

Abstract
Next-generation sequencing is a fast and cost-effective way to obtain sequence data for nonmodel organisms for many markers and for many individuals. We describe a protocol through which we obtain orthologous markers for the crested newts (Amphibia: Salamandridae: Triturus), suitable for analysis of interspecific hybridization. We use transcriptome data of a single Triturus species and design 96 primer pairs that amplify c. 180 bp fragments positioned in 3-prime untranslated regions. Next, these markers are tested with uniplex PCR for a set of species spanning the taxonomical width of the genus Triturus. The 52 markers that consistently show a single band of expected length at gel electrophoreses for all tested crested newt species are then amplified in five multiplex PCRs (with a plexity of ten or eleven) for 132 individual newts: a set of 84 representing the seven (candidate) species and a set of 48 from a presumed hybrid population. After pooling multiplexes per individual, unique tags are ligated to link amplicons to individuals. Subsequently, individuals are pooled equimolar and sequenced on the Ion Torrent next-generation sequencing platform. A bioinformatics pipeline identifies the alleles and recodes these to a genotypic format. Next, we test the utility of our markers. baps allocates the 84 crested newt individuals representing (candidate) species to their expected (candidate) species, confirming the markers are suitable for species delineation. newhybrids, a hybrid index and hiest confirm the 48 individuals from the presumed hybrid population to be genetically admixed, illustrating the potential of the markers to identify interspecific hybridization. We expect the set of markers we designed to provide a high resolving power for analysis of hybridization in Triturus.
Keywords: 3-prime untranslated region, genomics, hybridization, Ion PGM, nonmodel species
Introduction
To reliably infer interspecific gene flow, one requires dozens of single-copy markers that exhibit polymorphism and can be sequenced for closely related species (Twyford & Ennos 2012; McCormack et al. 2013). Next-generation sequencing provides large-scale sequencing data with relative ease and at a reasonable cost, making it possible to obtain many markers for a large number of individuals (Mardis 2011). This is not limited to ‘model’ species for which a reference genome is already available: genomic resources are now also within grasp for ‘nonmodel’ species (Garvin et al. 2010; Ekblom & Galindo 2011; Seeb et al. 2011). The vastness of the genomes in taxa such as salamanders (with an average information content of c. 34 Gb) presents an as yet unsurpassed barrier for whole genome sequencing (Orgel & Crick 1980; Calboli et al. 2011; Gregory 2013). Considering the advances in genomics, it should eventually be possible to pass this obstacle (Davey et al. 2011; McCormack et al. 2013), as foreshadowed by the recent publication of the draft assembly of the c. 20 Gb genome for the Norway spruce Picea abies (Nystedt et al. 2013). Fortunately, for the purpose of designing a suite of genetic markers spread across the genome and suitable for population genetics, it is not necessary to have a complete genome available (Davey et al. 2011), but what are the strategies for obtaining such markers?
In the absence of any genome-wide reference data, cloning of sheared genomic DNA can be used to obtain anonymous genetic markers (Jennings & Edwards 2005). Although proven reasonably successful for salamanders (Espregueira Themudo et al. 2009; Nadachowska & Babik 2009), this approach requires many resources for relatively few markers and has become redundant with the advent of next-generation sequencing. Another approach that does not require any existing genomic data, but takes advantage of next-generation sequencing, involves techniques that employ restriction enzyme digestion to reduce the complexity of target genomes (reviewed by Davey et al. 2011), particularly restriction-site-associated DNA sequencing (RAD-seq) (Baird et al. 2008) and reduced representation libraries (RRLs) (Lemmon & Lemmon 2012). These techniques produce copious amount of data that wildly vary in quality, posing a challenge for data filtering and analysis (Lemmon & Lemmon 2013). Also they are relatively expensive on a per individual basis unless your aim is to obtain hundreds of markers for thousands of individuals. The efficiency of these methods relatively quickly drops when genetic divergence between study species increases and the number of shared restriction sites decreases (Davey et al. 2011; Lemmon & Lemmon 2013). Application of restriction-enzyme-based sequencing methods to taxa with large genomes is unlikely to be straightforward (Davey et al. 2011; Lemmon & Lemmon 2012). A large genome implies many recognition sites, even for rare cutters, which increases the amount of sequencing required (this problem should be possible to overcome with double digest (dd) RAD-seq; Peterson et al. 2012). Additionally, the highly repetitive nature of noncoding parts of, for example, salamander genomes (Sun et al. 2012) may limit suitability of many restriction enzymes, because if the enzyme cuts within highly repetitive sequence, most recovered markers will not be single copy and hence a large fraction of the sequencing effort would be wasted. Considering the rapid progression of the field, the aforementioned barriers are unlikely to be insurmountable, but to the best of our knowledge, no restriction-enzyme-based sequencing methods have been applied to salamanders yet.
Another route is to first obtain genome-wide reference data and use these resources to identify regions expected to contain single-nucleotide polymorphisms (SNPs; Garvin et al. 2010). When the sheer size of the genome makes directly obtaining informative genomic DNA inefficient, a reduced representation of the genome should be obtained. A promising source is conserved or ultraconserved elements which, because they are flanked by more variable regions, are also applicable to evolutionary questions involving shallow evolutionary time scales (Lemmon et al. 2012; Smith et al. 2014). This way many markers can be obtained, perhaps much more than actually required (Lemmon & Lemmon 2013), and the approach is dependent on the costly sequence capture technique (see below).
