

Abstract
Objective
The number of confirmed rheumatoid arthritis (RA) loci currently stands at 32, but many lines of evidence indicate that expansion of existing genome-wide association studies (GWAS) enhances the power to detect additional loci. This study was undertaken to extend our previous RA GWAS in a UK cohort, adding more independent RA cases and healthy controls, with the aim of detecting novel association signals for susceptibility to RA in a homogeneous UK cohort.
Methods
A total of 3,223 UK RA cases and 5,272 UK controls were available for association analyses, with the extension adding 1,361 cases and 2,334 controls to the original GWAS data set. The genotype data for all RA cases were imputed using the Impute program version 2. After stringent quality control thresholds were applied, 3,034 cases and 5,271 controls (1,831,729 single-nucleotide polymorphisms [SNPs]) were available for analysis. Association testing was performed using Plink software.
Results
The analyses indicated a suggestive association with susceptibility to RA (P < 0.0001) for 6 novel RA loci that have been previously found to be associated with other autoimmune diseases; these 6 SNPs were genotyped in independent samples. Two of the associated loci were validated, one of which was associated with RA at genome-wide levels of significance in the combined analysis, identifying a novel RA locus at 22q12 (P = 6.9 × 10−9). In addition, most of the previously known RA susceptibility loci were confirmed to be associated with RA, and for 16 of the loci, the strength of the association was increased.
Conclusion
This study identified a new RA locus mapping to 22q12. These results support the notion that increasing the power of GWAS enhances novel gene discovery.
Understanding the genetic component of susceptibility to rheumatoid arthritis (RA) will increase our knowledge of the disease process and has the potential to inform new approaches to disease management. For example, the identification of disease-associated genetic variations, which are presumed to cause modified immune responses and precede the onset of disease symptoms, could inform stratification of patients into more phenotypically homogeneous subgroups and provide testable hypotheses regarding response to treatment. The use of genome-wide association studies (GWAS) has been remarkably successful in locating novel genetic loci associated with RA, and there are now more than 30 gene regions that have been confirmed as RA susceptibility loci, but, in total, they account for fewer than 50% of the total genetic heritability (1,2). It is likely that there are more common variants of small effect size that could be identified by increasing the power of the GWAS through the use of larger sample sizes. Indeed, this approach has been used successfully in a number of autoimmune diseases, such as type 1 diabetes (3) and inflammatory bowel disease (4,5), resulting in a more complete picture of the genetic background.
In the present study, we used the data set from an RA GWAS in the UK (6) and increased the sample sizes of the RA cases and healthy controls by 75% and 80%, respectively. With this extended data set, together with the data obtained in a validation study of the UK cohort, we were able to discover potential novel RA risk loci in the UK population. This study constitutes the largest UK-only GWAS to date, and the results will enhance the power to investigate whether population heterogeneity exists, i.e., whether different genes are associated with RA in different populations.
PATIENTS AND METHODS
Genotype data were available for 1,862 RA cases and 2,938 controls from the original Wellcome Trust Case Control Consortium (WTCCC) study (6); these genotypes were obtained using the Affymetrix 500K array. Together with this data set, we added GWAS genotype data for a further 2,334 UK controls and 1,361 UK RA cases (sample call rate >95%). The additional samples were from 5 different studies, with genotypes obtained using a range of GWAS arrays. Members of the WTCCC, UK Rheumatoid Arthritis Genetics Group, and Biologics in Rheumatoid Arthritis Genetics and Genomics Study Syndicate Consortiums and details on the platforms used are given in the Supplementary Materials and Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38196/abstract.
Since each GWAS array contains different single-nucleotide polymorphisms (SNPs), the first stage of the current study was to impute genotypes to generate data for a common set of SNPs, thus allowing combined analysis of the data from the 5 different studies. The genotypes of the patients in each RA case cohort were imputed using the Impute program (version 2) (7), based on 2 reference panels, the 1000 Genomes Project pilot data and HapMap3. The genotypes of control subjects were imputed using the same 1000 Genomes Project reference panel, with imputation performed using MaCH software (8). Stringent quality control (QC) thresholds were applied to individual cohorts and then to the merged data set (see Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38196/abstract). Self-reported information on each subject’s ethnicity was used to exclude non-Caucasians from the analyses.
