

Abstract

A palladium-catalyzed, enantioselective, aryl–aryl cross-coupling reaction using 1-naphthyldimethylsilanolates and chiral bis-hydrazone ligands has been developed. A family of glyoxal bis-hydrazone ligands containing various 2,5-diarylpyrrolidine groups was prepared to evaluate the influence of ligand structure on the rate and enantioselectivity of the cross-coupling. New synthetic routes to the 1-amino-2,5-diarylpyrrolidines were developed to enable the structure/reactivity–selectivity studies. Role reversal experiments of aryldimethylsilanolates and aryl bromides result in biaryl products with the same configuration and similar enantioselectivities implying that reductive elimination is the stereodetermining step. The origin of stereoselectivity is rationalized through computational modeling of diarylpalldium(II) complex which occurs through a conrotatory motion for the two aryl groups undergoing C–C bond formation.
1. Introduction
The importance of chiral biaryl compounds cannot be overstated; this motif is the central feature of many ligands and catalysts.1 For example, phosphoramidites derived from BINOL have been applied to asymmetric conjugate addition and allylic amination reactions (Figure 1).1b A large family of binaphthyl-based ammonium salts has been employed for asymmetric phase-transfer catalysis.1c The chiral biaryl structural motif is also found in numerous natural products2 such as michellamine B,3 steganacin,4 and vancomycin.5
Figure 1.

Examples of ligands and natural products containing a chiral biaryl motif.
The importance of this structural motif has led to the development of many methods for the preparation of chiral biaryls.2 Some common methods that have been applied to the synthesis of natural product include (1) oxidative coupling of arylcyanocuprates using a chiral tether derived from tartaric acid,6 (2) Ullmann coupling or SNAr reaction mediated by an o-oxazoline derived from valinol,7 (3) dynamic kinetic resolution of configurationally labile lactones,8 (4) oxidative homocoupling of naphthol derivatives catalyzed by a diaza-cis-decalin copper(II) complex,2c,9 and (5) chiral Brønsted acid catalyzed [3,3]-sigmatropic rearrangement of diarylhydrazines.10
Palladium-catalyzed cross-coupling reactions are arguably the most widely used method for the construction of aryl–aryl bond. However, the corresponding enantioselective reaction has not reached maturity as evident in their limited application in total synthesis.2b,11 Notable examples include the formation of the chiral biaryl units in (−)-steganone, vancomycin and rupensamines A and B which have been prepared by the coupling of chiral tricarbonylchromium complex of aryl halides in high diastereoselectivity.12
Recent years have witnessed a growing interest in the design of chiral ligands to facilitate catalytic enantioselective biaryl coupling. The key developments and mechanistic studies of catalytic enantioselective cross-coupling are briefly discussed in the following section.
2. Background
2.1. Evolution of Catalytic Asymmetric Aryl–Aryl Coupling
The use of chiral ligands to control enantioselective aryl–aryl coupling was first reported in 1975 using aryl Grignard reagents, aryl halides, and nickel catalysts.13 After more than a decade, the enantioselectivity of the reaction was substantially improved by tuning the reaction conditions and using monodentate phosphine ligand 1 (Figure 2) with a ferrocene backbone.14
Figure 2.

Representative ligands that have been used in enantioselective aryl–aryl coupling reactions.
The first, catalytic, enantioselective Suzuki–Miyaura coupling, was reported in 2000.15 The dimethylamino-substituted ferrocenyl phosphine 2 is more efficient at stereocontrol than methoxy analogue 1. The KenPhos system reported in the same year also features the dimethylamino functionality.16 Importantly, aryl substrates with polar functionalities such as phosphonate and nitro at the ortho position are demonstrated for the first time. In addition, non-naphthyl-derived substrates such as 2-substituted phenylboronic acids and halides became competent coupling partners. The scope of this method has been subsequently expanded to 2-halobenzamides.17 Comparable or improved selectivity for the preparation of biarylphosphonates has been reported with the use of monophosphine ligands 3 and 4.18
In 2008, Fernández et al. reported C2-symmetric bis-hydrazone ligand 5 for the cross-coupling of arylboronic acids.19 Excellent enantioselectivities are achieved for a number of biaryls when the reactions are conducted at 20 °C, albeit at the expense of reaction time (7 days). The conversion can be accelerated at 80 °C (<17 h) with some erosion in selectivity. Noting the relatively low catalytic activity at room temperature and the limited scope in this work, the same group designed a novel P/N-hybrid ligand 6 derived from C2-symmetric 2,5-bis(isopropylamino)pyrrolidine.20
Catalytic systems that maintain high enantioselectivity for both polar and nonpolar coupling partners are rare. One notable success employs polymer-supported chiral imidazoindole phosphine 7 (Figure 2).21 For example, the highly hindered but nonpolar 2,2′-dimethylbinaphthalene is obtained in 95% yield and 97:3 er. The less hindered but more polar 2-methyl-1-(2-nitrophenyl)naphthalene is obtained in 96% yield and 96:4 er.
Catalytic, enantioselective cross-coupling reactions have been accomplished for arylboron,18−22 -zinc,23 and -indium24 reagents. Although many systems involve phosphine-derived chiral ligands, diene,22i oxazoline,22g,25 bis-hydrazone,19 hydrazone–phosphine hybrid20 ligands and helical polymers22l have also been employed. Other important developments that have emerged in this research area include a recyclable chiral catalyst on a polymer support,21 catalysis by palladium nanoparticles,22h and C–H activation.25
2.2. Mechanistic Hypotheses of the Stereodetermining Step and the Origin of Enantioselectivity
Despite the preparative advances in asymmetric biaryl, insights required to decipher the stereodetermining step (SDS) are usually not available. As such, the knowledge behind the stereocontrol imposed by chiral ligands often remains speculative except in a few cases.17,18a,20 Hayashi et al. employed the role reversal experiment of cross-coupling partners to probe the SDS. In the preparation of 2-methyl-1,1′-binaphthalene a very different stereochemical outcome is observed when the 2-methyl substituent arises from the Grignard reagent rather than from the bromide (Scheme 1).14 Under the assumption that the diarylnickel(II) intermediate does not undergo racemization, the SDS is thought to be transmetalation.
Scheme 1.

In a Suzuki–Miyaura coupling using (R)-BINAP as the chiral ligand, transmetalation is also suggested to be the SDS.22c Altering the identity of the boron reagent from pinacol ester to ethylene glycol ester leads to 2,2′-dimethylbinaphthalene with the opposite configuration as the major product, although the enantioselectivity in both reactions are low. It should be mentioned that the interpretation of SDS can be complicated by the palladium/ligand ratio.22a
The SDS for the coupling promoted by phosphine–hydrazone hybrid ligand 6 was inferred from the configuration stability of oxidative addition complex (Scheme 2).20 The 2-methoxy-1-naphthylpalladium bromide complex exists as a 7:1 mixture of atropisomers. Treatment of this complex with 1-naphthylboronic acid affords the coupled product in 80:20 er at room temperature similar to that obtained from the catalytic reaction (83:17). The stereochemical outcome of this coupling appears to be mainly controlled by oxidative addition. In contrast, free C–Pd bond rotation is observed for the less hindered 1-naphthylpalladium bromide complex. These results imply that the stereodetermining step may be substrate dependent in this system.
Scheme 2.

A detailed computational study on the origin of enantioselectivity promoted by KenPhos (Figure 2) has been carried out by Buchwald et al.17 These authors assumed that reductive elimination is the SDS and then calculated a number of diarylpalladium transition structures from the crystallographic coordinates of the corresponding oxidative addition adduct. The most stable transition structure correctly predicts the configuration of the major biaryl product.
Despite the significant progress of catalytic, enantioselective, biaryl coupling reactions in the past two decades, a general cross-coupling process to access a range of chiral biaryls has yet to be developed. In continuation of our interest in the use of organosilanols and their derivatives in cross-coupling reactions, we considered the use of arylsilicon reagents for this purpose. The competency of an aryldimethylsilanolate unit to transfer a variety of aromatic groups to palladium has been reported from these laboratories.26 In addition, the ability to isolate silanolate complexes of palladium has facilitated our understanding of the catalytic cycle focusing on the transmetalation step.27 Given the ability to isolate the pretransmetalation intermediate, it was of interest to systematically examine the ligand effects on the enantioselectivity of the coupling and correlate those trends with structural variations. In addition to experimental interrogation, we also planned a computational study to investigate the origin of enantioselectivity.
3. Results
3.1. Preparation of Bis-hydrazone Ligands
3.1.1. Glyoxal-Derived Bis-hydrazones
To date, only a limited number of chiral glyoxal bis-hydrazones have been prepared (Figure 3).28 Among these variants, ligand 5 featuring (2S,5S)-diphenylpyrrolidine moiety has shown promise in enantioselective biaryl coupling.19 Despite the report of this ligand and its potential modularity almost a decade ago,28d no analogues with other 2,5-disubstituted pyrrolidines have appeared. A possible explanation for this lack of development is the challenge associated with the synthesis of the requisite building blocks, an analysis of which follows.
Figure 3.

Chiral glyoxal bis-hydrazone ligands.
The first route considered was that reported for the synthesis of (2S,5S)-diphenylpyrrolidine bis-hydrazone 5 (Scheme 3, route a).28d The key step is the enantio- and diastereoselective reduction of 1,4-diphenyl-1,4-butanedione by the Corey–Itsuno protocol.29 The scope of the diketone amenable to this reduction has not been extensively explored, and it is not applicable to the 2-naphthyl substituted diketone.29 Although these issues may be addressed by the use of a chiral cobalt catalyst,30 the reproducibility has been questioned.31
Scheme 3.

Noting the inability to install a 2-naphthyl moiety at the 2- and 5-positions of the pyrrolidine in a stereoselective manner, an approach based on sequential allylic amination and ring-closing metathesis (RCM) has been introduced (route b).31 Mild, albeit specialized, conditions are needed for the reduction of 2,5-diaryl-2,5-dihydro-1H-pyrrole to avoid cleavage of allylic C–N bond. It is unclear if this route can be generalized for other aromatic variants as only one example is reported.
The third disconnection is based on the α-arylation of protected pyrrolidine, which consists of three transformations: enantioselective deprotonation, transmetalation, and sp2–sp3 coupling (route c).32 This strategy has been applied to the preparation of a number of chiral phosphoramidite ligands.33
Although the most expedient approach to build up pyrrolidine ring is through [3 + 2]-cycloaddition (route d and e), a reliable and stereoselective method for the preparation of 2,5-diaryl substituted analogues has not been developed.34
This analysis suggests that α-arylation (route c) is the only approach that has demonstrated generality for the synthesis of nonracemic 2,5-diarylpyrrolidines.33 However, the critical bis-hydrazone ligand precursor, 1-amino-2,5-diarylpyrrolidine 8 cannot be obtained directly; therefore, this intermediate needs to be accessed through one of the three following methods: Hofmann-type rearrangement of the 1-carbamoylpyrrolidine,35 direct amination of pyrrolidine,36 or reduction of 1-nitrosopyrrolidine (Scheme 4).37
Scheme 4.

Because of the short synthesis by route (a) and the generality of α-arylation by route (c), these two synthetic plans were investigated for the preparation of chiral glyoxal bis-hydrazone ligands with 2,5-diarylpyrrolidine substituents.
3.1.1.1. Ligand Preparation via Corey–Itsuno Reduction of 1,4-Diketones
The investigation of route (a) began with Corey–Itsuno reduction of 1,4-diaryl-1,4-butanedione 9 (Table 1, method A).38 Guided by the protocol reported by Steel et al.,29 the preparation of chiral diols 10 with various aromatic substituents was evaluated. Excellent enantioselectivities were observed for electron-rich and moderately electron-deficient substrates, whereas the diastereoselectivities were moderate (entries 1–3). The results for 3,5-bis(trifluoromethyl)phenyl variant 9d were less satisfactory in both categories (entry 4). The best diastereoselectivity was obtained for the reduction of diphenyl dione 9e (entry 5).29
Table 1. Stereoselective Reduction of 1,4-Diaryl-1,4-butanediones 9.

| method A |
method B |
|||||
|---|---|---|---|---|---|---|
| entry | diol | R | dra | era | dra,b | era |
| 1 | 10a | 4-MeOC6H4 | 78:22 | >99:1 | 93:7, [99:1] | 99:1 |
| 2 | 10b | 4-t-BuC6H4 | 80:20 | 99:1 | 90:10, [87:13] | 99:1 |
| 3 | 10c | 4-F3CC6H4 | 84:16 | >99:1 | 89:11, [98:2] | >99:1 |
| 4 | 10d | 3,5-(F3C)2C6H3 | 63:37c | 87:13c | 79:21, [>99:1]c | >99:1c |
| 5 | 10e | Ph | 91:9 | >99:1 | n/a, n/a | n/a |
Diastereomeric ratios (chiral:meso) and enantiomeric ratios were determined by CSP-SFC.
The values in brackets are ratios after trituration or recrystallization.
Determined through dibenzoate derivative.
In view of the moderate diastereoselectivities, a modified protocol that employs tin(II) chloride and sodium borohydride was evaluated for the same set of diketones (Table 1, method B).39 Despite the higher reaction temperature, excellent enantioselectivities were maintained and higher diastereoselectivities were uniformly observed. Notably, electron-deficient diol 10d was obtained in greater than 99:1 enantiopurity and the diastereoselectivity was improved from 63:37 to 79:21 (entry 4). In general, the dr could be upgraded to at least 98:2 by recrystallization except in the case of diol 10b, in which the meso isomer was more crystalline. Analysis of the mother liquor showed a dr of 95:5.
With the 1,4-diols 10a, 10c ,and 10d available in high diastereo- and enantiomeric purities, their activation for subsequent reaction with hydrazine was studied (Scheme 5). Electron-rich dimesylate 11a could not be obtained because of polymerization. Partial decomposition and epimerization of dimesylate 11b were observed,40 whereas electron-deficient mesylate 11c was isolated in crystalline form and 11d was obtained as an oil. Displacement of the latter two dimesylates with hydrazine occurred smoothly to afford 1-aminopyrrolidines 8c and 8d. These compounds appeared to be oxygen sensitive and unstable to storage at ambient temperature, and were used shortly after their preparation. The condensation of 8c and 8d with glyoxal was carried out in two separate steps to provide the electron-deficient bis-hydrazone ligands 13c and 13d. A slightly elevated temperature was required to ensure condensation of 12d in a timely fashion. Malondialdehyde-derived ligand 15 was also prepared using this route.
Scheme 5.

3.1.1.2. Ligand Preparation via α-Arylation of N-Boc-pyrrolidine
In view of the inability to reduce 1,4-bis(2-naphthyl)-1,4-dione29 and the instability of electron-rich dimesylates 11a and 11b, a new synthetic approach to a broader range of bis-hydrazone ligands was needed. The preparation of ligands with monoaryl substituted pyrrolidine was studied initially to probe the accessibility of the 1-aminopyrrolidines from the parent heterocycle (Scheme 4). The investigation began with 4-methoxyphenyl- and 2-naphthyl-substituted N-Boc-(R)-2-arylpyrrolidines 16a and 16f (Scheme 6). These substrates were prepared by α-arylation of N-Boc-pyrrolidine through a sequence of enantioselective deprotonation mediated by (−)-sparteine, transmetalation to zinc chloride, and palladium-catalyzed sp3–sp2 coupling of aromatic bromides.32,33 The Boc protecting group was easily removed from 16f using an excess of trifluoroacetic acid. Electron-rich substrate 16a decomposed under these conditions but could be deprotected by treatment with a slight excess of iodotrimethylsilane at 0 °C to afford the deprotected 2-arylpyrrolidine 17a in 93% yield.41 Nitrosation of 17a with nitrosonium tetrafluoroborate42 took place in 1.5 h at 0 °C, but a higher temperature was required for the 2-naphthyl analogue 17f. The enantiomeric purity of 1-nitroso-2-arylpyrrolidine can be confirmed at this stage by CSP-SFC analysis; 16a of 92:8 er was converted to 18a of 91:9 er. The enantiomeric purity of 16a was improved to 96:4 by careful control of the reaction temperature of α-arylation during the deprotonation step. The reduction of 1-nitrosopyrrolidines 18a and 18f by LiAlH4 in THF proceeded smoothly at 0 °C. The two-step condensation protocol with glyoxal afforded the target bis-hydrazone ligands 21a and 21f.
Scheme 6.

With the successful preparation of bis-hydrazones 21a and 21f, the focus next shifted to analogues featuring C2-symmetric 2,5-diarylpyrrolidine moieties. The α-arylation protocol developed by Campos et al.32 and subsequently modified by Trost et al.33 was adopted in two consecutive steps from N-Boc-pyrrolidine to provide N-Boc-(2R,5R)-diarylpyrrolidines 22 (Table 2).
Table 2. Preparation of N-Boc-2-arylpyrrolidine 16 and N-Boc-2,5-diarylpyrrolidine 22.

| entry | R | refa | yieldb (%) | erc | refa | yieldd (%) |
|---|---|---|---|---|---|---|
| 1 | 4-MeOC6H4 | 16a | 71 | 96:4 | 22a | 18 |
| 2 | 2-naphthyl | 16f | 61 | 96:4 | 22f | 37g |
| 3 | 3,5-Me2C6H3 | 16g | 56 | 96:4 | 22g | 21 |
| 4 | 3,5-Ph2C6H3 | 16h | 61 | 96:4 | 22h | 18 |
| 5 | 2-tolyl | 16i | 54 | 95:5 | 22i | 17 |
| 6 | 5-Me-2-thienyl | 16j | ∼47e | 95:5 | 22j | 13 |
| 7 | 3,5-(F3C)2C6H3 | 16d | 55f | 96:4f | 22d | trace |
| 8 | 1-naphthyl | 16k | 48 | 96:4 | 22k | 0 |
Reference number for compound.
Isolated yield after column chromatography and Kugelrohr distillation if necessary.
Determined by CSP-SFC.
Isolated yield after column chromatography and recrystallization if necessary.
Cannot completely purify by chromatography and Kugelrohr distillation.
Determined through 3,5-dinitrobenzoate derivative.
Contains about 6% of the corresponding pyrroline.
In general, the N-Boc-(R)-2-arylpyrrolidines 16 were obtained in acceptable yields considering that three transformations are involved in each step. The enantioselectivity of this process is high, and ratios of 95:5 to 96:4 were achieved reproducibly.
The second arylation was significantly more challenging, and N-Boc-(2R,5R)-2,5-diarylpyrrolidines 22 were obtained in less satisfactory yields (Table 2). A significant amount of the starting material was not consumed. For example, 16a was recovered in 64% yield. The low yields were also attributed to the difficulty in purification from unconsumed 16 or removal of a small amount of the corresponding dehydropyrrolidine. Nevertheless, this procedure provided a range of Boc-protected diarylpyrrolidines 22 without a detectable amount of the meso-diastereomer. The second α-arylation could not provide 1-naphthyl- and 3,5-bis(trifluoromethyl)phenyl-substituted pyrrolidines 22d and 22k (entries 7 and 8).
With a number of N-Boc pyrrolidines 22 in hand, the stage was set to reveal the unprotected C2-symmetric diarylpyrrolidines 23. Standard reaction conditions using 20 equiv of TFA afforded 23f, 23g, and 23h in 81–93% yield (Table 3, entries 2–4). The reaction temperature and time required were dependent on the bulk of the aromatic group. The TMSI-assisted deprotection was applied to substrates that were incompatible with acid-promoted deprotection (entries 1 and 5). The reaction was slower for hindered 2-tolyl substituted 22i and was conducted for 2 h. For 5-methyl-2-thienyl-substituted pyrrolidine 22j, condition A also failed to provide the desired 2,5-diarylpyrrolidine 23j cleanly. A number of other deprotection conditions including HCl/Et2O, Ce(NH4)2(NO3)6, ZnBr2, or BF3Et2O were also not fruitful.43 Eventually, heat-promoted decarboalkoxylation (200 °C) allowed the isolation of desired product, although further optimization of the reaction time is required to improve the product yield.
Table 3. Preparation of 2,5-Diarylpyrrolidine 23 and 1-Nitroso-2,5-diarylpyrrolidine 24.

| entry | R | refa | temp (°C) | time (h) | yieldb (%) | refa | temp (°C) | time (h) | yieldb (%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 4-MeOC6H4 | 23a | 0c | 1 | 82 | 24a | 0 | 2 | 96 |
| 2 | 2-naphthyl | 23f | 0d | 6 | 81 | 24f | 22 | 2 | 92 |
| 3 | 3,5-Me2C6H3 | 23g | 0d | 2 | 86 | 24g | 22 | 20 | 96 |
| 4 | 3,5-Ph2C6H3 | 23h | 0d | 6 | 93 | 24h | 22 | 12 | 93 |
| 5 | 2-tolyl | 23i | 0c | 2 | 97f | 24i | 22 | 12 | 95 |
| 6 | 5-Me-2-thienyl | 23j | 200e | 18 | ∼64g | 24j | 0 | 0.25 | 57h |
Reference number for compound.
Yield of chromatographed product.
Reaction condition A (TMSI) was used.
Reaction condition B (TFA) was used.
Boc group was removed by heating at 200 °C under argon.
Yield of crude product.
Cannot completely purify by chromatography.
Yield after two steps.
An excess amount of nitrosonium tetrafluoroborate (2 equiv) was used in the nitrosation step (Table 3) because of the increased steric demands from two flanking aromatic substituents in 23 compared to one in 2-arylpyrrolidine 17. For the relatively unhindered bis(4-methoxyphenyl)pyrrolidine 23a, nitrosation proceeded at 0 °C in 2 h to provide 24a. In contrast, full consumption for the 2-naphthyl substituted analogue 23f was achieved at 22 °C. The bulkier substrates required extended reaction times (12–20 h) (entries 3–5). In general, the desired nitroso-2,5-diarylpyrrolidines 24 were obtained in excellent yields.
The reduction of nitroso group was particularly challenging. Unlike the clean reduction of the monoaryl 1-nitrosopyrrolidines 18a and 18f by LiAlH4 (Scheme 6), no reaction was observed for nitroso-2,5-di(2-naphthyl)pyrrolidine 24f at 0 °C, and extensive decomposition occurred at 22 °C. Other reagents known to effect N–N bond cleavage (titanium trichloride,44 zinc/acetic acid, sodium borohydride, borane or LiEt3BH) did not yield any (diarylamino)pyrrolidine 8f.
The most promising results were obtained using DIBAL-H in dichloromethane for 2 h.45 Longer reaction times led to a decrease of desired product 8f and an increase in the amount of byproducts 1,2-di(2-naphthyl)cyclobutane 25 and 2-ethenylnaphthalene 26. Doubling the amount of the reductant marginally improved the conversion but complicated the workup because of greater amount of gelatinous aluminate. The conversion of 24f in THF was slower and gave 25 as the dominant product. No N–N cleavage product 23f was observed in any case.
Although a perfect solution was not found for the preparation of (2,5-diarylamino)pyrrolidine 8,46 DIBAL-H reduction of 2,5-diaryl-1-nitrosopyrrolidines delivered the most promising results. Optimization of this route involved stopping the reaction at 2 h to minimize decomposition of the product 8, and the unconsumed nitrosopyrrolidine 24 can be recycled (Table 4). For example, reduction of 24f with DIBAL-H at ambient temperature in methylene chloride afforded a 51% yield of the desired aminopyrrolidine 8f (Table 4, entry 2). The unconsumed 24f was recovered in 38% yield and was subjected to a second round of reduction (52%). Other (diarylamino)pyrrolidines were prepared successfully with the same procedur procedure except for 2-tolyl analogue 8i. The two-step condensation proceeded well in general at room temperature, affording various chiral bis-hydrazone ligands 13 featuring the diaryl-substituted C2-symmetric pyrrolidine scaffold.
Table 4. Preparation of 2,5-Diarylpyrrolidine-Based Bis-hydrazone Ligands 13 from 1-Nitroso- 2,5-diarylpyrrolidine 24.
| entry | R | refa | yieldb (%) | refa | yieldb (%) | refa | yieldb (%) |
|---|---|---|---|---|---|---|---|
| 1 | 4-MeOC6H4 | 8a | 47 | 12a | 99 | 13a | 84 |
| 2 | 2-naphthyl | 8f | 51 | 12f | 94 | 13f | 87 |
| 3 | 3,5-Me2C6H3 | 8g | 58 | 12g | 84 | 13g | 83 |
| 4 | 3,5-Ph2C6H3 | 8h | 43 | 12h | ∼87d | 13h | 59e |
| 5 | 2-tolyl | 8i | 78c | 12i | ∼88d | 13i | 43 |
| 6 | 5-Me-2-thienyl | 8j | 47 | 12j | 99 | 13j | 82f |
Reference number for compound.
Yield of chromatographed product.
Estimated yield; reduction was carried out by a Zn/HCl protocol; see Scheme 7.
A small amount of hexanes adsorbed.
8h partially decomposed and so 12h was not consumed.
Reaction time was 21 h.
The reduction of 2,5-di(2-tolyl)-1-nitrosopyrrolidine 24i could not be effected by either DIBALH or LiAlH4. Fortunately, the reduction with Zn/HCl provided the 22i as the major product accompanied by over-reduced diarylpyrrolidine 23i (83:17) (Scheme 7). Because of the difficulty in separating these amines, this mixture was subjected directly to the stepwise condensation with glyoxal to furnish bis-hydrazone 13i.
Scheme 7.

In summary, synthetic route (a) (Scheme 3) allowed the preparation of bis-hydrazone ligands 5, 13c, 13d, and 15 with electron-deficient and -neutral aromatic substituents. The scope was complemented and expanded to include electron-rich and sterically hindered analogues 13a,f–i via synthetic route (c).
3.1.1.3. (R,R)-Bis(α-methylbenzyl)amine-Derived Glyoxal Bis-hydrazone Ligand
The preparation of acyclic bis-hydrazone 29 (Scheme 8) commenced with nitrosation of commercially available (R)-bis((R)-1-phenylethyl)amine.47 Sodium–metal reduction of nitrosamine 27 in ethanol at reflux afforded the desired 1,1-bis((R)-1-phenylethyl)hydrazine and the over-reduced product in 84:16 ratio.47 This mixture was subjected to a substoichiometric amount of glyoxal to provide chiral bis-hydrazone 29 in a one-step preparation.
Scheme 8.

3.1.1.4. Binaphthalene-Based Glyoxal Bis-hydrazone Ligands
Inspired by the structural feature of binaphthalene-based phosphine ligands such as BINAP, a novel type of bis-hydrazone ligand was proposed (Scheme 9). Adopting the reported synthetic route for Maruoka’s phase-transfer catalysts, hydrazones 33a, 33b, and 34 were prepared.48 Double displacement of the dibromide 30 with excess hydrazine provided aminoazepine 31. Stepwise condensation of 31 with glyoxal completed the synthesis of 33. The exclusion of oxygen is important for the second condensation to minimize decomposition of aminoazepine 31. The reaction of 32a with picolinaldehyde yielded hydrazone/pyridine hybrid ligand 34.
Scheme 9.

3.2. Structure–Activity and Structure–Selectivity Relationship Studies
With a number of chiral bis-hydrazone ligands in hand, the study of asymmetric biaryl coupling of aryldimethylsilanolates was initiated. The optimal reaction conditions for ligand survey was established using bis-hydrazone 5 bearing two (2S,5S)-diphenylpyrrolidine units (Table 5). Good conversion and enantioselectivity were observed at 90 °C in 1 h (entry 1). Lowering the reaction temperature improved the selectivity at the expense of conversion, except at 70 °C (entries 2–4). The loading of silanolate K+35a– was decreased to 1.5 equiv without detrimental impact on the product yield (entry 6). Doubling the ligand loading did not improve the enantioselectivity (entry 8). Although the use of (MeCN)2PdCl2 as palladium source did not affect the selectivity, the reaction was considerably slower (entry 9). The reaction in 1,4-dioxane was less efficient than in toluene in all aspects (entries 5 and 10). The optimal reaction conditions involved the use of 1.5 equiv of silanolate at 70 °C in toluene with 2.5 mol % of [allylPdCl]2 and 5 mol % of ligand 5 (entries 6 and 7).
Table 5. Reaction Optimization for the Cross-Coupling of 2-Methylnaphthylsilanolate (K+35a–) Using Bis-hydrazone Ligand 5.

| entry | K+35a– (equiv) | temp (°C) | time (h) | 37a (%) | 38b (%) | er, 37 (R/S)c |
|---|---|---|---|---|---|---|
| 1 | 1.75 | 90 | 1 | 85 | 2.4 | 92:8 |
| 2 | 1.75 | 70 | 1 | 85 | 2.3 | 95:5 |
| 3 | 1.75 | 50 | 4 | 71 | 3.4 | 96:4 |
| 4 | 1.75 | 21 | 7 d | 43 | 5.4 | 98:2 |
| 5d | 1.75 | 70 | 24 | 59 | 10 | 90:10 |
| 6 | 1.5 | 70 | 2 | 85 | 3.3 | 94:6 |
| 7 | 1.5 | 70 | 2 | 90e | 2.5 | 95:5 |
| 8f | 1.5 | 70 | 1 | 84 | 2.3 | 96:4 |
| 9g | 1.5 | 70 | 10 | 79 | 2.3 | 95:5 |
| 10d | 1.5 | 70 | 7.5 | 70 | 4.3 | 90:10 |
Yield of chromatographed product taking into account of 1,1′-binaphthalene.
Percentage of 1,1′-binaphthalene in the chromatographed product estimated by CSP-SFC.
Ratio of (R)-37:(S)-37 determined by CSP-SFC.
Reaction was conducted in 1,4-dioxane.
0.5 mmol scale.
10 mol % of ligand.
(MeCN)2PdCl2 (5 mol %) was used as the palladium source.
For the purpose of establishing structure–activity and −selectivity relationships (SAR and SSR) and further improving the enantioselectivity of the cross coupling, pyrrolidine-based bis-hydrazones 13 bearing various aromatic substituents were studied using the optimized reaction conditions (Table 6). Increasing the steric bulk of the aromatic group as in 13g and 13h had a negative impact on the yield and the enantiomeric purity of the coupled product (entries 2 and 3). In one extreme case, the 2-tolyl-substituted ligand 13i yielded a near-racemic mixture of 37 (entry 5). The use of 2-naphthyl- (13f) or 4-methoxy- (13a) substituted ligand restores the selectivity of the coupling process compared to that of the parent ligand (5) (cf. entries 6, 7, and 1). On the contrary, electron-deficient ligands 13c and 13d were less selective, and the reaction was significantly slower for 13d (entries 8 and 9). The cross-coupling stalled when 5-methyl-2-thienyl-substituted hydrazone 13j was used (entry 10). Replacing the palladium source with (MeCN)2PdCl2 did not affect the enantioselectivity but resulted in a sluggish reaction (cf. entries 4 and 3). The cross-coupling with the more hindered electrophile 1-bromo-2-methylnaphthalene was possible, although higher temperature was required (110 °C) to produced even a modest amount of 40, but gratifyingly with enantioselectivity in this preliminary experiment (entry 11).
Table 6. SAR and SSR Studies of Bis-hydrazone Ligands for the Coupling of 2-Methylnaphthylsilanolate (K+35a–).

| entry | ligand | configa | aryl | time (h) | product | yieldb (%) | yield, 38c (%) | product, er (R/S)d |
|---|---|---|---|---|---|---|---|---|
| 1 | 5 | S,S,S,S | Ph | 2 | 37 | 90e | 2.5 | 95:5 |
| 2 | 13g | R,R,R,R | 3,5-Me2C6H3 | 3 | 37 | 75 | 3.6 | 8:92 |
| 3 | 13h | R,R,R,R | 3,5-Ph2C6H3 | 3 | 37 | 73 | 4.1 | 13:87 |
| 4f | 13h | R,R,R,R | 3,5-Ph2C6H3 | 3 | 37 | 12 | 3.0 | 12:88 |
| 5 | 13i | R,R,R,R | 2-tolyl | 12 | 37 | 56 | 9.7 | 44:56 |
| 6 | 13f | R,R,R,R | 2-naphthyl | 2 | 37 | 86 | 1.4 | 5:95 |
| 7 | 13a | R,R,R,R | 4-MeOC6H4 | 2 | 37 | 81 | 2.0 | 6:94 |
| 8 | 13c | S,S,S,S | 4-F3CC6H4 | 2 | 37 | 81 | 3.8 | 90:10 |
| 9 | 13d | S,S,S,S | 3,5-(F3C)2C6H3 | 18 | 37 | 52 | 8.3 | 71:29 |
| 10 | 13j | R,R,R,R | 5-Me-thienyl | 7 | 37 | 45 | 5.8 | 20:80 |
| 11g | 5 | S,S,S,S | Ph | 19 | 40 | 24h | n/a | 92:8i |
| 12j | 5 | S,S,S,S | Ph | 7 | 37 | 72 | 1.2 | 95:5 |
Configuration of the bis-hydrazone ligand.
Yield of chromatographed product taking into account of 1,1′-binaphthalene.
Percentage of 1,1′-binaphthalene in the chromatographed product estimated by CSP-SFC.
Enantiomeric ratio of product determined by CSP-SFC.
0.5 mmol scale.
(MeCN)2PdCl2 (5 mol %) was used as the palladium source.
K+35a– and 39 were used in the experiment.
Yield of 2,2′-dimethyl-1,1′-binaphthalene; reaction was conducted at 110 °C.
The configuration was assigned based on the sense of induction for 2-methyl-1,1′-binaphthalene (R)-37 and optical rotation.
K+35b– and 39 were used in the experiment.
In an effort to probe the stereodetermining step, the donor/acceptor role of the two coupling substrates was reversed. The reaction was slower when 1-naphthyldimethylsilanolate K+35b– was used as the nucleophile and 1-bromo-2-methylnaphthalene was used as the electrophile (entry 12). Interestingly, the major enantiomer also possessed the (R)-configuration, and the enantiomeric ratios of the products from these reactions are the same (cf. entries 1 and 12).
Further SAR studies revealed that the presence of substituents at both the 2- and 5-positions on the pyrrolidine moiety was critical for high enantioselectivity (Table 7, entries 3 and 4). Bis-hydrazone 29, an analogue of 5 that lacks the C(3)–C(4) bond, was ineffective at inducing selective coupling (entry 5). The introduction of dimethylmethide linkage to the bis-hydrazone ligand was also detrimental; this reaction stalled after 2 h and a racemic product was obtained (entry 6). A novel type of bis-hydrazone ligand featuring the dinaphthylazepine backbone was also tested, which provided the biaryl product in 81:19 er (entry 7). Intriguingly, the reaction employing 3- and 3′-phenyl-substituted variant 33b favors the formation of the enantiomer (32:68 er), albeit with moderate yield after a prolonged reaction time (entry 8). The hydrazone–pyridine hybrid loses the ability to provide stereocontrol (entry 9).
Table 7. SAR and SSR Studies of Hydrazone Ligands for the Coupling of 2-Methylnaphthylsilanolate (K+35a–).

| entry | ligand | time (h) | 37a (%) | 38b (%) | er, 37 (R/S)c |
|---|---|---|---|---|---|
| 1 | 13a | 2 | 81 | 2.0 | 6:94 |
| 2 | 13f | 2 | 86 | 1.4 | 5:95 |
| 3 | 21a | 4 | 70 | 3.5 | 40:60 |
| 4 | 21f | 6 | 76 | 2.8 | 43:57 |
| 5 | 29 | 4 | 78 | 6.6 | 48:52 |
| 6 | 15 | 2d | ∼21e | 12.8 | 50:50 |
| 7 | 33a | 2 | 84 | 3.0 | 19:81 |
| 8 | 33b | 18 | 59f | 9.0 | 68:32 |
| 9 | 34 | 24 | 34 | 8.8 | 52:48 |
Yield of chromatographed product taking into account of 1,1′-binaphthalene.
Percentage of 1,1′-binaphthalene in the chromatographed product estimated by CSP-SFC.
Ratio of (R)-37:(S)-37 determined by CSP-SFC.
Reaction stalled.
Contains a contaminant.
0.1 mmol scale.
To evaluate the effect of the substituent next to the silanolate on the rate and selectivity of the coupling, the reaction of a more electron-rich, but less sterically hindered nucleophile, 2-methoxynaphthylsilanolate (K+35c–) was investigated (Table 8). Under standard conditions, the progress of the reaction was indicated by the observable changes in the physical state of the reaction mixture. Potassium silanolate K+35c–, has limited solubility in toluene even at elevated temperature (70 °C). The gradual consumption of this nucleophile was manifest by its dissolution and the darkening of the reaction mixture. Using 5, biaryl product 41 was obtained in 76% yield (GC) and 79:21 er (entry 1). Although silanolate K+35c– was fully converted in 3 h (GC analysis), a small amount of 1-bromonaphthlene was not consumed. Further purification of the product to remove siloxanes led to a slightly enhanced er (entry 2). Under “ligandless” conditions, a significant amount of the coupling product was also observed (56%) (entry 3). Whereas the employment of 2-naphthyl substituted ligand 13f marginally improved the enantioselectivity (entry 4), the reaction was slower. To improve the enantiomeric purity of the coupling product, bulkier ligands bearing 3,5-Me2C6H3 (13g), 3,5-Ph2C6H3 (13h), and 2-tolyl (13i) substituents were examined (entries 5–7). Disappointingly, the results were less satisfactory in both yield and er. The use of (MeCN)2PdCl2 as an alternative palladium source improved the enantioselectivity, albeit at a significant expense of conversion (cf. entries 8 and 1).
Table 8. SAR and SSR Studies of Bis-hydrazone Ligands for the Coupling of 2-Methoxynaphthylsilanolate (K+35c–).

| entry | ligand | ligand config | aryl | time (h) | R1 | R2 | yielda (%) | er (S/R)b |
|---|---|---|---|---|---|---|---|---|
| 1 | 5 | S,S,S,S | Ph | 3 | OMe | H | 76 | 79:21 |
| 2 | 5 | S,S,S,S | Ph | 3 | OMe | H | 60c | 84:16 |
| 3 | none | 3 | OMe | H | 56 | |||
| 4 | 13f | R,R,R,R | 2-naphthyl | 3 | OMe | H | 53 | 16:84 |
| 5 | 13g | R,R,R,R | 3,5-Me2C6H3 | 3 | OMe | H | 61 | 31:69 |
| 6 | 13h | R,R,R,R | 3,5-Ph2C6H3 | 3 | OMe | H | 38 | 26:74 |
| 7 | 13i | R,R,R,R | 2-tolyl | 3 | OMe | H | 66 | 44:56 |
| 8d | 5 | S,S,S,S | Ph | 3 | OMe | H | 41 | 88:12 |
| 9 | 5 | S,S,S,S | Ph | 24 | H | OMe | 60c | 84:16 |
| 10 | 13f | R,R,R,R | 2-naphthyl | 24 | H | OMe | 46c | 18:82 |
Determined by GC using biphenyl as the internal standard.
Determined by CSP-SFC of chromatographed product.
Isolated yield on a 0.25 mmol scale.
(MeCN)2PdCl2 (5 mol %) was used as the palladium source.
To probe the stereodetermining step, the donor/acceptor role of the two coupling substrates was reversed (entries 9 and 10). The reactions were considerably slower; both 1-naphthyldimethylsilanolate K+35b– and 2-methoxy-1-bromonaphthalene were not consumed within 12 h. The reactions were quenched after 24 h, at which point no more silanol was detected. The major enantiomer had the same configuration as before (cf. entry 1 and 9, and entry 4 and 10), although the enantiomeric composition differed slightly.
4. Discussion
4.1. Effect of Ligand on the Rate and Selectivity of the Cross-Coupling
4.1.1. 2-Methylnaphthylsilanolate
During the initial stage of the ligand survey, a high temperature (110 °C) was found to be necessary for the coupling of 2-methylnaphthylsilanolate K+8a– when chiral bidentate phosphine ligands were used (see the Supporting Information for the initial ligand survey). This is not surprising since partial ligand dissociation is required to generate an empty coordination site for transmetalation. Consistent with this notion, the monodentate, MOP-type ligands allowed the couplings to be conducted at 90 °C. Good reactivities were also observed with diene and bis-hydrazone ligands at this temperature because of facile ligand-dissociation from palladium that originates from their weaker coordinating abilities compared to bisphosphines.49
In the initial reaction optimization using bis-hydrazone ligand 5, toluene was found to be a superior solvent than 1,4-dioxane with respect to reaction time, yield, and enantioselectivity (Table 5, entries 2 and 5). This observation may be rationalized by the ability of polar solvent to interact with palladium intermediates. The dioxane molecule could potentially occupy a coordination site competitively because of its high concentration. As a consequence, transmetalation is slower, and the chelation of the chiral hydrazone ligand is disrupted.
The choice of palladium source had a more dramatic impact on the rate of the reaction. The cross-coupling employing (MeCN)2PdCl2 proceeded slowly possibly because of the difficulty in double transmetalations from two aryldimethylsilanolates, which provides diarylpalladium complex necessary for the formation of active Pd(0) species by reductive elimination (entry 9). In contrast, Pd(0) is generated readily from [allylPdCl]2 by nucleophilic attack on the allyl moiety by the silanolate.50
The structure–activity relationship revealed that the use of bulkier bis-hydrazone ligands generally correlates with longer reaction times and lower product yields (Table 6). The increased steric encumbrance likely raises the energy barriers for both oxidative addition and transmetalation steps, thus resulting in a more sluggish reaction.
Further SAR analyses have revealed the critical features of bis-hydrazone ligands (Figure 4). Aromatic substituents at both the 2- and 5-positions of the pyrrolidine are necessary for high enantioselectivity. The rigidity of the pyrrolidine ring is required because no asymmetric induction was observed if the C(3)–C(4) linkage is missing. The 2,6-diphenylpiperidine analogue also provides lower selectivity.19 A five-membered palladacycle formed from the chelation by the bis-hydrazone ligand is a much superior catalyst than the six-membered analogue. The presence of each chiral hydrazone unit is important as demonstrated by the coupling using pyridine–hydrazone hybrid 34 (52:48 er) (Table 7, entry 9).
Figure 4.

Critical features of 2,5-diaryl-based bis-hydrazone ligands to facilitate high enantioselectivity in the catalytic, asymmetric biaryl coupling.
4.1.2. 2-Methoxynaphthylsilanolate
The selectivity and reactivity for the coupling of 2-methoxynaphthylsilanolate (K+35c–) mimic those of 2-methylnaphthylsilanolate (K+35a–), a bis-hydrazone ligand with a bulkier aromatic substituent than phenyl correlates with a lower enantioselectivity and product yield. Nevertheless, further discussion can be made by scrutinizing the data in Table 8.
The results from entries 1–3 warrant comment. First, the enantiomeric purity of the chromatographed product (entry 1) is lower than that of the further purified product after removal of residual siloxanes (entry 2). This situation arises by accidental enantiomeric enrichment during purification. The solvent used to wash away siloxanes from the chromatographed product was analyzed, and the small amount of dissolved product showed a substantially lower er (33:67).
Second, a significant amount of background reaction was detected (entry 3). It is intriguing that coupling product was produced in substantial amount (56%) in the absence of hydrazone ligand. This phenomenon has been documented previously from these laboratories.50 Presumably, the product generated from the reduction of palladium(II) is noninnocent (Scheme 10). A small portion of the silanolate reacts with the allyl group of the allylpalladium chloride dimer, and the resulting silyl allyl ether 42 can, in principle, stabilize palladium(0) to some extent by acting as a π-ligand.51 Therefore, it should be noted that this is not a true “ligandless” condition.52 To support the above hypothesis, (MeCN)2PdCl2 was used in place of [allylPdCl]2 as the precatalyst to avoid formation of silyl allyl ether 42. Indeed, an increase of enantiomeric ratio of the biaryl product from 21:79 to 12:88 was observed (Table 8, entries 1 and 8).
Scheme 10.

Considering the extent of background reaction, it is remarkable that meaningful enantioenrichment can be observed when a phenyl- or naphthyl-substituted ligand is present (entries 1 and 4). This observation implies that palladium associates to the bis-hydrazone ligand much more strongly than to allyl silyl ether 42. However, such speculation may not be applied to the bulkier bis-hydrazone analogues in which lower enantioselectivities or near racemic biaryl products were obtained (entries 5–7). The increased steric bulk may hinder the coordination of these ligands to palladium effectively resulting in competitive background reaction.
The coupling of 2-methoxynaphthylsilanolate K+35c–, was more sluggish than the less electron-rich 2-methylnaphthylsilanolate K+35a– (Scheme 11). These results are somewhat surprising given that a Hammett study revealed an electron-rich nucleophile should accelerate the rate of the reaction by stabilizing a partial positive charge of a transition state.27b Additionally, the steric hindrance of a 2-methyl substituent should further retard the rate of transmetalation. This conundrum may be reconciled by the difference in the solubility of the silanolates. Whereas the 2-methyl-substituted silanolate K+35a– can be characterized in benzene-d6, the 2-methoxy congener K+35c– is almost completely insoluble, and its characterization was conducted in THF-d8. At 70 °C, silanolate K+35a– almost completely dissolved in toluene after 10 min, whereas silanolate K+35c– did not. Consequently, the lower concentration of silanolate in the solution may result in a slower reaction, because the activated transmetalation is the major pathway in the catalytic cycle and it is concentration dependent.27a Contrariwise, the minor, thermal transmetalation pathway does not depend on a second equivalent of the silanolate for the transfer of aryl group. The possibility of unproductive coordination of the methoxy group to palladium over π-coordination prior to transmetalation cannot be excluded (Scheme 11). The moderate yield of the biaryl product can potentially be improved by simply increasing the amount of silanolate K+35c– to ensure full consumption of the 1-bromonaphthalene in the cross-coupling reaction and to compensate for unproductive siloxane formation.
Scheme 11.

As has been noted previously for the role-reversal experiments, the reaction is significantly slower when the electron-donating group is on the aromatic bromide rather than on the aromatic silanolate (Table 8, cf. entries 1, 9, 4 and 10). This observation can be rationalized by examining the electronic and steric influence of the substituent on the catalytic cycle.27b Slower oxidative addition is expected for a more electron-rich aryl bromide such as 2-methoxy-1-bromonaphthalene compared to 1-bromonaphthalene.53 Additionally, the 2-methoxy substituent could engender some steric hindrance toward the site of C–Br bond breakage. These two factors can also manifest in the transmetalation step. Since the palladium(II) center is less electron-deficient but more sterically hindered, the transfer of aromatic moiety from the dimethylsilyl unit would be less efficient (Figure 5).
Figure 5.