A commonly applied workaround to obtain genome-wide reference data is sequencing the transcriptome (Ekblom & Galindo 2011; Everett et al. 2011). Based on transcriptome data, SNPs or markers that are expected to be positioned across the genome can be identified. One strategy would be to obtain transcriptome data for more than one species, align them and screen them for polymorphism (Geraldes et al. 2011). After this SNP discovery step, individuals can be genotyped for SNP variants using a SNP genotyping assay (Garvin et al. 2010). A disadvantage of determining SNPs in advance is that an ascertainment bias is introduced, where differences within the subset of species used for marker design are inflated compared to the remaining species (Garvin et al. 2010). An application of a SNP assay for a species with a large genome generated two SNP genotyping arrays that together target almost 17 000 SNPs for the white spruce Picea glauca (Pavy et al. 2013). As SNP genotyping assays are project–specific, they only become economic for large-scale projects, where a large number of SNPs are determined (Garvin et al. 2010). For small-scale projects focusing on dozens of markers, a SNP genotyping assay is not the most logical choice.
Another approach is to obtain transcriptome data for a single species and identify regions within the transcriptome that are expected a priori to contain a sufficient level of phylogenetic information. Reducing the number of transcriptomes sequenced (and hence the number of adequately stored samples required) keeps down costs. A particular advantage of not targeting a set of predetermined polymorphisms is that an ascertainment bias is avoided (Garvin et al. 2010). Data can be obtained using sequence capture techniques (also referred to as hybrid enrichment), where probes are designed that ‘capture’ the regions of the genome that are the target for sequencing (Ekblom & Galindo 2011; McCormack et al. 2013). This technique requires a large initial financial investment in terms of equipment and reagents and therefore better suits large-scale projects aiming to sequence thousands of markers (Lemmon & Lemmon 2013). For smaller-scale projects focusing on dozens of markers, targets for sequencing can be obtained via PCR, where cost increases linearly with the number of samples analyzed (Ekblom & Galindo 2011). The workload can be greatly reduced using multiplex PCR (Zieliński et al. in press), and individuals can be sequenced in parallel by labelling individual's amplicons with unique tags (Bybee et al. 2011). Parallel tagged amplicon sequencing of transcriptome-derived markers has been successfully applied to groups of species of Ambystoma (O'Neill et al. 2013) and Lissotriton (Zieliński et al. in press) salamanders and is the route we follow here.
We focus on a group of aquatic salamanders known as the crested newt Triturus cristatus superspecies. Crested newts are a group of six recognized and a seventh candidate species with parapatric ranges, separated by narrow hybrid zones (Arntzen 2003; Wielstra et al. 2013c). We aim to design dozens of markers that show genetic differences between species and that can be sequenced on a large scale with next-generation sequencing methods. In brief, we (i) design markers based on taxonomically narrow transcriptome data; (ii) test which of these markers work for a taxonomically broad set of species; (iii) amplify the markers that pass this check with multiplex PCR for representatives of each (candidate) species and attach unique tags to be able to recognize the product belonging to each individual; (iv) sequence amplicons on the Ion Torrent next-generation sequencing platform; and (v) process the output with a bioinformatics pipeline that filters out poor quality reads, identifies alleles and converts data to a format directly usable for population genetic analysis. We test the utility of our methodological approach with empirical data.
Material and methods
Primer design from transcriptome data
Obtaining transcriptome data
Transcriptome data were obtained from four embryos raised in the laboratory, produced by parents collected under permission from an introduced Triturus carnifex population (Noorderheide, The Netherlands, N52.305, E05.834). The transcriptome was sequenced commercially by ZF Screens, Leiden, on the Illumina HiSeq 2000 platform. Data of all embryos were pooled, and transcriptome-based gene models were assembled with Trinity (Grabherr et al. 2011). However, Trinity can reconstruct alternatively spliced forms and, occasionally, diverged alleles as separate contigs, rendering the assembly a redundant representation of the transcribed part of the genome. Therefore, redundancy in the Trinity assembly was reduced using the bioinformatics pipeline of Stuglik et al. (in press). This pipeline reconstructs transcriptome-based gene models, that is, nonredundant representations of transcribed genomic sequences, by collapsing alleles from the same locus and merging alternatively spliced transcripts.
Primer design
Our protocol for designing primers closely follows Zieliński et al. (in press). We designed primers in 3-prime untranslated regions (3′ UTRs), the rationale being that because 3′UTRs do not code for proteins, they are relatively free to mutate (Makałowski & Boguski 1998) and hence likely to show genetic differentiation between closely related species. We focused on relatively long transcriptome-based gene models (5400–6500 bp) as these are expected to possess long 3′ UTRs. To identify 3′UTRs in our transcriptome-based gene models, we blasted them against human and Xenopus transcripts. We only focused on transcriptome-based gene models that produced unambiguous hits to a single gene, to increase the chance that we focus on orthologous, single-copy genes. Based on the alignment with the reference gene, we identified the starting position of the 3′ UTR in our transcriptome-based gene models by determining where the coding region on the last exon (if present) stops. We continued this procedure until we identified 96 3′UTRs (Table S1, Supporting information). This number was used for practical reasons as primers can be ordered in 96-well plates (prediluted, one with forward and one with reverse primers), which facilitates subsequent laboratory work. Within the 96 3′UTR regions, primers were designed with batchprimer3 1.0 (You et al. 2008), setting product length to 180 ± 5 bp and primer size to 20 ± 1 bp, and leaving the remaining settings at default (Table S1, Supporting information). In the absence of a genetic map, it was impossible to know the distribution of markers on the genome, but given that we designed markers without prior information, we considered the chance of them being positioned physically close negligible.
Marker testing
We screened the markers for their potential to cross-amplify for the whole genus Triturus by testing them for five crested and one marbled newt species (Table S2, Supporting information), spanning the taxonomical width of the genus (Wielstra & Arntzen 2011). We used the QIAGEN Taq Master Mix Kit. We conducted PCR in a total volume of 25 μL, containing 12.5 μL QIAGEN Taq PCR Master Mix, 2.5 μL primer mix (0.5 μm end concentration in the PCR), 9 μL Milli-Q water and 1 μL of template DNA. We used a single PCR programme: denaturation at 95 °C for 30 s, 35 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 45 s, and a final extension at 72 °C for 5 min. We tested whether a single product of expected length was amplified by gel electrophoresis using the E-Gel Precast Agarose Gel system (Life Technologies).