Identity-by-descent analysis was performed in Plink statistical software. Pairs of samples with genome identity (pi-hat values) greater than 0.9 were reported to be duplicate samples, and those with pi-hat values greater than 0.2 were considered to be closely related. Case and control genotype data were compared using logistic regression, performed in Plink (9). Novel SNPs not previously found to be associated with RA in the UK population were selected for validation if the association P value was less than 0.0001, and if the region had been previously found to be associated with another autoimmune disease. The SNPs satisfying these criteria were genotyped in independent UK samples (4,726 cases and 2,625 controls), using Sequenom technology. Additional control data were available using non–autoimmune disease cases (n = 7,670) from the WTCCC study (6).
A regional association plot for the 22q12 locus was created using LocusZoom (http://csg.sph.umich.edu/locuszoom/). Bioinformatics analysis was performed using publicly available functional annotation files (http://genome.ucsc.edu/) and our own custom query SQL-code (Assimilator; available at http://assimilator.mhs.manchester.ac.uk/cgi-bin/assimilator.pl) (10).
RESULTS
Forty-two duplicate samples were removed after identity-by-descent analysis. There were 14 pairs of individual samples with pi-hat values greater than 0.2, and thus 1 sample from each pair was removed due to possible relatedness. A total of 1,831,729 SNPs passed QC and were included in the association analysis. In the expanded GWAS, 3,034 RA cases and 5,271 controls passed QC.
The genetic inflation factor lambda (λGC) was 1.06, implying that there was a low possibility of false-positive associations attributable to population stratification, genotyping errors, or other artifacts. For most of the previously known RA susceptibility loci (2), we confirmed their association with RA in the present study (Table 1). Interestingly, when compared to the original WTCCC study (6), the extended RA GWAS showed that the strength of the association with RA susceptibility was increased for 16 of the loci in the UK population (TNFAIP3, STAT4, PTPN22, HLA–DRB1, PTPRC, IL2/IL21, C5orf30, CD247, CTLA4, RBPJ, PRDM1, CCR6, IRF5, TRAF6, KIF5A, and CD28). For example, the association of TNFAIP3 with RA risk reached genome-wide levels of significance in this expansion of the WTCCC analysis, and the robustly validated locus STAT4, for which no significant association had been found in the original WTCCC study, was found to be associated with RA in the present study (Figure 1).
Table 1.
Previously confirmed rheumatoid arthritis (RA) loci association results in the original WTCCC association analysis and the expanded UK RA GWAS*
| Chr. | SNP | Gene | WTCCC study | Expanded UK RA GWAS | ||||
|---|---|---|---|---|---|---|---|---|
| Proxy | P | OR (95% CI) | Proxy | P | OR (95% CI) | |||
| 1 | rs3890745 | TNFRSF14 | 8.47 × 10−6 | 0.82 (0.75–0.89) | 1.43 × 10−6 | 0.85 (0.79–0.91) | ||
