 82.Elliott P, Chambers JC, Zhang W, Clarke R, Hopewell JC, Peden JF, Erdmann J, Braund P, Engert JC, Bennett D, et al. Genetic Loci associated with C-reactive protein levels and risk of coronary heart disease. JAMA. 2009;302:37–48. doi: 10.1001/jama.2009.954. PM:19567438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vogel U. Commentary. Atherosclerosis. 2013;228:324. doi: 10.1016/j.atherosclerosis.2012.11.014. PM:23219221. [DOI] [PubMed] [Google Scholar]
- 84.Barregard L, Sallsten G, Gustafson P, Andersson L, Johansson L, Basu S, Stigendal L. Experimental exposure to wood-smoke particles in healthy humans: effects on markers of inflammation, coagulation, and lipid peroxidation. Inhal Toxicol. 2006;18:845–853. doi: 10.1080/08958370600685798. PM:16864402. [DOI] [PubMed] [Google Scholar]
- 85.Allen RW, Carlsten C, Karlen B, Leckie S, van ES, Vedal S, Wong I, Brauer M. An air filter intervention study of endothelial function among healthy adults in a woodsmoke-impacted community. Am J Respir Crit Care Med. 2011;183:1222–1230. doi: 10.1164/rccm.201010-1572OC. PM:21257787. [DOI] [PubMed] [Google Scholar]
- 86.Brauner EV, Forchhammer L, Moller P, Barregard L, Gunnarsen L, Afshari A, Wahlin P, Glasius M, Dragsted LO, Basu S, et al. Indoor particles affect vascular function in the aged: an air filtration-based intervention study. Am J Respir Crit Care Med. 2008;177:419–425. doi: 10.1164/rccm.200704-632OC. PM:17932377. [DOI] [PubMed] [Google Scholar]
- 87.Ohlson CG, Berg P, Bryngelsson IL, Elihn K, Ngo Y, Westberg H, Sjogren B. Inflammatory markers and exposure to occupational air pollutants. Inhal Toxicol. 2010;22:1083–1090. doi: 10.3109/08958378.2010.520356. PM:21029032. [DOI] [PubMed] [Google Scholar]
- 88.Huttunen K, Siponen T, Salonen I, Yli-Tuomi T, Aurela M, Dufva H, Hillamo R, Linkola E, Pekkanen J, Pennanen A, et al. Low-level exposure to ambient particulate matter is associated with systemic inflammation in ischemic heart disease patients. Environ Res. 2012;116:44–51. doi: 10.1016/j.envres.2012.04.004. PM:22541720. [DOI] [PubMed] [Google Scholar]
- 89.Mills NL, Robinson SD, Fokkens PH, Leseman DL, Miller MR, Anderson D, Freney EJ, Heal MR, Donovan RJ, Blomberg A, et al. Exposure to concentrated ambient particles does not affect vascular function in patients with coronary heart disease. Environ Health Perspect. 2008;116:709–715. doi: 10.1289/ehp.11016. PM:18560524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Channell MM, Paffett ML, Devlin RB, Madden MC, Campen MJ. Circulating factors induce coronary endothelial cell activation following exposure to inhaled diesel exhaust and nitrogen dioxide in humans: evidence from a novel translational in vitro model. Toxicol Sci. 2012;127:179–186. doi: 10.1093/toxsci/kfs084. PM:22331494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Robertson S, Colombo ES, Lucas SN, Hall PR, Febbraio M, Paffett ML, Campen MJ. CD36 mediates endothelial dysfunction downstream of circulating factors induced by O3 exposure. Toxicol Sci. 2013;134:304–311. doi: 10.1093/toxsci/kft107. PM:23650127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Baranova IN, Bocharov AV, Vishnyakova TG, Kurlander R, Chen Z, Fu D, Arias IM, Csako G, Patterson AP, Eggerman TL. CD36 is a novel serum amyloid A (SAA) receptor mediating SAA binding and SAA-induced signaling in human and rodent cells. J Biol Chem. 2010;285:8492–8506. doi: 10.1074/jbc.M109.007526. PM:20075072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang X, Chai H, Wang Z, Lin PH, Yao Q, Chen C. Serum amyloid A induces endothelial dysfunction in porcine coronary arteries and human coronary artery endothelial cells. Am J Physiol Heart Circ Physiol. 2008;295:H2399–H2408. doi: 10.1152/ajpheart.00238.2008. PM:18931033. [DOI] [PMC free article] [PubMed] [Google Scholar]