Ion Torrent sequencing of tagged amplicons
Sampling
We amplified the markers that passed the marker testing stage for two sets of newts. The first set contained 84 newts representing the seven (candidate) crested newt species (Fig.1a; Table S3, Supporting information). For each (candidate) species, we included three individuals from four populations positioned throughout their range. To minimize adverse effects of interspecific gene flow, our sampling scheme avoided localities positioned close to contact zones between species and populations containing introgressed mtDNA (Wielstra et al. 2013b). The second set contained 48 newts from a population positioned in the contact zone of Triturus ivanbureschi and Triturus macedonicus that was, based on throat and belly patterning (BW and JWA, personal observation), suspected to contain genetically admixed individuals.
Fig 1.
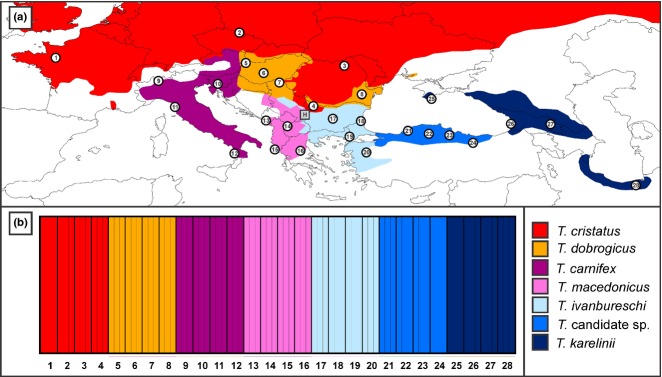
The potential of our methodological approach to delineate species. The map (a) shows the distribution of the seven crested newt (candidate) species, represented by different colours, and the geographical position of four populations sampled for each (numbered 1–28; n = 3 individuals per population). The grey square labelled ‘H’ is a genetically admixed population in the contact zone of Triturus ivanbureschi and Triturus macedonicus. Sampling details can be found in Table2 and Table S3, Supporting information. The baps plot (b), created with distruct (Rosenberg 2004), shows that each individual from populations 1–28 is allocated to its respective species.
Marker amplification using multiplex PCR
We reduced the workload by amplifying all markers in five multiplex PCRs (Table1). Primer combinations were chosen that showed a low risk of primer dimer formation, as determined with a macro written in Microsoft Excel. We used the QIAGEN Multiplex PCR Kit. We conducted PCR in a total volume of 12.5 μL, containing 7.5 μL QIAGEN Multiplex PCR Master Mix, 2.5 μL primer mix (0.08 μm end concentration in the PCR), 1.75 μL RNase-free water and 1 μL of template DNA. The PCR programme used was: initial denaturation at 95 °C for 15 min, 35 cycles of denaturation at 95 °C for 30 s, annealing at 55 °C for 60 s, and extension at 72 °C for 60 s, and a final extension step at 60 °C for 30 min. We confirmed the success of PCR reactions by gel electrophoresis using the E-Gel Precast Agarose Gel system (Life Technologies). We made a pool for each individual containing 5 μL of each multiplex PCR. These pools were cleaned up with Agencourt AMPure XP beads (Beckman Coulter).
Table 1.
Primer composition of the five multiplexes in which 52 markers were PCR amplified
| Multiplex | Included markers |
|---|---|
| I | abl, ace, asxl, chic, cnppd, col18, cri, gak, ngef, samdb, amot |
| II | fam178, ibtk, nisc, opa, phf, scap, sre, trab, plekhg1, pros1 |
| III | syncrip, tram, wiz, edc4, smo, msantd4, ccdc88a, wdr26, taf8, dbf4 |
| IV | arh, upf1, limch1, eif4ebp2, hmp19, bcor, myo18a, clasp2, ssh2, ddx17 |
| V | agl, dnaj, gys, kdm3, slc25, usp, ccdc124, supth6h, gcn1 l1, ganab, phip |
Marker abbreviations correspond to Table S1 (Supporting information).
Table 2.