Electronic and steric environments of the palladium(II) center of the pretransmetalation intermediate.
4.2. Stereodetermining Step
Preliminary mechanistic insight of the stereodetermining step is gained by the donor–acceptor reversal of the coupling partners (Scheme 12, eqs 1 and 2). Interestingly, 2-methyl-1,1′-binaphthalene is obtained as the (R)-configured isomer from both reactions with the same enantioselectivity (95:5 er). Further comparison with the results from the Suzuki-coupling (eq 3)19 indicates the stereodetermining step is also independent of the element of the transmetalating unit (Si vs B). This statement is also true for the coupling of 2-methoxynaphthyl and naphthyl moieties (see the Supporting Information). These observations strongly suggest a common intermediate and the same elementary step in the catalytic cycle is involved in the determination of product configuration.
Scheme 12.

Analysis of the proposed catalytic cycle (Scheme 13) reveals that Pd(II) complexes C and C′ are the common intermediates involved in the equations in Scheme 12; the aryl substituents on the palladium have no memory of their origins, either from the donor or the acceptor. In contrast, complexes A, A′ and B, B′ are directly associated with the identity of aryl bromide and transmetalating agent, respectively. On the basis of these preliminary findings, reductive elimination is hypothesized to be the stereodetermining step.
Scheme 13.

4.3. Origin of Enantioselectivity
To aid the understanding of the origin of enantioselectivity and the effects of ligand in the reductive elimination step, the possible isomers of the diarylpalladium(II) intermediates (C and C′, Scheme 13) and the reductive elimination transition states were computed using density functional theory (DFT). Reductive elimination from palladium(II) complexes, an important step in many cross-coupling reactions, has been studied thoroughly with computations.54 However, only a few computational studies on asymmetric biaryl coupling have been reported.17 In the present study, we investigated the origins of enantioselectivities with chiral bis-hydrazone ligands and the effects of substituents on the ligands. Calculations with (R,R,R,R)-5 were carried out first as the representative bis-hydrazone ligand. Geometry optimizations were performed with B3LYP55 and a mixed basis set of SDD for Pd and 6-31G(d) for other atoms. Single-point calculations were performed with M0656 and a mixed basis set of SDD for Pd and 6-311+G(d,p) and the SMD57 solvation model in toluene. All calculations were performed with Gaussian 09.58 This combination of methods has been widely tested in the studies of mechanisms and selectivities in various transition metal-catalyzed reactions.59,60 Computations employing a similar level of theory reproduced the experimental barrier of C–C reductive elimination from a phenylpalladium(II) N-heterocyclic carbene complex.61
The optimized geometry of the lowest energy conformer of the diarylpalladium-bishydrazone complex (5)PdRR′ (R = 1-naphthyl; R′ = 2-methyl-1-naphthyl) is shown in Figure 6 (see the Supporting Information for other higher energy isomers). For a clearer view of the spatial environment created by the ligand, the two front naphthyl moieties have been temporarily removed in Figure 6c. Because of the C2-symmetry of the ligand, the NW and SE quadrants are both occupied by phenyl groups on the ligand. On the contrary, empty spaces can be seen in the SW and NE quadrants. These chiral pockets are expected to dictate the orientation of the substrates. In the most stable conformer of the diarylpalladium intermediate, the two empty quadrants are occupied by the naphthyl rings, and the relatively less hindered MeC(2) and HC(2) substituents are positioned in the more congested quadrants. The two naphthyl groups are not aligned but are tilted in a propeller sense. The naphthyl B-rings, which are located on the unoccupied quadrants, are tilted toward the ligand, while the MeC(2) and HC(2) groups on the filled quadrants are tilted outward to avoid repulsions with the phenyl groups on the ligand.
Figure 6.

Optimized structure of the most stable conformer of the diarylpalladium intermediate with chiral ligand (R,R,R,R)-5: (a) top view; (b) side view; (c) side view with R and R′ groups omitted.
To facilitate discussion, a further-simplified picture is provided to depict the steric environment created by the ligand (red squares) and the position of the naphthyl group (letter B, Figure 6b). The shaded red block represents filled quadrant and the label B represents the location of naphthyl B-ring.
As was suggested by Fernández et al., arene–arene interaction may favor an alternative orientation of the two naphthyl groups with respect to the ligand such that the B-rings are in the same quadrants as the phenyl groups of the ligand.19 With this consideration, three more conformers of the diarylpalladium complexes can be formulated (Figure 7, B–D); one of which has two potential arene–arene interactions (B). These complexes are only about 1 kcal/mol less stable than the most stable isomer (5)PdRR′-A.
Figure 7.

Numbers of potential arene–arene interaction for diarylpalladium complexes. Gibbs free energies of the isomers are with respect to the most stable isomer (5)PdRR′-A.
Each of the four conformers of the diarylpalladium intermediate may lead to either (S)- or (R)-coupling product via the C–C bond forming transition state accompanied by either clockwise or counterclockwise conrotatory motion of both naphthyl groups about the aryl–Pd bond. Such conrotatory motion is necessary to avoid head-on collision between C(2)/(8) and C(2′)/(8′) substituents. All eight possible reductive elimination transition states were computed (Figure 8), four of which simulate clockwise conrotatory reductive elimination (TS-A, TS-B′, TS-C′, and TS-D), and the other four simulate counterclockwise conrotatory reductive elimination (TS-A′, TS-B, TS-C, TS-D′). In all transition states, the conformation of the bis-hydrazone ligand remains relatively rigid, with minimal changes from the intermediate complex (see Figure S1, Supporting Information, for an overlay of the intermediate and transition-state geometries). The relative energies of the transition states are determined by steric repulsions between the ligand and the aryl groups and between the C(2)/C(8) and C(2′)/C(8′) substituents about the forming aryl–aryl bond. Both types of steric repulsions are minimized in the most stable transition structure TS-A, which leads to the formation of biaryl (S)-37. The clockwise motion of the aryl substituents tilts the naphthyl B-rings toward the unoccupied NE and SW quadrants, away from the C(2)-Me and C(2′)-H groups. In contrast, transition structure TS-A′, which arises from counterclockwise motion of the aryl substituents and would lead to the (R)-product, has significantly higher energy. The counterclockwise motion in TS-A′ rotates the naphthyl B-rings toward the C(2)-Me and C(2′)-H groups, causing significant steric repulsion between Me-C(2)/HC(8′) (H–H distance of 1.99 Å) and HC(8)/HC(2′) (H–H distance of 1.86 Å). In TS-A′, the geometry of palladium is considerably distorted from square planar to minimize steric strain.
Figure 8.

Reductive elimination transition structures for diarylpalladium complex of phenyl substituted bis-hydrazone ligand (R,R,R,R)-5. Repulsions between the two naphthyl moieties are indicated by the distances between two atoms, when such distance is less than 80% of the sum of van der Waals radii. The H–H and H–C distances and the distances between the centroids of the B-rings are shown in black, green, and blue, respectively.
The second most stable transition structure TS-B has two B-rings at the NW and SE quadrants. The minor enantiomer (R)-product is expected from this transition state. The counterclockwise motion in TS-B rotates the naphthyl B-rings toward the occupied quadrants of the ligand. To minimize steric interaction between substrates and phenyl groups of the ligand, the palladium is again distorted from the perfect square-planar geometry. Although relieved from the substrate–ligand repulsion, the clockwise conrotatory motion implied by TS-B′ engenders significant steric strain between naphthyl B-rings and C(2)-Me/C(2′)-H groups observed similarly for TS-A′; the C–H distance between C(2)/H–C(8′) (2.26 Å) and the H–H between H–C(2′)/H–C(8) (1.86 Å) are both much shorter than the sum of the van der Waals radii (2.9 and 2.4 Å for C–H and H–H, respectively).
The two naphthyl moieties in transition structures TS-C, TS-C′, TS-D, and TS-D′ have a syn relationship. These structures all have unfavorable interaction between HC(8)/C(8′) or C(8)/HC(8′). Therefore, energies higher than those for TS-A and TS-B were found.
On the basis of the energy difference (2.9 kcal/mol) between the most stable transition structures TS-A and TS-B leading to the enantiomeric products, the predicted er (99:1) is comparable to observed er (95:5) at 70 °C.
The electron-deficient ligand 13c was similarly analyzed for the two lowest energy transition structures, TS-E and TS-F (Figure 9), which have the same conformation as TS-A and TS-B, respectively. This exercise revealed a smaller energy difference between the two transition structures (1.4 kcal/mol), which may be attributed to the increased arene–arene dispersion interaction between substrates (B-ring) and the more electron-deficient aromatic ligand (4-trifluoromethylphenyl) in TS-F. The distances between the centroids of the two π-systems are 3.96 and 4.47 Å in TS-F, noticeably shorter than the corresponding distances in the phenyl hydrazone-ligated TS-B (4.27 and 4.60 Å, respectively). See Figure S2 (Supporting Information) for an overlay of the geometries of TS-A/TS-E and TS-B/TS-F. These results are consistent with a stronger arene–arene interaction between electron-rich and electron-deficient π-system than between two electron-rich π-systems.62 In either case, a longer distance is measured between the π-systems located at the NW quadrant than at the SE because the 2-methyl substituent is repelled by the adjacent naphthyl moiety. The computed er (91:9) based on the energy difference between TS-E and TS-F closely approximates the observed er (90:10) at 70 °C.
Figure 9.

Transition structures with 4-(trifluoromethyl)phenyl-substituted bis-hydrazone ligand (R,R,R,R)-13c.
5. Outlook and Future Development
The modeling studies provided much insight into the origin and magnitude of enantioselectivity. Nevertheless, a few issues remained to be addressed. First, the conclusions from the calculations can be tested by using ligands such as 3,5-dimethyl- and 3,5-bis(trifluoromethyl)-substituted bis-hydrazones. Second, the interconversion barriers between diarylpalladium complexes A–D need to be estimated. High energy barriers would suggest that the reductive-elimination step does not determine the enantioselectivity and the product composition is a consequence of the ratio of A/B/C/D. Preliminary calculations at the PM6 semiempirical level indicates that interconversion is unlikely if the hydrazone ligand is bound to palladium in bidentate mode. However, interconversion may be possible if the ligand is bound in a monodentate mode leaving palladium with an empty coordination site.63 Circumstantial evidence indicates that conversion between diarylpalladium complexes A–D is a possibility through partial ligand dissociation.64 Importantly, the results from the donor/acceptor reversal experiments support reductive elimination as the stereodetermining step implying that diastereomeric complexes A–D are in equilibrium.
The empty coordination site required for the transmetalation event necessitates the partial dissociation of the bidentate ligand (Scheme 14). The use of tri-tert-butylphosphine in the preparative cross-coupling reactions26 ensures a tricoordinate palladium intermediate to facilitate transmetalation. Bidentate phosphine ligands, stronger chelating ligands than bis-hydrazones, were found to be generally less effective in the cross-coupling reaction of aryldimethylsilanolate. Transmetalation to a tetracoordinate palladium is unfavorable based on prior calculations.63 Therefore, an opportunity exists for the equilibration between diarylpalladium A–D through tricoordinate palladium after transmetalation and before reassociation of hydrazone ligand.
Scheme 14.

6. Conclusions
A variety of diarylpyrrolidine-based chiral bis-hydrazone ligands have been evaluated in the asymmetric cross-coupling reaction of aryldimethylsilanolates. Ligands with electron-rich/neutral and unhindered aromatic substituents on the 2,5-positions of the pyrrolidine ring generally correlate with higher enantioselectivities and reactivities. Preliminary mechanistic studies indicate that reductive elimination is likely the stereodetermining step. The interpretation of the origin of enantioselectivity has been facilitated by computational modeling. The lowest energy reductive elimination transition state features a propeller alignment of the aryl substituents, which tilts the naphthyl B-rings toward the unoccupied quadrants of the ligand. The arene–arene interaction has been identified as the potential culprit of lower selectivity observed for ligands with electron-deficient aromatic substituent. This result implies that eliminating the π-density on the ligand should enhance the enantioselectivity by raising the transition state energy toward the minor enantiomer.
7. Experimental Section
Stereoselective Reduction of 1,4-Diaryl-1,4-butanediones 9 (Table 1)
Asymmetric Reduction of 1,4-Diaryl-1,4-butanedione Using Borane Dimethyl Sulfide in the Presence of Methylborate Complex of (S)-(−)-α,α-Diphenyl-2-pyrrolidinemethanol19General Procedure 1

To an oven-dried Schlenk reaction flask equipped with a stir bar and a septum was added a methylborate complex of (S)-(−)-α,α-diphenyl-2-pyrrolidinemethanol (0.2 equiv), THF, and borane·dimethyl sulfide (2 equiv) under argon. A solution of 1,4-diarylbutane-1,4-dione (1 equiv) in THF was added by a syringe pump (0.5 mmol/h) at room temperature. After complete addition of the diketone, stirring was continued for 1–4 h. The reaction was quenched with MeOH, and the mixture was stirred until no bubbling was observed (5–10 min). The volatiles were evaporated, and the crude product was purified by column chromatography to provide 1,4-diarylbutane-1,4-diol. The diastereomeric and enantiomeric purities were analyzed by CSP-SFC analysis.
(1R,4R)-1,4-Bis(4-methoxyphenyl)butane-1,4-diol (10a) (Entry 1)
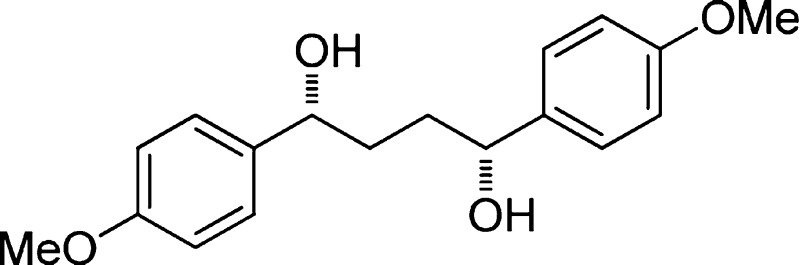
Following general procedure 1, a mixture of catalyst (7.8 mg, 26 μmol, 0.17 equiv), THF (0.16 mL), borane·dimethyl sulfide (28.5 μL, 0.3 mmol, 2 equiv), and a solution of 1,4-bis(4-methoxyphenyl)butane-1,4-dione (45 mg, 0.15 mmol, 1 equiv) in THF (3 mL) were combined in a 5 mL Schlenk reaction flask. Within 4 h, TLC showed complete consumption of the diketone. The reaction was quenched with MeOH (1.5 mL) and stirred for 5 min. Purification by column chromatography (SiO2, 1 × 8 cm, petroleum ether/EtOAc, gradient elution, 65/35, 50/50, then 0/100) afforded 36 mg (82%) of 10a as a white solid. The spectroscopic data matched those from the literature.20 Data for 10a: 1H NMR (500 MHz, CDCl3) 7.25 (d, J = 8.5 Hz, 4 H), 6.87 (d, J = 8.5 Hz, 4 H), 4.73–4.64 (m, 2 H), 3.80 (s, 6 H), 1.90–1.70 (m, 4 H); TLC Rf 0.13 (EtOAc/petroleum ether, 35/65) [silica gel, p-anisaldehyde]; SFC (S,S)-10a, tR 20.5 min (0.4); meso-10a, tR 25.8 min (22.1%); (R,R)-10a, 29.0 min (77.5%) (Chiralpak AD, 200 bar, 2 mg/mL, 10% MeOH in CO2, 2 mL/min, 220 nm, 40 °C).
(1R,4R)-1,4-Bis(4-tert-butylphenyl)butane-1,4-diol (10b) (Entry 2)

Following general procedure 1, a mixture of catalyst (6.2 mg, 20 μmol, 0.2 equiv), THF (0.25 mL), borane·dimethyl sulfide (19 μL, 0.2 mmol, 2 equiv), and a solution of 1,4-bis(4-tert-butylphenyl)butane-1,4-dione (35 mg, 0.1 mmol, 1 equiv) in THF (0.75 mL) were combined in a 5 mL Schlenk reaction flask. After 1 h, reaction was quenched with MeOH (1.5 mL), stirred for 5 min, and then concentrated. Purification by column chromatography (SiO2, 1 × 8.5 cm, petroleum ether/EtOAc, gradient elution, 65/35) afforded 29 mg (83%) of 10b as a white solid. The spectroscopic data matched those from the literature.21 Data for 10b: 1H NMR (500 MHz, CDCl3) 7.36 (d, J = 8.4 Hz, 4 H), 7.27 (d, J = 8.4 Hz, 4 H), 4.75–4.67 (m, 2 H), 2.37 (br s, 2 H), 1.99–1.79 (m, 4 H), 1.31 (s, 18 H); TLC Rf 0.22 (EtOAc/petroleum ether, 35/65) [silica gel, CAM]; SFC (S,S)-10b, tR 3.3 min (below detection limit); meso-10b, tR 4.2 min (20%); (R,R)-10b, 5.6 min (80%) (Chiralpak OB, 200 bar, 2 mg/mL, 10% MeOH in CO2, 2 mL/min, 220 nm, 40 °C).
(1R,4R)-1,4-Bis(4-(trifluoromethyl)phenyl)butane-1,4-diol (10c) (Entry 3)

Following general procedure 1, a mixture of catalyst (6.2 mg, 20 μmol, 0.2 equiv), THF (0.25 mL), borane·dimethyl sulfide (19 μL, 0.2 mmol, 2 equiv), and a solution of 1,4-bis(4-trifluoromethylphenyl)butane-1,4-dione (37 mg, 0.1 mmol, 1 equiv) in THF (0.75 mL) were combined in a 5 mL Schlenk reaction flask. After 4 h, the reaction was quenched with MeOH (2 mL), stirred for 5 min, and then concentrated. Purification by column chromatography (SiO2, 1 × 8 cm, petroleum ether/EtOAc, gradient elution, 65/35, 50/50, then 0/100) afforded 32 mg (85%) of 10c as a white solid. Data for 10c: 1H NMR (500 MHz, CDCl3) 7.60 (d, J = 8.2 Hz, 4 H), 7.45 (d, J = 8.1 Hz, 4 H), 4.82 (app br s, 2 H), 2.78 (d, J = 2.9 Hz, 2 H), 1.95–1.82 (m, 2 H, H2C(1)); TLC Rf 0.30 (EtOAc/petroleum ether, 1/1) [silica gel, CAM]; SFC (S,S)-10c, tR 9.2 min (below detection limit); (R,R)-10c, tR 10.2 min (84%); meso-10c, 11.7 min (26%) (Chiralpak AD, 200 bar, 3 mg/mL, 5% MeOH in CO2, 2 mL/min, 220 nm, 40 °C).
(1R,4R)-1,4-Bis(3,5-bis(trifluoromethyl)phenyl)butane-1,4-diol (10d) (Entry 4)

To an oven-dried, 5 mL, one-necked, round-bottom flask equipped with a stir bar and an argon inlet adaptor with a septum was added (S)-(−)-α,α-diphenyl-2-pyrrolidinemethanol (8.7 mg, 34 μmol, 0.17 equiv). After two cycles of evacuation/argon fill, THF (0.22 mL) and trimethyl borate (5.0 μL, 44 μmol, 0.22 equiv) were added. The colorless solution was stirred at room temperature for 1 h, and borane dimethyl sulfide (41 μL, 0.42 mmol, 2 equiv) was added. A solution of 1,4-bis(3,5-bis(trifluoromethyl)phenyl)butane-1,4-dione (102 mg, 0.2 mmol, 1 equiv) in THF (1 mL) was added at a rate of 0.5 mmol/h, and stirring was continued at 22 °C for 1 h (the diketone was not consumed). The reaction was cooled to 0 °C and quenched slowly with HCl (1 M, 1.5 mL). The aqueous phase was extracted with EtOAc (5 mL × 3), dried over Na2SO4, filtered, and concentrated. Purification by column chromatography (SiO2, 3 × 10 cm, CH2Cl2, then hexanes/EtOAc, 1/1) afforded 48 mg (47%) of 10d as a white solid. The diastereomeric and enantiomeric purities were analyzed through dibenzoate derivative. Data for 10d: 1H NMR (500 MHz, CD3OD) 7.92 (s, 4 H, HC(4)), 7.82 (s, 2 H, HC(6)), 4.91–2.83 (m, 2 H, HC(2)), 1.87–1.78 (m, 4 H, H2C(1)); TLC Rf 0.57 (hexanes/EtOAc, 1/1) [silica gel, KMnO4]; SFC (S,S)-10d, tR 4.5 min (9%); meso-10d, tR 5.5 min (27%); (R,R)-10d, 6.6 min (64%) ((R,R)-Whelk-O1, 200 bar, 2% MeOH in CO2, 2 mL/min, 220 nm, 40 °C).
Asymmetric Reduction of 1,4-Diaryl-1,4-butanedione Using Sodium Borohydride and Tin(II) Chloride in the Presence of (S)-(−)-α,α-Diphenyl-2-pyrrolidinemethanol (Table 1).20 General Procedure 2

To an oven-dried, one-necked, round-bottom flask equipped with a stir bar, a short reflux-condenser, and an argon inlet adaptor with a septum was added NaBH4 (2.4 equiv). After the flask was purged with argon, THF was added. Under a positive argon pressure and with rapid stirring, the septum was removed temporarily to allow the addition of SnCl2 (1.2 equiv), immediately resulting in gas evolution and the formation of a gray mixture. After the mixture was stirred at room temperature for 1 h, (S)-(−)-α,α-diphenyl-2-pyrrolidinemethanol (0.2 equiv) was added. The mixture was heated at reflux for 0.5 h, and a solution of 1,4-diaryl-1,4-butanedione (1 equiv) in THF was added by a syringe pump (0.5 mmol/h) under mild reflux. After complete addition of diketone, the reaction was stirred for another 10 min. The reaction was cooled to room temperature and then quenched with MeOH at 0 °C. After bubbling had subsided, the ice/water bath was removed, and stirring was continued at room temperature for 10 min. The gray solid was removed by filtration through a pad of Celite (2 cm deep, medium-porosity fritted funnel), eluted with Et2O or EtOAc, and concentrated to give the crude product. Purification by column chromatography, recrystallization, or trituration provided 1,4-diarylbutane-1,4-diol. The diastereomeric and enantiomeric purities were analyzed by CSP-SFC analysis.
(1R,4R)-1,4-Bis(4-methoxyphenyl)butane-1,4-diol (10a) (Entry 1)

Following general procedure 2, a mixture of NaBH4 (188 mg, 5.0 mmol, 2.4 equiv), THF (21 mL), SnCl2 (472 mg, 2.5 mmol, 1.2 equiv), (S)-(−)-α,α-diphenyl-2-pyrrolidinemethanol (106 mg, 0.41 mmol, 0.2 equiv), and a solution of 1,4-bis(4-methoxyphenyl)butane-1,4-dione (622 mg, 2.1 mmol, 1 equiv) in THF (41 mL) were combined in a 250 mL round-bottom flask equipped with a short reflux condenser. After complete addition of the diketone (0.5 mmol/h) at mild reflux, stirring was continued for 35 min. The reaction was cooled to 0 °C and quenched with MeOH (40 mL). The mixture was filtered and concentrated to give a white solid. Trituration of this solid with CH2Cl2 (∼4 mL) followed by addition of hexanes (∼15 mL) and filtration afforded 435 mg (69%) of 10a as a white solid. The spectroscopic data matched those from the literature.20 Data for 10a: 1H NMR (500 MHz, CDCl3) 7.25 (d, J = 8.5 Hz, 4 H), 6.87 (d, J = 8.5 Hz, 4 H), 4.73–4.64 (m, 2 H), 3.80 (s, 6 H), 1.90–1.70 (m, 4 H); TLC Rf 0.13 (EtOAc/petroleum ether, 35/65) [silica gel, p-anisaldehyde]; SFC Before trituration: (S,S)-10a, tR 20.5 min (below detection limit); meso-10a, tR 25.8 min (7%); (R,R)-10a, 29.0 min (93%). After trituration: (S,S)-10a, tR 20.5 min (below detection limit); meso-10a, tR 25.8 min (1%); (R,R)-10a, 29.0 min (99%) (Chiralpak AD, 200 bar, 2 mg/mL, 10% MeOH in CO2, 2 mL/min, 220 nm, 40 °C).
(1R,4R)-1,4-Bis(4-tert-butylphenyl)butane-1,4-diol (10b) (Entry 2)

Following general procedure 2, a mixture of NaBH4 (182 mg, 4.8 mmol, 2.4 equiv), THF (20 mL), SnCl2 (455 mg, 2.4 mmol, 1.2 equiv), (S)-(−)-α,α-diphenyl-2-pyrrolidinemethanol (101 mg, 0.4 mmol, 0.2 equiv), and a solution of 1,4-bis(4-tert-butylphenyl)butane-1,4-dione (703 mg, 2 mmol, 1 equiv) in THF (15 mL) were combined in a 100 mL round-bottom flask equipped with a short reflux condenser. After complete addition of the diketone (0.5 mmol/h) at mild reflux, stirring was continued for 30 min. The reaction was cooled to 0 °C and quenched with MeOH (30 mL). The mixture was filtered, concentrated, and purified by column chromatography (SiO2, 4 × 11 cm, petroleum ether/EtOAc, gradient elution, 65/35) to afford 651 mg (92%) of 10b as a white solid. Recrystallization from hexanes/CH2Cl2 (2.5/1) afforded 350 mg (49%) of 10b as a white solid. The spectroscopic data matched those from the literature.21 Data for 10b: 1H NMR (500 MHz, CDCl3) 7.36 (d, J = 8.4 Hz, 4 H), 7.27 (d, J = 8.4 Hz, 4 H), 4.75–4.67 (m, 2 H), 2.37 (br s, 2 H), 1.99–1.79 (m, 4 H), 1.31 (s, 18 H); TLC Rf 0.22 (EtOAc/petroleum ether, 35/65) [silica gel, CAM]; SFC before recrystallization: (S,S)-10b, tR 5.2 min (1%); meso-10b, tR 7.1 min (10%); (R,R)-10b, 9.8 min (89%), after recrystallization: (S,S)-10b, tR 5.2 min (below detection limit); meso-10b, tR 7.1 min (13%); (R,R)-10b, 9.8 min (87%) (Chiralpak OB, 200 bar, 2 mg/mL, 7.5% MeOH in CO2, 2 mL/min, 220 nm, 40 °C).
(1R, 4R)-1,4-Bis(4-(trifluoromethyl)phenyl)butane-1,4-diol (10c)

To an oven-dried, 100 mL, one-necked, round-bottom flask equipped with a stir bar, a short reflux-condenser, and an argon inlet adaptor with a septum was added sodium borohydride (182 mg, 4.8 mmol, 2.4 equiv). After three cycles of evacuation/argon fill, THF (18 mL) was added. Under a positive argon pressure and with rapid stirring, the septum was removed temporarily to allow the addition of tin(II) chloride (457 mg, 2.4 mmol, 1.2 equiv), immediately resulting in gas evolution and the formation of gray mixture. Additional THF (2 mL) was added to rinse all the reagents down the wall of the condenser. After the mixture was stirred at room temperature for 1 h, (S)-(−)-α,α-diphenyl-2-pyrrolidinemethanol (103 mg, 0.4 mmol, 0.2 equiv) was added. The mixture was heated to reflux for 0.5 h, and a solution of 1,4-bis(4-(trifluoromethyl)phenyl)butane-1,4-dione (749 mg, 2 mmol, 1 equiv) in THF (20 mL) was added by a syringe pump (0.5 mmol/h) under mild reflux. The reaction was stirred for another 0.5 h after complete addition of diketone. The reaction was cooled to room temperature and then quenched with MeOH (30 mL) at 0 °C. After bubbling subsided (∼5 min), the ice/water bath was removed and stirred at room temperature for 10 min. The gray solid was removed by filtration through a pad of Celite (2 cm deep, 30 mL size, medium-porosity fritted funnel), eluted with EtOAc (30 mL), concentrated to give 830 mg of 10c as a white solid (>99:1 er, 89:11 dr). Recrystallization from dichloromethane afforded 560 mg (74%) of 10c as colorless needles (>99:1 er, 98:2 dr). Data for 10c: 1H NMR (500 MHz, CD3OD) 7.49 (d, J = 8.2 Hz, 4 H, HC(5)), 7.39 (d, J = 8.2 Hz, 4 H, HC(4)), 4.62 (t, J = 4.5 Hz, 2 H, HC(2)), 1.80–1.69 (m, 2 H, H2C(1)), 1.67–1.58 (m, 2 H, H2C(1)); 13C NMR (126 MHz, CD3OD) 151.1 (C(3)), 130.3 (q, J = 32.0 Hz, C(6)), 126.1 (q, J = 3.9 Hz, C(5)), 125.8 (q, J = 270.6 Hz, C(7)), 74.0 (C(2)), 36.2 (C(1)); 19F NMR (470 MHz, CD3OD) −63.64; IR (Nujol) 3236 (m, broad), 1335 (s), 1166 (m), 1114 (s), 1085 (w), 1069 (m), 1040 (w), 1016 (w), 946 (w), 904 (w), 842 (w); MS (ESI) 517.0 (19, M + 2NaCl), 459.1 (28, M + NaCl), 401.1 (M + Na, 32), 361.1 (100), 343.1 (29), 254.2 (20), 236.1 (39); HRMS (ESI) calcd for C18H16O2F6Na 401.0952, found 401.0957; TLC Rf 0.16 (EtOAc/hexanes, 3/1) [silica gel, KMnO4]; [α]D24 +19.0 (c = 0.1, CHCl3); SFC Before recrystallization: (S,S)-10c, tR 9.2 min (below detection limit); (R,R)-10c, tR 10.2 min (89%); meso-10c, 11.7 min (11%). After recrystallization: (S,S)-10c, tR 9.2 min (below detection limit); (R,R)-10c, tR 10.2 min (98%); meso-10c, 11.7 min (2%) (Chiralpak AD, 200 bar, 3 mg/mL, 5% MeOH in CO2, 2 mL/min, 220 nm, 40 °C).
(1R, 4R)-1,4-Bis(3,5-bis(trifluoromethyl)phenyl)butane-1,4-diol (10d)

To an oven-dried, 250 mL, one-necked, round-bottom flask equipped with a stir bar, a short reflux-condenser, and an argon inlet adaptor with a septum was added sodium borohydride (217 mg, 5.64 mmol, 2.4 equiv). After three cycles of evacuation/argon fill, THF (24 mL) was added. Under a positive argon pressure and with rapid stirring, the septum was removed temporarily to allow the addition of tin(II) chloride (546 mg, 2.82 mmol, 1.2 equiv), immediately resulting in gas evolution and the formation of gray mixture. After the mixture was stirred at room temperature for 1 h, (S)-(−)-α,α-diphenyl-2-pyrrolidinemethanol (120 mg, 0.47 mmol, 0.2 equiv) was added. The mixture was heated to reflux for 0.5 h, and a solution of 1,4-bis(3,5-bis(trifluoromethyl)phenyl)butane-1,4-dione (1.20 g, 2.35 mmol, 1 equiv) in THF (18 mL) was added by a syringe pump (0.5 mmol/h) under mild reflux. The reaction was stirred for another 0.5 h after complete addition of diketone. The reaction was cooled to room temperature and then quenched with MeOH (40 mL) at 0 °C. After bubbling subsided (∼5 min), ice/water bath was removed and stirred at room temperature for 10 min. The gray solid was removed by filtration through a pad of Celite (2 cm deep, 30 mL size, medium-porosity fritted funnel), eluted with EtOAc (40 mL), concentrated. The white solid was taken up with EtOAc (∼5 mL) and filtered through a pad of silica (2 cm deep, 30 mL size, medium-porosity fritted funnel), eluted with EtOAc (80 mL), concentrated to give 10d as a white powder, 1.20 g (>99:1 er, 83:17 dr). Recrystallization twice from hexanes/dichloromethane (4/1, 0.04 g/mL then 0.03 g/mL, >99:1 er, 88:12 dr, 1.04 g), and once from hexanes/dichloromethane (3/1, 0.035 g/mL) afforded 490 mg (40%) of 10d as white needles (>99:1 er, >99:1 dr). Note that the third recrystallization was not cooled to 0 °C. Data for 10d: 1H NMR (500 MHz, CD3OD) 7.92 (s, 4 H, HC(4)), 7.82 (s, 2 H, HC(6)), 4.91–4.83 (m, 2 H, HC(2)), 1.87–1.78 (m, 4 H, H2C(1)); 13C NMR (126 MHz, CD3OD) 150.3 (C(3)), 132.6 (q, J = 33.2 Hz, C(5)), 127.4 (app d, J = 2.8 Hz, C(4)), 124.9 (q, J = 271.6 Hz, C(7)), 121.8 (m, C(6)), 72.9 (C(2)), 36.1 (C(1)); 19F NMR (470 MHz, CD3OD) −64.14; IR (Nujol) 3413 (w, broad), 3182 (w, broad), 1623 (w), 1347 (m), 1279 (s), 1170 (s), 1125 (s), 1040 (w), 904 (m), 844 (w), 710 (m), 682 (m); MS (ESI, no acid) 515.1 (M + H, 8), 497.1 (100), 479.1 (7); HRMS (ESI, no acid) calcd for C20H15O2F12 [M + H] 515.0880, found 515.0881; TLC Rf 0.74 (EtOAc/hexanes, 1/1) [silica gel, KMnO4]; [α]D24 +26.4 (c = 0.2, methanol); HPLC before recrystallization, (S,S)-10d, tR 5.6 min (below detection limit); meso-10d, tR 6.8 min (21%); (R,R)-10d, tR 7.6 min (79%), after recrystallization, (S,S)-10d, tR 5.6 min (below detection limit); meso-10d, tR 6.8 min (below detection limit); (R,R)-10d, tR 7.6 min (>99%) (Chiralpak AD-H, 200 bar, 1.5 mg/mL, 3.5% i-PrOH in hexanes, 1 mL/min, 22 °C)
Preparation of (2S,5S)-Diarylpyrrolidine-Based Bis-hydrazone Ligand by the Corey–Itsuno Reduction Route (Scheme 5)
(1R,4R)-1,4-Bis(methanesulfonyloxy)-1,4-bis(4-trifluoromethylphenyl)butane (11c)

An oven-dried, 50 mL, three-necked, round-bottom flask equipped with a stir bar, two septa, an argon inlet, and an internal temperature probe was evacuated and backfilled with argon (three cycles). Dichloromethane (2.6 mL) and methanesulfonyl chloride (210 μL, 2.7 mmol, 2.6 equiv) were added, and the flask was submerged to a −20 °C bath. A solution of (1R,4R)-1,4-bis(4-(trifluoromethyl)phenyl)butane-1,4-diol 10c (390 mg, 1.0 mmol, 1.0 equiv) in dichloromethane (2.6 mL) and triethylamine (435 μL, 3.1 mmol, 3.0 equiv) was cannulated to the methanesulfonyl chloride solution at a rate that the temperature did not exceed −10 °C. The resulting heterogeneous mixture was stirred at −20 °C for 1.5 h and then quenched with a saturated, aqueous solution of NH4Cl (4 mL) with vigorous stirring. The biphasic layers were poured into a separatory funnel containing EtOAc (20 mL), and the organic layer was washed sequentially with a 1:2:1 solution of H2O–brine-saturated NaHCO3 (7 mL × 4) and saturated NaHCO3 (7 mL × 2). The organic layer was dried over Na2SO4, filtered, and concentrated to the 7 mL mark. The solution was cooled to 0 °C, and hexanes (10 mL) was added dropwise to the flask with occasional swirling. After aging at 0 °C for 0.5 h, the colorless, fine crystals were collected by filtration and washed with hexanes (5 mL) to afford 352 mg (64%) of 11c. A second crop of the product was obtained by concentrating the mother liquor to the 7 mL mark, cooled to 0 °C for 5 min, filtered, and hexanes washed (5 mL) to afford 95 mg (17%) of 11c as a white powder. The product should not be dried under high vacuum because of its tendency to decompose and polymerize. Data for 11c: 1H NMR (500 MHz, C6D6) 7.25 (d, J = 8.1 Hz, 4 H, HC(5)), 7.09 (d, J = 8.1 Hz, 4 H, HC(4)), 5.76 (d, J = 10.4 Hz, 2 H, HC(2)), 1.90 (s, 6 H, HC(8)), 1.87–1.75 (m, 4 H, H2C(1)); 13C NMR (126 MHz, CDCl3) 142.2 (C(3)), 131.3 (q, J = 32.1 Hz, C(6)), 126.6 (C(4)), 126.1 (q, J = 3.9 Hz, C(5)), 123.7 (q, J = 271.4 Hz, C(7)), 81.1 (C(2)), 38.9 (C(8)), 33.1 (C(1)); 19F NMR (470 MHz, CDCl3) −62.91; MS (ESI) 557.0 (10, M + Na), 343.1 (100), 159.0 (18); HRMS (ESI) calcd for C20H20O6F6S2Na [M + Na] 557.0503, found 557.0499, calcd for C20H21O6F6S2 535.0684, found 535.0692.
(1R,4R)-1,4-Bis(methanesulfonyloxy)-1,4-bis(3,5-bis(trifluoromethyl)phenyl)butane (11d)

An oven-dried, 25 mL, three-necked, round-bottom flask equipped with a stir bar, two septa, an argon inlet, and an internal temperature probe was evacuated and backfilled with argon (three cycles). Dichloromethane (2.1 mL) and methanesulfonyl chloride (160 μL, 2.1 mmol, 2.6 equiv) were added, and the flask was submerged to a −20 °C bath. A solution of (1R,4R)-1,4-bis(3,5-bis(trifluoromethyl)phenyl)butane-1,4-diol 10d (400 mg, 0.77 mmol, 1.0 equiv) in dichloromethane (2.1 mL) and triethylamine (325 μL, 2.3 mmol, 3.0 equiv) was cannulated to the methanesulfonyl chloride solution at a rate that the temperature did not exceed −10 °C. The resulting heterogeneous mixture was stirred at −20 °C for 1.5 h and then quenched with a saturated, aqueous solution of NH4Cl (3.5 mL) with vigorous stirring. The biphasic layers were poured into a separatory funnel containing EtOAc (20 mL), and the organic layer was washed sequentially with a 1:2:1 solution of H2O–brine–saturated NaHCO3 (6 mL × 4) and saturated NaHCO3 (6 mL × 2). The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was freed from EtOAc by repeated azeotrope with hexanes (10 mL × 4) under reduced pressure. The pale yellow gum thus obtained was used without further purification. The crude product was not dried under high vacuum to avoid potential decomposition and polymerization. Data for 11d: 1H NMR (400 MHz, CDCl3) 7.90 (s, 6 H, HC(4) and HC(6)), 6.00–5.92 (m, 2 H, HC(2)), 2.95 (s, 6 H, HC(8)), 2.20–2.10 (m, 4 H, H2C(1)); 19F NMR (376 MHz, CDCl3) −63.33.
(2S,5S)-1-Amino-2,5-bis(4-trifluoromethylphenyl)pyrrolidine (8c)

To a 10 mL Schlenk reaction flask equipped with a stir bar and a septum was added (1R,4R)-1,4-bis(methanesulfonyloxy)-1,4-bis(4-trifluoromethylphenyl)butane 11c (396 mg, 0.74 mmol, 1.0 equiv). The flask was purged with argon, and 2-propanol (1.5 mL) was added to give a suspension. Anhydrous hydrazine (0.47 mL, 15 mmol, 20 equiv) was added, and the flask was submerged to a 40 °C oil bath. After being stirred at this temperature for 14 h, the suspension disappeared. The reaction was cooled to room temperature, and taken up with Et2O (30 mL). The mixture was washed with saturated NaHCO3 (10 mL × 2) and then with brine (10 mL). The organic layer was dried over Na2SO4, filtered, and concentrated to afford 249 mg (90%) of 8c as a white, waxy solid. Data for 8c: 1H NMR (500 MHz, CDCl3) 7.64 (d, J = 8.1 Hz, 4 H, HC(5)), 7.49 (d, J = 8.1 Hz, 4 H, HC(4)), 4.19 (t, J = 6.5 Hz, 2 H, HC(1)), 2.64–2.33 (m, 4 H, HC(2) and NH2), 2.12–1.94 (m, 2 H, HC(2)); 13C NMR (126 MHz, CDCl3) 145.5 (C(3)), 129.8 (q, J = 32.5 Hz, C(6)), 128.6 (C(4)), 125.4 (q, J = 3.8 Hz, C(5)), 124.1 (q, J = 272.5 Hz, C(7)), 69.0 (C(1)), 30.6 (C(2)); 19F NMR (470 MHz, CDCl3) −62.92; IR (CHCl3 film) 2954 (w), 2914 (w), 2815 (w), 1618 (m), 1594 (w), 1470 (w), 1421 (m), 1325 (s) 1167 (s), 1124 (s), 1067 (s), 1017 (m), 932 (m), 833 (m); MS (ESI) 375.1 (100, M + H), 358.1 (6); HRMS (ESI) calcd for C18H17N2F6 [M + H] 375.1296, found 375.1294; TLC Rf 0.14 (CH2Cl2/MeOH, 98/2) [silica gel, UV, KMnO4].
(2S,5S)- 1-Amino-2,5-bis(3,5-bis(trifluoromethyl)phenyl)pyrrolidine (8d)

To a 50 mL, round-bottom flask containing crude (1R,4R)-1,4-bis(methanesulfonyloxy)-1,4-bis(3,5-bis(trifluoromethyl)phenyl)butane 11d obtained above was added 2-propanol (7.7 mL) and swirled to give a homogeneous and colorless solution. A stir bar was added, and an argon inlet adaptor equipped with a septum was attached. The flask was purged with argon, and anhydrous hydrazine (0.48 mL, 15 mmol, 20 equiv) was added. The flask was submerged to a 40 °C oil bath and stirred for 14 h. The reaction was cooled to room temperature and taken up with Et2O (60 mL). The mixture was washed with saturated NaHCO3 (20 mL × 2), then with brine (20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated. Purification by column chromatography (SiO2, 3.0 × 16 cm, hexanes/CH2Cl, 1/1) afforded 313 mg (80%, two steps from 10d) of 8d as a viscous, colorless oil. Data for 8d: 1H NMR (500 MHz, CDCl3) 7.84 (s, 2 H, HC(6)), 7.83 (s, 4 H, HC(4)), 4.27 (dd, J = 7.1, 4.5 Hz, 2 H, HC(1)), 2.68–2.56 (m, 2 H, H2C(2)), 2.55 (s, 2 H, NH2), 2.10–2.01 (m, 2 H, H2C(2)); 13C NMR (126 MHz, CDCl3) 143.9 (C(3)), 131.9 (q, J = 33.3 Hz, C(5)), 128.3 (app d, J = 4.0 Hz, C(4)), 123.3 (q, J = 272.9 Hz, C(7)), 121.7 (m, C(6)), 68.8 (C(1)), 30.5 (C(2)); 19F NMR (470 MHz, CDCl3) −63.19; IR (neat) 1322 (w), 2962 (m), 2926 (m), 2871 (w), 2794 (w), 1810 (w), 1623 (m), 1465 (m), 1377 (s), 1353 (s), 1321 (m), 1279 (s), 1129 (s), 1033 (w), 994 (w), 896 (s), 843 (s), 792 (w), 708 (s), 682 (s); MS (ESI) 511.1 (100); HRMS (ESI) calcd for C20H15N2F12 [M + H] 511.1044, found 511.1036; TLC Rf 0.16 (hexanes/CH2Cl2, 1/1) [silica gel, PMA].
(2S,5S)-(2,5-Bis(4-trifluoromethylphenyl)pyrrolidine)-N-iminoacetaldehyde (12c)

To a 25 mL, one-necked, round-bottom flask equipped with a stir bar was added an aqueous solution of glyoxal (40% w/w, 720 μL, 6.3 mmol, 20 equiv). A solution of N-amino-(2S,5S)-2,5-bis(4-trifluoromethylphenyl)pyrrolidine 8c (118 mg, 0.32 mmol, 1.0 equiv) in THF (4.2 mL) was added at 0 °C. The ice/water bath was removed, and the flask was capped with a glass stopper. After the mixture was stirred at 22 °C for 1 h, THF was evaporated under reduced pressure. The residue was taken up by dichloromethane (10 mL) and basified with saturated NaHCO3 (15 mL) in a separatory funnel. The organic layer was saved, and the aqueous layer was extracted further with dichloromethane (10 mL × 2). The combined organic extract was dried over Na2SO4, filtered, and concentrated to give a very light yellow oil. Purification by column chromatography (SiO2, 1.5 × 16 cm, CH2Cl2 with 1% Et3N) afforded 111 mg (85%) of 12c as a white foam. Data for 12c: 1H NMR (500 MHz, C6D6, 60 °C) 9.49 (d, J = 7.5 Hz, 1 H, HC(9)), 7.39 (d, J = 8.0 Hz, 4 H, HC(5)), 6.71 (d, J = 7.9 Hz, 4 H, HC(4)), 6.35 (app d, J = 7.4 Hz, 1 H, HC(8)), 4.44 (app br s, 2 H, HC(1)), 1.85–1.75 (m, 2 H, HC(2)), 1.25–1.13 (m, 2 H, HC(2)); 13C NMR (126 MHz, C6D6, 60 °C) 188.9 (C(9)), 144.9 (C(3), broad), 134.0 (C(8)), 130.2 (q, J = 32.2 Hz, C(6)), 126.6 (C(4)), 126.0 (d, J = 3.9 Hz, C(5)), 124.7 (q, J = 272.4 Hz, C(7)), 65.4 (C(1), broad), 30.9 (C(2)); 19F NMR (470 MHz, CDCl3) −62.60. IR (CH2Cl2 film) 3052 (w), 2983 (w), 2940 (w), 2815 (w), 1923 (w), 1673 (s), 1620 (m), 1529 (s), 1450 (w), 1417 (m), 1388 (s), 1324 (s), 1247 (m), 1210 (w), 1128 (s), 1067 (s), 1016 (s), 896 (w), 837 (s), 765 (w), 737 (w), 715 (w); MS (ESI) 415.1 (100, M + H); HRMS (ESI) calcd for C20H17N2OF6 [M + H] 415.1245, found 415.1238; TLC Rf 0.37 (CH2Cl2 with 1% Et3N) [silica gel, UV, KMnO4].
(2S,5S)-(2,5-Bis(3,5-bis(trifluoromethyl)phenyl)pyrrolidine)-N-iminoacetaldehyde (12d)