| 1 | rs2476601 | PTPN22 | rs6679677 | 2.60 × 10−25 | 1.90 (1.68–2.15) | 4.87 × 10−33 | 1.77 (1.61–1.95) | |
| 1 | rs11586238 | CD2, CD58 | 4.13 × 10−4 | 1.19 (1.08–1.31) | rs4271251 | 6.47 × 10−3 | 1.11 (1.03–1.19) | |
| 1 | rs12746613 | FCGR2A | 0.04 | 1.14 (1.01–1.29) | 0.03 | 1.11 (1.01–1.22) | ||
| 1 | rs10919563 | PTPRC | 0.003 | 0.82 (0.72–0.94) | 7.18 × 10−5 | 0.82 (0.74–0.9) | ||
| 2 | rs13031237 | REL | None | rs13031721 | 0.29 | 1.04 (0.97–1.11) | ||
| 2 | rs934734 | SPRED2 | 0.10 | 0.93 (0.86–1.01) | 0.11 | 0.95 (0.89–1.01) | ||
| 2 | rs10865035 | AFF3 | rs9653442 | 5.48 × 10−4 | 1.16 (1.07–1.26) | rs1160542 | 1.37 × 10−5 | 1.15 (1.08–1.23) |
| 2 | rs7574865 | STAT4 | rs11893432 | 0.02 | 1.13 (1.02–1.25) | rs10181656 | 6.64 × 10−4 | 1.14 (1.06–1.23) |
| 2 | rs1980422 | CD28 | 4.80 × 10−3 | 1.15 (1.04–1.26) | 1.75 × 10−4 | 1.15 (1.07–1.24) | ||
| 2 | rs3087243 | CTLA4 | 0.09 | 0.93 (0.86–1.01) | 0.02 | 0.92 (0.87–0.99) | ||
| 3 | rs13315591 | PXK | 0.20 | 1.10 (0.95–1.26) | None | |||
| 4 | rs874040 | RBPJ | None | rs6448432 | 3.88 × 10−7 | 1.19 (1.11–1.28) | ||
| 4 | rs6822844 | IL2, IL21 | None | rs62322744 | 6.42 × 10−3 | 1.18 (1.05–1.32) | ||
| 5 | rs6859219 | ANKRD55, IL6ST | 5.5 × 10−6 | 0.78 (0.70–0.87) | None | |||
| 5 | rs26232 | C5orf30 | rs556560 | 2.46 × 10−4 | 0.85 (0.78–0.93) | rs556560 | 9.64 × 10−5 | 0.88 (0.82–0.94) |
| 6 | rs6910071 | HLA–DRB1 | rs6457617 | 3.49 × 10−79 | 0.44 (0.40–0.48) | rs3763309 | 1.50 × 10−124 | 2.3 (2.14–2.46) |
| 6 | rs548234 | PRDM1 | 0.01 | 1.12 (1.02–1.22) | 1.24 × 10−3 | 1.12 (1.04–1.19) | ||
| 6 | rs6920220 | TNFAIP3 | 6.11 × 10−6 | 1.25 (1.13–1.37) | 3.11 × 10−8 | 1.23 (1.14–1.32) | ||
| 6 | rs394581 | TAGAP | 5.86 × 10−3 | 0.88 (0.80–0.96) | None | |||
| 6 | rs3093023 | CCR6 | rs6907666 | 0.05 | 1.09 (1–1.18) | rs3093024 | 1.88 × 10−3 | 1.11 (1.04–1.18) |
| 7 | rs10488631 | IRF5 | rs12531711 | 0.03 | 1.16 (1.02–1.31) | 3.10 × 10−3 | 1.16 (1.05–1.28) | |
| 8 | rs2736340 | BLK | 8.01 × 10−3 | 1.14 (1.03–1.25) | 0.05 | 1.07 (1–1.15) | ||
| 9 | rs2812378 | CCL21 | 1.14 × 10−3 | 1.15 (1.06-1.26) | None | |||
| 9 | rs3761847 | TRAF1, C5 | 0.80 | 0.99 (0.91–1.07) | 0.19 | 1.04 (0.98–1.11) | ||
| 10 | rs2104286 | IL2RA | 7.06 × 10−6 | 0.81 (0.73–0.89) | 1.46 × 10−6 | 0.84 (0.78–0.9) | ||
| 10 | rs4750316 | PRKCQ | 5.22 × 10−5 | 0.80 (0.72–0.89) | 1.55 × 10−4 | 0.85 (0.78–0.93) | ||
| 11 | rs540386 | TRAF6 | 0.04 | 0.88 (0.77–0.99) | rs1046864 | 0.01 | 0.89 (0.8–0.98) | |
| 12 | rs1678542 | KIF5A | 2.81 × 10−5 | 0.83 (0.76–0.91) | 9.75 × 10−8 | 0.83 (0.78–0.89) | ||
| 20 | rs4810485 | CD40 | 0.07 | 0.91 (0.83–1.01) | rs1569723 | 0.22 | 0.95 (0.89–1.03) | |
| 22 | rs3218253 | IL2RB | 1.88 × 10−4 | 1.19 (1.09–1.31) | 2.51 × 10−4 | 1.15 (1.07–1.23) | ||
WTCCC = Wellcome Trust Case Control Consortium; GWAS = genome-wide association study; Chr. = chromosome; SNP = single-nucleotide polymorphism; OR = odds ratio; 95% CI = 95% confidence interval.
Figure 1.