Details on sampled crested newt (Triturus cristatus superspecies) populations
| Population code | Species | Locality | Sample size | Latitude | Longitude |
|---|---|---|---|---|---|
| 1 | Triturus cristatus | France: Mayenne | 3 | 48.300 | −0.617 |
| 2 | Triturus cristatus | Poland: Tłumaczów quarry | 3 | 50.558 | 16.434 |
| 3 | Triturus cristatus | Romania: Br˘d˘tel | 3 | 47.491 | 26.178 |
| 4 | Triturus cristatus | Bulgaria: Montana | 3 | 43.416 | 23.222 |
| 5 | Triturus dobrogicus | Austria: Tadten | 3 | 47.767 | 17.000 |
| 6 | Triturus dobrogicus | Hungary: Alap | 3 | 46.800 | 18.683 |
| 7 | Triturus dobrogicus | Serbia: Senta | 3 | 45.917 | 20.100 |
| 8 | Triturus dobrogicus | Romania: Giurgeni | 3 | 44.742 | 27.868 |
| 9 | Triturus carnifex | Switzerland: Locarno | 3 | 46.167 | 8.800 |
| 10 | Triturus carnifex | Slovenia: Kramplje | 3 | 45.733 | 14.500 |
| 11 | Triturus carnifex | Italy: Pisa | 3 | 43.717 | 10.400 |
| 12 | Triturus carnifex | Italy: Fuscaldo | 3 | 39.417 | 16.033 |
| 13 | Triturus macedonicus | Montenegro: Bjeloši | 3 | 42.374 | 18.907 |
| 14 | Triturus macedonicus | Macedonia: Gostivar | 3 | 41.817 | 20.899 |
| 15 | Triturus macedonicus | Greece: Kounoupena | 3 | 39.683 | 19.764 |
| 16 | Triturus macedonicus | Greece: Kerameia | 3 | 39.562 | 22.081 |
| 17 | Triturus ivanbureschi | Bulgaria: Alexandrovo | 3 | 42.601 | 25.093 |
| 18 | Triturus ivanbureschi | Bulgaria: Alepu | 3 | 42.348 | 27.714 |
| 19 | Triturus ivanbureschi | Turkey: Keşan | 3 | 40.917 | 26.633 |
| 20 | Triturus ivanbureschi | Turkey: Bigadiç | 3 | 39.351 | 28.217 |
| 21 | Triturus candidate species | Turkey: Karakoç | 3 | 41.487 | 32.142 |
| 22 | Triturus candidate species | Turkey: Cebeci | 3 | 41.201 | 34.036 |
| 23 | Triturus candidate species | Turkey: Kavak | 3 | 41.110 | 36.017 |
| 24 | Triturus candidate species | Turkey: Şebinkarahisar | 3 | 40.286 | 38.126 |
| 25 | Triturus karelinii | Ukraine: Nikita | 3 | 44.538 | 34.243 |
| 26 | Triturus karelinii | Georgia: Kobuleti | 3 | 41.822 | 41.814 |
| 27 | Triturus karelinii | Georgia: Telavi | 3 | 41.903 | 45.475 |
| 28 | Triturus karelinii | Iran: Alandan | 3 | 36.233 | 53.467 |
| H | Triturus ivanbureschi - Triturus macedonicus admixed | Serbia: Vlasi | 48 | 42.999 | 22.638 |
Population codes correspond to Fig.1.
Tagging and sequencing
Library preparation followed the manual of the NEBNext Fast DNA Library Prep Set for Ion Torrent kit (BioLabs Inc.), with the exception of using Ion Xpress Barcode Adapters (Life technologies) as tags. In brief, we conducted the following steps. We started with an end repair to ensure each molecule has 5′-phosphorylated blunt ends. Next, we ligated unique tags to each of the 132 pools, to allow our eventual sequence reads to be linked to the targeted individuals. We then conducted a dual bead-based size selection step with Agencourt AMPure XP beads, to remove fragments that are either too large or too small. The product was amplified by PCR and cleaned up with Agencourt AMPure XP beads. The tagged 132 pools were quantified on the QIAxcel system and pooled equimolar. After measuring the molarity of this single pool on a 2100 Bioanalyzer using the DNA High sensitivity chip (Agilent Technologies), it was diluted according to the calculated template dilution factor to target 10–30% of all positive Ion Sphere Particles. Template preparation and enrichment were carried out with the Ion One Touch 200 Template kit v2 DL (user guide revision 5.0) with the use of the Ion One Touch System. Quality control of the Ion One Touch 200 Ion Sphere Particles was conducted with the Ion Sphere Quality Control Kit using a Qubit 2.0 (Life Technologies). The enriched Ion Spheres were sequenced with a Life Technologies Ion Torrent Personal Genome Machine (Ion PGM) sequencer (Rothberg et al. 2011) on one-and-a-half Ion-318-chips (Ion PGM 200 Sequencing Kit).
Bioinformatics pipeline
Reads were automatically partitioned by the Ion Torrent software (torrent suite 3.6.2) into different FASTQ files for each individual (i.e. 132), based on their tags (reads without tags were discarded).
A first custom script filtered the raw reads, removing those with a length less than 100 bp and an average quality of less than Q20. Reads for each individual were mapped against the 52 reference sequences (excluding the primer sites) taken from the transcriptome data (Table S1, Supporting information) using bwa v0.7.3 (Li & Durbin 2009). On the resulting alignments, SNP/InDel calling was performed with samtools v0.1.18 (Li et al. 2009). Only the SNP/InDels with a samtools quality score over Q60 were retained.
A second custom script reconstructed the alleles for individual marker combinations. For marker-individual combinations with at least ten reads (those with less were considered to have failed), the combination of SNP/InDels present in each read was determined. Those SNP/InDel combinations that occurred in at least 25% of the reads were saved in an Excel table (i.e. SNP/InDels present in only one or a few reads were considered erroneous and were ignored). If more than two variants were found for a marker-individual combination (which could suggest paralogs), this combination was considered to have failed. Based on the saved SNP/InDel combinations, consensus sequences were created for each individual by substituting the relevant SNPs/InDels in the reference sequence. This resulted in two alleles, which may or may not be identical, for each individual for each marker. The two alleles for each marker for each individual were saved as FASTA files (i.e. one FASTA file per marker).
A third custom script converted the data into a genotypic data format (i.e. recoding the different allelic variants for each marker to a unique integer, resulting in two integers per individual per marker). This format can be directly fed into the program create (Coombs et al. 2008), which generates input files for a wide range of population genetics programmes.
Population genetics analyses
First, we converted the data of the 84 newts representing the seven (candidate) crested newt species to GENEPOP format and analyzed the data in baps v.5.3 (Corander et al. 2008). baps assigns individuals to distinct gene pools probabilistically, based upon multilocus genetic data, where each individual allele is coded as a haplotype (two alleles per marker, which may or may not belong to the same haplotype). We used ten replicates and tested for admixture between gene pools. We enabled fixed-k clustering and set the number of expected populations (k) to seven, the rationale being that if our methodology works, baps should be able to allocate all individual newts to the (candidate) species they belong to.
