

Abstract
Intravenous immunoglobulin (IVIG) is widely used in autoimmune neuromuscular diseases whose pathogenesis is undefined. Many different effects of IVIG have been demonstrated in vitro, but few studies actually identify the mechanism(s) most important in vivo. Doses and treatment intervals are generally chosen empirically. Recent studies in Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy show that some effects of IVIG are readily reversible and highly dependent on the serum IgG level. This suggests that in some autoantibody-mediated neuromuscular diseases, IVIG directly competes with autoantibodies that reversibly interfere with nerve conduction. Mechanisms of action of IVIG which most likely involve direct competition with autoantibodies include: neutralization of autoantibodies by anti-idiotypes, inhibition of complement deposition, and increasing catabolism of pathologic antibodies by saturating FcRn. Indirect immunomodulatory effects are not as likely to involve competition and may not have the same reversibility and dose-dependency. Pharmacodynamic analyses should be informative regarding most relevant mechanism(s) of action of IVIG as well as the role of autoantibodies in the immunopathogenesis of each disease. Better understanding of the role of autoantibodies and of the target(s) of IVIG could lead to more efficient use of this therapy and better patient outcomes.
Keywords: anti-idiotypes, autoimmune neuromuscular diseases, chronic inflammatory demyelinating polyneuropathy, Guillain-Barré syndrome, intravenous immunoglobulin, multifocal motor neuropathy
Introduction
Intravenous immunoglobulin (IVIG) is employed in a staggeringly wide spectrum of diseases, and can act by many diverse mechanisms. Different proposed mechanisms are not mutually exclusive – multiple mechanisms likely contribute to the overall therapeutic effect. In order to identify the action(s) of IVIG that are most important in any particular disease, we must understand the pathogenesis of that disease. Conversely, discerning the mechanism(s) of action of IVIG which are most important in different diseases may provide useful insights into their pathogenesis. Analysis of the pharmacodynamics (PD) of IVIG can thus not only reveal important insights into its mechanisms of action, but should also be informative as to the pathogenic mechanism(s) it is affecting. Better understanding of the pathologic process(es) targeted by IVIG in any given disease may allow more effective and efficient use of this therapy.
Pharmacokinetics of IVIG
The initial dose of IVIG used for most autoimmune/inflammatory diseases is 2 g/kg given over 2–5 days, followed by maintenance doses of 1–2 g/kg every 3–4 weeks. This regimen is based on the serendipitous 1981 observation that five consecutive daily repetitions of the monthly dose of IVIG for immune deficiencies at that time (0.4 g/kg) normalized the platelet counts in immune deficient patients who also had immune thrombocytopenia (Imbach et al., 1981). Neither measurements of autoantibodies nor in vitro determinations of the ability of IVIG to neutralize them are used to select or adjust the dose of IVIG or to set a target therapeutic “level” in autoimmune diseases. In contrast, recent studies in primary immune deficiency diseases (PIDD) suggest individualizing the IgG dose and treatment interval to achieve the “biological serum IgG level” necessary to keep that individual patient free from infection (Bonagura et al., 2008; Lucas et al., 2010). A wide variability in target levels, doses (on a mg/kg/month basis) and intervals are necessary in different patients (Lucas et al., 2010).
Infusion of 2 g/kg of IVIG increases the serum IgG level >4-fold, from pretreatment means of 700–1,060 mg/dl to peaks well over 3,000 mg/dl (Reinhart and Berchtold, 1992). The levels then drop by about 50% over 48–72 h, as IgG is distributed into the total extracellular fluid volume, of which only about 50% is intravascular (Waldmann and Strober, 1969; Pirofsky, 1984). After this rapid equilibration, the IgG is catabolized with first-order kinetics and a half-life of 21–30 days, so it is usually repeated monthly (Bonilla, 2008). In contrast, PD effects may be highly variable. Many neuromuscular disease patients note that the effects of IVIG “wear-off” long before the predicted half-life. On the other hand, in some diseases, the effects last much longer than the expected half-life of IgG. These observations suggest complex relationships between the PD and pharmacokinetics (PK) in different situations. Responses with a rapid onset and short duration are most consistent with therapeutic mechanisms that require high serum concentrations of therapeutic IgG in order to compete with pathologic autoantibodies, and are the subject of this review. Ultimately, detailed studies correlating changes in the serum IgG levels with the clinical response at frequent (even daily) time points are necessary to achieve a better understanding of what PD results are telling us about the mechanisms of action of IgG.