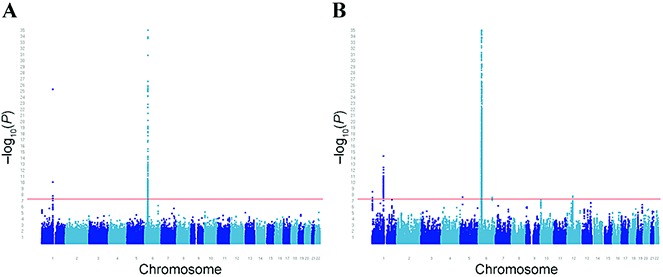
Manhattan plots showing P values for genetic association with rheumatoid arthritis (RA) in the original Wellcome Trust Case Control Consortium genome-wide association study (GWAS) (a) and the expanded UK GWAS (b). Panels are truncated at a −log10P value of 35. Single-nucleotide polymorphisms having an association with RA at genome-wide levels of significance are shown above the horizontal red line.
Similar to the findings in the original WTCCC study, some of the known RA loci (CD40, REL, SPRED2, BLK, and TRAF1/C5) were not found to have an association with RA in the present study. Finally, no proxies were available for 4 of the known RA loci.
Six of the SNPs with suggestive evidence of association with RA in the present study and with additional evidence of association in other studies were selected for validation analysis (Table 2). Evidence of an association with susceptibility to RA was found for rs1043099 (on chromosome 22q12) in this validation study (P = 2.7 × 10−3), and the association exceeded genome-wide significance thresholds in the combined analysis (P = 6.9 × 10−9) (Figure 2). A SNP on chromosome 11, near CXCR5 and in strong linkage disequilibrium (LD) (r2 > 0.9) with a SNP previously associated with RA at genome-wide significance levels (11), was also associated with RA in the validation cohort (P = 0.02), and there was suggestive evidence of association in the combined analysis (P = 1.8 × 10−5).
Table 2.
Candidate rheumatoid arthritis (RA) susceptibility loci previously associated with other autoimmune diseases and showing suggestive evidence of association with RA in the expanded UK RA GWAS*
| Chr. | SNP | Locus | Assoicated autoimmune disease | Expanded UK RA GWAS | Validation study | Combined analysis | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MAF | P†‡ | OR (95% CI)‡ | Proxy/comment | MAF | P | OR (95% CI) | ||||||||
| RA | Controls | RA | Controls | P | OR (95% CI) | |||||||||
| 7 | rs1047022 | SNX10 | T1D, IBD | 0.15 | 0.19 | 6.4 × 10−7 | 0.80 (0.74–0.88) | Failed QC | ||||||
| 11 | rs6421571 | CXCR5 | PBC | 0.17 | 0.19 | 6.3 × 10−5 | 0.84 (0.78–0.92) | 0.18 | 0.19 | 0.02 | 0.90 (0.82–0.98) | 1.8 × 10−5 | 0.88 (0.83–0.93) | |
| 12 | rs11181399 | YAF2 | MS | 0.40 | 0.45 | 2.5 × 10−7 | 0.8 (0.79–0.90) | rs7954523 | 0.39 | 0.39 | 0.96 | 0.99 (0.93–1.08) | 7.01 × 10−5 | 0.91 (0.87–0.95) |
| 16 | rs7187962 | CNTAP4 | T1D | 0.27 | 0.31 | 8.6 × 10−6 | 0.85 (0.79–0.91) | rs6564350§ | 0.30 | 0.31 | 0.40 | 0.97 (0.89–1.05) | ||
| 19 | rs6417247 | CDC37 | IBD | 0.30 | 0.33 | 9.1 × 10−5 | 0.87 (0.82–0.93) | rs8112449 | 0.32 | 0.33 | 0.20 | 0.95 (0.88–1.03) | 1.5 × 10−3 | 0.93 (0.88–0.97) |
| 22 | rs1043099 | GATSL3 | CD, T1D | 0.18 | 0.21 | 7.8 × 10−6 | 0.83 (0.77–0.90) | 0.19 | 0.21 | 2.70 × 10−3 | 0.87 (0.80–0.95) | 6.9 × 10−9 | 0.84 (0.79–0.89) | |
Associations were identified at the significance level of P < 0.0001. Chr. = chromosome; MAF = minor allele frequency; T1D = type 1 diabetes; IBD = inflammatory bowel disease; QC = quality control; PBC = primary biliary cirrhosis; MS = multiple sclerosis; CD = celiac disease.
Corrected P values (corrected for genetic inflation factor lambda) were as follows: for rs1047022, P = 1.42 × 10−6; for rs6421571, P = 1.07 × 10−4; for rs11181399, P = 5.86 × 10−7; for rs7187962, P = 1.628 × 10−5; for rs6417247, P = 1.50 × 10−4; for rs1043099, P = 1.49 × 10−5.