Next, we converted data for the 24 individuals of T. ivanbureschi and T. macedonicus taken from the set of 84 newts, together with those of the 48 newts representing the presumed genetically admixed population from their contact zone, to the input format for newhybrids 1.1 (Anderson & Thompson 2002). newhybrids infers for every individual the probability with which it belongs to a purebred (either of the two parental species under consideration) or a hybrid class (F1, F2 or a backcross with one of the two parental species). The 12 T. ivanbureschi and 12 T. macedonicus individuals were set to belong to the two parental classes a priori using the z option in newhybrids. We ran the programme five separate times with a burn-in and formal run of 10 000 iterations each.
Subsequently, we determined the hybrid index of the 48 newts from the presumed admixed population. We checked for which of the 52 markers nonoverlapping allele variants are present in the reference populations for T. ivanbureschi and T. macedonicus and established the proportion of these alleles in the 48 newts from the presumed admixed population. Finally, we coded for each marker-individual combination how many T. ivanbureschi alleles were present, that is, 0, 1 or 2, and noted those with missing data or alleles not present in the reference individuals as not available (NA). We analyzed these data with hiest, an R package that determines and visualizes the genomic composition of hybrids based on ancestry, the fraction of alleles derived from each parental species, and heterozygosity, the fraction of loci heterozygous for alleles from each parental species (Fitzpatrick 2012). The newhybrids, hybrid index and hiest analyses should, if our methodology is successful, reveal the usefulness of our markers for detecting genetic admixture.
Results
Of the 96 markers tested, 52 produced a single clear band of expected size on agarose gel for all five tested crested newt species (Table S2, Supporting information). Forty-one of these additionally worked for the included marbled newt species (multiplexes I–IV, Table1). Our Ion Torrent runs for the 132 crested newt individuals delivered 6 192 014 tagged raw reads. After filtering, we were left with 3 688 557 alignable reads (Table S4, Supporting information). The mean number of reads per individual per marker in 132 × 52 = 6864 combinations was 537.4 ± 9.3 (standard error). One hundred and twenty-two marker-individual combinations (1.8%) produced <10 reads and were considered as failed; for five markers, dropout for particular populations/species was observed, suggesting the presence of null alleles (Table S4, Supporting information). One marker-individual combination (0.015%) had more than two variants and was considered failed (Table S4, Supporting information). The SNP report used to create consensus sequences can be found in Table S5, Supporting information. Fifty-one of 52 markers were polymorphic (Table S6, Supporting information). The number of alleles per marker ranged from 1 to 25, with a mean of 9.4 ± 5.3 (standard deviation). The data recoded in genotypic format are available in Table S7 (Supporting information).
The baps analysis allocated all 84 crested newt individuals representing the seven (candidate) species to their respective (candidate) species with full support (P-value of 1.0 in the admixture analysis; Fig.1b). newhybrids showed mixed results: four runs identified the 48 individuals from the presumed hybrid population as pure Triturus ivanbureschi, whereas a fifth run allocated these individuals with equal probability to both T. ivanbureschi and F2 hybrid class (Table S8, Supporting information). Of the 52 tested markers, 27 possess nonoverlapping allele variants in the reference populations for T. ivanbureschi and Triturus macedonicus (Table S9, Supporting information). The proportion of these alleles in the 48 individuals from the presumed hybrid population is shown in Fig.2a. Although a considerable number of T. macedonicus alleles are found, the majority of alleles derives from T. ivanbureschi. This is also apparent from the hiest analyses: the 48 individuals form a ‘hybrid smear’ positioned in the T. ivanbureschi corner of the triangle plot in Fig.2b. The position near the right side of the plot reflects that the majority of alleles in individuals is derived from T. ivanbureschi (i.e. backcrossing with T. ivanbureschi). The position near the bottom of the plot reflects that markers for individuals are often represented by either T. macedonicus or T. ivanbureschi alleles but not both (i.e. a heterozygote deficit).
Fig 2.
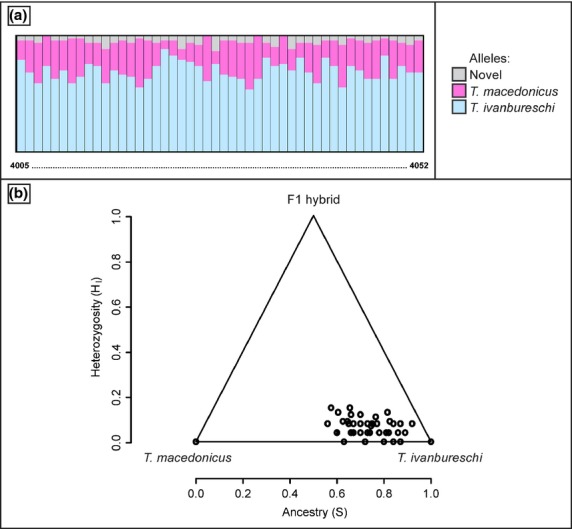
The potential of our methodological approach to detect genetic admixture. For a genetically admixed population positioned in the contact zone of Triturus ivanbureschi and Triturus macedonicus (the grey square labelled ‘H’ in Fig.1a), we show the proportion of alleles derived from the two parent species (a) and heterozygosity and ancestry as determined with hiest (b).
Discussion
We obtain a set of markers, informative at the level of closely related species, for a nonmodel system with large, unknown genomes (Triturus newts). For marker design, we use transcriptome data as a reduced representation of the genome, as the vastness of genomes in taxa such as salamanders still remains a challenge for whole genome sequencing. We employ transcriptome data of only one of our focal species, reducing costs and avoiding ascertainment bias. For successful marker design, the assembled transcriptome can be far from perfect (Stuglik et al. in press). We target 3′ UTR regions, which are typically contained entirely within a single exon (Hong et al. 2006), meaning there are no introns hampering PCR amplification. Because 3′ UTR regions are less evolutionary constrained than protein-coding markers (Makałowski & Boguski 1998), they are a priori expected to be informative.