Mechanisms of Action of IVIG
Many effects of IVIG have been demonstrated in vitro (reviewed by Ballow, 2011; Lehmann and Hartung, 2011; Gelfand, 2012; Schwab and Nimmerjahn, 2013), but it is rarely clear which effects are most important in any given disease in vivo. We hypothesize that PD results characterized by a rapid onset of action, strong dose dependency, and short duration suggest direct competition between IVIG and putative pathologic autoantibodies. In turn, these characteristics imply that the autoantibodies bear primary responsibility for the nerve dysfunction, and that decreasing the autoantibodies ameliorates the symptoms. Other immunomodulatory effects of IVIG involving networks of lymphocytes and/or antigen-presenting cells might be less likely to have this type of time course or dose-dependency. In this review, we focus on those mechanisms of action of IVIG which most likely involve direct competition with autoantibodies and explore the evidence for a direct role of autoantibodies in the pathophysiology of conditions with those PD characteristics. In the long run, this type of analysis should increase our understanding of IgG therapy and how it can be optimized. Of all the proposed mechanisms of action of IVIG, three stand out as the most likely to involve direct competition with pathologic autoantibodies, and these are discussed below.
Anti-idiotypic binding
Each person’s immune system makes approximately 1012 different antibody specificities. This remarkable diversity arises by splicing together multiple DNA segments to make a single antibody gene (Tonegawa, 1983; Schroeder and Cavacini, 2010). The heavy and light chains of each antibody molecule contain small “hyper-variable regions” that together form the antigen-binding site (Fig. 1). Upon exposure to an antigen, a B-cell with a complementary binding site is selected, and stimulated to secrete its particular antibody. That antibody may, in turn, seem “new” to the body, and can serve as an antigen itself. Each unique antigen-binding site is called an “idiotype”, and an antibody which recognizes or blocks the antigen-binding site of another antibody is called an “anti-idiotype” [anti-id] (Schroeder and Cavacini, 2010; Ballow, 2011). Some anti-ids may closely mimic the original antigen and fit into its binding site, while others may block the antigen-binding site by “steric hindrance” (Fig. 1).
Figure 1.
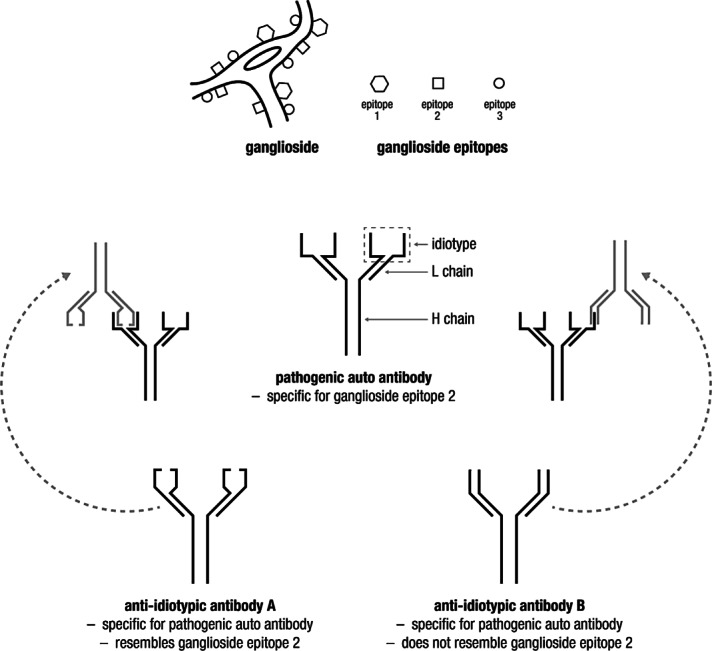
Structure of IgG molecule illustrating antigen-binding sites and showing binding of anti-idiotypic antibodies that resemble target antigen of the autoantibody (left) or block its antigen-binding site by “steric hindrance.”