Association results in the original Wellcome Trust Case Control Consortium study were as follows: for rs1047022, P = 1.106 × 10−6, odds ratio (OR) 0.74 (95% confidence interval [95% CI] 0.66–0.84); for rs6421571, P = 0.02, OR 0.88 (95% CI 0.79–0.98); for rs11181399, P = 3.98 × 10−5, OR 0.83 (95% CI 0.76–0.91); for rs7187962, P = 7.07 × 10−6, OR 0.80 (95% CI 0.73–0.88); for rs6417247, P = 2.31 × 10−3, OR 0.87 (95% CI 0.79–0.95); for rs1043099, P = 1.65 × 10−4, OR 0.81 (95% CI 0.72–0.90).
Single-nucleotide polymorphism (SNP) rs6564350 was not present in the expanded UK RA genome-wide association study (GWAS) data set, and therefore combined analysis of this SNP could not be performed.
Figure 2.
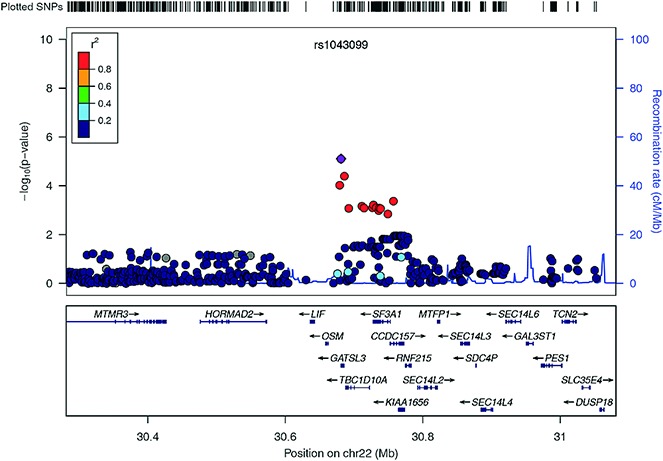
Regional plot of association with rheumatoid arthritis (RA) at chromosome 22q12. The P values for association (−log10 values) of each single-nucleotide polymorphism (SNP) are plotted against their physical position on chromosome 22 (top panel). Estimated recombination rates from the 1000 Genomes Project population show the local linkage disequilibrium (LD) structure (middle panel). Different colors indicate the LD of each SNP with rs1043099, based on pairwise r2 values from the 1000 Genomes Project. Gene annotations are shown in the lower panel.
In further analyses, we interrogated publicly available functional annotation data (10,12) and found evidence to indicate that rs1043099 and its correlated SNPs (r2 > 0.8) may have regulatory activity in RA. First, the SNP alleles were found to be associated with expression levels of the SF3A1 gene (which encodes subunit 1 of the splicing factor 3a protein complex) (http://eqtl.uchicago.edu/cgi-bin/gbrowse/eqtl/) (Mangravite LM, et al: unpublished observations). Second, several of the SNPs map to sites of transcription factor binding, histone modification, and open chromatin (Table 3), suggesting that these SNPs could influence gene transcription.
Table 3.