We design markers in a narrow size range and only use those that are verified to cross-amplify for a taxonomically broad set of species under the same PCR protocol. The highly standardized conditions minimize variation in concentrations of individual markers after multiplex PCR, increasing the likelihood that all markers will be sequenced. In our case, no optimization of our multiplex PCR protocol is required, and the separate multiplexes for individuals do not have to be diluted to equal concentration, saving time and resources. Furthermore, the preselection of markers likely to cross-amplify for all individuals reduces the presence of null alleles. We observe few null alleles, and for the markers where they are present, they affect only a subset of populations/species (i.e. the markers are still usable in a setting where these populations/species are not included). Tagging amplicons means that no marker-specific barcoded PCR primers are required, further reducing costs, labour and logistic complexity (Zieliński et al. in press). The Ion Personal Genome Machine is a rapid, compact and economical sequencer, easily implemented in any laboratory as a bench-top machine. As we focus on short sequences (180 bp including primers), most reads span the entire length of the target sequence, and allelic phases are directly available. Our bioinformatics pipeline uses standard, open-source bioinformatic tools to produce phase-resolved data into a format directly usable for population genetic analysis.
We demonstrate the utility of our set of markers to delineate species by sequencing a dozen individuals from each crested newt (candidate) species. The populations from which these individuals are sampled are positioned throughout the range of each (candidate) species. As illustrated in Fig.1, all individuals are assigned to the proper species. We show the usefulness of our markers for detecting genetic admixture by analyzing 48 individuals from a hybrid population. An interesting aspect is that, for our dataset, newhybrids fails to acknowledge genetic admixture in part of the runs, despite alleles of both parental species being present, as shown in Fig.2. This reflects the restricted set of discrete categories of admixture tested for, despite the myriad of recombinant genotypes that many generations of hybridization and backcrossing give rise to (Fitzpatrick 2012). A complex setting of genetic admixture over a large number of generations is revealed to apply to our tested hybrid population by hiest. The observed heterozygote deficit mirrors that found for allozymes (J. W. Arntzen, B. Wielstra & G. P. Wallis, submitted ) and can be explained by alleles being lost by drift over time (Excoffier & Ray 2008; Fitzpatrick 2012). The observation of allelic variants in the hybrid population not observed in the set of 84 newts representing the seven (candidate) species is in line with the ‘rare allele phenomenon’ (Lammers et al. 2013).
The methodology presented in this paper is successful in obtaining single copy, phased, informative markers that are efficiently and cost-effectively amplified and sequenced. We settle for 52 markers, but our procedure can be easily scaled up to get hundreds of markers, without increasing the costs of library preparation. We anticipate the current set of markers to provide high resolving power for our studies on Triturus hybrid zones. For example, we can use the markers to test the depth of hybridization (Arntzen et al. 2009), whether genetic footprints were left after hybridizing species displaced one another (Wielstra & Arntzen 2012), and the validity of hypothesized morphologically cryptic species (Wielstra et al. 2013a). Although we particularly focus on Triturus in this paper, the methodological framework described is broadly applicable to the study of gene flow between closely related species.
Acknowledgments
W. Babik and P. Zieliński provided helpful comments. M. Stuglik performed the Trinity assemblage. K. van der Gaag provided the Microsoft Excel macro and sheet to compose multiplexes. M. Eurlings and F. Stokvis helped with Ion Torrent sequencing. BW is a Newton International Fellow. PL was supported with a Martin fellowship from Naturalis Biodiversity Center. Obtaining the transcriptome data was funded by NWO (ALW project: 817.02.027) and by the NBC Naturalis scheme ‘ultra high-throughput sequencing 2010’ (PS2010.07).
Biography
B.W. and J.W.A. conceived and designed the project with input from E.D. and Y.L. J.M.Z. provided the transcriptome data. B.W. designed the markers. B.W., P.L. and W.R.M.M. tested and amplified the markers. E.D. conducted the sequencing. Y.L. conducted the bioinformatics. B.W. analyzed the data. B.W. drafted the manuscript. All authors contributed to and approved the final manuscript.
Data Accessibility
Triturus transcriptome-based gene models in FASTA formats, Excel sheet and macro to compose multiplexes, Raw Ion Torrent reads in FASTQ format, BWA alignments in SAM format, Raw SNP reports in VCF format, FASTA files of reconstructed sequences, input files for baps, newhybrids and hiest, the three scripts associated with the bioinformatics pipeline: Dryad Digital Repository entry doi:10.5061/dryad.36775.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
The 96 transcriptome-based gene models used for primer development, primers and reference sequences.
Table S2. Testing of 96 primer pairs for cross-amplification for the genus Triturus.
Table S3. Sampling details.
Table S4. Number of reads per individual per marker.
Table S5. Filtered SNP report used to construct consensus sequences.
Table S6. Allelic variants per marker.
Table S7. Data for 132 individual crested newts in genotypic format.
Table S8. Results of the newhybrids analysis.
Table S9. Distribution of 27 markers distinguishing T. ivanbureschi and T. macedonicus in the presumed hybrid population.