To a 10 mL, one-necked, round-bottom flask equipped with a stir bar was added an aqueous solution of glyoxal (40% w/w, 370 μL, 3.2 mmol, 20 equiv). A solution of N-amino-(2S,5S)-2,5-bis(3,5-bis(trifluoromethyl)phenyl)pyrrolidine 8d (82 mg, 0.16 mmol, 1.0 equiv) in THF (2.1 mL) was added at 0 °C. The ice/water bath was removed, and the flask was capped with a glass stopper. After the mixture was stirred at 22 °C for 1 h, THF was evaporated under reduced pressure. The residue was taken up by dichloromethane (10 mL) and basified with saturated NaHCO3 (15 mL) in a separatory funnel. The organic layer was saved, and the aqueous layer was extracted further with dichloromethane (10 mL × 2). The combined organic extract was dried over Na2SO4, filtered, and concentrated to give a semisolid. Purification by column chromatography (SiO2, 1.5 × 12 cm, hexanes/CH2Cl2, 1/1, with 1% Et3N) afforded 82 mg (93%) of 12d as a white solid. Data for 12d: 1H NMR (500 MHz, CDCl3) 9.20 (d, J = 7.4 Hz, 1 H, HC(9)), 7.87 (s, 2 H, HC(6)), 7.61 (s, 4 H, HC(4)), 6.41 (d, J = 7.4 Hz, 1 H, HC(8)), 5.34 (app br s, 2 H, HC(1)), 2.75–2.61 (m, 2 H, HC(2)), 2.13–1.99 (m, 2 H, HC(2)); 13C NMR (126 MHz, CDCl3) 190.1 (C(9)), 134.1 (C(8)), 132.7 (app d, J = 32.3 Hz, C(5)), 126.2 (br s C(4)), 123.0 (q, J = 273.0 Hz, C(7)), 122.2 (C(4)), 31.4 (br s, C(2)); C(1) and C(3) were not observed due to hindered rotation; 19F NMR (470 MHz, CDCl3) −63.33; IR (CDCl3 film) 3059 (w), 2989 (w), 2947 (w), 1885 (w), 1816 (w), 1681 (s), 1625 (m), 1531 (s), 1468 (m), 1378 (s), 1340 (m), 1320 (m), 1279 (s), 1130 (s), 1032 (w), 989 (w), 894 (s), 846 (m), 707 (m), 682 (s); MS (ESI) 551.1 (100, M + H); HRMS (ESI) calcd for C22H15N2OF12 [M + H] 551.0993, found 551.0988; TLC Rf 0.14 (hexanes/CH2Cl2, 1/1 with 0.1% Et3N) [silica gel, UV, KMnO4].
N,N′-(Ethane-1,2-diylidene)bis((2S,5S)-(2,5-bis(4-(trifluoromethyl)phenyl)pyrrolidin-1-amine)) (13c)

To an oven-dried, 15 mL, one-necked, round-bottom flask equipped with a stir bar, an argon gas inlet, and a septum were added (2S,5S)-(2,5-bis(4-(trifluoromethyl)phenyl)pyrrolidine)-N-iminoacetaldehyde 12c (109 mg, 0.27 mmol, 1.0 equiv) and Na2SO4 (19 mg, 0.13 mmol, 0.5 equiv). A solution of N-amino-(2S,5S)-2,5-bis(4-(trifluoromethyl)phenyl)pyrrolidine 8c (110 mg, 0.29 mmol, 1.1.0 equiv) in dichloromethane (1.0 mL) was added to give a yellow solution. After the flask was purged with argon, the reaction was stirred at 22 °C for 24 h. The solution was filtered and concentrated to give a lightly yellow solid. Purification by column chromatography (SiO2, 1.5 × 16 cm, hexanes/EtOAc, 9/1, with 1% Et3N) afforded 183 mg (90%) of 13c as a white powder. Data for 13c: 1H NMR (500 MHz, CDCl3) 7.58 (d, J = 8.0 Hz, 8 H, HC(5)), 7.22 (d, J = 8.0 Hz, 8 H, HC(4)), 6.55 (s, 2 H, HC(8)), 5.06 (d, J = 7.1 Hz, 4 H, HC(1)), 2.53–2.34 (m, 4 H, HC(2)), 1.80–1.65 (m, 4 H, HC(2)); 13C NMR (126 MHz, CDCl3) 146.9 (C(3)), 134.3 (C(8)), 129.3 (q, J = 32.2 Hz, C(6)), 126.5 (C(4)), 125.5 (app d, J = 3.7 Hz, C(5)), 124.2 (q, J = 271.9 Hz, C(7)), 64.4 (C(1)), 30.9 (C(2)). 19F NMR (470 MHz, CDCl3) −62.81; IR (CDCl3 film) 2984 (w), 2947 (w), 2878 (w), 1618 (m), 1546 (m), 1448 (w), 1417 (m), 1324 (s), 1224 (m), 1167 (s), 1123 (s), 1067 (s), 1016 (s), 870 (w), 836 (s), 732 (m); MS (ESI) 771.2 (100, M + H); HRMS (ESI) calcd for C38H31N4F12 [M + H] 771.2357, found 771.2354; TLC Rf 0.45 (hexanes/EtOAc, 9/1 with 0.1% Et3N) [silica gel, UV, KMnO4]; [α]D24 −291.3 (c = 0.15, chloroform).
N,N′-(Ethane-1,2-diylidene)bis((2S,5S)-(2,5-Bis(3,5-bis(trifluoromethyl)phenyl)pyrrolidin-1-amine)) (13d)

To an oven-dried, one-piece, 5 mL round-bottom flask and a reflux condenser equipped with a stir bar, an argon gas inlet and a septum were added Na2SO4 (10 mg, 0.07 mmol, 0.5 equiv) and a solution of (2S,5S)-(2,5-bis(3,5-bis(trifluoromethyl)phenyl)pyrrolidine)-N-iminoacetaldehyde 12d (80 mg, 0.15 mmol, 1.0 equiv) and N-amino-(2S,5S)-2,5-bis(4-trifluoromethylphenyl)pyrrolidine 8d (82 mg, 0.16 mmol, 1.1.0 equiv) in dichloromethane. The solvent was carefully removed under reduced pressure and CH2Cl2 (0.73 mL) was added to give a yellow solution. After purging the flask with argon, the reaction was stirred at reflux for 20 h. The solution was filtered and concentrated. Purification by column chromatography (SiO2, 1.5 × 20 cm, hexanes/CH2Cl2, 4/1, with 0.5% Et3N) afforded 138 mg (91%) of 13d as a white solid. Data for 13d: 1H NMR (500 MHz, CDCl3) 7.79 (s, 4 H, HC(6)), 7.52 (s, 8 H, HC(4)), 6.50 (s, 2 H, HC(8)), 5.26 (dd, J = 7.0, 4.5 Hz, 4 H, HC(1)), 2.53–2.43 (m, 4 H, H2C(2)), 1.87–1.80 (m, 4 H, H2C(2)); 13C NMR (126 MHz, CDCl3) 144.7 (C(3)), 134.7 (C(8)), 132.1 (q, J = 33.4 Hz, C(5)), 126.2 (C(4)), 123.1 (q, J = 272.7 Hz, C(7)), 121.4 (s, C(6)), 64.0 (C(1)), 30.9 (C(2)). 19F NMR (470 MHz, CDCl3) −63.45; IR (CDCl3 film) 2989 (w), 1624 (w), 1552 (m), 1466 (w), 1377 (m), 1336 (m), 1319 (m), 1279 (s), 1171 (s), 1129 (s), 987 (w), 894 (m), 846 (m), 707 (m); MS (ESI) 1043.2 (M + H, 100); HRMS (ESI) calcd for C42H27N4F24 [M + H] 1043.1853, found 1043.1853; TLC Rf 0.34 (hexanes/CH2Cl2, 4/1, with 0.5% Et3N) [silica gel, UV, KMnO4]; [α]D24 −196.7 (c = 0.15, chloroform).
Preparation of N,N′-(2,2-Dimethylpropane-1,3-diylidene)bis((2R,5R)-2,5-diphenylpyrrolidin-1-amine) (Scheme 8)
2,2-Dimethylmalonaldehyde65

To an oven-dried, 100 mL, three-necked, round-bottom flask equipped with a stir bar, an argon inlet adaptor, and two septa was charged 2,2-dimethyl-1,3-propanediol (208 mg, 2.0 mmol, 1.0 equiv). After three cycles of evacuation/argon fill, dichloromethane (20 mL) was added and the mixture stirred for 5 min to obtain a homogeneous solution. Dess–Martin periodinane (2.54 g, 6 mmol, 3.0 equiv) was added in one portion with rapid stirring. The cloudy solution was stirred at room temperature for 1 h. The insoluble particulates were removed by filtering through a pad of Celite (1 cm deep, 15 mL size, medium-porosity fritted funnel), eluted with dichloromethane (2.5 mL). To the filtrate was added NaHCO3–Na2S2O3 (1:1, 15 mL) to give a murky mixture. After vigorous stirring for several minutes, both aqueous and organic layers became transparent and fizzing subsided. The organic layer was dried over Na2SO4 and filtered to afford a solution of the target dialdehyde and possibly some polymeric species. The solution was used directly without concentration to minimize polymerization.
(2R,5R)-((2,5-Diphenylpyrrolidin-1-yl)imino)-2,2-dimethylpropanal (14)

A solution of N-amino-(R,R)-2,5-bisphenylpyrrolidine 8e(28d) (48 mg, 0.2 mmol) in dichloromethane (3 mL) was added to a solution of 2,2-dimethylmalonaldehyde prepared by oxidation of the corresponding diol in the scale described in the procedure above. After the flask was purged with argon, the flask was capped with a stopper. After being stirred at 22 °C for 2.5 h, the solution was concentrated. Purification by column chromatography (SiO2, 1 × 20 cm, hexanes/CH2Cl2, 1/1, with 1% Et3N) afforded 33 mg of 14 as a colorless, viscous oil. The desired product is contaminated with some other aldehydes based on CHO chemical shifts in NMR analysis, which may arise from the condensation of aminopyrrolidine 8e with polymeric species of 2,2-dimethylmalonaldehyde. Data for 14: 1H NMR (500 MHz, CDCl3) 9.22 (s, 1 H, HC(11)), 7.34 (t, J = 7.6 Hz, 4 H, HC(5)), 7.25 (t, J = 7.4 Hz, 2 H, HC(6)), 7.18 (d, J = 7.2 Hz, 4 H, HC(4)), 5.92 (s, 1 H, HC(7)), 4.99 (d, J = 6.7 Hz, 2 H, HC(1)), 2.56–2.45 (m, 2 H, H2C(2)), 1.87–1.79 (m, 2 H, H2C(2)), 1.01 (s, 3 H, HC(9)), 0.98 (s, 3 H, HC(10)); 13C NMR (126 MHz, CDCl3) 202.8 (C(11)), 143.0 (C(3)), 134.2 (C(7)), 128.3 (C(5)), 126.7 (C(6)), 126.3 (C(4)), 65.1 (C(1)), 49.4 (C(8)), 31.5 (C(2)), 20.5 (C(9)), 20.5 (C(10)); IR (CDCl3 film) 3080 (w), 3061 (m), 3027 (m), 2971 (s), 2870 (m), 2808 (m), 2704 (w), 1947 (w), 1878 (w), 1802 (w), 1727 (s), 1601 (m), 1580 (m), 1493 (m), 1451 (s), 1391 (m), 1360 (m), 1320 (m), 1303 (m), 1268 (m), 1213 (m), 1174 (m), 1139 (m), 1074 (m), 1028 (m), 999 (w), 980 (w), 955 (w), 909 (m), 862 (m), 795 (m), 751 (s), 700 (s), 677 (w); MS (ESI) 321.2 (100, M + H), 293.2 (15); HRMS (ESI) calcd for C21H25N2O [M + H] 321.1967, found 321.1963; TLC Rf 0.50 (CH2Cl2 with 1% Et3N) [silica gel, UV, KMnO4].
N,N′-(2,2-Dimethylpropane-1,3-diylidene)bis((2R,5R)-2,5-diphenylpyrrolidin-1-amine) (15)

To a 15 mL, one-necked, round-bottom flask equipped with a stir bar, an argon gas inlet, and a septum were added (2R,5R)-((2,5-diphenylpyrrolidin-1-yl)imino)-2,2-dimethylpropanal 14 (33 mg, 0.1 mmol, 1.0 equiv) and Na2SO4 (7.5 mg, 0.05 mmol, 0.5 equiv). A solution of (2R,5R)-1amino-2,5-diphenylpyrrolidine 8e (28 mg, 0.12 mmol, 1.2 equiv) in dichloromethane (1.5 mL) was added to give a colorless solution. After the flask was purged with argon, the reaction was stirred at 22 °C for 12 h. The solution was filtered and concentrated to give a colorless film. Purification by column chromatography (SiO2, 1 × 30 cm, hexanes/CH2Cl2, 1/1, with 0.2% Et3N) afforded 47 mg (84%) of 15 as a colorless, sticky gel (44% yield from condensation with 2,2-dimethylmalonaldehyde). Data for 15: 1H NMR (500 MHz, CDCl3) 7.29 (t, J = 7.4 Hz, 8 H, HC(5)), 7.22 (t, J = 7.3 Hz, 4 H, HC(6)), 7.11 (d, 8 H, HC(4)), 6.00 (s, 2 H, HC(7)), 4.80 (d, J = 6.6 Hz, 4 H, HC(1)), 2.50–2.38 (m, 4 H, H2C(2)), 1.81–1.71 (m, 4 H, H2C(2)), 0.74 (s, 6 H, H3C(9)); 13C NMR (126 MHz, CDCl3) 144.1 (C(3)), 141.4 (C(7)), 128.0 (C(5)), 126.5 (C(4)), 126.3 (C(6)), 65.0 (C(1)), 40.7 (C(8)), 31.5 (C(2)), 24.8 (C(9)); IR (CDCl3 film) 3085 (m), 3062 (m), 3026 (s), 2969 (s), 2871 (s), 1946 (w), 1874 (w), 1806 (w), 1727 (w), 1602 (m), 1494 (s), 1453 (s), 1385 (m), 1359 (s), 1303 (s), 1285 (m), 1214 (s), 1170 (s), 1126 (s), 1074 (m), 1052 (m), 1028 (m), 981 (m), 950 (w), 910 (s), 868 (w), 799 (w), 749 (s), 648 (m); MS (ESI) 541.3 (100, M + H), 291.2 (8); HRMS (ESI) calcd for C37H41N4 [M + H] 541.3331, found 541.3333; TLC Rf 0.66 (CH2Cl2 with 1% Et3N) [silica gel, UV, I2]; [α]D24 −184.8 (c = 0.15, chloroform).
Preparation of (2R)-Aryl- and (2R,5R)-Diarylpyrrolidine-Based Bis-hydrazone Ligands by the α-Arylation Route (Scheme 6 and Tables 2–4)
General Procedure 3: α-Arylation of N-Boc-pyrrolidine (Table 2)32,33

An oven-dried, 100 mL Schlenk reaction flask equipped with a stir bar, a septum, and an internal temperature probe was evacuated then filled with argon (three cycles). (−)-Sparteine (2.8 mL, 12 mmol, 1.2 equiv), N-Boc-pyrrolidine (2.1 mL, 12 mmol, 1.2 equiv), and TBME (30 mL) were added. The colorless solution was cooled to −75 °C, and s-BuLi (1.74 M in cyclohexanes, 6.9 mL, 12 mmol, 1.2 equiv) was added dropwise, keeping the temperature below −69 °C. (Note: s-BuLi should be added directly to the solution to avoid crystallization on the wall of the flask.) The resulting light yellow solution was stirred at −75 ± 1 °C for 3 h. A solution of ZnCl2 (1 M in Et2O, 7.2 mL, 7.2 mmol, 0.72 equiv) was then added to the reaction dropwise with rapid stirring, keeping the temperature below −69 °C. The cloudy solution was stirred at −75 ± 1 °C for 0.5 h and then warmed to 22 °C resulting in a heterogeneous mixture. After being stirred at 22 °C for 0.5 h, the septum was secured by copper wire and the flask was brought into a glovebox. Aryl bromide (10 mmol, 1.0 equiv), t-Bu3P-HBF4 (174 mg, 0.6 mmol, 0.06 equiv), and Pd(OAc)2 (110 mg, 0.48 mmol, 0.048 equiv) were added to the flask with rapid stirring. After the mixture was stirred at 22 °C for 18 h under argon, the reaction was quenched with an aqueous solution of NH4OH (30%, 1 mL), and the mixture was stirred at room temperature for 1 h. The resulting slurry was filtered through Celite (2 cm deep, 30 mL, medium-porosity fritted funnel) and eluted with 60 mL of TBME. The filtrate was washed with 50 mL of 1 M HCl and then with 50 mL of deionized water. The organic phase was dried over magnesium sulfate, filtered, and concentrated to give a brown oil. Purification by column chromatography provided N-Boc-(R)-2-arylpyrrolidine 16.
(−)-Sparteine was recovered by washing its acidic aqueous solution (in 1 M HCl) with Et2O (×6) to remove t-Bu3P(O) and then basified with an aqueous solution of NaOH (20%). The aqueous phase was extracted with Et2O ( × 4), and the combined organic phases were dried over K2CO3, filtered, and concentrated. The orange oil was stirred overnight over CaH2 then distilled under vacuum (bp: 115–120 °C/0.3 mmHg) to afford pure (−)-sparteine as a colorless oil.
Preparation of N-Boc-(R)-2-arylpyrrolidine
N-Boc-(R)-2-(4-methoxyphenyl)pyrrolidine (16a)

Following general procedure 3, (−)-sparteine (2.8 mL, 12 mmol, 1.2 equiv), N-Boc-pyrrolidine (2.1 mL, 12 mmol, 1.2 equiv) and TBME (30 mL), s-BuLi (1.74 M in cyclohexanes, 6.9 mL, 12 mmol, 1.2 equiv), ZnCl2 (1 M in Et2O, 7.2 mL, 7.2 mmol, 0.72 equiv), 4-bromoanisole (1.25 mL, 10 mmol, 1.0 equiv), t-Bu3P-HBF4 (174 mg, 0.6 mmol, 0.06 equiv), and Pd(OAc)2 (110 mg, 0.48 mmol, 0.048 equiv) were combined in a 100 mL Schlenk reaction flask. After the mixture was stirred at 22 °C for 18 h under argon, the reaction was quenched, filtered, subjected to aqueous washes, and then concentrated. Purification by column chromatography (SiO2, 4.5 × 26 cm, hexanes/EtOAc, gradient elution, 14/1, 9/1 then 7/1) afforded 1.61 g (71%) of 16a as a slightly tanned white solid (a rotameric mixture, 66:34, −20 °C). The spectroscopic data matched those from the literature.66 Data for 16a: 1H NMR (500 MHz, CDCl3, −20 °C) major 7.07 (d, J = 8.6 Hz, 2 H, HC(9)), 6.82 (d, J = 8.6 Hz, 2 H, HC(10)), 4.71 (dd, J = 7.7, 4.3 Hz, 1 H, HC(1)), 3.79 (s, 3 H, H3C(12)), 3.66–3.52 (m, 2 H, H2C(4)), 2.32–2.24 (m, 1 H, HC(2)), 1.92–1.74 (m, 3 H, HC(2) and H2C(3)), 1.18 (s, 9 H, H3C(7)), minor 7.10 (d, J = 8.5 Hz, 2 H, HC(9)), 6.83 (d, J = 8.5 Hz, 2 H, HC(10)), 4.89 (dd, J = 8.0, 2.9 Hz, 1 H, HC(1)), 3.77 (s, 3 H, H3C(12)), 3.50–3.44 (m, 2 H, H2C(4)), 2.25–2.19 (m, 1 H, HC(2)), 1.92–1.74 (m, 3 H, HC(2) and H2C(3)), 1.44 (s, 9 H, H3C(7)); 13C NMR (126 MHz, CDCl3, −20 °C), major 157.9 (C(11)), 154.6 (C(5)), 137.1 (C(8)), 126.5 (C(9)), 113.2 (C(10)), 79.0 (C(6)), 60.5 (C(1)), 55.2 (C(12)), 46.9 (C(4)), 36.0 (C(2)), 28.1 (C(7)), 23.0 (C(3)), minor 158.0 (C(11)), 154.4 (C(5)), 136.1 (C(8)), 126.3 (C(9)), 113.6 (C(10)), 79.1 C(6)), 59.9 (C(1)), 55.2 (C(12)), 47.2 (C(4)), 34.9 (C(2)), 28.4 (C(7)), 23.3 (C(3)); IR (CDCl3 film) 2975 (s), 2836 (m), 2058 (w), 1882 (w), 1693 (s), 1681 (s), 1651 (m), 1613 (s), 1586 (m), 1514 (s), 1454 (s), 1392 (s), 1246 (s), 1163 (s), 1113 (s), 1079 (m), 1036 (s), 971 (w), 917 (m), 901 (m), 875 (m), 828 (s), 768 (m); MS (EI, 70 eV) 277.2 (M+, 7), 220.2 (100), 176.2 (84), 148.1 (22); HRMS (ESI) calcd for C16H24NO3 [M + H] 278.1756, found 278.1765; TLC Rf 0.16 (hexanes/EtOAc, 9/1) [silica gel, UV, KMnO4]; [α]D24 +90.6 (c = 0.5, acetone); SFC (S)-16a, tR 3.8 min (4%); (R)-16a, tR 6.7 min (96%) ((R,R)-Whelk-O1, 200 bar, 5% MeOH in CO2, 2.5 mL/min, 220 nm, 40 °C).
N-Boc-(R)-2-(3,5-bis(trifluoromethyl)phenyl)pyrrolidine (16d)

Following general procedure 3, (−)-sparteine (2.4 mL, 10 mmol, 1.2 equiv), N-Boc-pyrrolidine (1.8 mL, 10 mmol, 1.2 equiv), TBME (26 mL), s-BuLi (1.74 M in cyclohexanes, 5.8 mL, 10 mmol, 1.2 equiv), ZnCl2 (1 M in Et2O, 6.1 mL, 6.1 mmol, 0.72 equiv), 1-bromo-3,5-bis(trifluoromethyl)benzene (1.45 mL, 8.4 mmol, 1.0 equiv), t-Bu3P-HBF4 (146 mg, 0.5 mmol, 0.06 equiv), and Pd(OAc)2 (90 mg, 0.4 mmol, 0.048 equiv) were combined in a 100 mL Schlenk reaction flask. After the mixture was stirred at 21 °C for 18 h, the reaction was quenched, filtered, subjected to aqueous washes, and then concentrated. Purification by column chromatography (SiO2, 4.5 × 23 cm, CH2Cl2/hexanes, 4/1) and Kugelrohr distillation afforded 1.73 g (54%) of 16d as a very pale yellow oil (a rotameric mixture, 64:36, −20 °C). This compound was derivatized to 3,5-dinitrophenylmethane for the assessment of enantiomeric purity (96:4 er). Data for 16d: bp 150 °C [0.1 mmHg, ABT]; 1H NMR (500 MHz, CDCl3, −20 °C) major 7.75 (s, 1 H, HC(11)), 7.62 (s, 2 H, HC(9)), 4.82 (dd, J = 7.6, 5.8 Hz, 1 H, HC(1)), 3.72–3.62 (m, 2 H, HC(4)), 2.45–2.42 (m, 1 H, HC(2)), 1.93–1.87 (m, 2 H, H2C(3)), 1.86–1.82 (m, 1 H, HC(2)), 1.14 (s, 9 H, H3C(7)), minor 7.72 (s, 1 H, HC(11)), 7.59 (s, 2 H, HC(9)), 5.01 (dd, J = 8.1, 3.5 Hz, 1 H, HC(1)), 3.58–3.52 (m, 2 H, HC(4)), 2.40–2.33 (m, 1 H, HC(2)), 1.93–1.87 (m, 2 H, H2C(3)), 1.81–1.75 (m, 1 H, HC(2)), 1.44 (s, 9 H, H3C(7)); 13C NMR (126 MHz, CDCl3, −20 °C) major 154.1 (C(5)), 147.7 (C(8)), 131.3 (q, J = 33.4 Hz, C(10)), 125.8 (C(9)), 123.2 (q, J = 272.6 Hz, C(12)), 120.5 (C(11)), 79.9 (C(6)), 60.8 (C(1)), 47.2 (C(4)), 36.1 (C(2)), 27.9 (C(7)), 23.4 (C(3)), minor 154.5 (C(5)), 146.6 (C(8)), 131.4 (q, J = 33.2 Hz, C(10)), 125.4 (C(9)), 123.3 (q, J = 272.6 Hz, C(12)), 120.5 (C(11)), 80.1 (C(6)), 60.2 (C(1)), 47.4 (C(4)), 34.8 (C(2)), 28.3 (C(7)), 23.5 (C(3)). 19F NMR (470 MHz, CDCl3) −63.31; IR (neat) 2978 (m), 2882 (m), 1699 (s), 1624 (w), 1479 (m), 1457 (m),1392 (s), 1278 (s), 1169 (s), 1133 (s), 1027 (w), 974 (w), 928 (w), 898 (m), 846 (w), 776 (w), 707 (m). MS (EI, 70 eV) 383.1 (M+, 7), 328.1 (100), 308.1 (50), 282.1 (48), 267.1 (49), 255.1 (48), 227.0 (34); HRMS (ESI) calcd for C17H20NO2F6 [M + H] 384.1398, found 384.1404; TLC Rf 0.26 (hexanes/CH2Cl2, 1/4) [silica gel, KMnO4]; [α]D24 +68.9 (c = 0.5, acetone).
(R)-2-(3,5-Bis(trifluoromethyl)phenyl)pyrrolidine (17d)

To an oven-dried, 10 mL, round-bottom flask equipped with a stir bar, an argon inlet adaptor, and a septum was added N-Boc-(R)-2-(3,5-bis(trifluoromethyl)phenyl)pyrrolidine 16d (61 mg, 0.16 mmol, 1.0 equiv). After three cycles of evacuation and argon fill, dichloromethane (1.6 mL) was added. The colorless solution was cooled to 0 °C, and TMSI (25 μL, 0.17 mmol, 1.05 equiv) was added. The resulting light burgundy solution was stirred at 0 °C for 1.5 h and then quenched with an aqueous of saturated NaHCO3/saturated Na2S2O3 (1:1, 3.2 mL). After vigorous stirring for a few minutes, the organic phase was saved and the aqueous phase was extracted with dichloromethane (1.5 mL × 3). The combined organic extracts was dried over Na2SO4, filtered, and concentrated. Purification by column chromatography (SiO2, 1 × 16 cm, CH2Cl2/MeOH, 98/2) afforded 40 mg (88%) of 17d as a pale yellow liquid. Data for 17d: 1H NMR (500 MHz, CDCl3) 7.86 (s, 2 H, HC(6)), 7.73 (s, 1 H, HC(8)), 4.28 (t, J = 4.3 Hz, 1 H, HC(1)), 3.22–3.17 (m, 1 H, HC(4)), 3.12–3.06 (m, 1 H, HC(4)), 2.31–2.24 (m, 1 H, HC(2)), 1.98 (s, 1 H, NH), 1.96–1.82 (m, 2 H, HC(3)), 1.67–1.59 (m, 1 H, HC(2)); 13C NMR (126 MHz, CDCl3) 148.4 (C(5)), 131.4 (q, J = 33.0 Hz, C(7)), 126.7 (C(6)), 123.5 (q, J = 272.6 Hz, C(9)), 120.6 (C(8)), 61.4 (C(1)), 47.0 (C(4)), 34.9 (C(2)), 25.5 (C(3)); 19F NMR (470 MHz, CDCl3) −63.21; IR (CDCl3 film) 2965 (w), 2871 (w), 1622 (w), 1464 (w), 1380 (m), 1352 (w), 1279 (s), 1171 (s), 1131 (s), 896 (m), 842 (m).
(R)-(2-(3,5-Bis(trifluoromethyl)phenyl)pyrrolidin-1-yl)(3,5-dinitrophenyl)methanone (17d-DNB)

To a 10 mL, one-necked round-bottom flask equipped with a stir bar, an argon inlet adaptor, and a septum was added (R)-2-(3,5-bis(trifluoromethyl)phenyl)pyrrolidine 17d (38 mg, 0.13 mmol, 1.0 equiv). After three quick cycles of evacuation and argon fill (17d is volatile), THF (1.4 mL), 3,5-dinitrobenzoyl chloride (35 mg, 0.15 mmol, 1.1.0 equiv) and Et3N (190 μL, 1.34 mmol, 10 equiv) were added to give a heterogeneous mixture. After the mixture was stirred at 21 °C for 4.5 h, THF was removed under reduced pressure. The crude product was taken up with Et2O (1.5 mL) and washed with 5% NaOH (3 mL). The organic phase was saved, and the aqueous phase was extracted with Et2O (1.5 mL × 3). The combined organic phases was dried over Na2SO4, filtered, and concentrated to give a brown sticky oil. Purification by column chromatography (SiO2, 1 × 21 cm, CH2Cl2/hexanes, 9/1) afforded 61 mg (94%) of 17d-DNB as a white, foamy liquid. Spectroscopic data for major conformer is shown. Data for 17d-DNB: 1H NMR (500 MHz, CDCl3) 9.14 (s, 1 H, HC(9)), 8.77 (s, 2 H, HC(7)), 7.80 (s, 1 H, HC(13)), 7.77 (s, 2 H, HC(11)), 5.36 (t, J = 7.0 Hz, 1 H, HC(1)), 3.96 (app q, J = 7.5 Hz, 1 H, HC(4)), 3.75–3.71 (m, 1 H, HC(4)), 2.64–2.58 (m, 1 H, HC(2)), 2.18–2.00 (m, 3 H, HC(2) and H2C(3)); 13C NMR (126 MHz, CDCl3) 165.2 (C(5)), 148.5 (C(Aryl)), 144.7 (C(Aryl)), 139.2 (C(Aryl)), 123.1 (q, J = 33.5 Hz, C(12)), 127.7 (C(Aryl)), 126.2 (C(Aryl)), 123.2 (q, J = 272.8 Hz, C(14)), 122.6 (C(Aryl)), 120.4 (C(Aryl)), 61.9 (C(1)), 51.2 (C(4)), 34.6 (C(2)), 25.6 (C(3)); 19F NMR (470 MHz, CDCl3) −63.23; IR (CDCl3 film) 3104 (m), 2981 (m), 2883 (m), 1818 (w), 1643 (s), 1591 (m), 1546 (s), 1467 (s), 1409 (s), 1380 (s), 1344 (s), 1278 (s), 1181 (s), 1132 (s), 968 (m), 870 (m), 846 (m), 836 (m), 706 (s). MS (EI, 70 eV) 478.1 (M + H, 100); HRMS (ESI) calcd for C19H14N3O5F6 [M + H] 478.0838, found 478.0835; TLC Rf 0.23 (CH2Cl2) [silica gel, UV]; SFC (S)-17d-DNB, tR 7.6 min (4%); (R)-17d-DNB, tR 18.5 min (96%) ((R,R)-Whelk-O1, 200 bar, 5% MeOH in CO2, 2.5 mL/min, 220 nm, 40 °C).
N-Boc-(R)-2-(2-naphthyl)pyrrolidine (16f)

Following general procedure 3, (−)-sparteine (2.8 mL, 12 mmol, 1.2 equiv), N-Boc-pyrrolidine (2.1 mL, 12 mmol, 1.2 equiv), TBME (30 mL), s-BuLi (1.74 M in cyclohexanes, 6.9 mL, 12 mmol, 1.2 equiv), ZnCl2 (1 M in Et2O, 7.2 mL, 7.2 mmol, 0.72 equiv), 2-bromonaphthalene (2.07 g, 10 mmol, 1.0 equiv), t-Bu3P-HBF4 (174 mg, 0.6 mmol, 0.06 equiv), and Pd(OAc)2 (110 mg, 0.48 mmol, 0.048 equiv) were combined in a 100 mL Schlenk reaction flask. After being stirred at 22 °C for 16 h, the reaction was quenched, filtered, subjected to aqueous washes, and then concentrated. Purification by column chromatography (SiO2, 4.5 × 20 cm, CH2Cl2/hexanes, gradient elution, 4/1, 9/1 then 1/0) afforded 1.82 g (61%) of 16f as a white solid (a rotameric mixture, 70:30, −20 °C). The spectroscopic data matched those from the literature.24 Data for 16f: 1H NMR (500 MHz, CDCl3, −20 °C) major 7.85–7.77 (m, 3 H, HC(Aryl)), 7.58 (s, 1 H, HC(8)), 7.50–7.40 (m, 2 H, HC(Aryl)), 7.35–7.30 (m, 1 H, HC(16) or HC(10)), 4.95 (dd, J = 7.9, 3.6 Hz, 1 H, HC(1)), 3.76–3.63 (m, 2 H, H2C(4)), 2.43–2.29 (m, 1 H, HC(2)), 1.97–1.83 (m, 3 H, HC(2) and H2C(3)), 1.13 (s, 9 H, H3C(7)), minor 7.85–7.77 (m, 3 H, HC(Aryl)), 7.59 (s, 1 H, HC(8)), 7.50–7.40 (m, 2 H, HC(Aryl)), 7.35–7.30 (m, 1 H, HC(16) or HC(10)), 5.11 (d, J = 6.5 Hz, 1 H, HC(1)), 3.59–3.55 (m, 2 H, H2C(4)), 2.43–2.29 (m, 1 H, HC(2)), 1.97–1.83 (m, 3 H, HC(2) and H2C(3)), 1.47 (s, 9 H, H3C(7)); 13C NMR (126 MHz, CDCl3, −20 °C) major 154.7 (C(5)), 142.2 (C(9)), 133.0 (C(12) or C(17)), 132.2 ((C(12) or C(17)), 127.9 (C(Aryl)), 127.5 (C(Aryl)), 127.5 (C(Aryl)), 126.0 (C(Aryl)), 125.3 (C(Aryl)), 124.1 (C(8)), 123.6 (C(8) or C(10)), 79.2 (C(6)), 61.2 (C(1)), 47.0 (C(4)), 35.7 (C(2)), 28.0 (C(7)), 23.0 (C(3)), minor 154.5 (C(5)), 141.3 (C(9)), 133.2 (C(12) or C(17)), 132.4 (C(12) or C(17)), 128.2 (C(Aryl)), 127.7 (C(Aryl)), 127.5 (C(Aryl)), 125.8 (C(Aryl)), 125.3 (C(Aryl)), 124.0 (C(8)), 123.5 (C(8) or C(10)), 79.3 C(6)), 60.7 (C(1)), 47.4 (C(4)), 34.7 (C(2)), 28.4 (C(7)), 23.4 (C(3)); IR (CDCl3 film) 3053 (m), 2973 (m), 2876 (m), 1693 (s), 1634 (w), 1602 (w), 1509 (w), 1478 (m), 1454 (m), 1392 (s), 1364 (s), 1255 (m), 1165 (s), 1109 (s), 1081 (w), 974 (w), 912 (w), 896 (w), 856 (w), 817 (m), 746 (m). MS (EI, 70 eV) 297.1 (M+, 20), 241.1 (80), 196.1 (100), 168.0 (34), 154.0 (23), 84.0 (23), 57.1 (75); HRMS (ESI) calcd for C19H24NO2 [M + H] 298.1807, found 298.1801; TLC Rf 0.11 (CH2Cl2) [silica gel, UV, KMnO4]; [α]D24 +117.7 (c = 0.5, acetone). SFC (S)-16f, tR 3.8 min (4%); (R)-16f, tR 11.6 min (96%) ((R,R)-Whelk-O1, 200 bar, 10% MeOH in CO2, 2.5 mL/min, 220 nm, 40 °C).
N-Boc-(R)-2-(3,5-dimethylphenyl)pyrrolidine (16g)

Following general procedure 3, (−)-sparteine (2.4 mL, 10 mmol, 1.2 equiv), N-Boc-pyrrolidine (1.8 mL, 10 mmol, 1.2 equiv), TBME (26 mL), s-BuLi (1.74 M in cyclohexanes, 5.8 mL, 10 mmol, 1.2 equiv), ZnCl2 (1 M in Et2O, 6.1 mL, 6.1 mmol, 0.72 equiv), 1-bromo-3,5-dimethylbenzene (1.15 mL, 8.4 mmol, 1.0 equiv), t-Bu3P-HBF4 (148 mg, 0.5 mmol, 0.06 equiv), and Pd(OAc)2 (92 mg, 0.4 mmol, 0.048 equiv) were combined in a 100 mL Schlenk reaction flask. After the mixture was stirred at 22 °C for 18 h, the reaction was quenched, filtered, subjected to aqueous washes, and then concentrated. Purification by column chromatography (SiO2, 4.5 × 23 cm, CH2Cl2/hexanes, 9/1) and Kugelrohr distillation afforded 1.30 g (56%) of 16g as a colorless oil (a rotameric mixture, 66:34, −20 °C). Data for 16g: bp 175 °C [0.02 mmHg, ABT]; 1H NMR (500 MHz, CDCl3, −20 °C) major 6.84 (s, 1 H, HC(11)), 6.76 (s, 2 H, HC(9)), 4.68 (dd, J = 7.7, 4.2 Hz, 1 H, HC(1)), 3.65–3.56 (m, 2 H, HC(4)), 2.31–2.20 (m, 1 H, HC(2)), 2.28 (s, 6 H, HC(12)), 1.93–1.79 (m, 3 H, H2C(3) and HC(2)), 1.17 (s, 9 H, H3C(7)), minor 6.84 (s, 1 H, HC(11)), 6.76 (s, 2 H, HC(9)), 4.88–4.84 (m, 1 H, HC(1)), 3.51–3.46 (m, 2 H, HC(4)), 2.31–2.20 (m, 1 H, HC(2)), 2.28 (s, 6 H, HC(12)), 1.93–1.72 (m, 3 H, H2C(3) and HC(2)), 1.45 (s, 9 H, H3C(7)); 13C NMR (126 MHz, CDCl3, −20 °C), major 154.6 (C(5)), 144.8 (C(10)), 137.4 (C(8)), 127.9 (C(11)), 123.3 (C(9)), 79.3 (C(6)), 61.0 (C(1)), 46.9 (C(4)), 35.8 (C(2)), 28.0 (C(7)), 23.1 (C(3)), 21.3 (C(12)), minor 154.4 (C(5)), 144.0 (C(10)), 137.7 (C(8)), 128.3 (C(11)), 122.9 (C(9)), 79.1 (C(6)), 60.5 (C(1)), 47.3 (C(4)), 34.9 (C(2)), 28.4 (C(7)), 23.4 (C(3)), 21.4 (C(12)). IR (neat) 3373 (w), 2972 (s), 1693 (s), 1681 (s), 1651 (m), 1605 (s), 1454 (s), 1392 (s), 1255 (s), 1163 (s), 1112 (s), 1036 (m), 1009 (m), 972 (m), 951 (m), 918 (m), 897 (m), 843 (m), 774 (m), 730 (m), 704 (s). MS (EI, 70 eV) 275.2 (M+, 7), 219.1 (100), 204.1 (24), 174.1 (99), 160.1 (56), 147.1 (28), 132.1 (74); HRMS (ESI) calcd for C17H26NO2 [M + H] 276.1964, found 276.1971; TLC Rf 0.17 (CH2Cl2) [silica gel, UV, KMnO4]; [α]D24 +88.3 (c = 0.51, acetone); SFC (S)-16g, tR 3.1 min (4%); (R)-16g, tR 5.6 min (96%) ((R,R)-Whelk-O1, 200 bar, 5% MeOH in CO2, 2.5 mL/min, 220 nm, 40 °C).
N-Boc-(R)-2-(5-phenylbiphenyl-3-yl)pyrrolidine (16h)

Following general procedure 3, (−)-sparteine (2.8 mL, 12 mmol, 1.2 equiv), N-Boc-pyrrolidine (2.1 mL, 12 mmol, 1.2 equiv), TBME (30 mL), s-BuLi (1.74 M in cyclohexanes, 6.9 mL, 12 mmol, 1.2 equiv), ZnCl2 (1 M in Et2O, 7.2 mL, 7.2 mmol, 0.72 equiv), 1-bromo-3,5-diphenylbenzene (3.10 g, 10 mmol, 1.0 equiv), t-Bu3P-HBF4 (175 mg, 0.6 mmol, 0.06 equiv), and Pd(OAc)2 (112 mg, 0.48 mmol, 0.048 equiv) were combined in a 100 mL Schlenk reaction flask. After the mixture was stirred at 22 °C for 18 h, the reaction was quenched, filtered, subjected to aqueous washes, and then concentrated. Purification by column chromatography (SiO2, 4.5 × 26 cm, CH2Cl2/hexanes, gradient elution, 6/4, 7/3, 8/2 then 9/1) afforded 2.44 g (61%) of 16h as a foamy, white solid (a rotameric mixture, 65:35, −20 °C). The spectroscopic data matched those from the literature.24 Data for 16h: 1H NMR (500 MHz, CDCl3, −20 °C) major 4.89 (dd, J = 7.6, 4.6 Hz, 1 H, HC(1)), 3.75–3.60 (m, 2 H, H2C(4)), 2.45–2.38 (m, 1 H, HC(2)), 2.04–1.86 (m, 3 H, HC(2) and H2C(3)), 1.20 (s, 9 H, H3C(7)), minor 5.12 (d, J = 6.6, 1 H, HC(1)), 3.57–3.51 (m, 2 H, H2C(4)), 2.38–2.30 (m, 1 H, HC(2)), 2.04–1.86 (m, 3 H, HC(2) and H2C(3)), 1.51 (s, 9 H, H3C(7)), aromatic protons for major and minor rotameric isomers 7.68–7.62 (m, 5 H), 7.50–7.45 (m, 4 H), 7.41–7.36 (m, 4 H); 13C NMR (126 MHz, CDCl3, −20 °C) major 154.5 (C(5)), 79.4 (C(6)), 61.4 (C(1)), 47.1 (C(4)), 36.1 (C(2)), 28.1 (C(7)), 23.3 (C(3)), minor 154.5 (C(5)), 79.3 C(6)), 60.5 (C(1)), 47.3 (C(4)), 34.9 (C(2)), 28.4 (C(7)), 23.3 (C(3)), aromatic carbons for major and minor rotameric isomers: 146.3, 144.8, 141.7, 141.5, 141.2, 140.9, 128.7, 128.6, 127.4, 127.3, 127.2, 127.1, 124.8, 124.4, 123.2, 123.0; IR (CDCl3 film) 3033 (w), 2973 (m), 2874 (w), 1693 (s), 1596 (m), 1577 (w), 1498 (m), 1477 (m), 1454 (m), 1435 (m), 1392 (s), 1365 (s), 1249 (w), 1165 (s), 1116 (m), 1081 (w), 1029 (w), 971 (w), 871 (w), 758 (s), 698 (s); MS (EI, 70 eV) 399.2 (M+, 20), 343.1 (72), 298.1 (100), 270.1 (45), 256.1 (65), 241.1 (22), 194.1 (19), 165.1 (10), 70.1 (31), 57.1 (84); HRMS (ESI) calcd for C27H30NO2 [M + H] 400.2277, found 400.2283; TLC Rf 0.15 (CH2Cl2/hexanes, 9/1) [silica gel, UV]; [α]D24 +83.1 (c = 0.5, acetone); SFC (S)-16h, tR 18.9 min (4%); (R)-16h, tR 21.0 min (96%) (Chiralpak-OD, 200 bar, 1–10% MeOH gradient in CO2 (30 min), 2 mL/min, 220 nm, 40 °C).
N-Boc-(R)-2-(2-tolyl)pyrrolidine (16i)