Potential regulatory role of the rheumatoid arthritis–associated single-nucleotide polymorphism (SNP) rs1043099 and its proxies*
| SNP | Relative location | Gene | Transcribed region | Histone modifications | TFBS | DNase I HS | FAIRE | CTCF binding | eQTL |
|---|---|---|---|---|---|---|---|---|---|
| rs2108093 | 3′ | GATSL3 | Yes | Yes | Yes | Yes | Yes | ||
| rs1043099 | Exonic, 3′-UTR | GATSL3 | Yes | Yes | Yes | Yes | Yes | ||
| rs4823085 | 5′ | GATSL3 | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| rs929454 | Intronic | TBC1D10A | Yes | Yes | Yes | Yes | Yes | ||
| rs4820831 | Intronic | TBC1D10A | Yes | Yes | Yes | Yes | Yes | Yes | |
| rs740219 | Intronic | TBC1D10A | Yes | Yes | Yes | Yes | Yes | ||
| rs5753071 | 3′ | SF3A1 | Yes | Yes | Yes | Yes | Yes | ||
| rs10376 | Exonic, 3′-UTR | SF3A1 | Yes | Yes | Yes | Yes | Yes | ||
| rs2041199 | Intronic | SF3A1 | Yes | Yes | Yes | Yes | |||
| rs4339043 | Intronic | SF3A1 | Yes | Yes | Yes | Yes | Yes | ||
| rs5749066 | Intronic | SF3A1 | Yes | Yes | Yes | Yes | Yes | ||
| rs5753080 | Intronic | SF3A1 | Yes | Yes | Yes | ||||
| rs10427610 | Intronic | SF3A1 | Yes | Yes | Yes | Yes | Yes | Yes | |
| rs4820008 | Intronic | SF3A1 | Yes | Yes | Yes | ||||
| rs737950 | Intronic | CCDC157 | Yes | Yes | Yes | ||||
| rs5749078 | Intronic | CCDC157 | Yes | Yes | Yes | Yes | |||
| rs9619104 | 3′ | CCDC157 | Yes | Yes | Yes | Yes |
Proxies of rs1043099 were correlated at r2 > 0.8. Results are the summary output from the Assimilator bioinformatics analysis. TFBS = transcription factor binding site; HS = hypersensitive sites; FAIRE = open chromatin by formaldehyde-assisted isolation of regulatory elements; CTCF = CCCTC binding factor; eQTL = expression quantitative trait loci; 3′-UTR = 3′-untranslated region.
We then stratified the expanded UK GWAS and validation data sets according to the presence or absence of anti–cyclic citrullinated peptide (anti-CCP) antibodies in RA patients. The proportion of anti-CCP–positive RA patients in the original WTCCC study, expanded UK GWAS, and validation study was 79%, 73.5%, and 67%, respectively. SNP rs1043099 was significantly associated with both anti-CCP–positive RA and anti-CCP–negative RA. In contrast, SNP rs6421571 was associated with anti-CCP–positive RA only (see Supplementary Table 2, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38196/abstract). Similar results were obtained when patients were stratified by the presence of rheumatoid factor (see Supplementary Table 3, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.38196/abstract).
DISCUSSION
In this study, we discovered a new RA risk locus on chromosome 22q12, rs1043099, for which the association with susceptibility to RA reached genome-wide levels of significance (combined P = 6.9 × 10−9). This locus has previously been associated with other autoimmune diseases, including type 1 diabetes (3) and inflammatory bowel disease (5,13). The SNP lies within a gene of unknown function, GATSL3. It will be interesting to better fine-map this region in RA, as well as in other autoimmune diseases, to help determine which gene is causal. Using bioinformatics analysis, we showed that rs1043099 and its correlated SNPs have potential regulatory activity. Interestingly, evidence suggests that these SNPs act as expression quantitative trait loci for the SF3A1 gene. SFA1 is involved in messenger RNA splicing, but the particular role that this gene might have in the pathogenesis of RA has not yet been explored. Further functional studies will be required to determine which of the SNPs is causal and to elucidate the mechanisms of action of each SNP.
Of the remaining SNPs tested in the validation samples, evidence of increased strength of the association with susceptibility to RA was found only for rs6421571 on chromosome 11q23 (combined P = 1.8 × 10−5). This SNP maps 5′ to the CXCR5 gene, which is a chemokine involved in B cell migration and localization, and has previously been associated with primary biliary cirrhosis (14). Different SNPs in the same region have been found to be associated with multiple sclerosis (15) and RA (11). The SNP identified as being significantly associated with RA and celiac disease in a combined analysis, rs10892279, is adjacent to the gene DDX6, and although it is >130 kb from rs6421571, the 2 SNPs are strongly correlated (r2 > 0.9). Therefore, which of these 2 strong candidate genes will ultimately be found to be causal requires further fine-mapping and functional studies.
The added power provided by this study strengthens the evidence for an association with RA for 16 loci that were already confirmed to be associated with RA in the UK population (2). The most striking findings supporting an association with RA within the UK population, as evidenced by an increase in the significance of the association from the original WTCCC study to the expanded GWAS, were seen for the loci at PTPRC (increasing from P = 0.003 to P = 7 × 10−5), TNFAIP3 (increasing from P = 6 × 10−6 to P = 3 × 10−8), and KIF5A (increasing from P = 2 × 10−5 to P = 9 × 10−8). RBPJ, not available for analysis in the original WTCCC study, showed evidence of a strong association with RA in this expanded GWAS (P = 3 × 10−7).