References
- Anderson EC, Thompson EA. A model-based method for identifying species hybrids using multilocus genetic data. Genetics. 2002;160:1217–1229. doi: 10.1093/genetics/160.3.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arntzen JW. Triturus cristatus Superspecies - Kammolch-Artenkreis (Triturus cristatus (Laurenti, 1768) - Nördlicher Kammolch, Triturus carnifex (Laurenti, 1768) - Italienischer Kammolch, Triturus dobrogicus (Kiritzescu, 1903) - Donau-Kammolch, Triturus karelinii (Strauch, 1870) - Südlicher Kammolch) In: Thiesmeier B, editor; Grossenbacher K, editor. Handbuch der Reptilien und Amphibien Europas. Schwanzlurche IIA. Wiebelsheim: Aula-Verlag; 2003. pp. 421–514. [Google Scholar]
- Arntzen JW, Jehle R, Bardakci F, Burke T, Wallis GP. Asymmetric viability of reciprocal-cross hybrids between crested and marbled newts (Triturus cristatus and T. marmoratus. Evolution. 2009;63:1191–1202. doi: 10.1111/j.1558-5646.2009.00611.x. [DOI] [PubMed] [Google Scholar]
- Baird NA, Etter PD, Atwood TS, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE. 2008;3:e3376. doi: 10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bybee SM, Bracken-Grissom H, Haynes BD, et al. Targeted amplicon sequencing (TAS): a scalable next-gen approach to multilocus, multitaxa phylogenetics. Genome Biology and Evolution. 2011;3:1312–1323. doi: 10.1093/gbe/evr106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calboli FCF, Fisher MC, Garner TWJ, Jehle R. The need for jumpstarting amphibian genome projects. Trends in Ecology & Evolution. 2011;26:378–379. doi: 10.1016/j.tree.2011.04.006. [DOI] [PubMed] [Google Scholar]
- Coombs JA, Letcher BH, Nislow KH. create: a software to create input files from diploid genotypic data for 52 genetic software programs. Molecular Ecology Resources. 2008;8:578–580. doi: 10.1111/j.1471-8286.2007.02036.x. [DOI] [PubMed] [Google Scholar]
- Corander J, Marttinen P, Siren J, Tang J. Enhanced Bayesian modelling in baps software for learning genetic structures of populations. BMC Bioinformatics. 2008;9:539. doi: 10.1186/1471-2105-9-539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey JW, Hohenlohe PA, Etter PD, et al. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nature Reviews Genetics. 2011;12:499–510. doi: 10.1038/nrg3012. [DOI] [PubMed] [Google Scholar]
- Ekblom R, Galindo J. Applications of next generation sequencing in molecular ecology of non-model organisms. Heredity. 2011;107:1–15. doi: 10.1038/hdy.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espregueira Themudo G, Babik W, Arntzen JW. A combination of techniques proves useful in the development of nuclear markers in the newt genus Triturus. Molecular Ecology Resources. 2009;9:1160–1162. doi: 10.1111/j.1755-0998.2009.02574.x. [DOI] [PubMed] [Google Scholar]
- Everett MV, Grau ED, Seeb JE. Short reads and nonmodel species: exploring the complexities of next-generation sequence assembly and SNP discovery in the absence of a reference genome. Molecular Ecology Resources. 2011;11:93–108. doi: 10.1111/j.1755-0998.2010.02969.x. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Ray N. Surfing during population expansions promotes genetic revolutions and structuration. Trends in Ecology & Evolution. 2008;23:347–351. doi: 10.1016/j.tree.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick B. Estimating ancestry and heterozygosity of hybrids using molecular markers. BMC Evolutionary Biology. 2012;12:131. doi: 10.1186/1471-2148-12-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvin MR, Saitoh K, Gharrett AJ. Application of single nucleotide polymorphisms to non-model species: a technical review. Molecular Ecology Resources. 2010;10:915–934. doi: 10.1111/j.1755-0998.2010.02891.x. [DOI] [PubMed] [Google Scholar]
- Geraldes A, Pang J, Thiessen N, et al. SNP discovery in black cottonwood (Populus trichocarpa) by population transcriptome resequencing. Molecular Ecology Resources. 2011;11:81–92. doi: 10.1111/j.1755-0998.2010.02960.x. [DOI] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory TR. Animal Genome Size Database. 2013. http://www.genomesize.com.
- Hong X, Scofield DG, Lynch M. Intron size, abundance, and distribution within untranslated regions of genes. Molecular Biology and Evolution. 2006;23:2392–2404. doi: 10.1093/molbev/msl111. [DOI] [PubMed] [Google Scholar]
- Jennings WB, Edwards SV. Speciational history of Australian grass finches (Poephila) inferred from thirty gene trees. Evolution. 2005;59:2033–2047. [PubMed] [Google Scholar]
- Lammers Y, Kremer D, Brakefield PM, et al. SNP genotyping for detecting the ‘rare allele phenomenon’ in hybrid zones. Molecular Ecology Resources. 2013;13:237–242. doi: 10.1111/1755-0998.12044. [DOI] [PubMed] [Google Scholar]
- Lemmon AR, Lemmon EM. High-throughput identification of informative nuclear loci for shallow-scale phylogenetics and phylogeography. Systematic Biology. 2012;61:745–761. doi: 10.1093/sysbio/sys051. [DOI] [PubMed] [Google Scholar]
- Lemmon EM, Lemmon AR. High-throughput genomic data in systematics and phylogenetics. Annual Review of Ecology, Evolution, and Systematics. 2013;44:99–121. [Google Scholar]
- Lemmon AR, Emme SA, Lemmon EM. Anchored hybrid enrichment for massively high-throughput phylogenomics. Systematic Biology. 2012;61:727–744. doi: 10.1093/sysbio/sys049. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makałowski W, Boguski MS. Evolutionary parameters of the transcribed mammalian genome: an analysis of 2,820 orthologous rodent and human sequences. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:9407–9412. doi: 10.1073/pnas.95.16.9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER. A decade's perspective on DNA sequencing technology. Nature. 2011;470:198–203. doi: 10.1038/nature09796. [DOI] [PubMed] [Google Scholar]