Following general procedure 3, (−)-sparteine (3.3 mL, 14.4 mmol, 1.2 equiv), N-Boc-pyrrolidine (2.6 mL, 14.4 mmol, 1.2 equiv), TBME (36 mL), s-BuLi (1.74 M in cyclohexanes, 8.3 mL, 12 mmol, 1.2 equiv), ZnCl2 (1 M in Et2O, 8.7 mL, 8.7 mmol, 0.72 equiv), 2-bromotoluene (1.5 mL, 12 mmol, 1.0 equiv), t-Bu3P-HBF4 (210 mg, 0.72 mmol, 0.06 equiv), and Pd(OAc)2 (132 mg, 0.58 mmol, 0.048 equiv) were combined in a 100 mL Schlenk reaction flask. After the mixture was stirred at 22 °C for 18 h, the reaction was quenched, filtered, subjected to aqueous washes, and then concentrated. Purification by column chromatography (SiO2, 4.5 × 21 cm, hexanes/EtOAc, 19/1) and Kugelrohr distillation afforded 1.69 g (54%) of 16i as a colorless oil (a rotameric mixture, 66:34, −20 °C). N-Boc-pyrrolidine was removed at 80–90 °C [0.02 mmHg, ABT]. The spectroscopic data matched those from the literature.32 Data for 16i: bp 125 °C [0.02 mmHg, ABT]; 1H NMR (500 MHz, CDCl3, −20 °C) major 7.17–7.09 (m, 3 H, HC(10), HC(11) and HC(12)), 7.07 (m, 1 H, HC(13)), 4.94 (dd, J = 7.9, 4.5 Hz, 1 H, HC(1)), 3.71–3.57 (m, 2 H, H2C(4)), 2.32 (s, 3 H, H3C(14)), 2.36–2.18 (m, 1 H, HC(2)), 1.98–1.82 (m, 2 H, H2C(3)), 1.75–1.65 (m, 2 H, H2C(3)), 1.13 (s, 9 H, H3C(7)), minor 7.17–7.09 (m, 3 H, HC(10), HC(11) and HC(12)), 7.03 (d, J = 7.1 Hz, 1 H, HC(13)), 5.11 (dd, J = 8.2, 1.8 Hz, 1 H, HC(1)), 3.71–3.57 (m, 1 H, H2C(4)), 3.50–3.45 (m, 1 H, H2C(4)), 2.34 (s, 3 H, H3C(14)), 2.36–2.18 (m, 1 H, HC(2)), 1.98–1.82 (m, 2 H, HC(3)), 1.75–1.65 (m, 2 H, HC(3)), 1.15 (s, 9 H, H3C(7)); 13C NMR (126 MHz, CDCl3, −20 °C) major 154.3 (C(5)), 143.0 (C(8)), 133.8 (C(9)), 129.8 (C(10)), 126.1 (C(11) or C(12)), 125.8 (C(11) or C(12)), 124.2 (C(13)), 78.9 (C(6)), 57.7 (C(1)), 46.9 (C(4)), 33.9 (C(2)), 27.9 (C(7)), 23.0 (C(3)), 19.4 (C(14)), minor 154.3 (C(5)), 141.5 (C(8)), 134.0 (C(9)), 130.4 (C(10)), 126.4 (C(11) or C(12)), 125.7 (C(11) or C(12)), 123.7 (C(13)), 79.2 (C(6)), 57.8 (C(1)), 47.2 (C(4)), 32.5 (C(2)), 28.4 (C(7)), 22.9 (C(3)), 19.3 (C(14)). IR (neat) 2973 (s), 2874 (m), 1697 (s), 1605 (w), 1479 (m), 1455 (m), 1392 (s), 1364 (s), 1275 (m), 1246 (m), 1161 (s), 1121 (s), 1102 (m), 1078 (m), 1051 (w), 1032 (w), 971 (w), 921 (m), 875 (m), 771 (m), 752 (m), 725 (m); MS (EI, 70 eV) 261.2 (M+, 3), 205.1 (67), 190.1 (15), 160.1 (25), 144.1 (44), 133.1 (26), 114.1 (29), 91.1 (12), 70.1 (30), 57.1 (100); HRMS (ESI) calcd for C16H24NO2 [M + H] 262.1807, found 262.1798; TLC Rf 0.15 (hexanes/EtOAc, 19/1) [silica gel, KMnO4]; [α]D24 +70.9 (c = 0.5, acetone); SFC (S)-16i, tR 7.1 min (5%); (R)-16i, tR 10.1 min (95%) ((R,R)-Whelk-O1, 200 bar, 2% MeOH in CO2, 2 mL/min, 220 nm, 40 °C).
N-Boc-(R)-2-(5-methyl-2-thienyl)pyrrolidine (16j)

An oven-dried, three-necked, round-bottom flask equipped with a stir bar, a septum, an internal temperature probe, and a solid addition bulb containing t-Bu3P-HBF4 (273 mg, 0.94 mmol, 0.06 equiv) and Pd(OAc)2 (174 mg, 0.75 mmol, 0.048 equiv) was carefully evacuated and then filled with argon (two cycles). (−)-Sparteine (4.4 mL, 18.8 mmol, 1.2 equiv), N-Boc-pyrrolidine (3.3 mL, 18.8 mmol, 1.2 equiv), and TBME (47 mL) were added. The colorless solution was cooled to −75 °C, and s-BuLi (1.47 M in cyclohexanes, 12.9 mL, 18.8 mmol, 1.2 equiv) was added dropwise, keeping the temperature below −69 °C. (Note: s-BuLi should be added directly to the solution to avoid crystallization on the wall of the flask.) The resulting light yellow solution was stirred at −76 for 3 h. A solution of ZnCl2 (1 M in Et2O, 11.3 mL, 11.3 mmol, 0.72 equiv) was then added to the reaction dropwise with rapid stirring, keeping the temperature below −69 °C. The cloudy solution was stirred at −76 °C for 0.5 h and then warmed to 22 °C resulting in a heterogeneous mixture. After the solution was stirred at 21 °C for 0.5 h, 2-bromo-5-methylthiophene (1.83 mL, 15.7 mmol, 1.0 equiv), t-Bu3P-HBF4, and Pd(OAc)2 were added with rapid stirring (exothermed from 21 to 29 °C). After the mixture was stirred at 21 °C for 18 h under argon, the reaction was quenched with an aqueous solution of NH4OH (30%, 1.6 mL), and the mixture was stirred at room temperature for 1 h. The resulting slurry was filtered through Celite (2 cm deep, 60 mL, medium-porosity fritted funnel) and eluted with 80 mL of TBME. The filtrate was washed with 60 mL of 1 M HCl and then with 60 mL of deionized water. The organic phase was dried over magnesium sulfate, filtered, and concentrated to give an orange oil. Purification by column chromatography (SiO2, 6 × 30 cm, gradient elution, hexanes/EtOAc, 19/1 then 9/1) and Kugelrohr distillation afforded 1.99 g (∼40%) of 16j as a semisolid (a rotameric mixture, 61:39, 21 °C) with an unknown side product. N-Boc-pyrrolidine was removed at 80–90 °C [0.02 mmHg, ABT]. The contaminated product was used for the second α-arylation without further purification. Data for 16j: bp 130 °C [0.025 mmHg, ABT]; 1H NMR (500 MHz, CDCl3) 6.68–6.54 (m, 2 H, HC(7)), 5.14 (app br s, 0.39 H, HC(1)), 5.01 (app br s, 0.61 H, HC(1)), 3.60–3.28 (m, 2 H, H2C(2)), 2.45 (s, 3 H, H3C(11)), 2.42 (s, 3 H, H3C(7)), 2.23 (app br s, 1 H, HC(2) or HC(3)), 2.10–1.97 (m, 2 H, HC(2) or HC(3)), 1.94–1.88 (m, 1 H, HC(2) or HC(3)), 1.48 (s, ∼2.5 H, H3C(7)), 1.37 (s, ∼6.5 H, H3C(7)); IR (CDCl3 film) 2975 (s), 2878 (m), 1694 (s), 1478 (m), 1453 (m), 1392 (s), 1365 (s), 1271 (m), 1255 (m), 1225 (m), 1167 (s), 1108 (s), 1039 (w), 964 (w), 916 (w), 886 (m), 796 (m), 770 (m); MS (EI, 70 eV) 267.1 (M+, 34), 211.0 (100), 196.0 (43), 166.0 (94), 152.0 (36), 139.0 (38), 124.0 (25), 111.0 (29), 97.0 (17); HRMS (EI, 70 eV) calcd for C14H21NO2S 267.12930, found 267.12882; TLC Rf 0.32 (hexanes/EtOAc, 9/1) [silica gel, UV, KMnO4]; SFC (S)-16j, tR 5.4 min (6%); (R)-16j, tR 7.7 min (94%) ((R,R)-Whelk-O1, 200 bar, 3% MeOH in CO2, 2 mL/min, 220 nm, 40 °C).
N-Boc-(R)-2-(1-naphthyl)pyrrolidine (16k)

Following general procedure 3, (−)-sparteine (1.4 mL, 6 mmol, 1.2 equiv), N-Boc-pyrrolidine 1.02 g, 6 mmol, 1.2 equiv), TBME (14 mL), s-BuLi (1.66 M in cyclohexanes, 3.65 mL, 6 mmol, 1.2 equiv), ZnCl2 (1 M in Et2O, 3 mL, 3 mmol, 0.6 equiv), 1-bromonaphthalene (0.64 mL, 5 mmol, 1.0 equiv), t-Bu3P-HBF4 (88 mg, 0.3 mmol, 0.06 equiv), and Pd(OAc)2 (55 mg, 0.24 mmol, 0.048 equiv) were combined in a 50 mL Schlenk reaction flask. After the mixture was stirred at 20 °C for 20.5 h, the reaction was quenched, filtered, subjected to aqueous washes, and then concentrated. Purification by column chromatography (SiO2, 3 × 26 cm, CH2Cl2/hexanes, 9/1) afforded 0.71 g (48%) of 16k as a pale yellow solid (a rotameric mixture, 62:38). The spectroscopic data matched those from the literature.24 Data for 16k: 1H NMR (400 MHz, CDCl3) 8.00 (d, J = 8.1 Hz, 1 H), 7.89–7.83 (m, 1 H), 7.75–7.72 (m, 1 H), 7.52–7.43 (m, 2 H), 7.41 (t, J = 7.7 Hz, 1 H), 7.26–7.24 (m, 1 H), 5.77–5.75 (m, 0.38 H), 5.61–5.60 (m, 0.62 H), 3.79–3.58 (m, 2 H), 2.49–2.41 (m, 1 H), 1.92–1.81 (m, 3 H), 1.48 (s, 3 H), 1.10 (s, 6 H); TLC Rf 0.33 (CH2Cl2/hexanes, 9/1) [silica gel, UV, KMnO4]. SFC (R)-16k, tR 6.2 min (6%); (S)-16k, tR 7.1 min (94%) (Chiralpak OJ, 200 bar, 1 mg/mL, 1–10% MeOH in CO2, 2 mL/min, 220 nm, 40 °C).
General Procedure 4: α-Arylation of N-Boc-(R)-2-arylpyrrolidine (Table 2)32,33

An oven-dried, 50 mL Schlenk reaction flask equipped with a stir bar, a septum and an internal temperature probe was added N-Boc-(R)-2-arylpyrrolidine 16 (4.4 mmol, 1.2 equiv). After three cycles of evacuation/argon fill, (−)-sparteine (1.0 mL, 4.4 mmol, 1.2 equiv), TBME (11 mL) and toluene (2.2 mL) were added. The light yellow solution was cooled to −75 °C, and s-BuLi (1.74 M in cyclohexanes, 2.5 mL, 4.4 mmol, 1.2 equiv) was added dropwise, keeping the temperature below −69 °C. (Note: s-BuLi should be added directly to the solution to avoid crystallization on the wall of the flask.) The intensively colored solution was stirred at −75 ± 1 °C for 3 h. A solution of ZnCl2 (1 M in Et2O, 2.65 mL, 2.65 mmol, 0.72 equiv) was then added to the reaction dropwise with rapid stirring, keeping the temperature below −69 °C. The yellow solution was stirred at −75 ± 1 °C for 0.5 h and then warmed to 22 °C resulting in a heterogeneous mixture. After the mixture was stirred at 22 °C for 0.5 h, the septum was secured by copper wire and the flask was brought into a glovebox. Aryl bromide (3.7 mmol, 1.0 equiv), t-Bu3P-HBF4 (64 mg, 0.22 mmol, 0.06 equiv), and Pd(OAc)2 (41 mg, 0.18 mmol, 0.048 equiv) were added to the flask with rapid stirring. In a fume hood, the flask was submerged to a 60 °C oil bath and stirred under argon. Within 2 h, the mixture turned gray. After the mixture was stirred at 60 °C for a total of 14 h, the reaction was quenched with an aqueous solution of NH4OH (30%, 0.37 mL), and stirred at room temperature for 0.5 h. The resulting dark gray mixture was filtered through Celite (2 cm deep, 30 mL, medium-porosity fritted funnel) and eluted with 50 mL of TBME. The filtrate was washed with 40 mL of 1 M HCl and then with 40 mL of deionized water. The organic layer was dried over magnesium sulfate, filtered, and concentrated to give an orange oil. Purification by column chromatography provided N-Boc-(2R,5R)-2,5-diarylpyrrolidine 22. (−)-Sparteine can be recovered as described in experimental procedure I.
Preparation of N-Boc-(2R,5R)-2,5-diarylpyrrolidine
N-Boc-(2R,5R)-2,5-bis(4-methoxyphenyl)pyrrolidine (22a)

Following general procedure 4, N-Boc-(R)-2-(4-methoxyphenyl)pyrrolidine 16a (1.22 g, 4.4 mmol, 1.2 equiv), (−)-sparteine (1.0 mL, 4.4 mmol, 1.2 equiv), TBME (11 mL) and toluene (2.2 mL), s-BuLi (1.74 M in cyclohexanes, 2.5 mL, 4.4 mmol, 1.2 equiv), ZnCl2 (1 M in Et2O, 2.65 mL, 2.65 mmol, 0.72 equiv), 4-bromoanisole (0.46 mL, 3.7 mmol, 1.0 equiv), t-Bu3P-HBF4 (64 mg, 0.22 mmol, 0.06 equiv), and Pd(OAc)2 (41 mg, 0.18 mmol, 0.048 equiv) were combined in a 50 mL Schlenk reaction flask. After the mixture was stirred at 60 °C for 14 h under argon, the reaction was quenched, filtered, subjected to aqueous washes, and then concentrated to give an orange oil. Purification by column chromatography (SiO2, 4.5 × 28 cm, hexanes/EtOAc, gradient elution, 7/1 then 4/1) afforded 364 mg (26%) of 22a as a mixture of white solid and colorless oil. Recrystallization from hexanes afforded 259 mg (18%) of 22a as colorless, star-shaped crystals. Two sets of NMR signals for the 2,5-diarylpyrrolidine moiety were observed due to hindered rotation of Boc group. Data for 22a: 1H NMR (500 MHz, CDCl3, −20 °C) 7.17 (d, J = 8.6 Hz, 2 H, HC(7)), 7.13 (d, J = 8.6 Hz, 2 H, HC(7)), 6.88 (d, J = 8.7 Hz, 2 H, HC(8)), 6.86 (d, J = 8.6 Hz, 2 H, HC(8)), 5.25 (d, J = 7.2 Hz, 1 H, HC(1)), 5.09 (d, J = 7.1 Hz, 1 H, HC(1)), 3.82 (s, 3 H, HC(10)), 3.79 (s, 3 H, HC(10)), 2.49–2.30 (m, 2 H, HC(2)), 1.72–1.63 (m, 2 H, HC(2)), 1.14 (s, 9 H, H3C(5)); 13C NMR (126 MHz, CDCl3, −20 °C) 158.1 (C(9)), 157.9 (C(9)), 154.0 (C(3)), 137.2 (C(6)), 135.9 (C(6)), 126.2 (C(7)), 126.1 (C(7)), 113.7 (C(8)), 113.2 (C(8)), 79.3 (C(4)), 61.5 (C(1)), 60.8 (C(1)), 55.2 (C(10)), 32.1 (C(2)), 31.5 (C(2)), 28.0 (C(5)); IR (CDCl3 film) 2974 (m), 2836 (m), 1693 (s), 1612 (m), 1586 (m), 1513 (s), 1462 (m), 1294 (m), 1247 (s), 1208 (m), 1174 (s), 1119 (s), 1036 (s), 977 (w), 901 (m), 829 (s), 810 (m), 779 (m), 731 (m), 647 (w), 633 (w). MS (EI, 70 eV) 383.2 (M+, 7), 326.1 (100), 282.1 (27), 255.1 (54), 220.1 (23), 193.1 (84), 176.1 (24), 148.1 (25), 134.1 (73), 121.1 (18), 57.1 (54); HRMS (ESI) calcd for C23H30NO4 [M + H] 384.2175, found 384.2175; TLC Rf 0.38 (hexanes/EtOAc, 4/1) [silica gel, UV, KMnO4]; [α]D24 +155.6 (c = 0.2, acetone).
N-Boc-(2R,5R)-2,5-bis(2-naphthyl)pyrrolidine (22f)

Following general procedure 4, N-Boc-(R)-2-(2-naphthyl)pyrrolidine 16f (1.43 g, 4.8 mmol, 1.2 equiv), (−)-sparteine (1.1 mL, 4.8 mmol, 1.2 equiv), TBME (12 mL) and toluene (2.4 mL), s-BuLi (1.74 M in cyclohexanes, 2.75 mL, 4.8 mmol, 1.2 equiv), ZnCl2 (1 M in Et2O, 2.9 mL, 2.9 mmol, 0.72 equiv), 2-bromonaphthalene (828 mg, 4.0 mmol, 1.0 equiv), t-Bu3P-HBF4 (71 mg, 0.23 mmol, 0.06 equiv), and Pd(OAc)2 (44 mg, 0.19 mmol, 0.048 equiv) were combined in a 50 mL Schlenk reaction flask. After the mixture was stirred at 60 °C for 14 h under argon, the reaction was quenched, filtered, subjected to aqueous washes, and then concentrated to give an orange oil. Purification by column chromatography (SiO2, 5.5 × 22 cm, CH2Cl2/hexanes, gradient elution, 7/3, 8/2 then 9/1) afforded 619 mg (37%) of 22f as a white solid which is contaminated by ∼6% of the corresponding pyrroline. Two sets of NMR signals for the 2,5-diarylpyrrolidine moiety were observed due to hindered rotation of Boc group. The spectroscopic data matched those from the literature.24 Data for 22f: 1H NMR (500 MHz, CDCl3, −20 °C) 7.90–7.82 (m, 6 H, HC(Aryl)), 7.71 (s, 1 H, HC(6)), 7.69 (s, 1 H, HC(6)), 7.55–7.40 (m, 6 H, HC(Aryl)), 5.58 (d, J = 7.6 Hz, 1 H, HC(1)), 5.44 (d, J = 7.8 Hz, 1 H, HC(1)), 2.61–2.50 (m, 2 H, H2C(2)), 1.87–1.77 (m, 2 H, H2C(2)), 1.13 (s, 9 H, H3C(5)); 13C NMR (126 MHz, CDCl3, −20 °C) 154.2 (C(3)), 142.2 (C(7)), 141.0 (C(7)), 133.2 (C(10) or C(15)), 133.0 ((C(10) or C(15)), 132.5 (C(10) or C(15)), 132.2 ((C(10) or C(15)), 128.4 (C(Aryl)), 128.0 (C(Aryl)), 127.8 (C(Aryl)), 127.6 (C(Aryl)), 127.6 (C(Aryl)), 127.5 (C(Aryl)), 126.1 (C(Aryl)), 125.9 (C(Aryl)), 125.4 (C(Aryl)), 125.4 (C(Aryl)), 124.2 (C(Aryl)), 124.0 (C(Aryl)), 123.2 (C(Aryl)), 79.6 (C(4)), 62.3 (C(1)), 61.7 (C(1)), 31.9 (C(2)), 31.2 (C(2)), 28.0 (C(5)); IR (CDCl3 film) 3053 (m), 2975 (m), 2247 (w), 1694 (s), 1633 (m), 1601 (m), 1508 (m), 1477(m), 1454 (m), 1383 (s), 1320 (m), 1269 (m), 1255 (m), 1171 (m), 1128 (m), 1111 (m), 1049 (w), 1018 (w), 981 (w), 961 (w), 909 (m), 855 (m), 817 (m), 780 (m), 732 (s), 646 (m). MS (EI, 70 eV) 423.2 (M+, 4), 367.1 (6), 354.1 (9), 322.1 (6), 295.1 (11), 279.1 (7), 239.1 (13), 213.1 (16), 194.1 (33), 167.0 (24), 155.0 (60), 149.0 (36), 127.0 (47), 115.0 (10), 97.1 (12), 83.1 (14), 71.1 (19); HRMS (ESI) calcd for C29H30NO2 [M + H] 424.2277, found 424.2285; TLC Rf 0.26 (CH2Cl2/hexanes, 9/1) [silica gel, UV, KMnO4]; [α]D24 +146.6 (c = 0.2, acetone).
N-Boc-(2R,5R)-2,5-bis(3,5-dimethylphenyl)pyrrolidine (22g)

Following general procedure 4, N-Boc-(R)-2-(3,5-dimethyphenyl)pyrrolidine 16g (1.28 g, 4.7 mmol, 1.2 equiv), (−)-sparteine (1.1 mL, 4.7 mmol, 1.2 equiv), TBME (12 mL) and toluene (2.4 mL), s-BuLi (1.74 M in cyclohexanes, 2.7 mL, 4.7 mmol, 1.2 equiv), ZnCl2 (1 M in Et2O, 2.8 mL, 2.8 mmol, 0.72 equiv), 1-bromo-3,5-dimethylbenzene (0.54 mL, 3.9 mmol, 1.0 equiv), t-Bu3P-HBF4 (69 mg, 0.23 mmol, 0.06 equiv), and Pd(OAc)2 (45 mg, 0.19 mmol, 0.048 equiv) were combined in a 50 mL Schlenk reaction flask. After the mixture was stirred at 60 °C for 14 h under argon, the reaction was quenched, filtered, subjected to aqueous washes, and then concentrated to give an orange oil. Purification by column chromatography (SiO2, 4.5 × 23 cm, CH2Cl2/hexanes, gradient elution, 1/1 then 2/1) afforded 390 mg (27%) of 22g as a white solid. Recrystallization from hexanes afforded 311 mg (21%) of 22g as a light pink cube. Two sets of NMR signals for the 2,5-diarylpyrrolidine moiety were observed due to hindered rotation of Boc group. Data for 22g: 1H NMR (500 MHz, CDCl3) 6.86 (br s, 2 H, HC(6)), 6.83 (br s, 2 H, HC(4)), 6.81 (br s, 2 H, HC(4)), 5.22 (d, J = 7.1, 1 H, HC(1)), 5.05 (d, J = 7.1, 1 H, HC(1)), 2.45–2.41 (m, 2 H, H2C(2)), 2.31 (s, 12 H, H3C(10)), 1.72–1.67 (m, 2 H, HC(2)), 1.15 (s, 9 H, H3C(5)); 13C NMR (126 MHz, CDCl3) 154.1 (C(3)), 145.2 (C(6)), 144.0 (C(6)), 137.8 (C(8)), 137.5 (C(8)), 128.5 (C(9)), 128.0 (C(9)), 123.2 (C(7)), 123.0 (C(7)), 79.2 (C(4)), 62.3 (C(1)), 61.7 (C(1)), 32.2 (C(2)), 31.6 (C(2)), 28.1 (C(5)), 21.5 (C(10)), 21.3 (C(10)); IR (CDCl3 film) 2974 (m), 2918 (m), 1699 (s), 1604 (m), 1476 (m), 1455 (m), 1383 (s), 1268 (w), 1255 (w), 1172 (m), 1117 (m), 1055 (w), 957 (w), 845 (m), 775 (w), 703 (m); MS (ESI) 380.3 (M + H, 28), 324.2 (100), 218.1 (6); HRMS (ESI) calcd for C25H34NO2 [M + H] 380.2590, found 380.2594; TLC Rf 0.33 (hexanes/CH2Cl2, 9/1) [silica gel, UV]; [α]D24 142.0 (c = 0.2, acetone).
N-Boc-(2R,5R)-2,5-bis(5-phenylbiphenyl-3-yl)pyrrolidine (22h)

Following general procedure 4, N-Boc-(R)-2-(5-phenylbiphenyl-3-yl)pyrrolidine 16h (2.33 g, 5.8 mmol, 1.2 equiv), (−)-sparteine (1.35 mL, 5.8 mmol, 1.2 equiv), TBME (15 mL) and toluene (3.0 mL), s-BuLi (1.74 M in cyclohexanes, 3.35 mL, 5.8 mmol, 1.2 equiv), ZnCl2 (1 M in Et2O, 3.5 mL, 3.5 mmol, 0.72 equiv), 1-bromo-3,5-diphenylbenzene (1.50 g, 4.9 mmol, 1.0 equiv), t-Bu3P-HBF4 (85 mg, 0.29 mmol, 0.06 equiv), and Pd(OAc)2 (54 mg, 0.23 mmol, 0.048 equiv) were combined in a 50 mL Schlenk reaction flask. After the mixture was stirred at 60 °C for 14 h under argon, the reaction was quenched, filtered, subjected to aqueous washes, and then concentrated to give an orange oil. Purification by column chromatography (SiO2, 4.5 × 25 cm, hexanes/EtOAc, 14/1) afforded 633 mg of 22h and contaminants. Further purification by column chromatography (SiO2, 3.5 × 25 cm, hexanes/CH2Cl2, 1/1) afforded 540 mg (18%) of 22h as a fine white powder. Two sets of NMR signals for the 2,5-diarylpyrrolidine moiety were observed due to hindered rotation of Boc group. The spectroscopic data matched those from the literature.24 Data for 22h: 1H NMR (500 MHz, CDCl3) 7.72–7.70 (m, 1 H, HC(Aryl)), 7.68–7.64 (m, 9 H, HC(Aryl)), 7.51–7.44 (m, 12 H, HC(Aryl)), 7.42–7.36 (m, 4 H, HC(Aryl)), 5.53 (d, J = 7.7 Hz, 1 H, HC(1)), 5.32 (d, J = 7.9 Hz, 1 H, HC(1)), 2.68–2.52 (m, 2 H, H2C(2)), 1.87–1.77 (dd, J = 12.7, 5.9 Hz, 2 H, HC(2)), 1.21 (s, 9 H, H3C(5)); 13C NMR (126 MHz, CDCl3) 154.1 (C(3)), 146.4 (C(6)), 144.8 (C(6)), 142.1 (C(Aryl)), 141.9 (C(Aryl)), 141.5 (C(Aryl)), 141.1 (C(Aryl)), 128.8 (C(Aryl)), 128.7 (C(Aryl)), 127.5 (C(Aryl)), 127.4 (C(Aryl)), 127.3 (C(Aryl)), 127.2 (C(Aryl)), 125.0 (C(Aryl)), 124.6 (C(Aryl)), 123.2 (C(Aryl)), 123.1 (C(Aryl)), 79.7 (C(4)), 62.6 (C(1)), 61.8 (C(1)), 32.3 (C(2)), 31.9 (C(2)), 28.2 (C(5)); IR (CDCl3 film) 3032 (w), 2974 (m), 1695 (s), 1596 (m), 1576 (m), 1497 (m), 1477 (w), 1454 (m), 1434 (m), 1385 (s), 1365 (m), 1272 (w), 1159 (m), 1120 (m), 1075 (w), 1048 (w), 1029 (w), 909 (m), 875 (m), 757 (s), 741 (s), 698 (s). MS (EI, 70 eV) 627.4 (M+, 6), 570.3 (34), 526.3 (46), 499.3 (38), 446.2 (60), 315.2 (66), 271.2 (100), 256.1 (80), 241.1 (21), 56.1 (78); HRMS (ESI) calcd for C45H42NO2 [M + H] 628.3216, found 628.3221; TLC Rf 0.12 (hexanes/CH2Cl2, 1/1) [silica gel, UV]; [α]D24 +49.6 (c = 0.2, acetone).
N-Boc-(2R,5R)-2,5-bis(2-tolyl)pyrrolidine (22i)

Following general procedure 4, N-Boc-(R)-2-(2-tolyl)pyrrolidine 16i (1.68 g, 6.4 mmol, 1.2 equiv), (−)-sparteine (1.5 mL, 6.4 mmol, 1.2 equiv), TBME (16 mL) and toluene (3.2 mL), s-BuLi (1.74 M in cyclohexanes, 3.7 mL, 6.4 mmol, 1.2 equiv), ZnCl2 (1 M in Et2O, 3.9 mL, 3.9 mmol, 0.72 equiv), 2-bromotoluene (0.64 mL, 5.4 mmol, 1.0 equiv), t-Bu3P-HBF4 (95 mg, 0.32 mmol, 0.06 equiv) and Pd(OAc)2 (59 mg, 0.26 mmol, 0.048 equiv) were combined in a 100 mL Schlenk reaction flask. After the mixture was stirred 60 °C for 14 h under argon, the reaction was quenched, filtered, subjected to aqueous washes, and then concentrated to give an orange oil. Purification by column chromatography (SiO2, 4.5 × 30 cm, hexanes/EtOAc, 92/8) afforded 413 mg (22%) of 22i as a white solid. Recrystallization from hexanes afforded 320 mg (17%) of 22i as a light pink cube. Two sets of NMR signals for the 2,5-diarylpyrrolidine moiety were observed due to hindered rotation of Boc group. Data for 22i: 1H NMR (500 MHz, CDCl3) 7.23–7.14 (m, 8 H, HC(Aryl)), 5.50 (d, J = 8.0 Hz, 1 H, HC(1)), 5.37 (d, J = 8.2 Hz, 1 H, HC(1)), 2.52–2.36 (m, 2 H, H2C(2)), 2.42 (s, 3 H, H3C(12)), 2.39 (s, 3 H, H3C(12)), 1.64 (dd, J = 12.1, 6.3 Hz, 2 H, H2C(2)), 1.13 (s, 6 H, H3C(5)), 13C NMR (126 MHz, CDCl3) 153.7 (C(3)), 143.0 (C(Aryl)), 141.5 (C(Aryl)), 134.2 (C(Aryl)), 133.7 (C(Aryl)), 130.8 (C(Aryl)), 130.1 (C(Aryl)), 126.6 (C(Aryl)), 126.4 (C(Aryl)), 125.8 (C(Aryl)), 125.7 (C(Aryl)), 124.3 (C(Aryl)), 123.7 (C(Aryl)), 79.2 (C(4)), 59.1 (C(1)), 58.9 (C(1)), 30.0 (C(2)), 29.5 (C(2)), 28.0 (C(5)), 19.3 (C(12)), 19.3 (C(12)); IR (CDCl3 film) 3066 (w), 3017 (w), 2973 (m), 2871 (w), 1698 (m), 1604 (w), 1485 (w), 1461 (w), 1383 (m), 1285 (w), 1250 (w), 1180 (m), 1159 (m), 1128 (m), 1101 (w), 976 (w), 792 (w), 752 (w). MS (EI, 70 eV) 351.2 (M+, 5), 295.2 (100), 223.2 (71), 164.1 (57), 119.1 (69); HRMS (ESI) calcd for C23H30NO2 [M + H] 352.2277, found 352.2278; TLC Rf 0.33 (hexanes/EtOAc, 92/8) [silica gel, UV, KMnO4]; [α]D24 +126.4 (c = 0.2, acetone).
N-Boc-(2R,5R)-2,5-bis(5-methyl-2-thienyl)pyrrolidine (22j)

Following general procedure 4, N-Boc-(R)-2-(5-methyl-2-thienyl)pyrrolidine 16j (1.94 g, 7.3 mmol, 1.2 equiv), (−)-sparteine (1.7 mL, 7.3 mmol, 1.2 equiv), TBME (18 mL) and toluene (3.6 mL), s-BuLi (1.465 M in cyclohexanes, 5.0 mL, 7.3 mmol, 1.2 equiv), ZnCl2 (1 M in Et2O, 4.35 mL, 4.35 mmol, 0.72 equiv), 2-bromo-5-methylthiophene (0.59 mL, 6.1 mmol, 1.0 equiv), t-Bu3P-HBF4 (108 mg, 0.36 mmol, 0.06 equiv), and Pd(OAc)2 (68 mg, 0.29 mmol, 0.048 equiv) were combined in a 100 mL Schlenk reaction flask. After the mixture was stirred 60 °C for 18 h under argon, the reaction was quenched, filtered, subjected to aqueous washes and then concentrated to give an orange oil. Purification by column chromatography (SiO2, 4.5 × 30 cm, CH2Cl2/hexanes, 3/2) afforded 440 mg (20%) of 22j as a tanned solid. Recrystallization from hexanes afforded 291 mg (13%) of 22j as a white fluffy needle. Two sets of NMR signals for the 2,5-diarylpyrrolidine moiety were observed due to hindered rotation of Boc group. Data for 22j: 1H NMR (500 MHz, CDCl3) 6.68 (d, J = 3.2 Hz, 1 H, HC(7)), 6.61 (d, J = 3.2 Hz, 1 H, HC(7)), 6.57–6.54 (m, 2 H, HC(8)), 5.32 (d, J = 6.9 Hz, 1 H, HC(1)), 5.15 (d, J = 6.9 Hz, 1 H, HC(1)), 2.59–2.49 (m, 2 H, H2C(2)), 2.44 (s, 3 H, H3C(7)), 2.42 (s, 3 H, H3C(7)), 1.94–1.84 (m, 2 H, H2C(2)), 1.26 (s, 9 H, H3C(5)); 13C NMR (126 MHz, CDCl3) 153.7 (C(3)), 146.3 (C(6)), 145.4 (C(6)), 137.6 (C(9)), 137.5 (C(9)), 124.7 (C(8)), 124.1 (C(8)), 123.0 (C(7)), 122.9 (C(7)), 79.7 (C(4)), 57.6 (C(1)), 56.9 (C(1)), 32.9 (C(2)), 31.9 (C(2)), 28.2 (C(5)), 15.3 (C(10)); IR (CDCl3 film) 3059 (w), 2977 (m), 2920 (w), 2871 (w), 1692 (s), 1478 (w), 1441 (w), 1374 (s), 1363 (s), 1298 (w), 1272 (m), 1227 (w), 1174 (m), 1112 (m), 1049 (w), 965 (w), 893 (w), 795 (m). MS (EI, 70 eV) 363.3 (M+, 14), 306.2 (26), 209.2 (28), 183.1 (68), 164.1 (37), 124.1 (44), 54.9 (100); HRMS (ESI) calcd for C19H26NO2S2 [M + H] 364.1405, found 364.1407; TLC Rf 0.24 (hexanes/CH2Cl2, 4/6) [silica gel, UV, KMnO4]; [α]D24 +148.2 (c = 0.2, acetone).
General Procedure 5: Iodotrimethylsilane-Assisted Boc Removal (Table 3)41a,41b

To an oven-dried, 25 mL, round-bottom flask equipped with a stir bar, an argon inlet adaptor, and a septum was added N-Boc-(2R,2R)-2,5-diarylpyrrolidine 22 (0.67 mmol, 1.0 equiv). After three cycles of evacuation and argon fill, dichloromethane (6.7 mL) was added. The colorless solution was cooled to 0 °C, and iodotrimethylsilane (105 μL, 0.7 mmol, 1.05 equiv) was added. The resulting light yellow solution was stirred at 0 or 22 °C for 1 or 2 h and then cannulated into an aqueous solution prepared from saturated NaHCO3/saturated Na2S2O3 (1:1, 20 mL). After vigorous stirring for 10 min, the aqueous layer was extracted with dichloromethane (10 mL × 4). The combined organic extract was dried over Na2SO4, filtered, and concentrated. Purification by column chromatography afforded (2R,2R)-2,5-diarylpyrrolidine 23.
General Procedure 6: Acid-Assisted Boc Removal (Table 3)24

To an oven-dried, 50 mL, one-necked, round-bottom flask, equipped with a stir bar, an argon inlet adaptor, and a septum was added N-Boc-(2R,2R)-2,5-diarylpyrrolidine 22 (0.77 mmol, 1.0 equiv). After three cycles of evacuation and argon fill, dichloromethane (7.7 mL) was added to give a slightly colored solution. Trifluoroacetic acid (1.2 mL, 15.6 mmol, 20 equiv) was added at 0 °C to give an orange solution, and the reaction mixture was stirred under argon at 0 or 22 °C for 2 to 6 h. The solvent was removed under reduced pressure. The brown oil was taken up by EtOAc (20 mL) and washed a 2 M solution of NaOH (20 mL). The organic phase was saved, and the aqueous layer was extracted with EtOAc (20 mL × 2). The combined organic extracts was dried over Na2SO4, filtered, and concentrated to give a lightly tanned oil. Purification by column chromatography afforded (2R,2R)-2,5-diarylpyrrolidine 23.
Preparation of (R)-2-Arylpyrrolidine
(R)-2-(4-Methoxyphenyl)pyrrolidine (17a)

To an oven-dried, 25 mL Schlenk reaction flask equipped with a stir bar, a septum, and an internal temperature probe was added N-Boc-(R)-2-(4-methoxyphenyl)pyrrolidine (16a) (330 mg, 1.2 mmol, 1.0 equiv). After three cycles of evacuation and argon fill, dichloromethane (6 mL) was added. The colorless solution was cooled to 0 °C, and TMSI (185 μL, 1.26 mmol, 1.05 equiv) was added dropwise. The resulting light brown solution was stirred at 0 °C for 1 h and then cannulated into an aqueous solution prepared from saturated NaHCO3/saturated Na2S2O3 (1:1, 20 mL). After vigorous stirring for 10 min, the aqueous layer was extracted with dichloromethane (10 mL × 3). The combined organic extract was dried over Na2SO4, filtered, and concentrated to give 228 mg (93%) of 17a as a light yellow oil with good purity based on 1H NMR analysis. The crude product was used without further purification. The spectroscopic data matched those from the literature.67 Data for 17a: 1H NMR (500 MHz, CDCl3) 7.28 (d, J = 8.7 Hz, 2 H, HC(6)), 6.85 (d, J = 8.7 Hz, 2 H, HC(7)), 4.06 (dd, J = 8.7, 6.9 Hz, 1 H, HC(1)), 3.79 (s, 3 H, H3C(9)), 3.16 (ddd, J = 10.4, 7.9, 5.5 Hz, 1 H, HC(4)), 3.10 (br s, 1 H, NH), 2.96 (ddd, J = 10.4, 8.4, 6.5 Hz, 1 H, HC(4)), 2.15 (dddd, J = 12.6, 8.3, 6.9, 4.5 Hz, 1 H, HC(2)), 1.99–1.78 (m, 2 H, H2C(3)), 1.67 (dtd, J = 12.4, 9.1, 7.8 Hz, 1 H, HC(2)); 13C NMR (126 MHz, CDCl3) 158.6 (C(8)), 135.7 (C(5)), 127.7 (C(6)), 113.8 (C(7)), 62.1 (C(1)), 55.2 (C(9)), 46.6 (C(4)), 33.9 (C(2)), 25.4 (C(3)). IR (neat) 3331 (w), 2957 (m), 2871 (m), 2834 (m), 1612 (m), 1584 (w), 1512 (s), 1462 (m), 1441(m), 1396 (m), 1300 (m), 1246 (s), 1179 (m), 1106 (w), 1035 (m), 904 (w), 828 (m), 730 (w). MS (EI, 70 eV) 177.1 (M+, 42), 176.1 (60), 149.1 (43), 148.1 (100), 134.0 (23), 118.1 (11), 91.1 (7), 77.1 (8), 70.1 (16); HRMS (ESI) calcd for C11H16NO [M + H] 178.1232, found 178.1238.
(R)-2-(2-Naphthyl)pyrrolidine (17f)

To an oven-dried, 25 mL, two-necked, round-bottom flask, equipped with a stir bar, a septum, and an argon inlet was added N-Boc-(R)-2-(2-naphthyl)pyrrolidine 16f (356 mg, 1.2 mmol, 1.0 equiv). After three cycles of evacuation and argon fill, dichloromethane (1.8 mL) was added to give a pale yellow solution. Trifluoroacetic acid (460 μL, 6 mmol, 5 equiv) was added at 22 °C, and the reaction was stirred under argon for 8.5 h. The reaction was cooled to 0 °C and quenched with an aqueous solution of NH4OH (30%, 6 mL), resulting in the formation of white fume and white solid. After the solution was stirred at room temperature for 5 min, all solid dissolved. The organic phase was saved, and the aqueous phase was extracted with dichloromethane (3 mL × 4). The combined organic extract was dried over Na2SO4, filtered and concentrated to afford 230 mg (97%) of 17f as a pale yellow oil with good purity based on NMR analysis. The crude product was used without further purification. Data for 17f: 1H NMR (500 MHz, CDCl3) 7.83–7.80 (m, 4 H, HC(Aryl)), 7.51–7.42 (m, 3 H, HC(Aryl)), 4.29 (t, J = 7.7, 3.6 Hz, 1 H, HC(1)), 3.26 (ddd, J = 10.1, 7.8, 5.2 Hz, 1 H, HC(4)), 3.08 (ddd, J = 10.1, 8.3, 6.7 Hz, 1 H, HC(4)), 2.30–2.20 (m, 2 H, HC(2) and NH), 2.02–1.85 (m, 2 H, H2C(3)), 1.81–1.73 (m, 1 H, HC(2)); 13C NMR (126 MHz, CDCl3) 142.2 (C(6)), 133.4 (C(9) or C(14)), 132.6 ((C(14) or C(9)), 128.0 (C(Aryl)), 127.7 (C(Aryl)), 127.5 (C(Aryl)), 125.9 (C(Aryl)), 125.4 (C(Aryl)), 125.2 (C(5) or C(7)), 124.6 (C(5) or C(7)), 61.6 (C(1)), 47.0 (C(4)), 34.3 (C(2)), 25.6 (C(3)). IR (neat) 3344 (w), 3282 (w), 3053 (m), 2962 (m), 2869 (m), 1676 (w), 1632 (w), 1600 (w), 1508 (w), 1455 (w), 1441 (w), 1399 (w), 1320 (w), 1269 (w), 1240 (w), 1198 (w), 1174 (w), 1123 (w), 1099 (w), 1018 (w), 962 (w), 945 (w), 892 (w), 855 (m), 818 (m), 746 (m); MS (ESI) 198.1 (M + H, 100), 181.1 (73); HRMS (ESI) calcd for C14H16N [M + H] 198.1283, found 198.1289.
Preparation of (2R,5R)-2,5-Diarylpyrrolidines
(2R,5R)-2,5-Bis(4-methoxyphenyl)pyrrolidine (23a)

Following general procedure 5, a mixture of N-Boc-(2R,5R)-2,5-bis(4-methoxyphenyl)pyrrolidine 22a (256 mg, 0.67 mmol, 1.0 equiv), CH2Cl2 (6.7 mL), and iodotrimethylsilane (105 μL, 0.7 mmol, 1.05 equiv) was stirred in a 25 mL round-bottom flask at 0 °C for 1 h under argon and then quenched into a 1:1 mixture of saturated NaHCO3/saturated Na2S2O3. The product was extracted into CH2Cl2, dried and concentrated. Purification by column chromatography (SiO2, 3.5 × 21 cm, CH2Cl2/MeOH, 98/2) afforded 156 mg (82%) of 23a as a slightly tanned, fluffy, needle. Data for 23a: 1H NMR (500 MHz, CDCl3) 7.33 (d, J = 8.6 Hz, 4 H, HC(4)), 6.88 (d, J = 8.6 Hz, 4 H, HC(5)), 4.49 (d, J = 7.0 Hz, 2 H, HC(1)), 3.81 (s, 3 H, H3C(7)), 2.43–2.29 (m, 2 H, HC(2)), 2.18 (s, 1 H, NH), 1.97–1.80 (m, 2 H, HC(2)); 13C NMR (126 MHz, CDCl3) 158.5 (C(6)), 137.7 (C(3)), 127.4 (C(4)), 113.8 (C(5)), 61.6 (C(1)), 55.3 (C(7)), 35.5 (C(2)); IR (CDCl3 film) 3004 (w), 2959 (m), 2935 (m), 2873 (m), 2835 (m), 1611 (m), 1584 (m), 1512 (s), 1457 (m), 1443 (m), 1424 (m) 1398 (m), 1353 (w), 1277 (m), 1243 (s), 1178 (s), 1088 (s), 1031 (s), 815 (s), 783 (m), 693 (m), 663 (m). MS (EI, 70 eV) 283.2 (M+, 23), 255.1 (100), 240.1 (28), 148.1 (17), 134.1 (21), 121.1 (14), 91.1 (24), 77.1 (14); HRMS (ESI) calcd for C18H22NO2 [M + H] 284.1651, found 284.1656; TLC Rf 0.38 (CH2Cl2/MeOH, 95/5) [silica gel, KMnO4]; [α]D24 +121.4 (c = 0.2, chloroform).
(2R,5R)-2,5-Bis(2-naphthyl)pyrrolidine (23f)

Following general procedure 6, a mixture of N-Boc-(2R,5R)-2,5-bis(2-naphthyl)pyrrolidine 22f (409 mg, 0.97 mmol, 1.0 equiv), CH2Cl2 (9.7 mL), and trifluoroacetic acid (1.5 mL, 19.4 mmol, 20 equiv) was stirred in a 50 mL round-bottom flask at 0 °C for 6 h under argon. The solvent was removed under reduced pressure to give a brown oil and EtOAc (20 mL) was added to give a heterogeneous mixture. A 2 M solution of NaOH (20 mL) and Et3N (1 mL) was added and vigorously stirred until no solid was visible. The organic layer was saved, and the aqueous layer was extracted with EtOAc (20 mL × 2). The combined organic extracts were dried over Na2SO4, filtered, and concentrated to give a mixture of white and brown solid. This mixture was taken up by dichloromethane (15 mL), filtered through a pad of Celite (1 cm deep, 30 mL size, medium-porosity fritted funnel), eluted with dichloromethane (40 mL), and concentrated to give a tanned solid. The crude product was taken up by a minimal amount of dichloromethane and loaded onto a column packed with silica (SiO2, 3 × 29 cm) in hexanes. Elution with EtOAc/hexanes (9/1) afforded 255 mg (81%) of 23f as a white solid contaminated with a small amount of 2,5-di(2-naphthyl)-2,5-dihydro-1H-pyrrole31 (<3%) based on NMR analysis. The spectroscopic data matched those from the literature.33 Data for 23f: 1H NMR (500 MHz, CDCl3) 7.90–7.83 (m, 8 H, HC(Aryl)), 7.60 (dd, J = 8.5, 1.8 Hz, 2 H, HC(Aryl)), 7.52–7.44 (m, 4 H, HC(Aryl)), 4.81 (t, J = 6.8 Hz, 2 H, HC(1)), 2.59–2.47 (m, 2 H, H2C(2)), 2.42 (br s, 1 H, NH), 2.12–2.01 (m, 2 H, H2C(2)); 13C NMR (126 MHz, CDCl3) 143.2 (C(4)), 133.4 (C(7) or C(12)), 132.7 ((C(7) or C(12)), 128.3 (C(Aryl)), 127.8 (C(Aryl)), 127.6 (C(Aryl)), 126.0 (C(Aryl)), 125.5 (C(Aryl)), 125.0 (C(Aryl)), 124.5 (C(Aryl)), 62.5 (C(1)), 35.5 (C(2)); IR (CDCl3 film) 3355 (w), 3052 (m), 2960 (m), 2868 (m), 1632 (w), 1599 (m), 1507 (m), 1442 (m), 1399 (w), 1366 (w), 1314 (w), 1273 (w), 1173 (w), 1122 (m), 1085 (m), 1017 (w), 948 (w), 906 (m), 860 (m), 820 (s), 789 (w), 745 (s), 694 (w), 650 (w); MS (ESI) 324.2 (M + H, 100), 307.1 (4), 179.1 (8), 165.1 (7), 141.1 (4); HRMS (ESI) calcd for C24H22N [M + H] 324.1752, found 324.1758; TLC Rf 0.34 (hexanes/EtOAc, 4/1) [silica gel, UV, KMnO4]; [α]D24 +144.8 (c = 0.2, chloroform).
(2R,5R)-2,5-Bis(3,5-dimethylphenyl)pyrrolidine (23g)