Since each individual’s immune repertoire arises randomly, it is readily understandable that some individuals may respond to a pathogen like Campylobacter jejuni with antibodies that cross-react with self-antigens, while other individuals recognize different epitopes on the bacteria. Further, one can speculate that individuals who mount rigorous anti-id responses will rapidly bring a self-reactive response under control, while others whose anti-id response is weak or ineffective may continue to produce clinically significant amounts of autoantibodies (Oak et al., 2008). Since IVIG contains the antibodies from tens of thousands of donors, it follows that it likely contains many different anti-ids (Ballow, 2011).
Shortly after its introduction, IVIG was found to neutralize “inhibitors” of clotting Factor VIII (FVIII) in hemophilia patients receiving replacement therapy (Rossi et al., 1988). These “inhibitors” are in fact antibodies against FVIII. F(ab’) fragments of IVIG, which contain the antigen-binding sites, neutralized the inhibitors, indicating anti-id binding (Rossi et al., 1988). Subsequent studies have shown that IVIG contains a wide variety of anti-ids, suggesting that anti-id suppression of potential autoimmunity is actually quite common in normal physiology. The most important support for an anti-id mechanism of IVIG would be the observation that F(ab’) or F(ab’)2 fragments from IVIG neutralize the autoantibody in vitro or can remove it by affinity chromatography (Table 1). Other examples of anti-ids in IVIG include: antibodies that neutralize anti-DNA (Evans et al., 1991), anti-ganglioside antibodies (Lopez et al., 2000; Jacobs et al., 2003), antibodies to M3 acetylcholine receptors (AChRs) (Cavill et al., 2003; Smith et al., 2005), anti-GAD (Oak et al., 2008), anti-microsomal antibodies (Tandon et al., 1992), anti-thyroglobulin (Dietrich and Kazatchkine, 1990), and others. Although an early search for anti-ids to autoantibodies to nicotinic AChRs in myasthenia gravis (MG) patients’ sera was not fruitful (Vincent et al., 1988), subsequent papers have suggested that IVIG does contain anti-ids against AChR autoantibodies (Liblau et al., 1991).
Table 1.
Types of evidence suggesting anti-idiotype effect of intravenous immunoglobulin (IVIG)
| •Tissue dysfunction/pathology is independent from Fc receptor bearing effector cells |
| •Removal of pathologic antibodies by plasma exchange has rapid effects |
| •Effects of plasma exchange are qualitatively and rapidly reproduced by IVIG treatment |
| •Antibodies from patients’ sera bind to appropriate target in vitro, and this binding can be blocked in a competitive way by IVIG |
| •F(ab)’ and/or F(ab)’2 fragments of the IVIG have the same effect as IVIG in vitro |
FcRn saturation increases catabolism of endogenous antibodies
Catabolism of IgG is controlled by the same Fc receptor which transports IgG across the placenta, FcRn (Yu and Lennon, 1999). FcRn on endothelial cells binds and internalizes circulating IgG using a pathway which is protected from lysosomal degradation (Fig. 2). The IgG is recycled back to the cell surface and re-released into the plasma. This maintains the relatively high normal concentration and long half-life of IgG in the circulation (Yu and Lennon, 1999). In contrast, IgA and IgM do not bind to FcRn (Yu and Lennon, 1999) and have very short half-lives (Fahey and Sell, 1965). In FcRn knockout mice and in patients with FcRn mutations, the half-life of IgG is very short and its plasma concentration is quite low (Junghans and Anderson, 1996; Wani et al., 2006). Furthermore, it is difficult to passively transfer IgG-mediated pathology to FcRn knockout mice because the half-life of the pathologic IgG is too short (Li et al., 2005). By analogy, if FcRn is saturated by high doses of exogenous IVIG, the catabolism of endogenous pathologic IgG is greatly increased (Hansen and Balthasar, 2003). In wild-type mice, high dose IgG dramatically increases the catabolism of pathogenic IgG; while in FcRn knockout mice, high dose IgG does not further enhance the already rapid catabolism of exogenous pathologic IgG (Li et al., 2005). Thus, by saturating FcRn with normal antibodies, IVIG can increase degradation of pathogenic autoantibodies. This therapeutic effect therefore requires that the total IgG concentration exceeds the binding capacity of FcRn and depends on the ratio of the serum concentration of normal IgG to the concentration of pathogenic autoantibodies (Hansen and Balthasar, 2003).