- McCormack JE, Hird SM, Zellmer AJ, Carstens BC, Brumfield RT. Applications of next-generation sequencing to phylogeography and phylogenetics. Molecular Phylogenetics and Evolution. 2013;66:526–538. doi: 10.1016/j.ympev.2011.12.007. [DOI] [PubMed] [Google Scholar]
- Nadachowska K, Babik W. Divergence in the face of gene flow: the case of two newts (Amphibia: Salamandridae) Molecular Biology and Evolution. 2009;26:829–841. doi: 10.1093/molbev/msp004. [DOI] [PubMed] [Google Scholar]
- Nystedt B, Street NR, Wetterbom A, et al. The Norway spruce genome sequence and conifer genome evolution. Nature. 2013;497:579–584. doi: 10.1038/nature12211. [DOI] [PubMed] [Google Scholar]
- O'Neill EM, Schwartz R, Bullock CT, et al. Parallel tagged amplicon sequencing reveals major lineages and phylogenetic structure in the North American tiger salamander (Ambystoma tigrinum) species complex. Molecular Ecology. 2013;22:111–129. doi: 10.1111/mec.12049. [DOI] [PubMed] [Google Scholar]
- Orgel LE, Crick FHC. Selfish DNA: the ultimate parasite. Nature. 1980;284:604–607. doi: 10.1038/284604a0. [DOI] [PubMed] [Google Scholar]
- Pavy N, Gagnon F, Rigault P, et al. Development of high-density SNP genotyping arrays for white spruce (Picea glauca) and transferability to subtropical and nordic congeners. Molecular Ecology Resources. 2013;13:324–336. doi: 10.1111/1755-0998.12062. [DOI] [PubMed] [Google Scholar]
- Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE. 2012;7:e37135. doi: 10.1371/journal.pone.0037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg NA. distruct: a program for the graphical display of population structure. Molecular Ecology Notes. 2004;4:137–138. [Google Scholar]
- Rothberg JM, Hinz W, Rearick TM, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475:348–352. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- Seeb JE, Carvalho G, Hauser L, et al. Single-nucleotide polymorphism (SNP) discovery and applications of SNP genotyping in nonmodel organisms. Molecular Ecology Resources. 2011;11:1–8. doi: 10.1111/j.1755-0998.2010.02979.x. [DOI] [PubMed] [Google Scholar]
- Smith BT, Harvey MG, Faircloth BC, Glenn TC, Brumfield RT. Target capture and massively parallel sequencing of ultraconserved elements for comparative studies at shallow evolutionary time scales. Systematic Biology. 2014;63:83–95. doi: 10.1093/sysbio/syt061. [DOI] [PubMed] [Google Scholar]
- Stuglik MT, Babik W, Radwan J. Alternative reproductive tactics and sex-biased gene expression: the study of the bulb mite transcriptome. Ecology and Evolution. in press;4:623–632. [Google Scholar]
- Sun C, Shepard DB, Chong RA, et al. LTR retrotransposons contribute to genomic gigantism in Plethodontid salamanders. Genome Biology and Evolution. 2012;4:168–183. doi: 10.1093/gbe/evr139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twyford AD, Ennos RA. Next-generation hybridization and introgression. Heredity. 2012;108:179–189. doi: 10.1038/hdy.2011.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wielstra B, Arntzen JW. Unraveling the rapid radiation of crested newts (Triturus cristatus superspecies) using complete mitogenomic sequences. BMC Evolutionary Biology. 2011;11:162. doi: 10.1186/1471-2148-11-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wielstra B, Arntzen JW. Postglacial species displacement in Triturus newts deduced from asymmetrically introgressed mitochondrial DNA and ecological niche models. BMC Evolutionary Biology. 2012;12:161. doi: 10.1186/1471-2148-12-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wielstra B, Baird AB, Arntzen JW. A multimarker phylogeography of crested newts (Triturus cristatus superspecies) reveals cryptic species. Molecular Phylogenetics and Evolution. 2013a;67:167–175. doi: 10.1016/j.ympev.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Wielstra B, Crnobrnja-Isailović J, Litvinchuk SN, et al. Tracing glacial refugia of Triturus newts based on mitochondrial DNA phylogeography and species distribution modeling. Frontiers in Zoology. 2013b;10:13. doi: 10.1186/1742-9994-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wielstra B, Litvinchuk S, Naumov B, Tzankov N, Arntzen JW. A revised taxonomy of crested newts in the Triturus karelinii group (Amphibia: Caudata: Salamandridae), with the description of a new species. Zootaxa. 2013c;3682:441–453. doi: 10.11646/zootaxa.3682.3.5. [DOI] [PubMed] [Google Scholar]
- You F, Huo N, Gu Y, et al. batchprimer3: a high throughput web application for PCR and sequencing primer design. BMC Bioinformatics. 2008;9:253. doi: 10.1186/1471-2105-9-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zieliński P, Stuglik MT, Dudek K, Konczal M, Babik W. Development, validation and high throughput analysis of sequence markers in non-model species. Molecular Ecology Resources. in press;14:352–360. doi: 10.1111/1755-0998.12171. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The 96 transcriptome-based gene models used for primer development, primers and reference sequences.
Table S2. Testing of 96 primer pairs for cross-amplification for the genus Triturus.
Table S3. Sampling details.
Table S4. Number of reads per individual per marker.
Table S5. Filtered SNP report used to construct consensus sequences.
Table S6. Allelic variants per marker.
Table S7. Data for 132 individual crested newts in genotypic format.
Table S8. Results of the newhybrids analysis.
Table S9. Distribution of 27 markers distinguishing T. ivanbureschi and T. macedonicus in the presumed hybrid population.
Data Availability Statement
Triturus transcriptome-based gene models in FASTA formats, Excel sheet and macro to compose multiplexes, Raw Ion Torrent reads in FASTQ format, BWA alignments in SAM format, Raw SNP reports in VCF format, FASTA files of reconstructed sequences, input files for baps, newhybrids and hiest, the three scripts associated with the bioinformatics pipeline: Dryad Digital Repository entry doi:10.5061/dryad.36775.