Following general procedure 6, a mixture of N-Boc-(2R,5R)-2,5-bis(3,5-dimethylphenyl)pyrrolidine 22g (290 mg, 0.77 mmol, 1.0 equiv), CH2Cl2 (7.7 mL), and trifluoroacetic acid (1.2 mL, 15.4 mmol, 20 equiv) was stirred in a 50 mL round-bottom flask at 0 °C for 2 h under argon. The solvent was removed under reduced pressure. The brown oil was taken up by EtOAc (20 mL) and washed a 2 M solution of NaOH (20 mL). The organic phase was saved and the aqueous layer was extracted with EtOAc (20 mL × 2). The combined organic extracts was dried over Na2SO4, filtered and concentrated to give a lightly tanned oil. Purification by column chromatography (SiO2, 1.5 × 18 cm, CH2Cl2/MeOH, 98/2) afforded 183 mg (86%) of 23g as a pale yellow oil which became a slightly tanned solid over time. Data for 23g: 1H NMR (500 MHz, CDCl3) 7.14 (s, 4 H, HC(4)), 6.97 (s, 2 H, HC(6)), 4.58 (d, J = 7.6, 2 H, HC(1)), 2.50–2.43 (m, 2 H, H2C(2)), 2.43 (s, 12 H, H3C(7)), 2.15 (br s, 1 H, NH), 2.08–2.02 (m, 2 H, H2C(2)); 13C NMR (126 MHz, CDCl3) 145.7 (C(3)), 137.8 (C(5)), 128.3 (C(6)), 124.0 (C(4)), 62.1 (C(1)), 35.5 (C(2)), 21.3 (C(7)); IR (CDCl3 film) 3367 (w), 3011 (s), 2916 (s), 2863 (s), 1767 (w), 1731 (w), 1681 (s), 1604 (s), 1462 (s), 1406 (m), 1376 (m), 1334 (m), 1308 (m), 1253 (w), 1153 (m), 1097 (m), 1036 (m), 949 (w), 894 (w), 846 (s), 815 (m), 760 (m), 701 (s); MS (ESI) 280.2 (M + H, 100), 119.1 (6); HRMS (ESI) calcd for C20H26N [M + H] 280.2065, found 280.2072; TLC Rf 0.16 (CH2Cl2/MeOH, 98/2) [silica gel, KMnO4]; [α]D24 +108.5 (c = 0.2, chloroform).
(2R,5R)-2,5-Bis(5-phenylbiphenyl-3-yl)pyrrolidine (23h)

Following general procedure 6, a mixture of N-Boc-(2R,5R)-2,5-bis(5-phenylbiphenyl-3-yl)pyrrolidine 22h (316 mg, 0.5 mmol, 1.0 equiv), CH2Cl2 (5.0 mL), and trifluoroacetic acid (0.77 mL, 10 mmol, 20 equiv) was stirred in a 25 mL round-bottom flask at 0 °C for 6 h under argon. The solvent was removed under reduced pressure, and the orange oil was subjected to basic aqueous workup to give a foamy solid. Purification by column chromatography (SiO2, 3 × 15 cm, CH2Cl2/MeOH, gradient elution, 100/0 then 98/2) afforded 248 mg (93%) of 23h as a white solid after swirling in a small amount of hexanes (∼2 mL) and removal of the pale yellow liquid. The spectroscopic data matched those from the literature.33 Data for 23h: 1H NMR (500 MHz, CDCl3) 7.74–7.67 (m, 14 H, HC(4), HC(6), HC(8)), 7.50 (t, J = 7.6 Hz, 8 H, HC(9)), 7.40 (t, J = 7.4 Hz, 4 H, HC(9)), 4.78 (t, J = 6.8 Hz, 2 H, HC(1)), 2.60–2.51 (m, 2 H, H2C(2)), 2.19 (br s, 1 H, NH), 2.13–2.04 (m, 2 H, H2C(2)); 13C NMR (126 MHz, CDCl3) 147.0 (C(3)), 142.0 (C(5)), 141.2 ((C(7)), 128.7 (C(9)), 127.4 (C(10)), 127.3 (C(8)), 124.8 (C(6)), 124.2 (C(4)), 62.4 (C(1)), 35.7 (C(2)); IR (CDCl3 film) 3365 (w), 3033 (m), 2961 (w), 2864 (w), 1947 (w), 1882 (w), 1809 (w), 1764 (w), 1595 (s), 1576 (m), 1497 (m), 1455 (m), 1435 (m), 1410 (m), 1354 (w), 1309 (w), 1247 (w), 1180 (w), 1157 (w), 1105 (w), 1076 (m), 1029 (m), 908 (s), 878 (m), 758 (s), 731 (s), 698 (s); MS (ESI) 528.3 (M + H, 100); HRMS (ESI) calcd for C40H34N [M + H] 528.2691, found 528.2693; TLC Rf 0.67 (CH2Cl2/MeOH, 98/2) [silica gel, UV]; [α]D24 +73.3 (c = 0.2, chloroform).
(2R,5R)-2,5-Bis(2-tolyl)pyrrolidine (23i)

Following general procedure 5, a mixture of N-Boc-(2R,5R)-2,5-bis(2-tolyl)pyrrolidine 22i (312 mg, 0.89 mmol, 1.0 equiv), CH2Cl2 (8.9 mL), and iodotrimethylsilane (140 μL, 0.93 mmol, 1.05 equiv) was stirred in a 25 mL round-bottom flask at 0 °C for 2 h under argon and then quenched into a 1:1 mixture of saturated NaHCO3/saturated Na2S2O3. The product was extracted into CH2Cl2, dried and concentrated. Purification by column chromatography (SiO2, 1.5 × 16 cm, CH2Cl2/MeOH, 98/2) afforded 216 mg (97%) of 23i as a pale orange oil which became a pale orange solid in the freezer over time. Data for 23i: 1H NMR (500 MHz, CDCl3) 7.61 (d, J = 7.6 Hz, 2 H, HC(8)), 7.29–7.24 (m, 2 H, HC(7)), 7.21–7.16 (m, 4 H, HC(6) and HC(5)), 4.81 (t, J = 6.3 Hz, 2 H, HC(1)), 2.48–2.41 (m, 2 H, H2C(2)), 2.44 (s, 6 H, H3C(9)), 1.85–1.77 (m, 2 H, H2C(2)); 13C NMR (126 MHz, CDCl3) 144.0 (C(3)), 135.3 (C(4)), 130.3 (C(5) or C(6)), 126.4 (C(7)), 126.1 (C(8)), 124.6 (C(8)), 58.5 (C(1)), 33.7 (C(2)), 19.5 (C(9)). IR (neat) 3353 (w), 3059 (m), 3018 (m), 2959 (m), 2864 (m), 1602 (w), 1484 (m), 1460 (m), 1380 (m), 1348 (w), 1278 (w), 1213 (w), 1177 (w), 1157 (w), 1139 (w), 1115 (w), 1081 (m), 1048 (m), 945 (w), 873 (w), 754 (s), 723 (m); MS (ESI) 252.2 (M + H, 100), 235.1 (10), 105.1 (16); HRMS (ESI) calcd for C18H22N [M + H] 252.1752, found 252.1744; TLC Rf 0.37 (CH2Cl2/MeOH, 98/2) [silica gel, KMnO4]; [α]D24 +143.5 (c = 0.2, chloroform).
(2R,5R)-2,5-Bis(5-methyl-2-thienyl)pyrrolidine (23j)

To a 10 mL, 1-necked round-bottom flask attached to a Kugelrohr bulb and a Kugelrohr shaft was added N-Boc-(2R,5R)-2,5-bis(5-methyl-2-thienyl)pyrrolidine 22j (210 mg, 0.58 mmol). After two cycles of evacuation and backfill with argon, the flask was heated to 200 °C (ABT). After 18 h, the flask was cooled to room temperature. The product was rinsed into the round-bottom flask with Et2O then concentrated to give a brown oil. Purification by column chromatography (SiO2, 1.5 × 18 cm, CH2Cl2/MeOH, 98/2; then SiO2, 1.0 × 23 cm, CH2Cl2/MeOH, 98/2) afforded ∼97 mg (∼64%) of 23j as a brown oil with a contaminant. This material was used without further purification. Data for 23j: 1H NMR (500 MHz, CDCl3) 6.70 (d, J = 3.3 Hz, 2 H, HC(4)), 6.58–6.56 (m, 2 H, HC(5)), 4.66 (dd, J = 6.2, 4.7 Hz, 2 H, HC(1)), 2.45 (s, 6 H, H3C(7)), 2.42–2.35 (m, 2 H, H2C(2)), 1.98–1.89 (m, 2 H, H2C(2)); TLC Rf 0.17 (CH2Cl2/MeOH, 98/2) [silica gel, UV, KMnO4].
General Procedure 7: Nitrosation of (2R)-Arylpyrrolidine or (2R,5R)-Diarylpyrrolidine (Table 3)42
To a an oven-dried, 15 mL, one-necked, round-bottom flask equipped with a stir bar and an argon inlet adaptor was charged (2R,2R)-2,5-diarylpyrrolidine 23 (0.39 mmol, 1.0 equiv). After two cycles of evacuation/argon fill, dichloromethane (2.0 mL) and pyridine (64 μL, 0.78 mmol, 2 equiv) were added. The light yellow solution was cooled to 0 °C, and nitrosonium tetrafluoroborate (182 mg, 1.5 mmol, 2 equiv) was added. Under a slight positive argon pressure, the septum was temporarily removed to allow the addition of nitrosonium tetrafluoroborate (94 mg, 0.78 mmol, 2 equiv) in one portion. The resulting mixture was stirred at 0 or 22 °C for 2 to 20 h, and an aqueous solution of HCl (1 M, 2 mL) was added. After the mixture was stirred for 3 min, the aqueous layer was extracted with dichloromethane (1.5 mL × 4). The combined organic extract was dried over Na2SO4, filtered, and concentrated to give a light yellow solid. Purification by column chromatography afforded N-nitroso-(2R,5R)-diarylpyrrolidine 24.
Preparation of N-Nitroso-(R)-2-arylpyrrolidines
N-Nitroso-(R)-2-(4-methoxyphenyl)pyrrolidine (18a)

To a an oven-dried, 15 mL, one-necked, round-bottom flask equipped with a stir bar, an argon inlet adaptor, and a septum was charged (R)-2-(4-methoxyphenyl)pyrrolidine 17a (205 mg, 1.16 mmol, 1.0 equiv). After two cycles of evacuation/argon fill, dichloromethane (5.8 mL) was added. The light yellow solution was cooled to 0 °C, and pyridine (190 μL, 2.31 mmol, 2 equiv) was added. Under a slight positive argon pressure, the septum was temporarily removed to allow the addition of nitrosonium tetrafluoroborate (277 mg, 2.31 mmol, 2 equiv) in one portion. The resulting orange solution was stirred at 0 °C for 1.5 h and then slowly poured into ice-cold HCl (1 M, 20 mL). After fizzing subsided, the aqueous layer was extracted with dichloromethane (10 mL × 3). The combined organic extract was dried over Na2SO4, filtered, and concentrated. Purification by column chromatography (SiO2, 1.5 × 16 cm, hexanes/EtOAc, gradient elution, 9/1 then 4/1) afforded 148 mg (62%) of 18a as a yellow solid (a rotameric mixture, 76:24, 21 °C). Data for 18a: 1H NMR (500 MHz, CDCl3) major 7.15 (d, J = 8.7 Hz, 2 H, HC(6)), 6.89 (d, J = 8.7 Hz, 2 H, HC(7)), 5.60 (t, J = 6.3 Hz, 1 H, HC(1)), 3.85 (dt, J = 15.0, 7.6 Hz, 1 H, HC(4)), 3.80 (s, 3 H, H3C(9)), 3.73–3.64 (m, 1 H, HC(4)), 2.50–2.40 (m, 1 H, HC(2)), 2.18–2.10 (m, 1 H, HC(2)), 2.10–1.92 (m, 2 H, H2C(3)), minor 6.98 (d, J = 8.7 Hz, 2 H, HC(6)), 6.82 (d, J = 8.7 Hz, 2 H, HC(7)), 5.23 (t, J = 6.5 Hz, 1 H, HC(1)), 4.64–4.57 (m, 1 H, HC(4)), 4.41–4.35 (m, 1 H, HC(4)), 3.77 (s, 3 H, H3C(9)), 2.41–2.36 (m, 1 H, HC(2)), 2.18–1.92 (m, 3 H, HC(2) and H2C(3)); 13C NMR (126 MHz, CDCl3) major 159.2 (C(8)), 132.5 (C(5)), 127.6 (C(6)), 114.2 (C(7)), 64.1 (C(1)), 55.3 (C(9)), 45.9 (C(4)), 33.4 (C(2)), 20.8 (C(3)), minor 158.7 (C(8)), 131.6 (C(5)), 126.7 (C(6)), 114.0 (C(7)), 59.8 (C(1)), 55.3 (C(9)), 50.7 (C(4)), 33.3 (C(2)), 22.6 (C(3)); IR (CDCl3 film) 2956 (w), 2836 (w), 1611 (m), 1585 (w), 1514 (s), 1454 (w), 1410 (m), 1296 (s), 1249 (s), 1178 (m), 1113 (w), 1031 (m), 828 (m), 808 (w), 773 (w); MS (ESI) 229.1 (17, M + Na), 207.1 (100, M), 161.1 (57), 137.0 (66), 99.0 (22); HRMS (ESI) calcd for C11H15N2O2 [M + H] 207.1134, found 207.1143; TLC Rf 0.16 (hexanes/EtOAc, 4/1) [silica gel, UV, KMnO4]; [α]D24 +129.6 (c = 0.2, acetone).
N-Nitroso-(R)-2-(2-naphthyl)pyrrolidine (18f)

To a an oven-dried, 25 mL, one-necked, round-bottom flask equipped with a stir bar, an argon inlet adaptor, and a septum was charged (R)-2-(2-naphthyl)pyrrolidine 17f (230 mg, 1.17 mmol, 1.0 equiv). After three cycles of evacuation/argon fill, dichloromethane (5.8 mL) was added. The colorless solution was cooled to 0 °C, and pyridine (100 μL, 1.22 mmol, 1.05 equiv) was added. Under a slight positive argon pressure, the septum was temporarily removed to allow the addition of nitrosonium tetrafluoroborate (147 mg, 1.22 mmol, 1.05 equiv) in one portion. The resulting bright yellow solution was stirred at 22 °C for 1.5 h. The resulting cloudy mixture was cooled to 0 °C, and an aqueous solution of HCl (1 M, 6 mL) was added slowly. After the solution was stirred for 3 min, bubbling subsided. The organic layer was saved, and the aqueous layer was extracted with dichloromethane (5 mL × 3). The combined organic extract was dried over Na2SO4, filtered, and concentrated to give a mixture of yellow oil and solid. Purification by column chromatography (SiO2, 3 × 20 cm, hexanes/EtOAc, 4/1) afforded 203 mg (77%) of 18f as a fluffy, white solid (a rotameric mixture, 77:23, 21 °C). Data for 18f: 1H NMR (500 MHz, CDCl3) major 7.86 (d, J = 8.5 Hz, 1 H, HC(8)), 7.86–7.78 (m, 2 H, HC(10) and HC(13)), 7.62 (s, 1 H, HC(5)), 7.53–7.43 (m, 2 H, HC(11) and HC(12)), 7.34 (d, J = 8.5 Hz, 1 H, HC(7)), 5.85 (t, J = 6.1 Hz, 1 H, HC(1)), 3.95–3.88 (m, 1 H, HC(4)), 3.83–3.76 (m, 1 H, HC(4)), 2.58–2.49 (m, 1 H, HC(2)), 2.30–2.23 (m, 1 H, HC(2)), 2.14–1.98 (m, 2 H, H2C(3)), minor 7.86–7.78 (m, 2 H, HC(10) and HC(13)), 7.76 (d, J = 8.0 Hz, 1 H, HC(8)), 7.53–7.43 (m, 2 H, HC(11) and HC(12)), 7.45 (s, 1 H, HC(5)), 7.19 (d, J = 8.6 Hz, 1 H, HC(7)), 5.43 (t, J = 6.8 Hz, 1 H, HC(1)), 4.73–4.67 (m, 1 H, HC(4)), 4.52–4.46 (m, 1 H, HC(4)), 2.54–2.45 (m, 1 H, HC(2)), 2.21–2.13 (m, 1 H, HC(2)), 2.14–1.98 (m, 2 H, H2C(3)); 13C NMR (126 MHz, CDCl3), major 137.9 (C(6)), 133.1 (C(14)), 132.7 (C(9)), 128.8 (C(8)), 127.9 (C(13)), 127.6 (C(10)), 126.5 (C(11) or C(12)), 126.2 (C(11) or C(12)), 125.3 (C(15)), 123.9 (C(7)), 64.6 (C(1)), 46.2 (C(4)), 33.3 (C(2)), 20.8 (C(3)), minor 136.8 (C(6)), 133.2 (C(14)), 132.5 (C(9)), 128.6 (C(10) or C(13)), 127.7 (C(8)), 127.6 (C(10) or C(13)), 126.2 (C(11) or C(12)), 125.8 (C(5)), 123.9 (C(11) or C(12)), 123.6 (C(7)), 60.5 (C(1)), 50.9 (C(4)), 33.3 (C(2)), 22.7 (C(3)); IR (CDCl3 film) 3052 (w), 2959 (m), 2876 (w), 1632 (w), 1601 (w), 1507 (w), 1444 (m), 1398 (s), 1333 (s), 1269 (s), 1233 (m), 1204 (m), 1125 (w), 1027 (w), 977 (w), 956 (w), 903 (m), 869 (m), 822 (s), 762 (s) 656 (w). MS (EI, 70 eV) 226.1 (M+, 41), 209.1 (27), 196.1 (100), 167.1 (52), 154.1 (54), 141.1 (23), 127.0 (28), 115.1 (12), 82.9 (33); HRMS (ESI) calcd for C14H15N2O [M + H] 227.1184, found 227.1189; TLC Rf 0.21 (hexanes/EtOAc, 4/1) [silica gel, UV, KMnO4]; [α]D24 +170.2 (c = 0.2, acetone).
Preparation of N-Nitroso-(2R,5R)-2,5-diarylpyrrolidines
N-Nitroso-(2R,5R)-2,5-bis(4-methoxyphenyl)pyrrolidine (24a)

Following general procedure 7 a mixture of (2R,5R)-2,5-bis(4-methoxyphenyl)pyrrolidine 23a (111 mg, 0.39 mmol, 1.0 equiv), CH2Cl2 (2.0 mL), pyridine (64 μL, 0.78 mmol, 2 equiv), and nitrosonium tetrafluoroborate (94 mg, 0.78 mmol, 2 equiv) were combined in a 15 mL, one-necked, round-bottom flask. The mixture was stirred at 0 °C for 2 h under argon and then subjected to aqueous workup. Purification by column chromatography (SiO2, 1.5 × 31 cm, CH2Cl2/EtOAc, gradient elution, 99.5/0.5, 99/1, 98/2 then 95/5) afforded 118 mg (96%) of 24a as a white powder. Two sets of NMR signals were observed due to restricted rotation of nitroso group. Data for 24a: 1H NMR (500 MHz, CDCl3) 7.21 (d, J = 8.7 Hz, 2 H, HC(4)), 7.02 (d, J = 8.7 Hz, 2 H, HC(4)), 6.92 (d, J = 8.7 Hz, 2 H, HC(5)), 6.85 (d, J = 8.6 Hz, 2 H, HC(5)), 5.82 (dd, J = 7.6, 4.5 Hz, 1 H, HC(1)), 5.46 (dd, J = 8.4, 3.8 Hz, 1 H, HC(1)), 3.81 (s, 3 H, HC(7)), 3.78 (s, 3 H, HC(7)), 2.64–2.56 (m, 1 H, HC(2)), 2.54–2.45 (m, 1 H, HC(2)), 2.14–2.08 (m, 1 H, HC(2)), 1.97–1.90 (m, 1 H, HC(2)); 13C NMR (126 MHz, CDCl3) 159.2 (C(6)), 158.6 (C(6)), 133.0 (C(3)), 131.4 (C(3)), 128.0 (C(4)), 126.5 (C(4)), 114.1 (C(5)), 114.0 (C(5)), 65.0 (C(1)), 61.2 (C(1)), 55.3 (C(7)), 55.3 (C(7)), 31.9 (C(2)), 30.9 (C(2)); IR (CDCl3 film) 3020 (w), 2967 (m), 2942 (m), 2837 (w), 2361 (w), 1613 (m), 1585 (m), 1518 (s), 1462 (m), 1417 (m), 1371 (w), 1304 (m), 1285 (m) 1246 (s), 1174 (m) 1126 (m), 1108 (m), 1077 (w), 1026 (s), 827 (s), 770 (w). MS (EI, 70 eV) 312.1 (22), 134.1 (100); HRMS (ESI) calcd for C18H21N2O3 [M + H] 313.1552, found 313.1564; TLC Rf 0.23 (CH2Cl2/EtOAc, 99/1) [silica gel, UV, KMnO4]; [α]D24 +223.9 (c = 0.2, chloroform).
N-Nitroso-(2R,5R)-2,5-bis(2-naphthyl)pyrrolidine (24f)

Following general procedure 7, a mixture of (2R,5R)-2,5-bis(2-naphthyl)pyrrolidine 23f (246 mg, 0.76 mmol, 1.0 equiv), CH2Cl2 (7.6 mL), pyridine (125 μL, 1.5 mmol, 2 equiv), and nitrosonium tetrafluoroborate (182 mg, 1.5 mmol, 2 equiv) were combined in a 50 mL, one-necked, round-bottom flask. The mixture was stirred at 21 °C for 2 h under argon, and then an aqueous solution of HCl (1 M, 7.6 mL) was added at 0 °C. After vigorous stirring for 3 min, the two layers were allowed to separate. The organic layer was saved, and the aqueous layer was extracted with dichloromethane (4 mL × 3). The combined organic extract was dried over Na2SO4, filtered, and concentrated to give a yellow solid (259 mg). The solid was dissolved in dichloromethane (5 mL) and filtered through a pad of silica (2 cm deep, 30 mL, medium-porosity fritted funnel), eluted with dichloromethane (50 mL). The filtrate was concentrated to about 2 mL to give a mixture of yellow solution and solid. Hexanes (16 mL) was added while the flask was gently swirled to cause the formation of precipitate. The yellow solution was carefully removed, and the residue was dried under reduced pressure to afford 247 mg (92%) of 24f as a very lightly yellow powder. Two sets of NMR signals were observed due to restricted rotation of nitroso group. Data for 24f: 1H NMR (500 MHz, CDCl3) 7.91 (d, J = 8.6 Hz, 1 H, HC(Aryl)), 7.88–7.79 (m, 5 H, HC(Aryl)), 7.79 (s, 1 H, HC(Aryl)), 7.55–7.45 (m, 5 H, HC(Aryl)), 7.44 (dd, J = 8.5, 1.9 Hz, 1 H, HC(Aryl)), 7.28 (dd, J = 8.6, 1.9 Hz, 1 H, HC(Aryl)), 6.19 (dd, J = 7.8, 3.9 Hz, 1 H, HC(1)), 5.78 (dd, J = 8.4, 3.9 Hz, 1 H, HC(1)), 2.80–2.72 (m, 1 H, HC(2)), 2.67–2.59 (m, 1 H, HC(2)), 2.30–2.23 (m, 1 H, HC(2)), 2.10–2.04 (m, 1 H, HC(2)); 13C NMR (126 MHz, CDCl3) 138.4 (C(4)), 136.4 (C(4)), 133.2 (C(7) or C(12)), 133.2 ((C(7) or C(12)), 132.9 (C(7) or C(12)), 132.6 ((C(7) or C(12)), 128.9 (C(Aryl)), 128.8 (C(Aryl)), 128.0 (C(Aryl)), 127.8 (C(Aryl)), 127.7 (C(Aryl)), 127.6 (C(Aryl)), 126.6 (C(Aryl)), 126.4 (C(Aryl)), 126.3 (C(Aryl)), 125.9 (C(Aryl)), 125.7 (C(Aryl)), 124.3 (C(Aryl)), 123.8 (C(Aryl)), 123.6 (C(Aryl)), 65.7 (C(1)), 62.1 (C(1)), 31.8 (C(2)), 30.7 (C(2)); IR (CDCl3 film) 3052 (w), 2982 (w), 2947 (w), 1597 (w), 1508 (w), 1421 (m), 1358 (w), 1268 (m), 1240 (m), 1136 (w), 908 (w), 856 (w), 816 (m), 747 (m), 730 (m); MS (ESI) 375.1 (M + Na, 21), 353.2 (M + H, 100), 307.2 (13), 197.1 (41), 179.1 (36), 141.1 (50); HRMS (ESI) calcd for C24H21N2O [M + H] 353.1654, found 353.1658; TLC Rf 0.43 (CH2Cl2) [silica gel, UV, KMnO4]; [α]D24 +246.3 (c = 0.2, chloroform).
N-Nitroso-(2R,5R)-2,5-bis(3,5-dimethylphenyl)pyrrolidine (24g)

Following general procedure 7, a mixture of (2R,5R)-2,5-bis(3,5-dimethylphenyl)pyrrolidine 23g (180 mg, 0.64 mmol, 1.0 equiv), CH2Cl2 (3.2 mL), pyridine (105 μL, 1.3 mmol, 2 equiv), and nitrosonium tetrafluoroborate (153 mg, 1.3 mmol, 2 equiv) were combined in a 25 mL, one-necked, round-bottom flask. The mixture was stirred at 21 °C for 2 h under argon and then subjected to aqueous workup. Purification by column chromatography (SiO2, 3 × 16 cm, CH2Cl2/hexanes, 2/1) afforded 184 mg (96%) of 24g as a white solid. Two sets of NMR signals were observed due to restricted rotation of nitroso group. Data for 24g: 1H NMR (500 MHz, CDCl3) 6.97 (s, 1 H, HC(4)), 6.88 (s, 3 H, HC(4)), 6.68 (s, 2 H, HC(6)), 5.86 (dd, J = 7.7, 3.8 Hz, 1 H, HC(1)), 5.47 (dd, J = 8.3, 3.1 Hz, 1 H, HC(1)), 2.67–2.59 (m, 1 H, HC(2)), 2.54–2.46 (m, 1 H, HC(2)), 2.34 (s, 6 H, H3C(7)), 2.30 (s, 6 H, H3C(7)), 2.15–2.08 (m, 1 H, HC(2)), 1.95–1.89 (m, 2 H, HC(2)); 13C NMR (126 MHz, CDCl3) 141.2 (C(3)), 139.2 (C(3)), 138.3 (C(5)), 138.2 (C(5)), 129.4 (C(6)), 128.9 (C(6)), 124.4 (C(4)), 122.9 (C(4)), 65.5 (C(1)), 61.8 (C(1)), 31.8 (C(2)), 30.8 (C(2)), 21.3 (C(7)); IR (CDCl3 film) 3003 (w), 2982 (w), 2914 (m), 2850 (w), 1607 (m), 1469 (m), 1462 (m), 1451 (m), 1444 (m), 1407 (s), 1292 (m), 1277 (s), 1246 (s), 1194 (m), 1041 (m), 846 (s), 785 (m), 693 (m); MS (ESI) 331 (M + Na, 10), 309.2 (M + H, 100), 263.2 (10), 175.1 (9), 157.1 (23), 119.1 (29); HRMS (ESI) calcd for C20H25N2O [M + H] 309.1967, found 309.1974; TLC Rf 0.35 (CH2Cl2) [silica gel, UV]; [α]D24 202.5 (c = 0.2, acetone).
N-Nitroso-(2R,5R)-2,5-bis(5-phenylbiphenyl-3-yl)pyrrolidine (24h)

Following general procedure 7, a mixture of (2R,5R)-2,5-bis(5-phenylbiphenyl-3-yl)pyrrolidine 23h (335 mg, 0.64 mmol, 1.0 equiv), CH2Cl2 (3.2 mL), pyridine (105 μL, 1.3 mmol, 2 equiv), and nitrosonium tetrafluoroborate (154 mg, 1.3 mmol, 2 equiv) were combined in a 25 mL, one-necked, round-bottom flask. The mixture was stirred at 21 °C for 12 h under argon and then subjected to aqueous workup. Purification by column chromatography (SiO2, 3 × 16 cm, CH2Cl2/hexanes, 1/1) afforded 184 mg (93%) of 24h as a white powder. Two sets of NMR signals were observed due to restricted rotation of nitroso group. Data for 24h: 1H NMR (500 MHz, CDCl3) 7.79 (s, 1 H, HC(6)), 7.71 (s, 1 H, HC(6)), 7.66 (t, J = 8.7 Hz, 8 H, HC(9)), 7.52–7.47 (m, 10 H, HC(4) and HC(8)), 7.44–7.39 (m, 4 H, HC(10)), 7.30–7.29 (m, 2 H, HC(4)), 6.15 (dd, J = 7.9, 3.7 Hz, 1 H, HC(1)), 5.75 (dd, J = 8.5, 3.3 Hz, 1 H, HC(1)), 2.85–2.77 (m, 1 H, H2C(2)), 2.72–2.64 (m, 1 H, H2C(2)), 2.33–2.28 (m, 1 H, H2C(2)), 2.15–2.09 (m, 1 H, H2C(2)); 13C NMR (126 MHz, CDCl3) 142.8 (C(7)), 142.7 (C(7)), 142.6 (C(3)), 141.2 (C(5)), 141.0 (C(5)), 140.5 (C(3)), 129.2 (C(8)), 129.1 (C(8)), 128.0 (C(10)), 127.9 (C(10)), 127.6 (C(9)), 126.2 ((6)), 125.7 (C(6)), 124.7 (C(4)), 123.4 (C(4)), 65.9 (C(1)), 62.3 (C(1)); IR (CDCl3 film) 3052 (w), 3033 (w), 2982 (w), 2940 (w), 2871 (w), 1951 (w), 1885 (w), 1812 (w), 1764 (w), 1596 (s), 1577 (m), 1497 (m), 1455 (m), 1423 (m), 1344 (w), 1306 (w), 1265 (m), 1076 (w), 1028 (w), 983 (w), 874 (m), 788 (w), 758 (s), 613 (m); MS (ESI) 579.2 (M + Na, 7), 557.3 (M + H, 100), 511.2 (14), 415.2 (11), 281.1 (19); HRMS (ESI) calcd for C40H33N2O [M + H] 557.2593, found 557.2590; TLC Rf 0.39 (CH2Cl2/hexanes, 2/1) [silica gel, UV]; [α]D24 +122.3 (c = 0.2, acetone).
N-Nitroso-(2R,5R)-2,5-bis(2-tolyl)pyrrolidine (24i)

Following general procedure 7, a mixture of (2R,5R)-2,5-bis(2-tolyl)pyrrolidine 23i (213 mg, 0.85 mmol, 1.0 equiv), CH2Cl2 (4.2 mL), pyridine (140 μL, 1.7 mmol, 2 equiv), and nitrosonium tetrafluoroborate (202 mg, 1.7 mmol, 2 equiv) were combined in a 25 mL, one-necked, round-bottom flask. The mixture was stirred at 21 °C for 12 h under argon, and then subjected to aqueous workup. Purification by column chromatography (SiO2, 3 × 20 cm, CH2Cl2/hexanes, 4/1) afforded 227 mg (95%) of 24i as a white solid. Two sets of NMR signals were observed due to restricted rotation of nitroso group. Data for 24i: 1H NMR (500 MHz, CDCl3) 7.30–7.21 (m, 4 H, HC(5), HC(6) and HC(7)), 7.19 (t, J = 7.2 Hz, 1 H, HC(6)), 7.14 (t, J = 7.3 Hz, 1 H, HC(7)), 6.96 (d, J = 7.1 Hz, 1 H, HC(8)), 6.78 (d, J = 7.5 Hz, 1 H, HC(8)), 6.23 (d, J = 7.9 Hz, 1 H, HC(1)), 5.72 (d, J = 7.3 Hz, 1 H, HC(1)), 2.72–2.64 (m, 1 H, HC(2)), 2.58–2.48 (m, 1 H, HC(2)), 2.54 (s, 3 H, H3C(9)), 2.45 (s, 3 H, H3C(9)), 2.08–2.03 (m, 1 H, HC(2)), 1.87–1.82 (m, 1 H, HC(2)); 13C NMR (126 MHz, CDCl3) 139.8 (C(3)), 136.9 (C(3)), 134.8 (C(4)), 134.1 (C(4)), 131.1 (C(5)), 130.9 (C(5)), 127.6 (C(6)), 127.2 (C(6)), 126.2 (C(7)), 126.0 (C(7)), 125.6 (C(8)), 123.5 (C(8)), 62.7 (C(1)), 59.7 (C(1)), 29.9 (C(2)), 28.6 (C(2)), 19.5 (C(9)), 19.4 (C(9)). IR (nujol) 1487 (w), 1425 (m), 1344 (w), 1270 (m), 1248 (w), 1218 (w), 1102 (w), 1051 (w), 1017 (w), 976 (w), 870 (w), 775 (w), 765 (m), 725 (w); MS (ESI) 281.1 (M + H, 100), 143.1 (21), 105.1 (32); HRMS (ESI) calcd for C18H21N2O [M + H] 281.1654, found 281.1646; TLC Rf 0.24 (CH2Cl2/hexanes, 4/1) [silica gel, UV, KMnO4]; [α]D24 +223.0 (c = 0.2, acetone).
N-Nitroso-(2R,5R)-2,5-bis(5-methyl-2-thienyl)pyrrolidine (24j)

Following general procedure 7, a mixture of (2R,5R)-2,5-bis(5-methyl-2-thienyl)pyrrolidine 23j (90 mg, 0.34 mmol, 1.0 equiv), CH2Cl2 (1.7 mL), pyridine (55 μL, 0.68 mmol, 2 equiv), and nitrosonium tetrafluoroborate (84 mg, 0.68 mmol, 2 equiv) were combined in a 15 mL, one-necked, round-bottom flask. The mixture was stirred at 0 °C for 15 min under argon and then subjected to aqueous workup. Purification by column chromatography (SiO2, 1 × 25 cm, CH2Cl2/hexanes, 1/1 then 2/1) afforded 57 mg (57%) of 24j as a white solid. Two sets of NMR signals were observed due to restricted rotation of nitroso group. Data for 24j: 1H NMR (500 MHz, CDCl3) 6.83 (d, J = 3.4 Hz, 1 H, HC(4)), 6.67 (d, J = 3.4 Hz, 1 H, HC(4)), 6.62–6.61 (m, 1 H, HC(5)), 6.56–6.54 (m, 1 H, HC(5)), 5.95 (dd, J = 7.8, 2.7 Hz, 1 H, HC(1)), 5.66 (d, J = 8.1 Hz, 1 H, HC(1)), 2.82–2.73 (m, 1 H, HC(2)), 2.58–2.48 (m, 1 H, HC(2)), 2.45 (s, 3 H, H3C(7)), 2.42 (s, 3 H, H3C(7)), 2.33–2.26 (m, 1 H, HC(2)), 2.19–2.13 (m, 1 H, HC(2)); TLC Rf 0.35 (CH2Cl2/hexanes, 2/1) [silica gel, UV, KMnO4].
General Procedure 8: Reduction of N-Nitroso-(2R)-arylpyrrolidine or N-Nitroso-(2R,5R)-diarylpyrrolidine (Table 4)45

To an oven-dried, 25 mL, one-necked, round-bottom flask, equipped with a stir bar, an argon inlet, and a septum was added N-nitroso-(2R,5R)-2,5-diarylpyrrolidine 24 (0.54 mmol, 1.0 equiv). After three cycles of evacuation and argon fill, dichloromethane (5.4 mL) was added. The pale yellow solution was cooled to 0 °C, and a hexanes solution of diisobutylaluminum hydride (1 M, 1.625 mL, 1.625 mmol, 3.0 equiv) was added (no exotherm). The ice/water bath was removed, and the bright yellow solution was stirred at 21 °C for 2 h. The reaction was cooled to 0 °C and quenched slowly with an aqueous solution of NaOH (10%, 5.4 mL) with vigorous stirring. The resulting heterogeneous mixture was stirred for 5 min and filtered through a pad of Celite (1 cm deep, 15 mL, coarse, frit funnel), eluted with dichloromethane (15 mL). The organic layer was saved, and the aqueous layer was extracted with dichloromethane (3 mL × 3). The combined organic extract was dried over Na2SO4, filtered, and concentrated. Purification by column chromatography afforded (2R,5R)-1-amino-2,5-diarylpyrrolidine 8.
Preparation of (R)-1-Aminoarylpyrrolidines
(R)-1-Amino-2-(4-methoxyphenyl)pyrrolidine (19a)

To an oven-dried, 25 mL, one-necked, round-bottom flask equipped with a stir bar, an argon inlet adaptor and a septum was charged N-nitroso-(R)-2-(4-methoxyphenyl)pyrrolidine 18a (142 mg, 0.69 mmol, 1.0 equiv). After three cycles of evacuation/argon fill, THF (2.8 mL) was added. The light yellow solution was cooled to 0 °C, and a THF solution of LiAlH4 (1.19 M, 0.86 mL, 1.03 mmol, 1.5 equiv) was added dropwise, keeping the temperature below 2 °C. After the solution was stirred at 0 °C for 5 min, the ice/water bath was removed. The light yellow solution was stirred at 22 °C for 1 h and then cooled to 0 °C. With vigorous stirring, the reaction was quenched by dropwise addition of H2O (140 μL), maintaining the temperature below 10 °C. After 3 min, a solution of NaOH (10%, 280 μL) and H2O (140 μL) were added. The ice/water bath was removed, and the mixture was stirred at room temperature for 1 h. The mixture was filtered through a pad of Celite (1 cm deep) using a 15 mL, medium-porosity fritted funnel, eluted with dichloromethane (20 mL). The filtrate was diluted with H2O (15 mL), and the aqueous layer was extracted with dichloromethane (8 mL × 4). The combined organic extract was dried over Na2SO4, filtered and concentrated to give a light yellow liquid. Purification by column chromatography (SiO2, 1.5 × 16 cm, CH2Cl2/MeOH, gradient elution, 100/0, 98/2 then 95/5) afforded 105 mg (80%) of 19a as a viscous, light yellow oil. Data for 19a: 1H NMR (500 MHz, CDCl3) 7.27 (d, J = 8.6 Hz, 2 H, HC(6)), 6.89 (d, J = 8.6 Hz, 2 H, HC(7)), 3.78 (s, 3 H, H3C(9)), 3.39 (td, J = 8.6, 2.0 Hz, 1 H, HC(4)), 3.05 (dd, J = 9.5, 7.5 Hz, 1 H, HC(1)), 2.80 (br s, 2 H, NH2), 2.44 (q, J = 9.2 Hz, 1 H, HC(4)), 2.21–2.09 (m, 1 H, HC(2)), 1.97–1.85 (m, 1 H, HC(3)), 1.84–1.70 (m, 3 H, HC(2) and H2C(3)); 13C NMR (126 MHz, CDCl3) 159.0 (C(8)), 133.9 (C(5)), 128.7 (C(6)), 114.0 (C(7)), 75.4 (C(1)), 58.4 (C(4)), 55.2 (C(9)), 33.2 (C(2)), 22.6 (C(3)). IR (neat) 3335 (w), 2956 (s), 2875 (w), 2833 (m), 2803 (m), 1611 (s), 1585 (w), 1512 (s), 1461 (m), 1442 (w), 1369 (w), 1301 (m), 1245 (s), 1172 (m), 1104 (m), 1035 (s), 947 (m), 919 (w), 900 (w), 830 (s); MS (ESI) 193.1 (100, M + H), 176.1 (31); HRMS (ESI) calcd for C11H17N2O [M + H] 193.1341, found 193.1340; TLC Rf 0.29 (CH2Cl2/MeOH, 95/5) [silica gel, KMnO4].
(R)-1-Amino-2-(2-naphthyl)pyrrolidine (19f)

To an oven-dried, 25 mL, one-necked, round-bottom flask equipped with a stir bar, an argon inlet adaptor, and a septum was charged N-nitroso-(R)-2-(2-naphthyl)pyrrolidine 18f (197 mg, 0.87 mmol, 1.0 equiv). After three cycles of evacuation/argon fill, THF (3.5 mL) was added. The light yellow solution was cooled to 0 °C, and a THF solution of LiAlH4 (1.19 M, 1.1 mL, 1.3 mmol, 1.5 equiv) was added dropwise, keeping the temperature below 2 °C. After the solution was stirred at 0 °C for 5 min, the ice/water bath was removed. The light yellow solution gradually turned red. After the solution was stirred at 22 °C for 1 h, the flask was cooled to 0 °C. After vigorous stirring, the reaction was quenched by dropwise addition of H2O (200 μL), maintaining the temperature below 10 °C. After 3 min, a solution of NaOH (10%, 400 μL) and H2O (200 μL) was added. The ice/water bath was removed, and the resulting bright yellow mixture was stirred at room temperature for 1 h. The mixture was filtered through a pad of Celite (1 cm deep) using a 15 mL, medium-porosity fritted funnel, eluted with dichloromethane (25 mL). The filtrate was diluted with H2O (18 mL), the organic layer was saved, and the aqueous layer was extracted with dichloromethane (10 mL × 4). The combined organic extracts was dried over Na2SO4, filtered, and concentrated to afford 180 mg (97%) of 19f as a viscous, light yellow oil. The crude product is unstable to silica gel and was used without further purification. Data for 19f: 1H NMR (500 MHz, CDCl3) 7.86–7.80 (m, 3 H, HC(Aryl)), 7.81 (s, 1 H, HC(5)), 7.55 (d, J = 8.5, 1 H, HC(7)), 7.50–7.44 (m, 2 H, HC(Aryl)), 3.49 (t, J = 8.8, 1 H, HC(4)), 3.31 (t, J = 8.6 Hz, 1 H, HC(1)), 2.90 (s, 2 H, NH2), 2.55 (q, J = 9.0, 1 H, HC(4)), 2.34–2.23 (m, 1 H, H2C(2)), 2.05–1.94 (m, 1 H, HC(3)), 1.93–1.84 (m, 2 H, HC(2) and HC(3)); 13C NMR (126 MHz, CDCl3) 139.5 (C(6)), 133.4 (C(14)), 133.0 (C(9)), 128.4 (C(Aryl)), 127.7 (C(Aryl)), 127.6 (C(Aryl)), 126.5 (C(5)), 126.0 (C(Aryl)), 125.6 (C(Aryl)), 125.4 (C(5)), 75.9 (C(1)), 58.6 (C(4)), 33.2 (C(2)), 20.6 (C(3)). IR (neat) 3339 (w), 3053 (w), 2968 (m), 2800 (m), 1600 (m), 1507 (w), 1459 (w), 1350 (w), 1318 (w), 1270 (w), 1125 (w), 1101 (w), 1019 (w), 947 (w), 893 (w), 857 (m), 820 (m), 748 (m); MS (ESI) 213.1 (M + H, 100), 196.1 (32), 181.1 (7); HRMS (ESI) calcd for C14H17N2 [M + H] 213.1392, found 213.1400.
Preparation of (2R,5R)-1-Amino-2,5-diarylpyrrolidines
(2R,5R)-1-Amino-2,5-bis(4-methoxyphenyl)pyrrolidine (8a)

Following general procedure 8, a mixture of N-nitroso-(2R,5R)-2,5-bis(4-methoxyphenyl)pyrrolidine 24a (169 mg, 0.54 mmol, 1.0 equiv), dichloromethane (5.4 mL) and a hexanes solution of diisobutylaluminum hydride (1 M, 1.625 mL, 1.625 mmol, 3.0 equiv) were combined at 0 °C in a 25 mL, one-necked round-bottom flask. After the mixture was stirred 22 °C for 2 h under argon, the reaction was carefully quenched an aqueous solution of NaOH, filtered, and extracted with dichloromethane. The ratio of 24a:8a was 40:60 based on NMR analysis. Purification by column chromatography (SiO2, 1.5 × 20 cm, gradient elution in the order of CH2Cl2/Et2O (100/0, 80/20, 75/25, 50/50), CH2Cl2/MeOH (98/2, 95/5) afforded 76 mg (47%) of 8a as a white solid and recovered 58 mg (34%) of 24a as a white solid. Data for 8a: 1H NMR (500 MHz, CDCl3) 7.30 (d, J = 8.6 Hz, 4 H, HC(4)), 6.91 (d, J = 8.7 Hz, 4 H, HC(5)), 4.03 (t, J = 5.5 Hz, 2 H, HC(1)), 3.82 (s, 6 H, H3C(7)), 2.57–2.29 (m, 4 H, HC(2) and NH2), 2.04–1.97 (m, 2 H, HC(2)); 13C NMR (126 MHz, CDCl3) 158.9 (C(6)), 133.1 (C(3)), 129.6 (C(4)), 113.7 (C(5)), 68.4 (C(1)), 55.2 (C(7)), 30.2 (C(2)); IR (CDCl3 film) 2955 (w), 2835 (w), 1609 (m), 1511 (m), 1463 (w), 1302 (w), 1246 (m), 1178 (m), 1115 (w), 924 (w), 827 (m); MS (ESI) 299.2 (M + H, 100), 267.1 (56), 191.1 (23), 159.1 (11), 121.1 (12); HRMS (ESI) calcd for C18H23N2O2 [M + H] 299.1760, found 299.1766; TLC Rf 0.22 (CH2Cl2/MeOH, 98/2) [silica gel, KMnO4].
(2R,5R)-1-Amino-2,5-bis(2-naphthyl)pyrrolidine (8f)