There was little evidence of an association with other previously confirmed RA loci in this expanded UK data set, including FCGR2A, REL, SPRED2, CTLA4, BLK, TRAF1, and CD40. This could be attributed to a number of factors, but the most likely explanation is that the current study was underpowered to detect such associations. Although we performed a relatively large discovery study, the power to detect all of the associations was limited (average power across SNPs with a minor allele frequency >5% was 47% for an odds ratio [OR] of 1.1, and >90% for an OR of >1.2), and therefore failure to detect a signal could simply be the result of stochastic variations leading to a false-negative result. Indeed, it is well established that even the most strongly associated signals across multiple diseases are not found to be consistently associated in all cohorts, and often an accumulation of evidence is required in many thousands of samples. In this regard, a recent meta-analysis failed to detect the association between the 22q12 locus and RA that we identified in the current study (for rs929454, P = 0.63, r2 = 0.91 for LD with rs1043099) (2).
Interestingly, most of the confirmed RA loci for which we did not find any association with RA in the expanded UK data set (FCGR2A, REL, SPRED2, CTLA4, BLK, TRAF1, and CD40) were found to have an association with anti-CCP–positive RA, but not anti-CCP–negative RA, in a recent study, which was the largest anti-CCP–stratified RA sample size studied to date (16). We found very modest evidence of association with anti-CCP–positive RA for only 2 of the above-mentioned variants (P = 0.03 for FCGR2A, and P = 0.03 for BLK), and therefore it is likely that the present study was underpowered to detect serotype-specific effects at these RA loci.
This study has added to the argument that increasing the size, and consequently the power, of RA GWAS can prove fruitful in the discovery of novel genes. Increasing the number of known disease loci will facilitate the estimation of disease risk, potentially allowing early intervention in high-risk groups, possibly informing prognosis, and, ultimately, aiding in the discovery of novel targets for pharmacologic therapy. Future work will involve adding this new extended UK data to existing meta-analyses of RA case data with the aim of expanding our knowledge of the common genetic variants predisposing to RA.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Eyre had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Orozco, Wilson, Barton, Worthington, Eyre.
Acquisition of data. Orozco, Steer, Wordsworth, Hocking.
Analysis and interpretation of data. Orozco, Viatte, Bowes, Martin, Morgan, Barton, Eyre.
Acknowledgments
We would like to thank Edward Flynn for preparing and genotyping the UK Rheumatoid Arthritis Group samples. We thank Eleftheria Zeggini for providing the imputed data for healthy controls.
REFERENCES
- 1.Orozco G, Barton A. Update on the genetic risk factors for rheumatoid arthritis. Expert Rev Clin Immunol. 2010;6:61–75. doi: 10.1586/eci.09.72. [DOI] [PubMed] [Google Scholar]
- 2.Stahl EA, Raychaudhuri S, Remmers EF, Xie G, Eyre S, Thomson BP, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. 2010;42:508–14. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41:703–7. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson CA, Boucher G, Lees CW, Franke A, D’Amato M, Taylor KD, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43:246–52. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–25. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–34. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin P, Barton A, Eyre S. ASSIMILATOR: a new tool to inform selection of associated genetic variants for functional studies. Bioinformatics. 2011;27:144–6. doi: 10.1093/bioinformatics/btq611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhernakova A, Stahl EA, Trynka G, Raychaudhuri S, Festen EA, Franke L, et al. Meta-analysis of genome-wide association studies in celiac disease and rheumatoid arthritis identifies fourteen non-HLA shared loci. PLoS Genet. 2011;7:e1002004. doi: 10.1371/journal.pgen.1002004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–40. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 13.Imielinski M, Baldassano RN, Griffiths A, Russell RK, Annese V, Dubinsky M, et al. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat Genet. 2009;41:1335–40. doi: 10.1038/ng.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mells GF, Floyd JA, Morley KI, Cordell HJ, Franklin CS, Shin SY, et al. Genome-wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet. 2011;43:329–32. doi: 10.1038/ng.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–9. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Viatte S, Plant D, Bowes J, Lunt M, Eyre S, Barton A, et al. Genetic markers of rheumatoid arthritis susceptibility in anti-citrullinated peptide antibody negative patients. Ann Rheum Dis. 2012;71:1984–90. doi: 10.1136/annrheumdis-2011-201225. [DOI] [PMC free article] [PubMed] [Google Scholar]