Following general procedure 8, a mixture of N-nitroso-(2R,5R)-2,5-bis(2-naphthyl)pyrrolidine 24f (239 mg, 0.68 mmol, 1.0 equiv), dichloromethane (6.8 mL), and a hexanes solution of diisobutylaluminum hydride (1 M, 2.0 mL, 2.0 mmol, 3.0 equiv) were combined at 0 °C in a 25 mL, one-necked round-bottom flask. After the mixture was stirred 21 °C for 2 h under argon, the reaction was carefully quenched an aqueous solution of NaOH, filtered and extracted with dichloromethane. The ratio of 24f:8f was 40:60 based on NMR analysis. Purification by column chromatography (SiO2, 3 × 26 cm, gradient elution, CH2Cl2/MeOH (100/0, 98/2, then 95/5) afforded 117 mg (51%) of 8f as a slightly sticky, very pale yellow solid and recovered 73 mg (31%) of 24f as a very light yellow solid. The desired product 8f was further purified by swirling in 1.5 mL of hexanes, careful removal of hexanes, and drying in vacuo to give a free-flowing powder (114 mg, 50%). A mixture of 1,2-di(2-naphthyl)cyclobutane 25 (major) and 2-ethenylnaphthalene 26 (minor) was also isolated (23 mg). This mixture exhibited optical activity ([α]D24 +356.0, c = 0.5, CHCl3) suggesting a trans relationship between the two naphthyl groups in 25. A small multiplet at ∼4.28 ppm in 1H NMR may belong to the benzylic proton of meso-25. The spectral characterization of meso-25 and the enantiomeric purity of trans-25 were not established at this time. Data for 8f: 1H NMR (500 MHz, CDCl3) 7.90–7.83 (m, 8 H, HC(Aryl)), 7.60 (dd, J = 8.5, 1.8 Hz, 2 H, HC(Aryl)), 7.53–7.46 (m, 4 H, HC(Aryl)), 4.38 (t, J = 5.8 Hz, 2 H, HC(1)), 2.70–2.60 (m, 2 H, H2C(2)), 2.59 (br s, 2 H, NH2), 2.26–2.19 (m, 2 H, H2C(2)); 13C NMR (126 MHz, CDCl3) 138.8 (C(4)), 133.3 (C(7) or C(12)), 132.9 ((C(7) or C(12)), 128.2 (C(Aryl)), 127.9 (C(Aryl)), 127.6 (C(Aryl)), 127.4 (C(Aryl)), 126.4 (C(Aryl)), 126.1 (C(Aryl)), 125.8 (C(Aryl)), 69.6 (C(1)), 30.6 (C(2)); IR (CDCl3 film) 3053 (w), 3017 (w), 2957 (w), 2912 (w), 2871 (w), 2808 (w), 1628 (w), 1599 (w), 1507 (w), 1466 (w), 1438 (w), 1371 (w), 1326 (w), 1270 (w), 1174 (w), 1124 (w), 1017 (w), 907 (m), 856 (m), 813 (m), 745 (m); MS (ESI) 339.2 (M + H, 100), 322.2 (6); HRMS (ESI) calcd for C24H23N2 [M + H] 339.1861, found 339.1859; TLC Rf 0.74 (CH2Cl2/MeOH, 95/5) [silica gel, UV].
trans-1,2-Di(2-naphthyl)cyclobutane (25)

Data for 25: 1H NMR (500 MHz, CDCl3) 7.85–7.77 (m, 6 H, HC(Aryl)), 7.72 (br s, 2 H, HC(3)), 7.49–7.41 (m, 6 H, HC(Aryl)), 3.90–3.83 (m, 2 H, HC(1)), 2.52–2.43 (m, 2 H, H2C(2)), 2.26–2.19 (m, 2 H, H2C(2)); 13C NMR (126 MHz, CDCl3) 142.0 (C(4)), 133.5 (C(7) or C(12)), 132.2 ((C(7) or C(12)), 128.0 (C(Aryl)), 127.6 (C(Aryl)), 127.6 (C(Aryl)), 125.9 (C(Aryl)), 125.5 (C(Aryl)), 125.2 (C(Aryl)), 124.7 (C(Aryl)), 48.2 (C(1)), 25.9 (C(2)); HRMS (EI) calcd for C24H20 308.15650, found 308.15724.
2-Ethenylnaphthalene (26)68

Data for 26: 1H NMR (500 MHz, CDCl3) 6.91 (dd, J = 17.6, 10.9 Hz, 1 H, HC(1)), 5.90 (d, J = 17.6 Hz, 1 H, HtransC(2)), 5.37 (d, J = 10.9 Hz, 1 H, HcisC(2)); the aromatic signals of 2-ethenylnaphthalene (26) overlap with the aromatic signals of trans-1,2-di(2-naphthyl)cyclobutane (25); 13C NMR (126 MHz, CDCl3) 136.9 (C(1)), 135.0 (C(4)), 133.5 ((C(12)), 133.1 (C(7)), 128.1 (C(6)), 128.0 (C(11)), 127.7 (C(8)), 126.3 (C(3)), 126.2 (C(10)), 125.9 (C(9)), 123.2 (C(5)), 114.2 (C(2)). HRMS (EI) calcd for C12H10 154.07825, found 154.07748.
(2R,5R)-1-Amino-2,5-bis(3,5-dimethyphenyl)pyrrolidine (8g)

Following general procedure 8, a mixture of (2R,5R)-2,5-bis(3,5-dimethylphenyl)pyrrolidine 24g (182 mg, 0.59 mmol, 1.0 equiv), dichloromethane (5.9 mL), and a hexanes solution of diisobutylaluminum hydride (1 M, 1.8 mL, 1.8 mmol, 3.0 equiv) were combined at 0 °C in a 25 mL, one-necked round-bottom flask. After the mixture was stirred 21 °C for 2 h under argon, the reaction was carefully quenched with an aqueous solution of NaOH, filtered, and extracted with dichloromethane. The ratio of 24g:8g was 40:60 based on NMR analysis. Purification by column chromatography (SiO2, 1.5 × 17 cm, gradient elution in the order of CH2Cl2/hexanes (2/1) then CH2Cl2/MeOH (98/2, 95/5) afforded 101 mg (58%) of 8g as a viscous pale yellow oil and recovered 37 mg (20%) of 24g as a white solid. Data for 8g: 1H NMR (500 MHz, CDCl3) 7.03 (s, 4 H, HC(4)), 6.97 (s, 2 H, HC(6)), 4.10 (t, 2 H, HC(1)), 2.58 (br s, 2 H, NH2), 2.53–2.46 (m, 2 H, H2C(2)), 2.37 (s, 6 H, H3C(7)), 2.08–2.02 (m, 2 H, H2C(2)); 13C NMR (126 MHz, CDCl3) 141.3 (C(3)), 137.8 (C(5)), 129.0 (C(6)), 126.2 (C(4)), 69.3 (C(1)), 30.6 (C(2)), 21.3 (C(7)); IR (CDCl3 film) 3337 (w), 3303 (w), 3012 (m), 2948 (m), 2917 (s), 1605 (s), 1469 (m), 1376 (w), 1351 (w), 1313 (w), 1271 (w), 1219 (w), 1157 (w), 1115 (w), 1038 (w), 923 (w), 887 (m), 848 (s), 702 (m); MS (ESI) 295.2 (M + H, 100), 278.2 (6); HRMS (ESI) calcd for C20H27N2 [M + H] 295.2174, found 295.2175; TLC Rf 0.67 (CH2Cl2/MeOH, 95/5) [silica gel, UV, KMnO4].
(2R,5R)-1-Amino-2,5-bis(5-phenylbiphenyl-3-yl)pyrrolidine (8h)

Following general procedure 8, a mixture of (2R,5R)-2,5-bis(5-phenylbiphenyl-3-yl)pyrrolidine 24h (300 mg, 0.54 mmol, 1.0 equiv), dichloromethane (5.4 mL), and a hexanes solution of diisobutylaluminum hydride (1 M, 1.6 mL, 1.6 mmol, 3.0 equiv) were combined at 0 °C in a 25 mL, one-necked round-bottom flask. After the mixture was stirred 21 °C for 2 h under argon, the reaction was carefully quenched with an aqueous solution of NaOH, filtered and extracted with dichloromethane. The ratio of 24h:8h was 34:66 based on NMR analysis. Purification by column chromatography (SiO2, 1.5 × 17 cm, gradient elution, CH2Cl2 with 1% Et3N, then CH2Cl2/MeOH, 98/2 with 1% Et3N) afforded 127 mg (43%) of 8h as slightly sticky white solid. Data for 8h: 1H NMR (500 MHz, CDCl3) 7.78 (s, 2 H, HC(6)), 7.71 (d, J = 7.5 Hz, 8 H, HC(8)), 7.64 (s, 4 H, HC(4)), 7.50 (t, J = 7.6 Hz, 8 H, HC(9)), 7.40 (t, J = 7.3 Hz, 4 H, HC(10)), 4.38–4.35 (m, 2 H, HC(1)), 2.75 (s, 2 H, NH2), 2.68–2.61 (m, 2 H, H2C(2)), 2.24–2.18 (m, 2 H, H2C(2)); 13C NMR (126 MHz, CDCl3) 142.6 (C(3)), 142.0 (C(5)), 141.1 ((C(7)), 128.8 (C(9)), 127.4 (C(10)), 127.4 (C(8)), 126.2 (C(4)), 125.4 (C(6)), 69.6 (C(1)), 30.8 (C(2)); IR (CDCl3 film) 3337 (w), 3052 (w), 3033 (w), 2960 (w), 2912 (w), 1944 (w), 1888 (w), 1809 (w), 1595 (m), 1576 (m), 1497 (m), 1455 (m), 1434 (m), 1411 (w), 1358 (w), 1309 (w), 1247 (w), 1178 (w), 1153 (w), 1115 (w), 1075 (w), 1029 (w), 875 (m), 758 (s), 698 (s); MS (ESI) 543.3 (M + H, 100); HRMS (ESI) calcd for C40H35N2 [M + H] 543.2795, found 543.2796; TLC Rf 0.71 (CH2Cl2/MeOH, 98/2) [silica gel, UV, PMA].
(2R,5R)-1-Amino-2,5-bis(2-tolyl)pyrrolidine (8i)

To a 10 mL, 1-necked round-bottom flask equipped with a stir bar and a nitrogen inlet adaptor were added N-nitroso-(2R,5R)-2,5-bis(2-tolyl)pyrrolidine 24i (214 mg, 0.76 mmol, 1.0 equiv), THF (1.6 mL), and zinc powder (210 mg, 3.2 mmol, 4.0 equiv). The flask was cooled to 0 °C, and concd HCl (520 μL, 6.2 mmol, 8 equiv) was added to the mixture with vigorous stirring. After vigorous stirring at room temperature for 30 min, the reaction mixture was decanted into an ice-cold solution of NaOH (2 M, 20 mL) leaving consumed zinc powder in the flask. The aqueous phase was extracted with CH2Cl2 (15 mL × 3), and the combined organic phases was dried over Na2SO4, filtered over Celite (1 cm deep, 15 mL size, medium-porosity fritted funnel), eluted with CH2Cl2 (15 mL), and then concentrated to give 188 mg of 8i and 23i (83:17) as a mixture. The estimated yield for 8i was 158 mg (78%). The product was not stable toward silica gel chromatography and was used without further purification. Data for 8i: 1H NMR (500 MHz, CDCl3) 7.48 (d, J = 7.5 Hz, 2 H, HC(8)), 7.29–7.22 (m, 2 H, HC(7)), 7.20–7.14 (m, 4 H, HC(6) and HC(5)), 4.59 (dd, J = 6.0, 4.3 Hz, 2 H, HC(1)), 2.54–2.46 (m, 2 H, H2C(2)), 2.61 (br s, 2 H, NH2), 2.44 (s, 6 H, H3C(9)), 1.93–1.86 (m, 2 H, H2C(2)); MS (ESI) 267.2 (M + H, 100), 252.2 (20), 235.2 (9), 143.1 (6), 105.1 (8); HRMS (ESI) calcd for 8i, C18H23N2 [M + H] 267.1861, found 267.1863, calcd for 23i, C18H22N [M + H] 252.1752, found 252.1753.
(R,S)-1-Amino-2,5-bis(2-tolyl)pyrrolidine (meso-8i)

To a flame-dried, 5 mL Schlenk reaction flask equipped with a septum was charged N-nitroso-(2R,5R)-2,5-bis(2-tolyl)pyrrolidine 24i (42 mg, 0.15 mmol, 1.0 equiv). After three cycles of evacuation/argon fill, THF (1.5 mL) was added. The colorless solution was cooled to 0 °C, and a THF solution of LiAlH4 (1.19 M, 190 μL, 0.23 mmol, 1.5 equiv) was added dropwise. After the mixture was stirred 0 °C for 5 min, the ice/water bath was removed. The light yellow solution was stirred at room temperature for 2 h and then cooled to 0 °C. With vigorous stirring, the reaction was quenched by dropwise addition of H2O (150 μL), followed by a solution of NaOH (10%, 300 μL) and then H2O (150 μL). The mixture was filtered through a pad of Celite (0.6 cm deep, 15 mL size, medium-porosity fritted funnel), eluted with dichloromethane (20 mL). The organic phase from the filtrate was saved and the aqueous layer was extracted with dichloromethane (1.5 mL × 2). The combined organic extracts was dried over Na2SO4, filtered, and concentrated to give a colorless film. Purification by column chromatography (SiO2, 1.5 × 14 cm, CH2Cl2) afforded 12 mg (29%) of meso-8i as a light yellow film. Data for meso-8i: 1H NMR (500 MHz, CDCl3) 7.82 (d, J = 7.7 Hz, 2 H, HC(8)), 7.33–7.27 (m, 2 H, HC(7)), 7.21–7.14 (m, 4 H, HC(6) and HC(5)), 3.85 (t, J = 5.9 Hz, 2 H, HC (1)), 2.84 (br s, 2 H, NH2), 2.41 (s, 6 H, H3C(9)), 2.38–2.32 (m, 2 H, H2C(2)), 1.33–1.64 (m, 2 H, H2C(2)); TLC Rf 0.60 (CH2Cl2/MeOH, 98/2) [silica gel, KMnO4].
meso-2,5-Bis(2-tolyl)pyrrolidine (meso-23i)

To a 4 mL vial containing meso-1-amino-2,5-bis(2-tolyl)pyrrolidine meso-8i (12 mg, 44 μmol, 1.0 equiv) and a stir bar was added glacial acetic acid (44 μL, 2 mmol, 44 equiv) at 0 °C. Sodium nitrite (3.3 mg, 45 μmol, 1.0 equiv) was added, and the vial was sealed after purging with nitrogen. After vigorous stirring for 2 h, CH2Cl2 (1 mL) and 2 M NaOH (1 mL) were added at 0 °C. The two phases were thoroughly mixed and then allowed to settle. The organic phase was saved, and the aqueous phase was extracted with CH2Cl2 (1 mL × 2). The combined organic phases was dried over Na2SO4, filtered, and concentrated. Purification by column chromatography (SiO2, 1 × 12 cm, CH2Cl2/MeOH, 100/0 then 98/2) afforded 3.8 mg (35%) of meso-23i as a colorless crystal. Data for meso-23i: 1H NMR (500 MHz, CDCl3) 7.96 (d, J = 7.7 Hz, 2 H, HC(8)), 7.28–7.24 (m, 2 H, HC(7)), 7.18–7.14 (m, 4 H, HC(6) and HC(5)), 4.52 (app t, J = 5.6 Hz, 2 H, HC (1)), 2.39 (s, 6 H, H3C(9)), 2.35–2.28 (m, 2 H, H2C(2)), 1.73–1.63 (m, 3 H, H2C(2) and NH); TLC Rf 0.23 (CH2Cl2/MeOH, 98/2) [silica gel, KMnO4].
(R,S)-1-Benzyl-2,5-di(2-tolyl)pyrrolidine 43

To a 4 mL vial equipped with a PTFE septum cap and a stir bar was added meso-2,5-bis(2-tolyl)pyrrolidine meso-23i (3.8 mg, 15 μmol, 1.0 equiv). The vial was purged with argon and CH2Cl2 (0.2 mL), benzaldehyde (1.6 μL, 15 μmol, 1.0 equiv) and sodium triacetoxyborohydride (4.8 mg, 22 μmol, 1.4 equiv) were added. After the mixture was stirred room temperature for 14 h, CH2Cl2 (1 mL) and a saturated solution of NaHCO3 (1 mL) were added. The two phases were thoroughly mixed then allowed to settle. The organic phase was dried over Na2SO4, filtered, and concentrated. Purification by column chromatography (SiO2, 1 × 20 cm, hexanes/CH2Cl2, 20/1) afforded 3.7 mg (72%) of meso-43 as a colorless film. Data for meso-43: 1H NMR (500 MHz, CDCl3) 7.92 (d, J = 7.7 Hz, 2 H, HC(8)), 7.33–7.28 (m, 2 H, HC(7)), 7.21–7.17 (m, 7 H, HC(Aryl)), 6.82–6.80 (m, 2 H, HC(Aryl)), 3.90 (app t, J = 5.6 Hz, 2 H, HC (1)), 3.65 (s, 2 H, HC(10)), 2.34 (s, 6 H, H3C(9)), 2.16–2.09 (m, 2 H, H2C(2)), 1.63–1.56 (m, 2 H, H2C(2)); TLC Rf 0.22 (hexanes/CH2Cl2, 20/1) [silica gel, UV, KMnO4]. The splitting pattern of HC(10) is indicative of meso isomer.69
(2R,5R)-1-Amino-2,5-bis(5-methyl-2-thienyl)pyrrolidine (8j)

Following general procedure 8, a mixture of N-nitroso-(2R,5R)-2,5-bis(5-methyl-2-thienyl)pyrrolidine 24j (57 mg, 0.2 mmol, 1.0 equiv), dichloromethane (2.0 mL), and a hexanes solution of diisobutylaluminum hydride (1 M, 0.59 mL, 0.59 mmol, 3.0 equiv) were combined at 0 °C in a 15 mL, one-necked round-bottom flask. After the mixture was stirred 21 °C for 2 h under argon, the reaction was carefully quenched with an aqueous solution of NaOH, filtered, and extracted with dichloromethane. The ratio of 24j:8j was 33:67 based on NMR analysis. Purification by column chromatography (SiO2, 1 × 26 cm, CH2Cl2/MeOH, 98/2) afforded 26 mg (47%) of 8j as colorless oil. Data for 8j: 1H NMR (500 MHz, CDCl3) 6.77 (d, J = 3.6 Hz, 2 H, HC(4)), 6.63–6.61 (m, 2 H, HC(5)), 4.26 (dd, J = 7.2, 4.7 Hz, 2 H, HC(1)), 2.78 (s, 2 H, NH2), 2.49–2.42 (m, 2 H, H2C(2)), 2.47 (s, 6 H, H3C(7)), 2.03–1.95 (m, 2 H, H2C(2)); TLC Rf 0.16 (CH2Cl2/MeOH, 98/2) [silica gel, UV, KMnO4].
General Procedure 9: Glyoxal Condensation with (2R)-1-Amino-2-arylpyrrolidine or (2R,5R)-1-Amino-2,5-diarylpyrrolidine (Table 4)

To a 10 mL, one-necked, round-bottom flask equipped with a stir bar was added an aqueous solution of glyoxal (40% w/w, 360 μL, 3.15 mmol, 20 equiv). A solution of N-amino-(2R,5R)-2,5-diarylpyrrolidine 8 (0.16 mmol, 1.0 equiv) in THF (1.6 mL) was added at 0 °C. The flask was capped with a glass stopper and stirred at 0 °C for 0.5 or 1 h. The reaction was basified with a solution of saturated NaHCO3 (1.5 mL), vigorously stirred at 0 °C for 3 min. The organic phase was saved and the aqueous phase was extracted with dichloromethane (1 mL × 5). The combined organic phase was dried over Na2SO4, filtered, and concentrated to give a light yellow oil. Purification by column chromatography afforded (2R,5R)-(2,5-diarylpyrrolidine)-N-iminoacetaldehyde 12.
Preparation of (R)-(2-Arylpyrrolidine)-N-iminoacetaldehydes (20)
(R)-(2-(4-Methoxyphenyl)pyrrolidine)-N-iminoacetaldehyde (20a)

To a 10 mL, one-necked, round-bottom flask equipped with a stir bar was added an aqueous solution of glyoxal (40% w/w, 570 μL, 5.0 mmol, 20 equiv). A solution of N-amino-(R)-2-(4-methoxyphenyl)pyrrolidine 19a (47.6 mg, 0.25 mmol, 1.0 equiv) in THF (2.5 mL) was added at 0 °C. The ice/water bath was removed, and the flask was capped with a glass stopper. After the solution was stirred at 22 °C for 0.5 h, THF was evaporated under reduced pressure. The residue was taken up by dichloromethane (10 mL) and basified with saturated NaHCO3 (15 mL) in a separatory funnel. The organic layer was saved, and the aqueous layer was extracted further with dichloromethane (10 mL × 4). The combined organic extract was dried over Na2SO4, filtered, and concentrated to give a brown oil. Purification by column chromatography (SiO2, 1.5 × 16 cm, hexanes/EtOAc with 1% Et3N, gradient elution, 9/1, 4/1, 2/1 then 1/1) afforded 42 mg (74%) of 20a as a very viscous, red oil. Data for 20a: 1H NMR (500 MHz, CDCl3) 9.30 (d, J = 7.6 Hz, 1 H, HC(11)), 7.05 (d, J = 8.7 Hz, 2 H, HC(6)), 6.87 (d, J = 8.7 Hz, 2 H, HC(7)), 6.62 (app br s, 1 H, HC(10)), 4.78 (app br s, 1 H, HC(1)), 3.80 (s, 3 H, H3C(9)), 3.80–3.40 (br m, 2 H, HC(4)), 2.44 (dq, J = 14.9, 7.5 Hz, 1 H, HC(2)), 2.20–2.00 (m, 2 H, HC(2) and/or HC(3)), 2.21–2.02 (m, 1 H, HC(2) or HC(3)); 13C NMR (126 MHz, CDCl3) 190.7 (C(11)), 159.0 (C(8)), 131.0 (C(5)), 127.3 (C(6)), 114.2 (C(7)), 55.2 (C(9)), 34.1 (C(2), broad), 22.0 (C(3)); C(1), C(4) and C(10) were not observed due to hindered rotation; IR (neat) 3316 (w), 2954 (m), 2871 (m), 2836 (m), 2794 (m), 1734 (w), 1667 (s), 1610 (m), 1585 (w), 1513 (s), 1455 (m), 1394 (s), 1337 (m), 1304 (m), 1248 (s), 1138 (s), 1032 (m), 897 (w), 828 (m), 806 (w); MS (ESI) 255.1 (13, M + Na), 233.1 (100, M), 164.1 (12), 146.0 (18), 125.1 (12); HRMS (ESI) calcd for C13H17N2O2 [M + H] 233.1290, found 233.1289; TLC Rf 0.22 (hexanes/EtOAc, 2/1 with 1% Et3N) [silica gel, KMnO4].
(R)-(2-(2-Naphthyl)pyrrolidine)-N-iminoacetaldehyde (20f)

To a 25 mL, one-necked, round-bottom flask equipped with a stir bar was added an aqueous solution of glyoxal (40% w/w, 920 μL, 8.0 mmol, 20 equiv). A solution of (R)-1-amino-2-(2-naphthyl)pyrrolidine 19f (85 mg, 0.4 mmol, 1.0 equiv) in THF (4 mL) was added at 0 °C. The ice/water bath was removed, and the flask was capped with a glass stopper. After the solution was stirred at 22 °C for 0.5 h, THF was evaporated under reduced pressure. The residue was taken up by dichloromethane (20 mL) and basified with saturated NaHCO3 (20 mL) in a separatory funnel. The organic layer was saved, and the aqueous layer was extracted further with dichloromethane (15 mL × 4). The combined organic extract was dried over Na2SO4, filtered, and concentrated. Purification by column chromatography (SiO2, 1.5 × 28 cm, hexanes/EtOAc, 4/1, then 3/1, both with 1% Et3N) afforded 83 mg (82%) of 20f as a very viscous, yellow oil. Data for 20f: 1H NMR (500 MHz, CDCl3) 9.31 (d, J = 7.6 Hz, 1 H, HC(16)), 7.86–7.78 (m, 3 H, HC(Aryl)), 7.55 (s, 1 H, HC(5)), 7.52–7.45 (m, 2 H, HC(Aryl)), 7.26–7.23 (m, 1 H, HC(Aryl)), 6.69 (app br s, 1 H, HC(15)), 4.99 (app br s, 1 H, HC(1)), 3.95–3.40 (br m, 2 H, HC(4)), 2.58–2.48 (m, 1 H, HC(2) or HC(3)), 2.23–2.04 (m, 3 H, HC(2) and HC(3)); 13C NMR (126 MHz, CDCl3) 190.7 (C(16)), 133.2 (C(Aryl)), 132.8 (C(Aryl)), 131.2 (C(Aryl)), 128.9 (C(Aryl)), 127.8 (C(Aryl), 127.7 (C(Aryl)), 126.4 (C(Aryl)), 126.0 (C(Aryl)), 124.9 (C(Aryl)), 124.0 (C(Aryl)), 34.0 (C(2), broad), 22.0 (C(3)); C(1), C(4) and C(15) were not observed due to hindered rotation; IR (CDCl3 film) 3052 (w), 2975 (w), 2801 (w), 1667 (s), 1601 (w), 1514 (s), 1454 (w), 1393 (s) 1319 (m), 1272 (m), 1158 (s), 1137 (s), 894 (w), 818 (m), 752 (m); MS (ESI) 253.1 (M + H, 100), 235.1 (4), 184.1 (4), 118.1 (5); HRMS (ESI) calcd for C16H17N2O [M + H] 253.1341, found 253.1341; TLC Rf 0.28 (hexanes/EtOAc, 2/1 with 1% Et3N) [silica gel, UV, KMnO4].
Preparation of (2R,5R)-(2,5-Diarylpyrrolidine)-N-iminoacetaldehydes (12)
(2R,5R)-(2,5-Bis(4-methoxyphenyl)pyrrolidine)-N-iminoacetaldehyde (12a)

Following general procedure 9, to a 10 mL, one-necked, round-bottom flask equipped with a stir bar was added an aqueous solution of glyoxal (40% w/w, 360 μL, 3.15 mmol, 20 equiv). A solution of (2R,5R)-amino-2,5-bis(4-methoxyphenyl)pyrrolidine 8a (47 mg, 0.16 mmol, 1.0 equiv) in THF (1.6 mL) was added at 0 °C. The flask was capped with a glass stopper and stirred at 0 °C for 1 h. The reaction was basified with a solution of saturated NaHCO3 (1.5 mL), vigorously stirred at 0 °C for 3 min. The organic phase was saved, and the aqueous phase was extracted with dichloromethane (1 mL × 5). The combined organic phase was dried over Na2SO4, filtered and concentrated to give a very light yellow oil. Purification by column chromatography (SiO2, 1.5 × 18 cm, CH2Cl2 with 1% Et3N) afforded 52.7 mg (99%) of 12a as a waxy, ivory colored solid. Data for 12a: 1H NMR (500 MHz, C6D6, 60 °C) 9.61 (d, J = 7.6 Hz, 1 H, HC(9)), 6.85 (d, J = 8.2 Hz, 4 H, HC(4)), 6.79 (d, J = 8.1 Hz, 4 H, HC(5)), 6.64 (d, J = 7.5 Hz, 1 H, HC(8)), 4.66 (app br s, 2 H, HC(1)), 2.13–2.02 (m, 2 H, H2C(2)), 1.52–1.40 (m, 2 H, H2C(2)); 13C NMR (126 MHz, C6D6, 60 °C) 189.3 (C(9)), 159.6 (C(6)), 133.3 (C(3)), 127.5 (C(4)), 114.7 (C(5)), 65.9 (C(1), broad), 54.9 (C(7)), 31.7 (C(2)); C(3) and C(8) were not observed due to hindered rotation; IR (CDCl3 film) 2935 (w), 1666 (m), 1610 (m), 1512 (m), 1462 (w), 1390 (m), 1288 (w), 1248 (m), 1175 (m), 1139 (m), 1033 (m), 829 (m); MS (ESI) 361.1 (7, M + Na), 339.2 (100, M + H), 321.2 (6), 267.1 (3); HRMS (ESI) calcd for C20H23N2O3 [M + H] 339.1709, found 339.1704; TLC Rf 0.62 (CH2Cl2/MeOH, 98/2 with 1% Et3N) [silica gel, UV, KMnO4].
(2R,5R)-(2,5-Bis(2-naphthyl)pyrrolidine)-N-iminoacetaldehyde (12f)

Following general procedure 9 to a 15 mL, one-necked, round-bottom flask equipped with a stir bar was added an aqueous solution of glyoxal (40% w/w, 390 μL, 3.4 mmol, 20 equiv). A solution of (2R,5R)-1-amino-2,5-bis(2-naphthyl)pyrrolidine 8f (57 mg, 0.17 mmol, 1.0 equiv) in THF (1.7 mL) was added at 0 °C. The ice/water bath was removed, and the flask was capped with a glass stopper. After the solution was stirred at 21 °C for 0.5 h, TLC indicated the complete consumption of 8f. The reaction was basified with a solution of saturated NaHCO3 (7 mL) at 0 °C, vigorously stirred for 3 min. The organic phase was saved and the aqueous phase was extracted with dichloromethane (3 mL × 3). The combined organic phase was dried over Na2SO4, filtered, and concentrated to give a light yellow oil. Purification by column chromatography (SiO2, 1.5 × 18 cm, CH2Cl2 with 1% Et3N) afforded 60 mg (94%) of 12f as a white solid. Data for 12f: 1H NMR (500 MHz, CDCl3) 9.20 (d, J = 7.6 Hz, 1 H, HC(14)), 7.90 (d, J = 8.5 Hz, 2 H, HC(Aryl)), 7.88–7.79 (br m, 4 H, HC(Aryl)), 7.61 (br s, 2 H, HC(3)), 7.55–7.49 (m, 4 H, HC(Aryl)), 7.33 (br d, J = 8.1 Hz, 2 H, HC(Aryl)), 6.60 (d, J = 7.1 Hz, 1 H, HC(13)), 5.70–5.10 (br m, 4 H, HC(1)), 2.85–2.60 (br m, 2 H, H2C(2)), 2.15–1.97 (br m, 2 H, H2C(2)); 13C NMR (126 MHz, CDCl3) 190.7 (C(14)) 133.3 (C(Aryl)), 133.1 (C(Aryl)), 132.8 (C(Aryl)), 129.3 (C(Aryl)), 127.9 (C(Aryl)), 127.7 (C(Aryl)), 126.5 (C(Aryl)), 126.2 (C(Aryl)), 124.4 (C(Aryl)), 123.7 (C(Aryl)), 69.1 (C(1)), 64.2 (C(1)), 32.1 (C(2)), 30.8 (C(2)); due to hindered rotation, many signals were broadened, the molecule became unsymmetrical and some carbons were not observed; IR (CDCl3 film) 3055 (w), 2979 (w), 2802 (w), 1666 (s), 1601 (w), 1519 (s), 1444 (w), 1389 (s), 1312 (m), 1285 (m), 1247 (m), 1169 (m), 1137 (s), 855 (m), 817 (m); MS (ESI) 401.2 (M + Na, 3), 379.2 (M + H, 100); HRMS (ESI) calcd for C26H23N2O [M + H] 379.1810, found 379.1813; TLC Rf 0.20 (CH2Cl2 with 1% Et3N) [silica gel, UV].
(2R,5R)-(2,5-Bis(3,5-dimethyphenyl)pyrrolidine)-N-iminoacetaldehyde (12g)

Following general procedure 9, to a 15 mL, one-necked, round-bottom flask equipped with a stir bar was added an aqueous solution of glyoxal (40% w/w, 390 μL, 3.4 mmol, 20 equiv). A solution of (2R,5R)-1-amino-2,5-bis(3,5-dimethylphenyl)pyrrolidine 8g (50 mg, 0.17 mmol, 1.0 equiv) in THF (1.0 mL) was added at 0 °C. The ice/water bath was removed, and the flask was capped with a glass stopper. After the solution was stirred at 21 °C for 0.5 h, TLC indicated the complete consumption of 8g. The reaction was basified with a solution of saturated NaHCO3 (1 mL) at 0 °C and vigorously stirred for 3 min. The organic phase was saved, and the aqueous phase was extracted with dichloromethane (1 mL × 3). The combined organic phase was dried over Na2SO4, filtered, and concentrated to give a light yellow oil and solid. Purification by column chromatography (SiO2, 1.5 × 16 cm, CH2Cl2/hexanes, 4/1 with 1% Et3N) afforded 48 mg (84%) of 12g as a slightly tanned solid. Data for 12g: 1H NMR (500 MHz, CDCl3) 9.22 (d, J = 7.6, 1 H, HC(9)), 6.92 (s, 2 H, HC(6)), 6.74 (s, 4 H, HC(4)), 6.50 (d, J = 7.6, 4 H, HC(8)), 5.26 (app br s, 1 H, HC(1)), 4.90 (app br s, 1 H, HC(1)), 2.60 (app s, 2 H, H2C(2)), 2.33 (s, 12 H, H3C(7)), 1.92 (app s, 2 H, H2C(2)); 13C NMR (126 MHz, CDCl3) 190.9 (C(9)), 142.5 (C(5)), 138.7 (C(5)), 138.2 (C(5)), 128.2 (C(8)), 129.4 (C(6)), 129.1 (6), 124.1 (C(4)), 123.3 (C(4)), 68.8 (C(1)), 64.5 (C(1)), 32.1 (C(2)), 30.8 (C(2)), 21.4 (C(7)); IR (CDCl3 film) 3010 (m), 2975 (m), 2940 (m), 2918 (m), 2871 (m), 2804 (m), 1667 (s), 1604 (m), 1519 (s), 1454 (m), 1389 (s), 1320 (m), 1306 (m), 1281 (m), 1246 (m), 1188 (m), 1165 (m), 1137 (s), 1039 (w), 957 (w), 845 (s); MS (ESI) 335.2 (M + H, 100); HRMS (ESI) calcd for C22H27N2O [M + H] 335.2133, found 335.2131; TLC Rf 0.81 (CH2Cl2/MeOH, 98/2 with 1% Et3N) [silica gel, UV].
(2R,5R)-(2,5-Bis(5-phenylbiphenyl-3-yl)pyrrolidine)-N-iminoacetaldehyde (12h)

Following general procedure 9, to a 10 mL, one-necked, round-bottom flask equipped with a stir bar was added an aqueous solution of glyoxal (40% w/w, 230 μL, 2 mmol, 20 equiv). A solution of (2R,5R)-1-amino-2,5-bis(5-phenylbiphenyl-3-yl)pyrrolidine 8h (55 mg, 0.1 mmol, 1.0 equiv) in THF (1.0 mL) was added at 0 °C. The ice/water bath was removed, and the flask was capped with a glass stopper. After the solution was stirred at 21 °C for 0.5 h, TLC indicated the complete consumption of 8h. The reaction was basified with a solution of saturated NaHCO3 (7.5 mL) at 0 °C, vigorously stirred for 3 min. The organic phase was saved and the aqueous phase was extracted with dichloromethane (3 mL × 3). The combined organic phases was dried over Na2SO4, filtered and concentrated to give a light yellow oil and solid. Purification by column chromatography (SiO2, 1.5 × 21 cm, CH2Cl2/hexanes, gradient elution, 4/1 then 3/1 with 1% Et3N) afforded 51 mg (87%) of 12h as a white solid. Data for 12h: 1H NMR (500 MHz, CDCl3) 9.35 (d, J = 7.6 Hz, 1 H, HC(8)), 7.76 (s, 2 H, HC(6)), 7.65 (d, J = 7.4 Hz, 8 H, HC(8)), 7.51 (t, J = 7.6 Hz, 8 H, HC(9)), 7.42 (t, J = 7.4 Hz, 4 H, HC(10)), 7.37 (br s, 4 H, HC(4)), 6.70 (d, J = 7.6 Hz, 1 H, HC(7)), 5.56 (br s, 1 H, HC(1)), 5.20 (br s, 1 H, HC(1)), 2.78 (app br s, 2 H, H2C(2)), 2.17–2.09 (m, 2 H, H2C(2)); 13C NMR (126 MHz, CDCl3) 190.8 (C(8)), 133.3 (C(Aryl)), 128.9 (C(Aryl)), 127.7 (C(Aryl)), 127.3 (C(Aryl)); because of hindered rotation, many signals are not well-defined or not observed; IR (CDCl3 film) 3033 (w), 2923 (s), 2853 (m), 1951 (w), 1882 (w), 1668 (s), 1596 (m), 1576 (m), 1519 (s), 1498 (m), 1455 (m), 1434 (m), 1388 (m), 1304 (w), 1283 (m), 1249 (w), 1171 (m), 1138 (m), 1076 (w), 1029 (w), 875 (m), 844 (w), 758 (s), 698 (s); MS (ESI) 583.3 (M + H, 100), 526.3 (12), 338.3 (8); HRMS (ESI) calcd for C42H35N2O [M + H] 583.2749, found 583.2751; TLC Rf 0.22 (hexanes/CH2Cl2, 1/4 with 0.1% Et3N) [silica gel, UV].
(2R,5R)-(2,5-Bis(2-tolyl)pyrrolidine)-N-iminoacetaldehyde (12i)

Following general procedure 9, to a 10 mL, one-necked, round-bottom flask equipped with a stir bar was added an aqueous solution of glyoxal (40% w/w, 620 μL, 5.4 mmol, 20 equiv). A solution of (2R,5R)-1-amino-2,5-bis(2-tolyl)pyrrolidine 8i (72 mg, 0.27 mmol, 1.0 equiv) as a mixture with (2R,5R)-2,5-bis(2-tolyl)pyrrolidine 23i (14 mg, 0.06 mmol, 1.0 equiv) in THF (1.0 mL) was added at 0 °C. The ice/water bath was removed, and the flask was capped with a glass stopper. After the mixture was stirred 21 °C for 0.5 h, the reaction was basified with a solution of saturated NaHCO3 (2 mL) at 0 °C and vigorously stirred for 3 min. The organic phase was saved and the aqueous phase was extracted with dichloromethane (2 mL × 4). The combined organic phases was dried over Na2SO4, filtered, and concentrated to give a light yellow oil and solid. Purification by column chromatography (SiO2, 1.5 × 27 cm, hexanes/EtOAc, 9/1) afforded 73 mg (88%) of 12i as a slightly sticky white solid which adsorbs a small amount of hexanes. Data for 12i: 1H NMR (500 MHz, CDCl3) 9.23 (d, J = 7.6 Hz, 1 H, HC(11)), 7.28–7.12 (m, 6 H, HC(5), HC(6) and HC(7)), 6.92 (d, J = 6.9 Hz, 1 H, HC(8)), 6.84 (br s, 1 H, HC(8)), 6.34 (d, J = 7.6 Hz, 1 H, HC(10)), 5.53 (d, J = 5.8 Hz, 1 H, HC(1)), 5.12 (d, J = 6.2 Hz, 1 H, HC(1)), 2.65–2.52 (m, 2 H, H2C(2)), 2.42 (s, 3 H, H3C(9)), 2.40 (s, 3 H, H3C(9)), 1.92–1.80 (m, 2 H, H2C(2)); 13C NMR (126 MHz, CDCl3) 190.7 (C(11)), 140.6 (C(3)), 135.2 (C(3)), 134.7 (C(4)), 134.3 (C(4)), 132.8 (C(10)), 131.5 (C(5)), 130.9 (C(5)), 127.7 (C(7) or C(6)), 127.3 (C(7) or C(6)), 126.4 (C(6) or C(7)), 126.0 (C(6) or C(7)), 125.4 (C(8)), 124.6 (C(8)), 66.3 (C(3)), 61.7 (C(3)), 29.6 (C(2)), 28.7 (C(2)), 19.4 (C(9)), 19.2 (C(9)); IR (CDCl3 film) 3066 (w), 3021 (w), 2976 (w), 2871 (w), 2801 (w), 1668 (s), 1601 (w), 1518 (s), 1487 (m), 1460 (m), 1390 (m) 1348 (w), 1307 (m), 1287 (w), 1251 (m), 1220 (w), 1175 (m), 1147 (s), 1100 (w), 1052 (w), 1024 (w), 898 (w), 841 (w), 763 (m), 741 (m); MS (ESI) 329.2 (M + Na, 8), 307.2 (M + H, 100); HRMS (ESI) calcd for C20H23N2O [M + H] 307.1810, found 307.1816; TLC Rf 0.20 (hexanes/EtOAc, 9/1) [silica gel, UV, KMnO4].
(2R,5R)-(2,5-Bis(5-methyl-2-thienyl)pyrrolidine)-N-iminoacetaldehyde (12j)

Following general procedure 9, to a 4 mL vial equipped with a stir bar was added an aqueous solution of glyoxal (40% w/w, 100 μL, 0.88 mmol, 20 equiv). A solution of (2R,5R)-1-amino-2,5-bis(5-methyl-2-thienyl)pyrrolidine 8j (12 mg, 44 μmol, 1.0 equiv) in THF (0.66 mL) was added at 0 °C. The ice/water bath was removed, and the vial was sealed with a PTFE septum cap. After the solution was stirred at 21 °C for 0.5 h, TLC indicated the complete consumption of 8j. The reaction was basified with a solution of saturated NaHCO3 (1 mL) at 0 °C, vigorously stirred for 3 min. The aqueous phase was extracted with dichloromethane (1 mL × 5). The combined organic phases was dried over Na2SO4, filtered, and concentrated to give a light yellow oil and solid. Purification by column chromatography (SiO2, 1 × 25 cm, CH2Cl2/hexanes, 4/1, with 1% Et3N) afforded 14 mg (99%) of 12j as colorless flakes. Data for 12j: 1H NMR (500 MHz, CDCl3) 9.28 (d, J = 7.6 Hz, 1 H, HC(9)), 6.70–6.60 (br m, 3 H, HC(8) and HC(4)), 6.60–6.58 (br m, 2 H, HC(5)), 5.16 (app br s, 2 H, HC(1)), 2.72–2.62 (m, 2 H, H2C(2)), 2.44 (s, 6 H, H3C(7)), 2.15–2.05 (m, 2 H, H2C(2)); TLC Rf 0.16 (CH2Cl2/hexanes, 4/1) [silica gel, UV, KMnO4].
General Procedure 10: Second Condensation for the Preparation of Ligands 21 and 13 (Scheme 6 and Table 4)

To an oven-dried, 5 mL, Schlenk reaction flask equipped with a stir bar and a septum were added Na2SO4 (10.7 mg, 0.075 mmol, 0.5 equiv), a dichloromethane solution of (2R,5R)-(2,5-diarylpyrrolidine)-N-iminoacetaldehyde 12 (58 mg, 0.15 mmol, 1.0 equiv), and a dichloromethane solution of (2R,5R)-1-amino-2,5-diarylpyrrolidine 8 (0.17 mmol, 1.1.0 equiv). The solvent was carefully evaporated under reduced pressure assisted by vigorous stirring to give a light yellow solid. The flask was then filled with argon, and dichloromethane (0.75 mL) was added to give a light yellow solution. The reaction was stirred at 21 °C for 20 h, and the solution was filtered and concentrated to give a lightly yellow solid. Purification by column chromatography afforded the target ligand 13. We were unable to obtain melting points for bis-hydrazone ligands due to decomposition at elevated temperature.
Preparation of N,N′-(Ethane-1,2-diylidene)bis((2R)-(2-arylpyrrolidin-1-amines)) (21)
N,N′-(Ethane-1,2-diylidene)bis((R)-(2-(4-methoxyphenyl)pyrrolidin-1-amine)) (21a)

To a 15 mL, one-necked, round-bottom flask equipped with a stir bar, an argon gas inlet, and a septum were added (R)-(2-(4-methoxyphenyl)pyrrolidine)-N-iminoacetaldehyde 20a (40 mg, 0.17 mmol, 1.0 equiv) and Na2SO4 (13 mg, 0.09 mmol, 0.5 equiv). After two cycles of evacuation/argon fill, a solution of N-amino-(R)-2-(4-methoxyphenyl)pyrrolidine 19a (37 mg, 0.19 mmol, 1.1.0 equiv) in dichloromethane (0.86 mL) was added at room temperature to give a red solution. After the solution was stirred at 22 °C for 20 h, the solvent was evaporated under reduced pressure to give a red oil. Purification by column chromatography (SiO2, 1.5 × 16 cm, hexanes/EtOAc with 1% Et3N, gradient elution, 9/1, then 4/1) afforded 60 mg (86%) of 21a as a slightly pink, white solid. The target compound is very unstable in air and gradually turned into a purple sticky oil. Data for 21a: 1H NMR (500 MHz, CDCl3) 7.13 (d, J = 8.5 Hz, 4 H, HC(6)), 6.97 (s, 2 H, HC(10)), 6.83 (d, J = 8.5 Hz, 4 H, HC(7)), 4.43 (dd, J = 8.0, 5.2 Hz, 2 H, HC(1)), 3.78 (s, 6 H, HC(9)), 3.62 (ddd, J = 9.7, 7.6, 4.2 Hz, 2 H, HC(4)), 3.21 (q, J = 9.5, 2 H HC(4)), 2.28 (dq, J = 12.6, 7.9 Hz, 2 H, HC(2)), 2.04–1.83 (m, 4 H, HC(3)), 1.80–1.73 (m, 2 H, HC(2)); 13C NMR (126 MHz, CDCl3) 158.4 (C(8)), 135.9 (C(5)), 134.8 (C(10)), 127.3 (C(6)), 113.8 (C(7)), 65.6 (C(1)), 55.2 (C(9)), 51.2 (C(4)), 34.5 (C(2)), 22.1 (C(3)); IR (CDCl3 film) 2965 (m), 2834 (m), 1611 (m), 1585 (w), 1541 (m), 1511 (s), 1462 (m), 1338 (w), 1301 (w), 1246 (s), 1172 (m), 1126 (m), 1032 (m), 828 (m), 806 (w); MS (ESI) 407.2 (100, M + H); HRMS (ESI) calcd for C24H31N4O2 [M + H] 407.2447, found 407.2437; TLC Rf 0.39 (hexanes/EtOAc, 2/1 with 1% Et3N) [silica gel, UV, KMnO4]; [α]D24 +229.3 (c = 0.15, chloroform).
N,N′-(Ethane-1,2-diylidene)bis((R)-(2-(2-naphthyl)pyrrolidin-1-amine)) (21f)

To a 25 mL, one-necked, round-bottom flask equipped with a stir bar, an argon gas inlet and a septum were added (R)-(2-(2-naphthyl)pyrrolidine)-N-iminoacetaldehyde 20f (82 mg, 0.32 mmol, 1.0 equiv) and Na2SO4 (24 mg, 0.16 mmol, 0.5 equiv). After two cycles of evacuation/argon fill, a solution of N-amino-(R)-2-(2-naphthyl)pyrrolidine 19f (76 mg, 0.36 mmol, 1.1.0 equiv) in dichloromethane (1.65 mL) was added at room temperature to give a yellow solution. After the solution was stirred at 22 °C for 20 h, the solvent was evaporated. The crude product was purified by column chromatography (SiO2, 1.5 × 21 cm, CH2Cl2 with 1% Et3N) to afford 125 mg (86%) of 21f as a white solid. Data for 21f: 1H NMR (500 MHz, CDCl3) 7.81–7.76 (m, 4 H, HC(Aryl)), 7.65 (br s, 4 H, HC(5)), 7.47–7.39 (m, 4 H, HC(Aryl)), 7.35–7.32 (m, 2 H, HC(Aryl)), 7.03 (s, 0.36 H, HC(15)), 7.01 (s, 0.64 H, HC(15)), 4.65–4.61 (m, 2 H, HC(1)), 3.73–3.68 (m, 2 H, HC(4)), 3.31–3.24 (m, 2 H, HC(4)), 2.41–2.33 (m, 2 H, HC(2)), 2.06–1.91 (m, 4 H, H2C(3)), 1.90–1.83 (m, 2 H, HC(2)); 13C NMR (126 MHz, CDCl3) 141.3 (C(6)), 141.2 (C(6)), 134.9 (C(9) or C(14)), 134.9 (C(9) or C(14)), 133.4 (C(Aryl)), 132.6 (C(Aryl)), 128.2 (C(Aryl)), 127.8 (C(Aryl)), 127.8 (C(Aryl)), 127.5 (C(Aryl)), 125.9 (C(Aryl)), 125.4 (C(Aryl)), 125.3 (C(Aryl)), 124.8 (C(5) or (C(7)), 124.7 (C(5) or (C(7)), 66.5 (C(1)), 66.3 (C(1)), 51.5 (C(4)), 51.5 (C(4)), 34.3 (C(2)), 34.3 (C(2)), 22.2 (C(3)), 22.2 (C(3)); IR (CDCl3 film) 3053 (m), 2969 (m), 2868 (m), 1632 (w), 1600 (w), 1543 (s), 1507 (m), 1477 (w), 1444 (w), 1367 (m), 1334 (m), 1311 (m), 1270 (m), 1197 (s), 1126 (s), 1030 (w), 855 (m), 817 (s), 748 (s); MS (ESI) 447.3 (100, M + H); HRMS (ESI) calcd for C30H31N4 [M + H] 447.2549, found 447.2547; TLC Rf 0.73 (hexanes/EtOAc, 1/1 with 0.5% Et3N) [silica gel, UV, KMnO4]; [α]D24 +295.0 (c = 0.15, chloroform).
Preparation of N,N′-(Ethane-1,2-diylidene)bis((2R,5R)-(2,5-diarylpyrrolidin-1-amines)) (13)
N,N′-(Ethane-1,2-diylidene)bis((2R,5R)-(2,5-bis(4-methoxyphenyl)pyrrolidin-1-amine)) (13a)

Following general procedure 10, to an oven-dried, 10 mL Schlenk reaction flask equipped with a stir bar and a septum were added Na2SO4 (11 mg, 0.08 mmol, 0.5 equiv), a dichloromethane solution of (2R,5R)-(2,5-bis(4-methoxyphenyl)pyrrolidine)-N-iminoacetaldehyde 12a (51 mg, 0.15 mmol, 1.0 equiv), and a dichloromethane solution of (2R,5R)-1-amino-2,5-bis(4-methoxyphenyl)pyrrolidine 8a (50 mg, 0.17 mmol, 1.1.0 equiv). The solvent was carefully evaporated under reduced pressure assisted by vigorous stirring to give a light yellow solid. The flask was then filled with argon and dichloromethane (0.75 mL) was added to give a light yellow solution. The reaction was stirred at 22 °C for 20 h, and the solution was filtered and concentrated to give a lightly yellow solid. Purification by column chromatography (SiO2, 1.5 × 22 cm, CH2Cl2/hexanes, 9/1, with 1% Et3N) afforded 93 mg (84%) of 13a as a white solid. Data for 13a: 1H NMR (500 MHz, CDCl3) 7.02 (d, J = 8.4 Hz, 4 H, HC(4)), 6.83 (d, J = 8.7 Hz, 4 H, HC(5)), 6.62 (s, 2 H, HC(8)), 4.92 (d, J = 6.8 Hz, 4 H, HC(1)), 3.80 (s, 12 H, H3C(7)), 2.42–2.33 (m, 4 H, HC(2)), 1.67–1.57 (m, 4 H, HC(2)); 13C NMR (126 MHz, CDCl3) 158.4 (C(6)), 135.5 (C(3)), 134.4 (C(8)), 127.2 (C(4)), 113.8 (C(5)), 64.0 (C(1)), 55.3 (C(7)), 31.2 (C(2)); IR (CDCl3 film) 2982 (m), 2955 (m), 2898 (m), 2833 (m), 1610 (m), 1584 (m), 1542 (m), 1510 (s), 1461 (m), 1420 (w), 1352 (m), 1246 (s), 1172 (s), 1139 (m), 1107 (m), 1033 (m), 890 (w), 868 (w), 829 (m), 808 (m), 764 (w), 728 (m); MS (ESI) 619.3 (M + H, 100); HRMS (ESI) calcd for C38H43N4O4 [M + H] 619.3284, found 619.3277; TLC Rf 0.18 (CH2Cl2/hexanes, 9/1 with 0.5% Et3N) [silica gel, UV]; [α]D24 +467.4 (c = 0.15, chloroform).
N,N′-(Ethane-1,2-diylidene)bis((2R,5R)-(2,5-bis(2-naphthyl)pyrrolidin-1-amine)) (13f)

Following general procedure 10, to an oven-dried, 10 mL Schlenk reaction flask equipped with a stir bar and a septum were added Na2SO4 (10.9 mg, 0.075 mmol, 0.5 equiv), a dichloromethane solution of (2R,5R)-(2,5-bis(2-naphthyl)pyrrolidine)-N-iminoacetaldehyde 12f (58 mg, 0.15 mmol, 1.0 equiv) and a dichloromethane solution of (2R,5R)-1-amino-2,5-bis(2-naphthyl)pyrrolidine 8f (57 mg, 0.17 mmol, 1.1.0 equiv). The solvent was carefully evaporated under reduced pressure assisted by vigorous stirring to give a light yellow solid. The flask was then filled with argon and dichloromethane (0.75 mL) was added to give a light yellow solution. The reaction was stirred at 21 °C for 20 h, and the solution was filtered, concentrated to give a lightly yellow solid. Purification by column chromatography (SiO2, 1.5 × 22 cm, hexanes/CH2Cl2, 3/2, with 1% Et3N) afforded 93 mg (87%) of 13f as a white solid. Data for 13f: 1H NMR (500 MHz, CDCl3) 7.85–7.76 (m, 4 H, HC(Aryl)), 7.53–7.45 (m, 12 H, HC(Aryl)), 7.27–7.25 (m, 4 H, HC(Aryl)), 5.20 (d, J = 7.0 Hz, 4 H, HC(1)), 2.55–2.45 (m, 4 H, HC(2)), 1.79–1.72 (m, 4 H, HC(2)); 13C NMR (126 MHz, CDCl3) 140.7 (C(4)), 134.5 (C(13)), 133.4 (C(12) or C(7)), 132.6 (C(12) or C(7)), 128.3 (C(Aryl)), 128.0 (C(Aryl)), 127.6 (C(Aryl)), 126.0 (C(Aryl)), 125.5 (C(Aryl)), 124.8 (C(Aryl)), 124.7 (C(Aryl)), 64.8 (C(1)), 31.0 (C(2)); IR (CDCl3 film) 3053 (m), 2972 (m), 2940 (m), 2871 (m), 1672 (w), 1632 (w), 1599 (m), 1540 (m), 1508 (m), 1443 (w), 1369 (m), 1311 (m), 1268 (m), 1219 (s), 1161 (s), 1135 (s), 1052 (w), 1018 (w), 984 (w), 950 (w), 855 (m), 816 (s); MS (ESI) 699.3 (100, M + H); HRMS (ESI) calcd for C50H43N4 [M + H] 699.3488, found 699.3488; TLC Rf 0.30 (hexanes/CH2Cl2, 6/4 with 0.1% Et3N) [silica gel, UV, KMnO4]; [α]D24 +626.0 (c = 0.15, chloroform).
N,N′-(Ethane-1,2-diylidene)bis((2R,5R)-(2,5-bis(3,5-dimethyphenyl)pyrrolidin-1-amine)) (13g)

Following general procedure 10, to an oven-dried, 5 mL Schlenk reaction flask equipped with a stir bar and a septum were added Na2SO4 (10 mg, 0.07 mmol, 0.5 equiv), a dichloromethane solution of (2R,5R)-(2,5-bis(3,5-dimethyphenyl)pyrrolidine)-N-iminoacetaldehyde 12g (47 mg, 0.14 mmol, 1.0 equiv), and a dichloromethane solution of (2R,5R)-1-amino-2,5-bis(3,5-dimethyphenyl)pyrrolidine 8g (46 mg, 0.15 mmol, 1.1.0 equiv). The solvent was carefully evaporated under reduced pressure assisted by vigorous stirring to give a light yellow solid. The flask was then filled with argon, and dichloromethane (0.7 mL) was added to give a light yellow solution. The reaction was stirred at 21 °C for 20 h. Purification by column chromatography (SiO2, 1.5 × 21 cm, hexanes/CH2Cl2, 2/1, with 1% Et3N) afforded 77 mg (90%) of 13g as a white solid. Recrystallization from hexanes afforded 71 mg (83%) of 13g as a fluffy needle. Data for 13g: 1H NMR (500 MHz, CDCl3) 6.87 (s, 4 H, HC(6)), 6.74 (s, 8 H, HC(4)), 6.69 (s, 2 H, HC(8)), 4.95 (d, J = 7.1 Hz, 4 H, HC(1)), 2.47–2.43 (m, 4 H, H2C(2)), 2.31 (s, 24 H, H3C(7)), 1.71–1.63 (m, 4 H, H2C(2)); 13C NMR (126 MHz, CDCl3) 143.6 (C(3)), 137.8 (C(5)), 134.5 (C(8)), 128.3 (C(6)), 123.9 (C(4)), 64.6 (C(1)), 31.1 (C(2)), 21.4 (C(7)); IR (CDCl3 film) 2969 (m), 2917 (m), 2871 (m), 1602 (m), 1542 (m), 1455 (m), 1376 (w), 1316 (m), 1296 (w), 1267 (m), 1211 (s) 1155 (s), 1040 (w), 703 (m); MS (ESI) 611.4 (M + H, 100), 335.2 (43); HRMS (ESI) calcd for C42H51N4 [M + H] 611.4114, found 611.4125; TLC Rf 0.16 (hexanes/CH2Cl2, 2/1 with 0.1% Et3N) [silica gel, UV, KMnO4]; [α]D24 313.5 (c = 0.15, chloroform).
N,N′-(Ethane-1,2-diylidene)bis((2R,5R)-(2,5-bis(5-phenylbiphenyl-3-yl)pyrrolidin-1-amine)) (13h)

Following general procedure 10, to an oven-dried, 5 mL Schlenk reaction flask equipped with a stir bar and a septum were added Na2SO4 (6.2 mg, 0.044 mmol, 0.5 equiv), a dichloromethane solution of (2R,5R)-(2,5-bis(5-phenylbiphenyl-3-yl)pyrrolidine)-N-iminoacetaldehyde 12f (48 mg, 0.082 mmol, 1.0 equiv), and a dichloromethane solution of (2R,5R)-1-amino-2,5-bis(5-phenylbiphenyl-3-yl)pyrrolidine 8h (53 mg, 0.98 mmol, 1.2 equiv). The solvent was carefully evaporated under reduced pressure assisted by vigorous stirring to give a light yellow solid. The flask was then filled with argon, and dichloromethane (0.45 mL) was added to give a light yellow solution. The reaction was stirred at 21 °C for 20 h and then concentrated to give a bright yellow gel. Purification by column chromatography (SiO2, 1.5 × 30 cm, hexanes/CH2Cl2, 1/1, with 0.5% Et3N) afforded 54 mg (59%) of 13h as a white powder after trituration with hexanes. Data for 13h: 1H NMR (500 MHz, CDCl3) 7.67 (s, 4 H, HC(6)), 7.61–7.57 (m, 16 H, H2C(Aryl)), 7.38–7.30 (m, 32 H, H2C(Aryl)), 6.95 (s, 2 H, HC(7)), 5.22 (d, J = 6.9 Hz, 4 H, HC(1)), 2.63–2.54 (m, 4 H, H2C(2)), 1.86–1.79 (m, 2 H, H2C(2)); 13C NMR (126 MHz, CDCl3) 144.4 (C(3)), 142.0 (C(5)), 141.1 (C(Aryl)), 134.7 (7), 128.8 (C(Aryl)), 127.3 (C(Aryl)), 127.2 (C(Aryl)), 124.8 (C(6)), 124.0 (C(4)), 65.0 (br, C(1)), 31.3 (C(2)); IR (CDCl3 film) 3058 (m), 3032 (m), 2973 (m), 2940 (w), 2871 (w), 1947 (w), 1885 (w), 1596 (s), 1576 (m), 1542 (m), 1497 (m), 1454 (m), 1433 (m), 1410 (m), 1344 (w), 1316 (m), 1268 (m), 1215 (m), 1164 (m), 1143 (m), 1075 (w), 1029 (w), 980 (w), 875 (m), 757 (s), 698 (s); MS (ESI) 1107.5 (M + H, 100), 637 (24), 583 (47); HRMS (ESI) calcd for C82H67N4 [M + H] 1107.5366, found 1107.5365; TLC Rf 0.55 (hexanes/CH2Cl2, 1/1 with 0.5% Et3N) [silica gel, UV]; [α]D24 +444.0 (c = 0.15, chloroform).
N,N′-(Ethane-1,2-diylidene)bis((2R,5R)-(2,5-bis(2-tolyl)pyrrolidin-1-amine)) (13i)

To an oven-dried, 5 mL Schlenk reaction flask equipped with a stir bar and a septum were added Na2SO4 (16.2 mg, 0.11 mmol, 0.5 equiv), a dichloromethane solution of (2R,5R)-(2,5-bis(2-tolyl)pyrrolidine)-N-iminoacetaldehyde 12i (68 mg, 0.22 mmol, 1.0 equiv) and a dichloromethane solution of (2R,5R)-1-amino-2,5-bis(5-phenylbiphenyl-3-yl)pyrrolidine 8i (73 mg, 0.27 mmol, 1.2 equiv) as a mixture with (2R,5R)-2,5-bis(5-phenylbiphenyl-3-yl)pyrrolidine 23i (14 mg, 0.056 mmol). The solvent was carefully evaporated under reduced pressure assisted by vigorous stirring to give a bright yellow gel. The flask was then filled with argon and dichloromethane (0.5 mL) was added to give a light yellow solution. The reaction was stirred at 21 °C for 20 h. Purification by column chromatography (SiO2, 1.5 × 20 cm, hexanes/EtOAc, 19/1, with 0.5% Et3N) afforded 84 mg of 13i contaminated with 23i. Trituration with hexanes (∼2.5 mL) and careful removal of the solvent provided 54 mg (43%) of 13i as a white solid. Data for 13i: 1H NMR (500 MHz, CDCl3) 7.18–7.12 (m, 12 H, HC(5), HC(6) and HC(7)), 6.96–6.93 (m, 4 H, HC(8)), 6.48 (s, 2 H, HC(10)), 5.12 (d, J = 7.1 Hz, 4 H, HC(1)), 2.38–2.34 (m, 4 H, H3C(2)), 2.32 (s, 12 H, H2C(9)), 1.65–1.57 (m, 4 H, HC(2)); 13C NMR (126 MHz, CDCl3) 140.7 (C(3)), 134.5 (C(4)), 133.8 (C(10)), 130.7 (C(5)), 126.5 (C(7) or C(6)), 125.9 (C(8)), 125.7 (C(6) or C(7)), 62.3 (C(1)), 29.1 (C(2)), 19.3 (C(9)); IR (CDCl3 film) 3059 (w), 3018 (w), 2975 (m), 2942 (m), 2869 (w), 1603 (w), 1538 (m), 1483 (m), 1460 (m), 1443 (m), 1382 (w), 1351 (m), 1319 (w), 1280 (m), 1266 (m), 1231 (m), 1216 (m), 1194 (s), 1178 (s), 1146 (s), 1099 (w), 1049 (w), 1021 (m), 980 (w), 885 (w), 871 (w), 787 (w), 763 (s); MS (ESI) 555.3 (M + H, 100); HRMS (ESI) calcd for C38H43N4 [M + H] 555.3488, found 555.3494; TLC Rf 0.44 (hexanes/EtOAc, 9/1 with 0.1% Et3N) [silica gel, UV, KMnO4]; [α]D24 +343.4 (c = 0.15, chloroform).
N,N′-(Ethane-1,2-diylidene)bis((2R,5R)-(2,5-bis(5-methyl-2-thienyl)pyrrolidin-1-amine)) (13j)

Following general procedure 10, to an oven-dried, 4 mL reaction flask equipped with a stir bar and a PTFE septum cap were added Na2SO4 (6.2 mg, 0.044 mmol, 0.5 equiv), a solution of (2R,5R)-(2,5-bis(5-methyl-2-thienyl)pyrrolidine)-N-iminoacetaldehyde 12j (14.0 mg, 0.044 mmol, 1.0 equiv) in CH2Cl2 (60 μL), and a solution of (2R,5R)-1-amino-2,5-bis(5-methyl-2-thienyl)pyrrolidine 8j (13.3 mg, 0.48 mmol, 1.1.0 equiv) in CH2Cl2 (270 μL). The vial was flushed with argon, and the reaction was stirred at 22 °C. After 21 h, the mixture was filtered and concentrated. Purification by column chromatography (SiO2, 1 × 23 cm, hexanes/CH2Cl2, 65/35, with 1% Et3N) afforded 21 mg (82%) of 13j as a white solid. Data for 13j: 1H NMR (500 MHz, CDCl3) 6.88 (s, 2 H, HC(8)), 6.59 (d, J = 3.4 Hz, 4 H, HC(4)), 6.55–6.53 (m, 4 H, HC(5)), 4.99 (d, J = 6.9 Hz, 4 H, HC(1)), 2.57–2.45 (m, 4 H, H2C(2)), 2.42 (s, 12 H, H3C(7)), 1.90–1.82 (m, 4 H, H2C(2)); 13C NMR (126 MHz, CDCl3) 144.6 (C(3)), 138.2 (C(6)), 135.5 (C(8)), 124.7 (C(5)), 123.6 (C(4)), 60.3 (C(1)), 31.8 (C(2)), 15.3 (C(7)); IR (CDCl3 film) 3059 (w), 2972 (m), 2916 (m), 2857 (m), 1546 (m), 1483 (w), 1443 (m), 1354 (m), 1296 (m), 1269 (m), 1214 (m), 1167 (m), 1132 (m), 1040 (m), 1006 (w), 966 (w), 797 (m); MS (ESI) 579.2 (100, M + H); HRMS (ESI) calcd for C30H35N4S4 [M + H] 579.1745, found 579.1741; TLC Rf 0.63 (hexanes/CH2Cl2, 35/65 with 0.1% Et3N) [silica gel, UV, KMnO4]; [α]D24 +448.2 (c = 0.15, chloroform).
Preparation of 1,2-Bis(2,2-bis((R)-1-phenylethyl)hydrazono)ethanes (Scheme 8)
(−)-N-Nitroso-α,α′-dimethyldibenzylamine47 (27)

To an oven-dried, 25 mL, one-necked, round-bottom flask equipped with a stir bar, an argon inlet adaptor was charged (R,R)-bis(α-methylbenzyl)amine (460 μL, 2.0 mmol, 1.0 equiv). After three cycles of evacuation/argon fill, dichloromethane (10 mL) and pyridine (325 μL, 4 mmol, 2 equiv) solution were added. The colorless solution was cooled to 0 °C, and the septum was temporarily removed to allow the addition of nitrosonium tetrafluoroborate (94 mg, 0.78 mmol, 2 equiv) in one portion. The ice/water bath was removed, and mixture was stirred at 22 °C. After 12 h, a large amount of precipitates formed. The reaction was quenched with an aqueous solution of HCl (1 M, 15 mL) at 0 °C with vigorous stirring to dissolve the precipitates. The organic layer was saved, and the aqueous layer was extracted with dichloromethane (10 mL × 2). The combined organic extract was dried over Na2SO4, filtered, and concentrated to give a mixture of light yellow and colorless crystals. Purification by column chromatography (SiO2, 3 × 15 cm, hexanes/EtOAc, 9/1) afforded 482 mg (95%) of 27 as a light yellow solid. Data for 27: 1H NMR (500 MHz, CDCl3) 7.20–7.06 (m, 6 H, HC(5), HC(5′), HC(6) and HC(6′)), 6.98 (d, J = 7.4 Hz, 2 H, HC(4)), 6.91 (d, J = 7.4 Hz, 2 H, HC(4′)), 6.28 (q, J = 7.0 Hz, 1 H, HC(2)), 4.94 (q, J = 7.2 Hz, 1 H, HC(2′)), 1.86 (d, J = 7.2 Hz, 3 H, H3C(1′)), 1.55 (d, J = 7.0 Hz, 3 H, H3C(1)); 13C NMR (126 MHz, CDCl3) 141.1 (C(3′)), 136.9 (C(3)), 128.2 (C(4′)), 128.1 (C(Aryl)), 127.8 (C(Aryl)), 127.4 (C(Aryl)), 126.6 (C(Aryl)), 59.3 (C(2′)), 51.5 (C(2)), 24.0 (C(1′)), 15.2 (C(1)). IR (CHCl3 film) 3062 (m), 3031 (m), 2978 (m), 2935 (m), 1602 (w), 1495 (m), 1432 (s), 1386 (s), 1341 (m), 1170 (s), 1084 (s), 1027 (m), 991 (w), 913 (w), 825 (w), 790 (w), 697 (s); MS (ESI) 255.1 (100, M + H), 151.1 (23), 105.1 (75); HRMS (ESI) calcd for C16H19N2O [M + H] 255.1497, found 255.1493; TLC Rf 0.40 (hexanes/EtOAc, 9/1) [silica gel, UV].
(−)-N-Amino-α,α′-dimethyldibenzylamine47 (28)

A solution of (−)-N-nitroso-α,α′-dimethyldibenzylamine 27 (240 mg, 0.94 mmol, 1.0 equiv) in EtOH (2.2 mL) was cannulated into a 10 mL, one-piece, round-bottom flask and reflux condenser equipped with a stir bar, an argon inlet adaptor, and a septum. The solution was refluxed for 15 min, and sodium (∼480 mg, 21 mmol, 22 equiv) was added in 18 portions at this temperature under a slight positive argon pressure by temporary removal of the septum. Each addition caused vigorous bubbling. The reaction mixture gradually thickened, and additional amounts of EtOH (0.2 mL × 3) were added from time to time to keep the mixture stirring. The reaction was stirred for a further 15 min after complete addition of sodium before cooling to 0 °C. Ice-cold water (2.5 mL, degassed by nitrogen) was added slowly to the flask with swirling periodically, and the flask was then warmed to room temperature to give a mostly homogeneous solution. This solution was extracted with Et2O ( × 5), and the combined organic phase was dried with MgSO4, filtered, and concentrated to give a mixture of 28 and bis(α-methylbenzyl)amine (84:16) as a colorless oil (184 mg). This mixture was used without further purification for the glyoxal condensation. Data for 28: 1H NMR (500 MHz, CDCl3) 7.30–7.12 (m, 10 H, HC(Aryl), 3.62 (q, J = 6.7 Hz, 2 H, HC(2)), 1.30 (d, J = 6.7 Hz, 6 H, H3C(1)).
1,2-Bis(2,2-bis((R)-1-phenylethyl)hydrazono)ethane (29)

To a 5 mL Schlenk reaction flask equipped with a stir bar and a septum was added a solution of crude (−)-N-amino-α.α′-dimethyldibenzylamine 28 (156 mg, 0.65 mmol, 2.5 equiv) as a mixture with (−)-α.α′-dimethyldibenzylamine (0.12 mmol) in MeOH (0.85 mL). A solution of glyoxal (38 μL, 0.26 mmol, 1.0 equiv) was added at room temperature. The solution turned light green initially and eventually became light yellow after 12 h. Methanol was evaporated under reduced pressure, and the residue was taken up by EtOAc (4 mL), dried over Na2SO4, filtered and concentrated to give a yellow oil. Purification by column chromatography (SiO2, 1 × 25 cm, gradient elution, hexanes/EtOAc with 1% Et3N, 19/1, 14/1 then 9/1) afforded 84 mg (65%) of 29 as a light yellow, sticky oil. Data for 29: 1H NMR (500 MHz, CDCl3) 7.34 (s, 2 H, HC(1)), 7.24–7.16 (m, 20 H, HC(Aryl)), 4.63 (q, J = 6.8 Hz, 4 H, H2C(2)), 1.51 (q, J = 6.9 Hz, 12 H, H2C(3)); 13C NMR (126 MHz, CDCl3) 142.9 (C(4)), 135.5 (C(1)), 128.0 (C(5) or C(6)), 127.5 (C(5) or C(6)), 126.7 (C(7)), 58.3 (C(2)), 18.8 (C(3)); IR (CDCl3 film) 3059 (w), 3026 (m), 2973 (m), 2930 (m), 1947 (w), 1878 (w), 1805 (w), 1671 (w), 1600 (w), 1543 (m), 1492 (m), 1450 (m), 1371 (m), 1299 (w), 1276 (w), 1206 (m), 1156 (m), 1078 (m), 1034 (m), 981 (w), 789 (w), 758 (m); MS (ESI) 503.3 (M + H, 100); HRMS (ESI) calcd for C34H39N4 [M + H] 503.3175, found 503.3186; TLC Rf 0.55 (hexanes/EtOAc, 9/1 with 0.5% Et3N) [silica gel, UV]; [α]D24 −268.1 (c = 0.15, chloroform).
Preparation of Bis-hydrazone Ligands with Binaphthalene Scaffold (Scheme 9)
(S)-3,5-Dihydro-4H-dinaphtho[2,1-c:10,20-e]azepin-4-amine (31a)

To a 10 mL Schlenk reaction flask equipped a stir bar and a septum was added (S)-2,2′-bis(bromomethyl)-1,1′-binaphthalene48 (158 mg, 0.36 mmol, 1.0 equiv). After one cycle of evacuation/argon fill, THF (3.6 mL) was added to give a pale yellow solution. Anhydrous hydrazine (230 μL, 7.2 mmol, 20 equiv) was added at room temperature to give a cloudy mixture. After the mixture was stirred for 3 h, the solvent was evaporated under reduced pressure, and a saturated solution of NaHCO3 (15 mL) was added. The aqueous phase was extracted with CH2Cl2 (8 mL × 4), and the combined organic phases was dried over K2CO3, filtered, and concentrated to give 110 mg (99%) of 31a after trituration with pentane and evaporation of the solvent ( × 4). The crude product was used without further purification. The spectroscopic data matched those from the literature.70 Data for 31a: 1H NMR (500 MHz, CDCl3) 7.98 (d, J = 8.2 Hz, 2 H, HC(7)), 7.97 (d, J = 8.1 Hz, 2 H, HC(6)), 7.62 (d, J = 8.2 Hz, 2 H, HC(5)), 7.50–7.46 (m, 4 H, HC(8) and HC(10)), 7.30–7.26 (m, 2 H, HC(9)), 3.91 (d, J = 12.4 Hz, 2 H, H2C(1)), 3.64 (d, J = 12.3 Hz, 2 H, H2C(1)), 3.29 (s, 2 H, NH2); 13C NMR (126 MHz, CDCl3) 135.0 (C(Aryl)), 133.3 (C(Aryl)), 132.9 (C(Aryl)), 131.5 (C(11)), 128.5 (C(5)), 128.3 (C(6)), 127.5 (C(4)), 127.5 (C(7) or C(9)), 125.9 (C(8)), 125.6 (C(7) or C(9)), 61.7 (C(1)); MS (ESI) 311.2 (M + H, 100), 281.1 (13); HRMS (ESI) calcd for C22H19N2 [M + H] 311.1548, found 311.1554; TLC Rf 0.32 (CH2Cl2/MeOH, 95/5 with 1% Et3N) [silica gel, UV].
(S)-2-((3H-Dinaphtho[2,1-c:1′,2′-e]azepin-4(5H)-yl)imino)acetaldehyde (32a)

A solution of 1-aminoazepine 31a (50 mg, 0.16 mmol, 1.0 equiv) in THF (0.8 mL) was added to an aqueous solution of glyoxal (40% w/w, 370 μL, 3.2 mmol, 20 equiv) at 0 °C in a 10 mL, one-necked round-bottom flask equipped with a stir bar. The ice/water bath was removed, and the reaction was stirred at room temperature for 1 h. The solution was poured a saturated solution of NaHCO3 (15 mL) at 0 °C, and the aqueous phase was extracted with CH2Cl2 (8 mL × 4). The combined organic phases was dried over Na2SO4, filtered, and concentrated to give a yellow film. The product was loaded onto a silica gel column (SiO2, 2.5 × 20 cm) with hexanes/CH2Cl2 (1/1) and eluted with hexanes/EtOAc (9/1 with 1% Et3N) to afford 187 mg (86%) of 32a as a white powder. Purification by column chromatography (SiO2, 1.5 × 21 cm, CH2Cl2 with 1% Et3N) afforded 49 mg (∼87%) of 32a as a white solid with some hexanes adsorbed. Data for 32a: 1H NMR (500 MHz, CDCl3) 7.46 (d, J = 7.4 Hz, 1 H, HC(3)), 8.01 (d, J = 8.3 Hz, 2 H, HC(Aryl)), 7.97 (d, J = 8.2 Hz, 2 H, HC(Aryl)), 7.59 (d, J = 8.3 Hz, 2 H, HC(Aryl)), 7.53–7.49 (m, 2 H, HC(Aryl)), 7.44 (d, J = 8.6 Hz, 2 H, HC(Aryl)), 7.30 (ddd, J = 8.4, 6.8, 1.1 Hz, 1 H, HC(Aryl)), 6.86 (d, J = 7.4 Hz, 1 H, HC(2)), 4.65 (d, J = 13.2 Hz, 2 H, H2C(1)), 4.10 (br s, 2 H, H2C(1)); 13C NMR (126 MHz, CDCl3) 191.0 (C(3)), 133.4 (C(Aryl)), 131.6 (C(Aryl)), 130.7 (C(Aryl)), 129.5 (C(Aryl)), 128.4 (C(Aryl)), 127.4 (C(Aryl)), 127.0 (C(Aryl)), 126.4 (C(Aryl)), 126.2 (C(Aryl)), three carbon signals were not observed due to hindered rotation; IR (CHCl3 film) 3053 (m), 2928 (m), 2815 (m), 1672 (s), 1595 (w), 1519 (s), 1446 (m), 1395 (m), 1346 (m), 1326 (m), 1241 (m), 1139 (s), 1072 (m), 1029 (w), 989 (m), 918 (w), 866 (w), 817 (s), 701 (w); MS (ESI) 351.1 (M + H, 100); HRMS (ESI) calcd for C24H19N2O [M + H] 351.1497, found 351.1501; TLC Rf 0.21 (CH2Cl2 with 1% Et3N) [silica gel, UV].
(S,S)-N,N′-(Ethane-1,2-diylidene)bis(3H-dinaphtho[2,1-c:1′,2′-e]azepin-4(5H)-amine) (33a)

To an oven-dried 5 mL, one-necked, round-bottom flask equipped with a stir bar, an argon gas inlet, and a septum were added Na2SO4 (8.6 mg, 0.06 mmol, 0.5 equiv), a solution of 1-aminoazepine 31a (43 mg, 0.14 mmol, 1.1.0 equiv) in CH2Cl2 (0.4 mL), and a solution of 32a (44 mg, 0.12 mmol, 1.0 equiv) in CH2Cl2 (1.0 mL). The flask was purged with argo,n and the reaction was stirred at room temperature for 12 h. The pale yellow solution was filtered and concentrated to give a pale yellow solid. Purification by column chromatography (SiO2, 1.5 × 16 cm, hexanes/EtOAc with 1% Et3N, 19/1, then 2:1) afforded 67 mg of 33a as a pale yellow powder after azeotrope removal of EtOAc with hexanes. Data for 33a: 1H NMR (500 MHz, CDCl3) 7.93 (d, J = 8.1 Hz, 4 H, HC(6)), 7.91 (d, J = 8.3 Hz, 4 H, HC(5)), 7.55 (d, J = 8.3 Hz, 4 H, HC(4)), 7.49–7.46 (m, 8 H, HC(7) and HC(9)), 7.29–7.26 (m, 4 H, HC(8)), 7.23 (s, 2 H, HC(12)), 4.46 (d, J = 12.5 Hz, 2 H, H2C(1)), 3.73 (d, J = 12.4 Hz, 2 H, H2C(1)); 13C NMR (126 MHz, CDCl3) 136.1 (C(12)), 134.6 (C(Aryl)), 133.2 (C(Aryl)), 133.1 (C(Aryl)), 131.4 (C(11)), 128.8 (C(5)), 128.3 (C(6)), 127.4 (C(4)), 127.4 (C(7) or C(9)), 125.9 (C(8)), 125.7 (C(7) or C(9)), 56.3 (C(1)); IR (CDCl3 film) 3051 (m), 3004 (w), 2939 (m), 2875 (w), 2815 (w), 1594 (w), 1546 (m), 1508 (m), 1460 (m), 1439 (w), 1364 (m), 1325 (w), 1235 (m), 1144 (m), 1092 (m), 1055 (s), 1006 (w), 969 (s), 817 (s), 751 (s), 703 (w); MS (ESI) 643.3 (M + H, 100); HRMS (ESI) calcd for C46H35N4 [M + H] 643.2862, found 643.2848; TLC Rf 0.55 (hexanes/EtOAc, 2/1 with 0.5% Et3N) [silica gel, UV]; [α]D24 −412.3 (c = 0.5, chloroform).
(S)-N-(Pyridin-2-ylmethylene)-3H-dinaphtho[2,1-c:1′,2′-e]azepin-4(5H)-amine (34)

To a 10 mL, one-necked round-bottom flask were added 1-aminoazepine 31b (77 mg, 0.25 mmol, 1.0 equiv) and Na2SO4 (24 mg, 0.14 mmol, 0.7 equiv). The flask was evacuated and backfilled with argon followed by the addition of CH2Cl2 (1.75 mL) and picolinaldehyde (33 μL, 0.35 mmol, 1.4 equiv). The reaction was stirred at room temperature for 3 h, and the solution was filtered and concentrated to give a bright yellow oil. Purification by column chromatography (SiO2, 1.5 × 16 cm, hexanes/EtOAc with 1% Et3N, 4/1, then 2/1) afforded 81 mg (58%) of 34 as a white foam. Data for 34: 1H NMR (500 MHz, CDCl3) 8.47 (d, J = 4.6 Hz, 1 H, HC(17)), 7.96 (d, J = 8.2 Hz, 2 H, HC(6)), 7.95 (d, J = 8.0 Hz, 2 H, HC(7) or HC(10)), 7.89 (d, J = 8.0 Hz, 1 H, HC(14)), 7.64 (app t, J = 7.8 Hz, 1 H, HC(15)), 7.62 (d, J = 8.6 Hz, 2 H, HC(5)), 7.50–7.46 (m, 5 H, HC(1) and HC(Aryl)), 7.29 (app t, J = 7.8 Hz, 2 H, HC(8) or HC(9)), 7.09 (app t, J = 5.9 Hz, 1 H, HC(16)), 4.65 (d, J = 12.4 Hz, 2 H, H2C(2)), 3.89 (d, J = 12.5 Hz, 2 H, H2C(2)); 13C NMR (126 MHz, CDCl3) 155.8 (C(13)), 149.0 (C(17)), 136.1 (C(15)), 134.7 (C(Aryl)), 134.3 (C(1)), 133.2 (C(Aryl)), 133.1 (C(Aryl)), 131.5 (C(Aryl)), 128.9 (C(6)), 128.3 (C(7) or C(10)), 127.4 (C(7) or C(10)), 127.4 (C(5)), 126.0 (C(8) or C(9)), 125.8 (C(8) or C(9)), 121.8 (C(16)), 118.9 (C(14)), 56.4 (C(2)); IR (CDCl3 film) 3053 (m), 3006 (w), 2939 (w), 2815 (w), 1954 (w), 1913 (w), 1590 (m), 1567 (s), 1508 (m), 1467 (m), 1433 (m), 1367 (m), 1325 (w), 1295 (w), 1234 (m), 1144 (m), 1108 (m), 1064 (m), 1006 (w), 981 (m), 866 (w), 818 (s), 774 (m), 752 (s); MS (ESI) 400.2 (M + H, 100); HRMS (ESI) calcd for C28H22N3 [M + H] 400.1814, found 400.1804; TLC Rf 0.52 (hexanes/EtOAc, 1/1 with 1% Et3N) [silica gel, UV, KMnO4]; [α]D24 −428.7 (c = 0.15, chloroform).
(S)-2,6-Diphenyl-3H-dinaphtho[2,1-c:10,20-e]azepin-4(5H)-amine (31b)

To a 25 mL, one-necked, round-bottom flask equipped with a stir bar and an argon inlet adaptor with a septum was added (S)-3,3′-diphenyl-2,2′-bis(bromomethyl)-1,1′-binaphthalene48 (676 mg, 1.1 mmol, 1.0 equiv). After three cycles of evacuation/argon fill, THF (12 mL) was added to give a colorless solution. Anhydrous hydrazine (730 μL, 22 mmol, 20 equiv) was added at room temperature to give a cloudy mixture. After being stirred for 2 h, the mixture was poured into ice-cold H2O (25 mL) and extracted with TBME (30 mL × 3). The organic phase was dried over Na2SO4, filtered, and concentrated to give 602 mg of 31b with TMBE trapped in the solid. Repeated trituration with hexanes and then pentane followed solvent removal afforded 441 mg (84%) of 31b as a white solid containing a small amount of hydrocarbons. The spectroscopic data matched those from the literature.70 Data for 31b: 1H NMR (500 MHz, CDCl3) 7.98–7.96 (m, 4 H, HC(Aryl)), 7.62–7.60 (m, 4 H, HC(Aryl)), 7.53–7.46 (m, 8 H, HC(Aryl)), 7.43–7.39 (m, 2 H, HC(Aryl)), 7.32–7.28 (m, 2 H, H2C(Aryl)), 4.12 (d, J = 12.6 Hz, 2 H, H2C(1)), 3.25 (d, J = 12.7 Hz, 2 H, H2C(1)), 2.79 (br s, 2 H, NH2); 13C NMR (126 MHz, CDCl3) 141.1 (C(Aryl)), 140.5 (C(Aryl)), 136.3 (C(Aryl)), 132.7 (C(Aryl)), 130.9 (C(Aryl)), 129.9 (C(Aryl)), 129.9 (C(Aryl), 129.3 (C(Aryl)), 129.2 (C(Aryl)), 128.3 (C(Aryl)), 127.5 (C(Aryl)), 127.2 (C(Aryl)), 126.0 (C(Aryl)), 125.9 (C(Aryl)), 56.3 (C(1)); IR (CDCl3 film) 3054 (w), 2926 (w), 2808 (w), 1589 (w), 1494 (m), 1448 (w), 1328 (w), 1226 (w), 1063 (w), 1028 (w), 976 (w), 785 (m), 765 (m), 702 (m); MS (ESI) 463.2 (M + H, 100); HRMS (ESI) calcd for C34H27N2 [M + H] 463.2174, found 463.2167.
(S)-2-((2,6-Diphenyl-3H-dinaphtho[2,1-c:1′,2′-e]azepin-4(5H)-yl)imino)acetaldehyde (32b)

A 10 mL Schlenk reaction flask equipped with a stir bar and a septum was evacuated and backfilled with argon (×2). An aqueous solution of glyoxal (40% w/w, 990 μL, 8.7 mmol, 20 equiv) was added. The flask was cooled to 0 °C, and a solution of 1-aminoazepine 31b (200 mg, 0.43 mmol, 1.0 equiv) in THF (4.4 mL) was added. The ice/water bath was removed, and the reaction was stirred at room temperature for 1 h. The mixture was poured into a saturated solution of NaHCO3 (15 mL) and extracted with Et2O (15 mL × 3). The combined organic phases was dried over Na2SO4, filtered and concentrated. The product was loaded onto a silica gel column (SiO2, 2.5 × 20 cm) with hexanes/CH2Cl2 (1/1) and eluted with hexanes/EtOAc (9/1 with 1% Et3N) to afford 187 mg (86%) of 32b as a white powder. Data for 32b: 1H NMR (500 MHz, CDCl3) 9.29 (d, J = 7.4 Hz, 1 H, H2C(3)), 7.99–7.96 (m, 4 H, HC(Aryl)), 7.56–7.52 (m, 2 H, HC(Aryl)), 7.50–7.38 (m, 12 H, HC(Aryl)), 7.33 (ddd, = 8.4, 6.8, 1.2 Hz, 2 H, HC(Aryl)), 6.50 (d, J = 7.4 Hz, 1 H, H2C(2)), 4.90 (d, J = 13.3 Hz, 2 H, H2C(1)), 3.91 (app br s, 2 H, H2C(1)); 13C NMR (126 MHz, CDCl3) 191.0 (C(3)), 140.2 (C(Aryl)), 139.8 (C(Aryl)), 135.9 (C(Aryl)), 132.9 (C(Aryl)), 130.9 (C(Aryl)), 130.4 (C(2)), 130.1 (C(Aryl)), 129.9 (C(Aryl)), 129.6 (C(Aryl)), 128.4 (C(Aryl)), 128.4 (C(Aryl)), 127.7 (C(Aryl)), 127.6 (C(Aryl)), 126.7 (C(Aryl)), 126.3 (C(Aryl)), 51.9 (br s, C(1)); IR (CDCl3 film) 3055 (m), 2807 (m), 1673 (s), 1590 (w), 1524 (s), 1494 (m), 1383 (m), 1356 (w), 1337 (w), 1230 (m), 1208 (w), 1185 (m), 1148 (m), 1129 (m), 1075 (m), 1029 (w), 988 (m), 943 (w), 842 (w), 813 (w), 786 (m), 766 (m), 755 (m), 702 (s); MS (ESI) 503.2 (M + H, 100); HRMS (ESI) calcd for C36H27N2O [M + H] 503.2123, found 503.2125; TLC Rf 0.21 (hexanes/EtOAc, 9/1 with 1% Et3N) [silica gel, UV].
(S,S)-N,N′-(Ethane-1,2-diylidene)bis(2,6-diphenyl-3H-dinaphtho[2,1-c:1′,2′-e]azepin-4(5H)-amine) (33b)

To an oven-dried, 10 mL, one-necked, round-bottom flask equipped with a stir bar, an argon gas inlet, and a septum were added 1-aminoazepine 31b (130 mg, 0.28 mmol, 1.1.0 equiv) and Na2SO4 (18 mg, 0.13 mmol, 0.5 equiv). The flask was evacuated and backfilled with argon (×3). A solution of 32b (127 mg, 0.25 mmol, 1.0 equiv) in dichloromethane (0.6 mL) was added to give a light yellow solution. The reaction was stirred at room temperature for 12 h. The mixture was then filtered and concentrated. Purification by column chromatography (SiO2, 1.5 × 20 cm, hexanes/CH2Cl2, 1/1, with 1% Et3N) afforded 206 mg of 33b as a pale yellow solid. Trituration in hexanes/EtOAc (1/1, 0.5 mL) followed by removal of the light yellow liquid afforded 190 mg (80%) of 33b as a white solid. Data for 33b: 1H NMR (500 MHz, CDCl3) 7.98–7.92 (m, 8 H, HC(Aryl)), 7.53–7.49 (m, 8 H, HC(Aryl)), 7.40–7.37 (m, 8 H, HC(Aryl)), 7.34–7.10 (m, 16 H, HC(Aryl)), 6.58 (s, 2 H, H2C(2)), 4.60 (d, J = 12.5 Hz, 2 H, H2C(1)), 3.54 (d, J = 12.5 Hz, 2 H, H2C(1)); 13C NMR (126 MHz, CDCl3) 140.6 (C(Aryl)), 140.2 (C(Aryl)), 136.0 (C(Aryl)), 135.9 (C(2)), 132.7 (C(Aryl)), 131.3 (C(Aryl)), 130.8 (C(Aryl)), 129.7 (C(Aryl)), 129.5 (C(Aryl)), 128.3 (C(Aryl)), 127.5 (C(Aryl)), 127.2 (C(Aryl)), 126.2 (C(Aryl)), 125.9 (C(Aryl)), 51.6 (C(1)); IR (CDCl3 film) 3056 (m), 3024 (m), 2947 (w), 2823 (w), 1953 (w), 1589 (m), 1555 (m), 1494 (m), 1448 (m), 1397 (w), 1351 (m), 1228 (m), 1156 (m), 1102 (m), 1053 (m), 1028 (w), 1007 (m), 982 (m), 964 (m), 855 (w), 785 (m), 702 (s); MS (ESI) 947.4 (M + H, 100); HRMS (ESI) calcd for C70H51N4 [M + H] 947.4114, found 947.4092; TLC Rf 0.33 (hexanes/CH2Cl2, 1/1 with 1% Et3N) [silica gel, UV]; [α]D24 −45.6 (c = 0.15, chloroform).
Preparation of Aryldimethylsilanes, Silanols, and Silanolates
(2-Methylnaphthalen-1-yl)dimethylsilane

To a flame-dried, 3-necked, 250 mL, round-bottomed flask equipped with a 50 mL addition funnel, a septum, an internal temperature probe, a gas adapter, and a magnetic stir bar was charged 1-bromo-2-methylnaphthalene (∼85 mol % purity, ∼15 mol % dimethylnaphthalene, 6.2 g, 25 mmol, 1.0 equiv). After evacuation and backfilling with argon, Et2O (75 mL) was added. The solution was cooled to −78 °C, and t-butyllithium (1.53 M, 33 mL, 50 mmol, 2 equiv) was added via the addition funnel (internal temperature was never higher than −60 °C). The suspension was stirred at −75 °C for 1 h before being warmed to −45 °C, and the mixture was quickly transferred via cannula to the dimethylchlorosilane solution prepared below.
The solution of dimethylchlorosilane was prepared by addition of dimethylchlorosilane (3.6 mL, 33 mmol, 1.3.0 equiv) to Et2O (32 mL) in a flame-dried, 250 mL three-necked round-bottomed flask equipped two septa, an argon inlet, and a magnetic stir bar. This solution was cooled to 0 °C in an ice bath before the (2-methylnaphthalen-1-yl)lithium solution prepared above was added via cannula. After being stirred at room temperature for 4 h, the mixture was concentrated and treated with pentane (50 mL). The precipitate was filtered, and the filtrate was then concentrated to give a colorless liquid. Short-path distillation afforded 5.25 g of the target silane contaminated with isomers of dimethylnaphthalene (∼10 mol %). The yield for (2-methylnaphthalen-1-yl)dimethylsilane was estimated to be ∼4.83 g (∼96%). Data: bp 85 °C [0.025 mmHg]; 1H NMR (500 MHz, CDCl3) 8.28 (d, J = 8.5 Hz, 1 H, HC(8)), 7.84 (d, J = 8.0 Hz, 1 H, HC(5)), 7.80 (d, J = 8.4 Hz, 1 H, HC(4)), 7.50 (ddd, J = 8.4, 6.8, 1.4 Hz, 1 H, HC(7)), 7.46–7.42 (m, 1 H, HC(6)), 7.24 (d, J = 9.0 Hz, 1 H, HC(3)), 5.11 (hept, J = 4.0 Hz, 1 H, SiH), 2.72 (s, 3 H, H3C(11)), 0.58 (d, J = 4.1 Hz, 6 H, H3C(12)); 13C NMR (126 MHz, CDCl3) 143.2 (C(2)), 137.6 (C(10)), 132.3 (C(1)), 131.8 (C(9)), 129.7 (C(4)), 129.2 (C(3)), 128.8 (C(5)), 127.5 (C(8)), 125.6 (C(7)), 124.4 (C(6)), 24.3 (C(11)), −2.0 (C(12)). IR (neat) 3043 (m), 3003 (m), 2958 (m), 2919 (m), 2147 (s), 1617 (m), 1593 (m), 1550 (w), 1507 (s), 1443 (m), 1420 (m), 1313 (m), 1251 (s), 1166 (m), 1141 (m), 1035 (m), 1023 (m), 985 (m), 915 (s), 882 (s), 838 (s), 810 (s), 782 (s), 763 (s), 739 (m), 700 (m). MS (EI, 70 eV) 200.1 (M+, 100), 185.0 (92), 156.1 (61), 141.0 (74), 115.0 (24); HRMS (EI, 70 eV) calcd for C13H16Si 200.10213, found 200.10165.
(2-Methylnaphthalen-1-yl)dimethylsilanol (35a)

Following the reaction protocol developed by Lee et al.,71 to a 100 mL, round-bottomed flask containing a magnetic stir bar were added (2-methylnaphthalen-1-yl)dimethylsilane (∼10 mol % purity, 2.16 g, 10 mmol, 1.0 equiv) and acetonitrile (10 mL). To this solution was added [(COD)IrCl]2 (68 mg, 0.1 mmol, 0.01.0 equiv) resulting in a bright yellow solution. H2O (0.9 mL, 50 mmol, 5 equiv) was added in 30 s. After the solution was stirred at room temperature for 0.5 h, no more bubbling was observed. The orange solution was poured into a solution of H2O (40 mL) and brine (10 mL) and extracted with pentane (60 mL × 4). The combined organic layers were dried over Na2SO4, filtered, and concentrated to give a tanned, waxy solid. Purification by column chromatography (SiO2, 4.5 × 16 cm, hexanes then hexanes/EtOAc, 10/1) and Kugelrohr distillation afforded 1.87 g (87%) of 35a as a pale yellow solid. The color was removed by swirling in hexanes (∼2 mL) and careful removal of solvent to afford 1.653 g (76%) of 35a as a white solid. Data for 35a: mp 58–60 °C; 1H NMR (500 MHz, CDCl3) 8.46 (d, J = 8.4 Hz, 1 H, HC(8)), 7.80 (d, J = 8.0 Hz, 1 H, HC(5)), 7.77 (d, J = 8.4 Hz, 1 H, HC(4)), 7.46 (ddd, J = 8.5, 6.8, 1.5 Hz, 1 H, HC(7)), 7.41 (ddd, J = 7.8, 6.9, 1.1 Hz, 1 H, HC(6)), 7.27 (d, J = 8.4 Hz, 1 H, HC(3)), 2.68 (s, 3 H, H3C(11)), 2.01 (s, 1 H, OH), 0.64 (s, 6 H, H3C(12)); 13C NMR (126 MHz, CDCl3) 142.7 (C(2)), 137.4 (C(10)), 133.3 (C(1)), 131.9 (C(9)), 130.0 (C(4)), 129.6 (C(3)), 128.7 (C(5)), 127.7 (C(8)), 125.6 (C(7)), 124.4 (C(6)), 24.5 (C(11)), 4.4 (C(12)); IR (CDCl3 film) 3574 (w), 3307 (br m), 3042 (m), 2956 (m), 1618 (w), 1593 (w), 1550 (w), 1507 (m), 1450 (w), 1421 (m), 1378 (w), 1353 (w), 1301 (w), 1255 (s), 1168 (w), 1141 (m), 1023 (m), 984 (m), 839 (s), 811 (s), 782 (s), 740 (m); MS (EI, 70 eV) 216.0 (M+, 62), 201.0 (100), 183.0 (45), 141.0 (27), 115.0 (19), 75.0 (23); HRMS (EI, 70 eV) calcd for C13H16OSi: 216.09705, found 216.09692; TLC Rf 0.17 (hexanes/EtOAc, 9/1) [silica gel, UV, KMnO4].
Potassium (2-Methylnaphthalen-1-yl)dimethylsilanolate (K+35a–)

In a drybox, (2-methylnaphthalen-1-yl)dimethylsilanol 35a (1.3 g, 6 mmol, 1.0 equiv) was added dropwise over 5 min to a suspension of KH (290 mg, 7.2 mmol, 1.2 equiv) in Et2O (12 mL) in an oven-dried, 100 mL, 1-necked round-bottomed flask equipped with a stir bar. The resulting mixture was stirred for 30 min further and was filtered through a medium-porosity fritted funnel into a one-neck flask, containing a stir bar, fitted with a vacuum stopcock adaptor. The solvent was evaporated in vacuo to give a sticky, pale yellow oil. The residue was vigorous stirred in hexanes (15 mL) for 30 min, and the volatiles were once again evaporated. This sequence was repeated once more. The resulting solid was treated hexanes (20 mL), filtered through a medium-porosity fritted funnel. The collected solids were further washed with dry hexanes (10 mL × 2). The solids were placed in an oven-dried, 15 mL recovery flask equipped with a vacuum stopcock adapter, and any excess volatiles were removed in vacuo to give 1.45 g (95%) of K+35a– as a white powder. Data for K+35a–: 1H NMR (500 MHz, C6D6) 9.03 (d, J = 8.6 Hz, 1 H, HC(8)), 7.72 (d, J = 7.4 Hz, 1 H, HC(5)), 7.62 (d, J = 8.3 Hz, 1 H, HC(4)), 7.39 (ddd, J = 8.5, 6.7, 1.4 Hz, 1 H, HC(7)), 7.33–7.25 (m, 1 H, HC(6)), 7.22 (d, J = 8.4 Hz, 1 H, HC(3)), 2.66 (s, 3 H, H3C(11)), 0.34 (s, 6 H, H3C(12)); 13C NMR (126 MHz, C6D6) 142.2 (C(1)), 140.8 (C(2)), 139.0 (C(10)), 132.9 (C(9)), 130.4 (C(3)), 129.4 (C(5)), 128.7 (C(4)), 128.9 (C(8)), 125.0 (C(7)), 124.3 (C(6)), 25.3 (C(11)), 9.0 (C(12)).
(2-Methoxynaphthalen-1-yl)dimethylsilane

To a flame-dried, 3-necked, 250 mL round-bottomed flask equipped with two septa, an internal temperature probe, a gas adapter, and a magnetic stir bar was charged 1-bromo-2-methoxynaphthalene (1.54 g, 6.5 mmol, 1.0 equiv). After evacuation and backfilling with argon, Et2O (28 mL) was added. The solution was cooled to −76 °C, and n-butyllithium (2.38 M, 2.8 mL, 6.5 mmol, 1.0 equiv) was added to the suspension (internal temperature was never higher than −70 °C). After 5 min, the IPA/CO2 bath was replaced with an ice/water bath. The mixture was stirred at 0 °C for 1.5 h and then transferred via cannula to the dimethylchlorosilane solution prepared below.
The solution of dimethylchlorosilane was prepared by adding dimethylchlorosilane (0.95 mL, 8.5 mmol, 1.3.0 equiv) to Et2O (9 mL) in a flame-dried, 250 mL three-necked round-bottomed flask equipped two septa, an argon inlet, and a magnetic stir bar. This solution was cooled to 0 °C in an ice bath before the (2-methoxynaphthalen-1-yl)lithium solution prepared above was added via cannula. After being stirred at room temperature for 4 h, the mixture was concentrated and treated with pentane (50 mL). The precipitate was filtered, and the filtrate was then concentrated to give a light yellow oil. Short-path distillation afforded 1.28 g of the target silane contaminated with 2-methoxynaphthalene (∼6 mol %). The yield for (2-methoxynaphthalen-1-yl)dimethylsilane was estimated to be ∼1.22 g (∼87%). Data: bp 120 °C [0.025 mmHg, ABT]. 1H NMR (500 MHz, CDCl3) 8.34 (d, J = 8.6 Hz, 1 H, HC(8)), 7.88 (d, J = 9.0 Hz, 1 H, HC(4)), 7.79 (d, J = 8.1 Hz, 1 H, HC(5)), 7.46 (ddd, J = 8.4, 6.8, 1.3 Hz, 1 H, HC(7)), 7.33 (ddd, J = 7.9, 6.9, 0.9 Hz, 1 H, HC(6)), 7.24 (d, J = 9.0 Hz, 1 H, HC(3)), 5.05 (hept, J = 3.6 Hz, 1 H, SiH), 3.93 (s, 3 H, H3C(11)), 0.44 (d, J = 3.8 Hz, 6 H, H3C(12)); 13C NMR (126 MHz, CDCl3) 163.3 (C(2)), 138.2 (C(10)), 132.0 (C(4)), 129.4 (C(9)), 128.6 (C(5)), 127.0 (C(8)), 126.3 (C(7)), 123.2 (C(6)), 119.0 (C(1)), 112.8 (C(3)), 56.2 (C(11)), −3.0 (C(12)). IR (neat) 3054 (w), 2957 (m), 2903 (m), 2837 (m), 2126 (s), 1618 (m), 1588 (s), 1559 (m), 1506 (s), 1460 (s), 1440 (m), 1427 (m), 1354 (w), 1320 (s), 1263 (s), 1243 (s), 1175 (m), 1146 (m), 1138 (m), 1069 (s), 1025 (m), 994 (s), 916 (s), 881 (s), 839 (s), 809 (s), 783 (s), 747 (s), 707 (m); MS (EI, 70 eV) 216.1 (M+, 51), 201.1 (100), 171.0 (65), 141.1 (62), 115.0 (26), 89.1 (15); HRMS (EI, 70 eV) calcd for C13H16OSi 216.09705, found 216.09672.
Preparation of (2-Methoxynaphthalen-1-yl)dimethylsilanol (35c)

Following the reaction protocol developed by Lee et al.,71 to a 100 mL, round-bottomed flask containing a magnetic stir bar were added (2-methoxynaphthalen-1-yl)dimethylsilane (∼94 mol % purity, 1.22 g, 5.4 mmol, 1.0 equiv) and acetonitrile (5.4 mL). To this solution was added [(COD)IrCl]2 (36 mg, 0.054 mmol, 0.01.0 equiv) resulting in a bright yellow solution. H2O (480 μL, 27 mmol, 5 equiv) was added in 30 s. After the solution was stirrred at room temperature for 0.5 h, no more bubbling was observed. The purple solution was poured into a solution of H2O (40 mL) and brine (10 mL) and extracted with pentane (60 mL × 4). The combined organic layers were dried over Na2SO4, filtered, and concentrated to give a purple oil. Purification by column chromatography (SiO2, 4.5 × 16 cm, hexanes/EtOAc, 10/1) and Kugelrohr distillation afforded 1.07 g of 35c as a colorless oil contaminated with 1,1,3,3-tetramethyl-3-(2-methylnaphthalen-1-yl)disiloxan-1-ol (∼2 mol %). The yield for 35c was estimated to be 1.04 g (83%). Data for 35c: bp 170 °C [0.025 mmHg, ABT]; 1H NMR (500 MHz, CDCl3) 8.19 (d, J = 8.6 Hz, 1 H, HC(8)), 7.90 (d, J = 9.0 Hz, 1 H, HC(4)), 7.79 (d, J = 8.1 Hz, 1 H, HC(5)), 7.45 (ddd, J = 8.4, 6.8, 1.4 Hz, 1 H, HC(7)), 7.35 (ddd, J = 7.9, 6.8, 1.0 Hz, 1 H, HC(6)), 7.25 (d, J = 8.5 Hz, 1 H, HC(3)), 3.97 (s, 3 H, H3C(11)), 3.53 (s, 1 H, OH), 0.58 (s, 6 H, H3C(12)); 13C NMR (126 MHz, CDCl3) 162.5 (C(2)), 137.7 (C(10)), 132.4 (C(4)), 129.5 (C(9)), 128.7 (C(5)), 127.0 (C(8)), 126.4 (C(7)), 123.5 (C(6)), 120.0 (C(1)), 112.9 (C(3)), 56.5 (C(11)), 3.3 (C(12)); IR (neat) 3390 (bm), 3055 (w), 2957 (m), 2838 (w), 1617 (m), 1589 (m), 1558 (m), 1505 (s), 1461 (s), 1356 (w), 1317 (s), 1265 (s), 1242 (s), 1177 (m), 1147 (m), 1138 (m), 1066 (m), 1026 (m), 993 (m), 838 (s), 810 (s), 786 (s), 750 (m) 709 (w). MS (EI, 70 eV) 232.1 (M+, 65), 187.0 (100), 141.1 (98), 115.0 (27), 83.0 (38); HRMS (EI, 70 eV) calcd for C13H16O2Si: 232.09196, found 232.09227; TLC Rf 0.43 (hexanes/EtOAc, 4/1) [silica gel, UV, KMnO4].
Potassium (2-Methoxynaphthalen-1-yl)dimethylsilanolate (K+35c–)

In a drybox, a solution of (2-methoxynaphthalen-1-yl)dimethylsilanol 35c (98 mol % purity, 807 mg, 3.5 mmol, 1.0 equiv) in Et2O (9 mL) was added dropwise over 5 min to a suspension of KH (168 mg, 4.2 mmol, 1.2 equiv) in Et2O (8 mL) in an oven-dried, 50 mL, 1-necked round-bottomed flask equipped with a stir bar. After 10 min, a large amount of white solid formed and bubbling subsided. The resulting mixture was stirred for 1 h and was filtered through a medium-porosity fritted funnel into a one-neck flask fitted with a vacuum stopcock adaptor, eluted with THF (15 mL). The solvent was evaporated in vacuo to give a sticky, pale yellow oil. The residue was vigorous stirred in hexanes (10 mL) for 10 min, and the volatiles were once again evaporated. This sequence was repeated a total of three times to give a chunky white solid. The solid was treated with Et2O (2 mL) and hexanes (15 mL) with vigorous stirring to give a thick, white suspension, which was filtered through a medium-porosity fritted funnel. The collected solids were further washed with dry hexanes (10 mL × 2). The solids were placed in an oven-dried, 15 mL recovery flask equipped with a vacuum stopcock adaptor, and any excess volatiles were removed in vacuo to give 0.92 g (98%) of K+35c– as a white powder in 97% purity. Data for K+35c–: 1H NMR (500 MHz, d8-THF) 9.21 (d, J = 8.1 Hz, 1 H, HC(8)), 7.69 (d, J = 8.8 Hz, 1 H, HC(4)), 7.64 (d, J = 8.1 Hz, 1 H, HC(5)), 7.24–7.18 (m, 1 H, HC(7)), 7.21 (d, J = 8.6 Hz, 1 H, HC(3)), 7.08 (t, J = 7.2, 1 H, HC(6)), 3.81 (s, 3 H, H3C(11)), 0.26 (s, 6 H, H3C(12)); 13C NMR (126 MHz, d8-THF) 162.5 (C(2)), 140.8 (C(10)), 130.8 (C(4)), 130.4 (C(9)), 130.0 (C(1)), 129.4 (C(8)), 128.8 (C(5)), 125.7 (C(7)), 123.1 (C(6)), 114.5 (C(3)), 56.4 (C(11)), 7.6 (C(12)).
Cross Coupling of K+35a– and 1-Bromonaphthalene Using Bis-hydrazone Ligand 5 (Table 6, Entry 1)

To an oven-dried, 5 mL, round-bottomed flask equipped with a magnetic stir bar, reflux condenser, and three-way argon adapter were charged [allylPdCl]2 (4.6 mg, 12.5 μmol, 0.025 equiv) and bis-hydrazone ligand 5 (12.5 mg, 25 μmol, 0.05 equiv). The flask was brought into a drybox where toluene (0.25 mL) and 1-bromonaphthalene (71 μL, 0.5 mmol, 1.0 equiv) were added to give a bright yellow solution after stirring. Following the addition of aryldimethylsilanolate K+35a– (191 mg, 0.75 mmol, 1.5 equiv) and additional toluene (0.25 mL), the flask was sealed away from the atmosphere and removed to a hood. The flask was then placed into a preheated 70 °C oil bath and stirred at this temperature under argon for 2 h. The reaction was cooled to room temperature, and the brown solution was filtered through a pad of silica gel (1.5 cm deep, 30 mL size, medium-porosity fritted funnel), eluted with Et2O (30 mL), and concentrated to give a brown oil and some white solid. Purification by column chromatography (SiO2, 1.5 × 21 cm, hexanes) afforded 123 mg (92%) of 37 as a white solid containing ∼2.5% binaphthalene measured by SFC analysis. Data for 37: 1H NMR (500 MHz, CDCl3) 7.96 (d, J = 8.2 Hz, 2 H), 7.89 (dd, J = 8.3, 3.2 Hz, 2 H), 7.62 (dd, J = 8.2, 7.0 Hz, 1 H), 7.52–7.46 (m, 2 H), 7.44–7.37 (m, 2 H), 7.32–7.20 (m, 3 H), 7.16 (d, J = 8.3 Hz, 1 H), 2.12 (s, 3 H); 13C NMR (126 MHz, CDCl3) 137.5, 136.0, 134.4, 133.7, 133.4, 132.5, 131.9, 128.6, 128.2, 127.7, 127.7, 127.6, 127.5, 126.2, 126.1, 126.0, 125.9, 125.8, 125.6, 124.8, 20.5; MS (EI, 70 eV) 268.1 (M+, 100), 253.0 (49), 126.0 (17), 109.0 (23); HRMS (EI, 70 eV) calcd for C21H16: 268.12520, found 268.12526; [α]D24 −40.2 (c = 0.5, chloroform); SFC (R)-37, tR 12.5 min (95%); (S)-37, tS 26.6 min (5%) (Chiralpak OJ, 200 bar, 1 mg/mL, 5% MeOH in CO2, 2 mL/min, 220 nm, 40 °C); TLC Rf 0.32 (hexanes) [silica gel, UV].
Cross-Coupling of K+35c– and 1-Bromonaphthalene Using Bis-hydrazone Ligand 5 (Table 8, Entry 1 and 2)

To an oven-dried, 5 mL, round-bottomed flask equipped with a magnetic stir bar, reflux condenser, and three-way argon adapter were charged [allylPdCl]2 (2.3 mg, 6.3 μmol, 0.025 equiv), bis-hydrazone ligand (S,S,S,S)-5 (6.3 mg, 12.5 μmol, 0.05 equiv), and biphenyl (19.3 mg, internal standard, for GC analysis). The flask was brought into a drybox where toluene (0.125 mL) and 1-bromonaphthalene (36 μL, 0.25 mmol, 1.0 equiv) were added to give a bright yellow solution after stirring. Following the addition of aryldimethylsilanolate K+35c– (106 mg, 0.38 mmol, 1.5 equiv) and additional toluene (0.125 mL), the flask was sealed away from the atmosphere and removed to a hood. The flask was then placed into a preheated 70 °C oil bath and stirred at this temperature under argon for 3 h (GC yield: 76%). The reaction was cooled to room temperature, and the brown solution was filtered through a pad of silica gel (1.5 cm deep, 30 mL size, medium-porosity fritted funnel), eluted with Et2O (30 mL), and concentrated to give a brown oil. Purification by column chromatography (SiO2, 1.5 × 27 cm, hexanes/CH2Cl2, 4/1 then 2/1) afforded 57 mg of 41 (79:21 er) as a sticky solid contaminated with siloxanes. The solid was swirled in MeOH (1 mL), and the solvent was removed carefully using a syringe with a 25-gauged needle. This procedure was repeated with a smaller portion of MeOH (0.5 mL) go give 42 mg (60%) of 41 (84:16 er) as a white powder after drying in vacuo. The product in the combined methanol mother liquors has an enantiomeric ratio of 67:33. Data for 41: GC: 41, tR 10.0 min (76%); 1H NMR (500 MHz, CDCl3) 7.99 (d, J = 9.0 Hz, 1 H), 7.95 (dd, J = 8.0, 5.3 Hz, 2 H), 7.88 (d, J = 8.2 Hz, 1 H), 7.63 (dd, J = 7.6, 7.6 Hz, 1 H), 7.49–7.43 (m, 3 H), 7.36–7.21 (m, 4 H), 7.16 (d, J = 8.5 Hz, 1 H), 3.77 (s, 3 H); 13C NMR (126 MHz, CDCl3) 154.6, 134.5, 134.2, 133.7, 132.9, 129.4, 129.0, 128.4, 128.2, 127.8, 127.7, 126.3, 126.1, 125.8, 125.7, 125.5, 125.5, 123.5, 123.2, 113.8, 56.7. MS (EI, 70 eV) 283.9 (M+, 100), 268.9 (23), 252.9 (16), 238.9 (26), 119.4 (11); HRMS (EI, 70 eV) calcd for C21H16O 284.12012, found 284.11980; [α]D24 −19.4 (c = 0.5, chloroform); SFC (S)-41, tR 14.2 min (79%); (R)-41, tS 23.2 min (21%); (Chiralpak OJ, 200 bar, 1 mg/mL, 10% MeOH in CO2, 2 mL/min, 220 nm, 40 °C); TLC Rf 0.21 (hexanes/CH2Cl2, 4/1) [silica gel, UV].
Acknowledgments
We are grateful to the National Institutes of Health (GM R01 063167) and the National Science Foundation (NSF CHE-1151566, CHE-1059084) for generous financial support. W.-T.T.C. thanks Mr. Alex Geanes and Mr. Richard Thornbury for the preparation of some starting materials and Eli Lilly for a graduate fellowship. Calculations were performed on the Hoffman2 cluster at UCLA and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the NSF.
Supporting Information Available
Full experimental procedures for optimization experiments, NMR spectra for new compounds, and Cartesian coordinates of all computed structures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Pereira M. M.; Calvete M. J. F.; Carrilho R. M. B.; Abreu A. R. Chem. Soc. Rev. 2013, 42, 6990–7027. [DOI] [PubMed] [Google Scholar]; b Teichert J. F.; Feringa B. L. Angew. Chem., Int. Ed. 2010, 49, 2486–2528. [DOI] [PubMed] [Google Scholar]; c Hashimoto T.; Maruoka K. Chem. Rev. 2007, 107, 5656–5682. [DOI] [PubMed] [Google Scholar]; d Noyori R. Angew. Chem., Int. Ed. 2002, 41, 2008–2022. [PubMed] [Google Scholar]; e Yu M.; Ibrahem I.; Hasegawa M.; Schrock R. R.; Hoveyda A. H. J. Am. Chem. Soc. 2012, 134, 2788–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Baudoin O. Eur. J. Org. Chem. 2005, 4223–4229. [Google Scholar]; b Bringmann G.; Price Mortimer A. J.; Keller P. A.; Gresser M. J.; Garner J.; Breuning M. Angew. Chem., Int. Ed. 2005, 44, 5384–5427. [DOI] [PubMed] [Google Scholar]; c Kozlowski M. C.; Morgan B. J.; Linton E. C. Chem. Soc. Rev. 2009, 38, 3193–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Bringmann G.; Gulder T.; Gulder T. A. M.; Breuning M. Chem. Rev. 2010, 111, 563–639. [DOI] [PubMed] [Google Scholar]
- a Bringmann G.; Zagst R.; Schäffer M.; Hallock Y. F.; Cardellina J. H.; Boyd M. R. Angew. Chem., Int. Ed. 1993, 32, 1190–1191. [Google Scholar]; b Manfredi K. P.; Blunt J. W.; Cardellina J. H.; McMahon J. B.; Pannell L. L.; Cragg G. M.; Boyd M. R. J. Med. Chem. 1991, 34, 3402–3405. [DOI] [PubMed] [Google Scholar]
- Kupchan S. M.; Britton R. W.; Ziegler M. F.; Gilmore C. J.; Restivo R. J.; Bryan R. F. J. Am. Chem. Soc. 1973, 95, 1335–1336. [DOI] [PubMed] [Google Scholar]
- Williams D. H.; Bardsley B. Angew. Chem., Int. Ed. 1999, 38, 1172–1193. [DOI] [PubMed] [Google Scholar]
- a Lipshutz B. H.; Kayser F.; Liu Z.-P. Angew. Chem., Int. Ed. 1994, 33, 1842–1844. [Google Scholar]; b Lin G.-Q.; Zhong M. Tetrahedron Lett. 1997, 38, 1087–1090. [Google Scholar]
- a Meyers A. I.; Meier A.; Rawson D. J. Tetrahedron Lett. 1992, 33, 853–856. [Google Scholar]; b Meyers A. I. J. Heterocycl. Chem. 1998, 35, 991–1002. [Google Scholar]; c Nelson T. D.; Meyers A. I. Tetrahedron Lett. 1993, 34, 3061–3062. [Google Scholar]; d Meyers A. I.; Nelson T. D.; Moorlag H.; Rawson D. J.; Meier A. Tetrahedron 2004, 60, 4459–4473. [Google Scholar]
- a Bringmann G.; Menche D. Acc. Chem. Res. 2001, 34, 615–624. [DOI] [PubMed] [Google Scholar]; b Bringmann G.; Tasler S.; Pfeifer R.-M.; Breuning M. J. Organomet. Chem. 2002, 661, 49–65. [Google Scholar]
- a Xu Z.; Kozlowski M. C. J. Org. Chem. 2002, 67, 3072–3078. [DOI] [PubMed] [Google Scholar]; b Li X.; Yang J.; Kozlowski M. C. Org. Lett. 2001, 3, 1137–1140. [DOI] [PubMed] [Google Scholar]
- a Li G.-Q.; Gao H.; Keene C.; Devonas M.; Ess D. H.; Kürti L. J. Am. Chem. Soc. 2013, 135, 7414–7417. [DOI] [PubMed] [Google Scholar]; b De C. K.; Pesciaioli F.; List B. Angew. Chem., Int. Ed. 2013, 52, 9293–9295. [DOI] [PubMed] [Google Scholar]
- a Herrbach A.; Marinetti A.; Baudoin O.; Guénard D.; Guéritte F. o. J. Org. Chem. 2003, 68, 4897–4905. [DOI] [PubMed] [Google Scholar]; b Xu G.; Fu W.; Liu G.; Senanayake C. H.; Tang W. J. Am. Chem. Soc. 2014, 136, 570–573. [DOI] [PubMed] [Google Scholar]; c Nicolaou K. C.; Li H.; Boddy C. N. C.; Ramanjulu J. M.; Yue T.-Y.; Natarajan S.; Chu X.-J.; Bräse S.; Rübsam F. Chem.—Eur. J. 1999, 5, 2584–2601. [Google Scholar]; d Bringmann G.; Hamm A.; Schraut M. Org. Lett. 2003, 5, 2805–2808. [DOI] [PubMed] [Google Scholar]
- a Kamikawa K.; Watanabe T.; Daimon A.; Uemura M. Tetrahedron 2000, 56, 2325–2337. [Google Scholar]; b Monovich L. G.; Le Huérou Y.; Rönn M.; Molander G. A. J. Am. Chem. Soc. 1999, 122, 52–57. [Google Scholar]; c Uemura M.; Daimon A.; Hayashi Y. Chem. Commun. 1995, 1943–1944. [Google Scholar]; d Kamikawa K.; Tachibana A.; Sugimoto S.; Uemura M. Org. Lett. 2001, 3, 2033–2036. [DOI] [PubMed] [Google Scholar]; e Watanabe T.; Tanaka Y.; Shoda R.; Sakamoto R.; Kamikawa K.; Uemura M. J. Org. Chem. 2004, 69, 4152–4158. [DOI] [PubMed] [Google Scholar]
- a Tamao K.; Minato A.; Miyake N.; Matsuda T.; Kiso Y.; Kumada M. Chem. Lett. 1975, 4, 133–136. [Google Scholar]; b Tamao K.; Yamamoto H.; Matsumoto H.; Miyake N.; Hayashi T.; Kumada M. Tetrahedron Lett. 1977, 18, 1389–1392. [Google Scholar]
- Hayashi T.; Hayashizaki K.; Kiyoi T.; Ito Y. J. Am. Chem. Soc. 1988, 110, 8153–8156. [Google Scholar]
- Cammidge A. N.; Crépy K. V. L Chem. Commun. 2000, 1723–1724. [Google Scholar]
- Yin J.; Buchwald S. L. J. Am. Chem. Soc. 2000, 122, 12051–12052. [Google Scholar]
- Shen X.; Jones G. O.; Watson D. A.; Bhayana B.; Buchwald S. L. J. Am. Chem. Soc. 2010, 132, 11278–11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tang W.; Patel N. D.; Xu G.; Xu X.; Savoie J.; Ma S.; Hao M.-H.; Keshipeddy S.; Capacci A. G.; Wei X.; Zhang Y.; Gao J. J.; Li W.; Rodriguez S.; Lu B. Z.; Yee N. K.; Senanayake C. H. Org. Lett. 2012, 14, 2258–2261. [DOI] [PubMed] [Google Scholar]; b Wang S.; Li J.; Miao T.; Wu W.; Li Q.; Zhuang Y.; Zhou Z.; Qiu L. Org. Lett. 2012, 14, 1966–1969. [DOI] [PubMed] [Google Scholar]
- Bermejo A.; Ros A.; Fernández R.; Lassaletta J. M. J. Am. Chem. Soc. 2008, 130, 15798–15799. [DOI] [PubMed] [Google Scholar]
- Ros A.; Estepa B.; Bermejo A.; Álvarez E.; Fernández R.; Lassaletta J. M. J. Org. Chem. 2012, 77, 4740–4750. [DOI] [PubMed] [Google Scholar]
- Uozumi Y.; Matsuura Y.; Arakawa T.; Yamada Y. M. A. Angew. Chem., Int. Ed. 2009, 48, 2708–2710. [DOI] [PubMed] [Google Scholar]
- a Castanet A.-S.; Colobert F. o.; Broutin P.-E.; Obringer M. Tetrahedron: Asymmetry 2002, 13, 659–665. [Google Scholar]; b Jensen J. F.; Johannsen M. Org. Lett. 2003, 5, 3025–3028. [DOI] [PubMed] [Google Scholar]; c Cammidge A. N.; Crépy K. V. L Tetrahedron 2004, 60, 4377–4386. [Google Scholar]; d Mikami K.; Miyamoto T.; Hatano M. Chem. Commun. 2004, 2082–2083. [DOI] [PubMed] [Google Scholar]; e Genov M.; Almorín A.; Espinet P. Chem.—Eur. J. 2006, 12, 9346–9352. [DOI] [PubMed] [Google Scholar]; f Bronger R. P. J.; Guiry P. J. Tetrahedron: Asymmetry 2007, 18, 1094–1102. [Google Scholar]; g Takemoto T.; Iwasa S.; Hamada H.; Shibatomi K.; Kameyama M.; Motoyama Y.; Nishiyama H. Tetrahedron Lett. 2007, 48, 3397–3401. [Google Scholar]; h Sawai K.; Tatumi R.; Nakahodo T.; Fujihara H. Angew. Chem., Int. Ed. 2008, 47, 6917–6919. [DOI] [PubMed] [Google Scholar]; i Zhang S.-S.; Wang Z.-Q.; Xu M.-H.; Lin G.-Q. Org. Lett. 2010, 12, 5546–5549. [DOI] [PubMed] [Google Scholar]; j Grach G.; Pieters G.; Dinut A.; Terrasson V.; Medimagh R.; Bridoux A.; Razafimahaleo V.; Gaucher A.; Marque S.; Marrot J.; Prim D.; Gil R.; Planas J. G.; Vin̋as C.; Thomas I.; Roblin J.-P.; Troin Y. Organometallics 2011, 30, 4074–4086. [Google Scholar]; k Kamei T.; Sato A. H.; Iwasawa T. Tetrahedron Lett. 2011, 52, 2638–2641. [Google Scholar]; l Yamamoto T.; Akai Y.; Nagata Y.; Suginome M. Angew. Chem., Int. Ed. 2011, 50, 8844–8847. [DOI] [PubMed] [Google Scholar]; m Wu W.; Wang S.; Zhou Y.; He Y.; Zhuang Y.; Li L.; Wan P.; Wang L.; Zhou Z.; Qiu L. Adv. Synth. Catal. 2012, 354, 2395–2402. [Google Scholar]
- Genov M.; Fuentes B.; Espinet P.; Pelaz B. Tetrahedron: Asymmetry 2006, 17, 2593–2595. [Google Scholar]
- Mosquera Á.; Pena M. A.; Pérez Sestelo J.; Sarandeses L. A. Eur. J. Org. Chem. 2013, 2013, 2555–2562. [Google Scholar]
- Yamaguchi K.; Yamaguchi J.; Studer A.; Itami K. Chem. Sci. 2012, 3, 2165–2169. [Google Scholar]
- Denmark S. E.; Smith R. C.; Chang W.-T. T.; Muhuhi J. M. J. Am. Chem. Soc. 2009, 131, 3104–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Denmark S. E.; Smith R. C. J. Am. Chem. Soc. 2010, 132, 1243–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Denmark S. E.; Smith R. C.; Chang W.-T. T. Tetrahedron 2011, 67, 4391–4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Enders D.; Meiers M. Synthesis 2002, 2002, 2542–2560. [Google Scholar]; b Vázquez J.; Cristea E.; Díez E.; Lassaletta J. M.; Prieto A.; Fernández R. Tetrahedron 2005, 61, 4115–4128. [Google Scholar]; c Alcarazo M.; Roseblade S. J.; Alonso E.; Fernández R.; Alvarez E.; Lahoz F. J.; Lassaletta J. M. J. Am. Chem. Soc. 2004, 126, 13242–13243. [DOI] [PubMed] [Google Scholar]; d Lassaletta J. M.; Alcarazo M.; Fernández R. Chem. Commun. 2004, 298–299. [DOI] [PubMed] [Google Scholar]
- Aldous D. J.; Dutton W. M.; Steel P. G. Tetrahedron: Asymmetry 2000, 11, 2455–2462. [Google Scholar]
- Sato M.; Gunji Y.; Ikeno T.; Yamada T. Synthesis 2004, 2004, 1434–1438. [Google Scholar]
- Teichert J. F.; Feringa B. L. Synthesis 2010, 1200–1204. [Google Scholar]
- Campos K. R.; Klapars A.; Waldman J. H.; Dormer P. G.; Chen C.-y. J. Am. Chem. Soc. 2006, 128, 3538–3539. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Silverman S. M.; Stambuli J. P. J. Am. Chem. Soc. 2011, 133, 19483–19497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Stanley L. M.; Sibi M. P. Chem. Rev. 2008, 108, 2887–2902. [DOI] [PubMed] [Google Scholar]; b Pandey G.; Banerjee P.; Gadre S. R. Chem. Rev. 2006, 106, 4484–4517. [DOI] [PubMed] [Google Scholar]; c Pellissier H. Tetrahedron 2007, 63, 3235–3285. [Google Scholar]
- Enders D.; Fey P.; Kipphardt H. Org. Synth. 1987, 65, 173–178. [Google Scholar]
- Shen Y.; Friestad G. K. J. Org. Chem. 2002, 67, 6236–6239. [DOI] [PubMed] [Google Scholar]
- Overberger C. G.; Valentine M. E.; Anselme J. P. J. Am. Chem. Soc. 1969, 91, 687–694. [Google Scholar]
- Corey E. J.; Helal C. J. Angew. Chem., Int. Ed. 1998, 37, 1986–2012. [DOI] [PubMed] [Google Scholar]
- Li X.; Zhao G.; Cao W.-G. Chin. J. Chem. . 2006, 24, 1402–1405. [Google Scholar]
- Chen H.; Sweet J. A.; Lam K.-C.; Rheingold A. L.; McGrath D. V. Tetrahedron: Asymmetry 2009, 20, 1672–1682. [Google Scholar]
- a Jung M. E.; Lyster M. A. Chem. Commun. 1978, 315–316. [Google Scholar]; b Lott R. S.; Chauhan V. S.; Stammer C. H. Chem. Commun. 1979, 495–496. [Google Scholar]
- a Nagasawa H. T.; Fraser P. S.; Yuzon D. L. J. Med. Chem. 1973, 16, 583–585. [DOI] [PubMed] [Google Scholar]; b Olah G. A.; Olah J. A.; Overchuk N. A. J. Org. Chem. 1965, 30, 3373–3376. [Google Scholar]; c Romea P.; Urpi F.; Vilarrasa J. J. Org. Chem. 1989, 54, 3209–3211. [Google Scholar]
- Wuts P. G. M.; Greene T. W. In Greene’s Protective Groups in Organic Synthesis; John Wiley & Sons, Inc.: New York, 2006; p 696. [Google Scholar]
- Lunn G.; Sansone E. B.; Keefer L. K. J. Org. Chem. 1984, 49, 3470–3473. [Google Scholar]
- Auzzas L.; Palomba M.; Boatto G.; Manconi P.; Pau A.; Becciu A.; Cerri R.; Tullio V.; Roana J.; Carlone N. Pharmazie 2000, 55, 483–489. [PubMed] [Google Scholar]
- Electrophilic amination with O-(4-nitrobenzoyl)hydroxylamine36 as well as Hofmann-type rearrangement reported for the preparation of (S)-1-amino-2-methoxymethypyrrolidine (SAMP)35 were also unsuccessful.
- Overberger C. G.; Marullo N. P.; Hiskey R. G. J. Am. Chem. Soc. 1961, 83, 1374–1378. [Google Scholar]
- Ooi T.; Kameda M.; Maruoka K. J. Am. Chem. Soc. 2003, 125, 5139–5151. [DOI] [PubMed] [Google Scholar]
- a Dias P. B.; de Piedade M. E. M.; Simões J. A. M. Coord. Chem. Rev. 1994, 136, 737–807. [Google Scholar]; b Negishi E.-i. In Handbook of Organopalladium Chemistry for Organic Synthesis; John Wiley & Sons, Inc.: New York, 2003; pp 17, 47. [Google Scholar]; c Echavarren A. M.; Cárdenas D. J. In Metal-Catalyzed Cross-Coupling Reactions; Wiley-VCH: Weinheim, 2008; Chapter 1. [Google Scholar]; d Constable E. C. In Metals and Ligand Reactivity; Wiley-VCH: Weinheim, 2005; p 22. [Google Scholar]; e Defieber C.; Grützmacher H.; Carreira E. M. Angew. Chem., Int. Ed. 2008, 47, 4482–4502. [DOI] [PubMed] [Google Scholar]
- Denmark S. E.; Smith R. C. Synlett 2006, 2006, 2921–2928. [Google Scholar]
- Negishi E.-i. In Handbook of Organopalladium Chemistry for Organic Synthesis; John Wiley & Sons, Inc.: New York, 2003; Chapter II.3. [Google Scholar]
- Johnson J. B.; Rovis T. Angew. Chem., Int. Ed. 2008, 47, 840–871. [DOI] [PubMed] [Google Scholar]
- Dong Z. B.; Manolikakes G.; Shi L.; Knochel P.; Mayr H. Chem.—Eur. J. 2010, 16, 248–253. [DOI] [PubMed] [Google Scholar]
- For reviews on selected examples, see:; a Pérez-Rodríguez M.; Braga A. A. C.; Garcia-Melchor M.; Pérez-Temprano M. H.; Casares J. A.; Ujaque G.; de Lera A. R.; Álvarez R.; Maseras F.; Espinet P. J. Am. Chem. Soc. 2009, 131, 3650–3657. [DOI] [PubMed] [Google Scholar]; b Pérez-Rodríguez M.; Braga A. A. C.; de Lera A. R.; Maseras F.; Álvarez R.; Espinet P. Organometallics 2010, 29, 4983–4991. [Google Scholar]; c Xue L. Q.; Lin Z. Y. Chem. Soc. Rev. 2010, 39, 1692–1705. [DOI] [PubMed] [Google Scholar]; d García-Melchor M.; Braga A. A. C.; Lledós A.; Ujaque G.; Maseras F. Acc. Chem. Res. 2013, 46, 2626–2634and references cited therein. [DOI] [PubMed] [Google Scholar]
- a Becke A. D. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]; b Lee C.; Yang W.; Parr R. G. Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- a Zhao Y.; Truhlar D. G. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]; b Zhao Y.; Truhlar D. G. Acc. Chem. Res. 2008, 41, 157–167. [DOI] [PubMed] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. J. Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2010. [Google Scholar]
- For a recent benchmark study on transition-state geometries using B3LYP, see:Simón L.; Goodman J. M. Org. Biomol. Chem. 2011, 9, 689–700. [DOI] [PubMed] [Google Scholar]
- a Lin M.; Kang G.-Y.; Guo Y.-A.; Yu Z.-X. J. Am. Chem. Soc. 2012, 134, 398–405. [DOI] [PubMed] [Google Scholar]; b Zhou M.; Balcells D.; Parent A. R.; Crabtree R. H.; Eisenstein O. ACS Catal. 2012, 2, 208–218. [Google Scholar]; c Tang S.-Y.; Guo Q.-X.; Fu Y. Chem.—Eur. J. 2011, 17, 13866–13876. [DOI] [PubMed] [Google Scholar]; d Liu P.; Xu X.; Dong X.; Keitz B. K.; Herbert M. B.; Grubbs R. H.; Houk K. N. J. Am. Chem. Soc. 2012, 134, 1464–1467. [DOI] [PubMed] [Google Scholar]; e Dang Y.; Qu S.; Wang Z.-X.; Wang X. J. Am. Chem. Soc. 2014, 136, 986–998. [DOI] [PubMed] [Google Scholar]; f Green A. G.; Liu P.; Merlic C. A.; Houk K. N. J. Am. Chem. Soc. 2014, 136, 4575–4583. [DOI] [PubMed] [Google Scholar]
- Couzijn E. P. A.; Zocher E.; Bach A.; Chen P. Chem.—Eur. J. 2010, 16, 5408–5415. [DOI] [PubMed] [Google Scholar]
- Wheeler S. E. Acc. Chem. Res. 2012, 46, 1029–1038. [DOI] [PubMed] [Google Scholar]
- Weng C.-M.; Hong F.-E. Dalton Trans. 2011, 40, 6458–6468. [DOI] [PubMed] [Google Scholar]
- Several observations suggest that bis-hydrazones are weakly coordinating ligands: (1) failure to displace mono- and bidentate phosphines and diamines from complexes of palladium, (2) inefficient and incomplete displacement of cyclooctadiene from its PdCl2 complex with a piperidine-derived bis-hydrazone, (3) solution 1H NMR spectra of (S,S,S,S)-(5)·PdCl2 showed undefined broad peaks at −40 °C, implying rapid equilibration between bidentate and monodentate binding modes, and (4) byproduct 42 generated from activation of [allylPdCl]2 was found to slightly lower the product er by acting as a competitive ligand.
- Hulley E. B.; Wolczanski P. T.; Lobkovsky E. B. J. Am. Chem. Soc. 2011, 133, 18058–18061. [DOI] [PubMed] [Google Scholar]
- Chen F.; Ding Z.; Qin J.; Wang T.; He Y.; Fan Q.-H. Org. Lett. 2011, 13, 4348–4351. [DOI] [PubMed] [Google Scholar]
- Higashiyama K.; Inoue H.; Takahashi H. Tetrahedron 1994, 50, 1083–1092. [Google Scholar]
- Denmark S. E.; Butler C. R. Org. Lett. 2005, 8, 63–66. [DOI] [PubMed] [Google Scholar]
- Hill R. K.; Chang T.-H Tetrahedron 1965, 21, 2015–2019. [DOI] [PubMed] [Google Scholar]
- Widhalm M.; Abraham M.; Arion V. B.; Saarsalu S.; Maeorg U. Tetrahedron: Asymmetry 2010, 21, 1971–1982. [Google Scholar]
- Lee Y.; Seomoon D.; Kim S.; Han H.; Chang S.; Lee P. H. J. Org. Chem. 2004, 69, 1741–1743. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.