Figure 2.
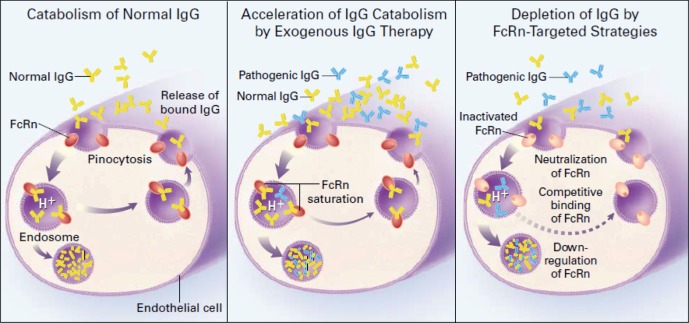
Function of endothelial FcRn in maintaining normal serum IgG concentration (left panel) and mechanism by which FcRn allows intravenous immunoglobulin (IVIG, yellow) to selectively increase degradation of pathogenic IgG (blue) (center panel). From Yu and Lennon (1999); reproduced with permission from the publisher.
Both of the above mechanisms would decrease the amount of pathologic autoantibody available for binding to its target. This is nicely illustrated by the correlation between clinical improvement and decreased levels of anti-glutamic acid decarboxylase (anti-GAD) antibodies before and after IVIG treatment in stiff-person syndrome (Fig. 3) (Dalakas et al., 2001; Shiraishi et al., 2002).
Figure 3.
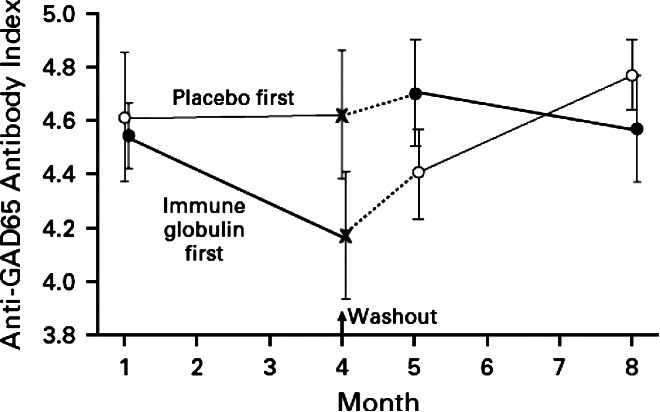
Anti-GAD 65 antibodies in patients with “stiff-person syndrome” during treatment with IVIG or placebo. From Dalakas et al. (2001); reproduced with permission from the publisher.
Complement scavenging
Complement components C4 and C3 contain a unique internal thioester bond, which upon activation, transfers covalently to a free amino or hydroxyl group on the target (Walport, 1969). The CH1 domain of IgG has particularly good acceptors for this transfer. For example, when complement is activated by IgG on pneumococcus, much of C3b attaches to the IgG rather than the bacteria itself (Brown et al., 1983). Therefore, we hypothesized that high concentrations of soluble IgG might compete with surface-bound IgG for newly activated C4b and C3b. At concentrations readily achieved during therapy, IVIG caused dose-dependent inhibition of 125I-C3b uptake onto antibody-coated erythrocytes, and inhibited complement-mediated lysis (Fig. 4) (Berger et al., 1985). This phenomenon, in which soluble “irrelevant” IgG competes with bound IgG on the target for attachment of C3b is termed “complement scavenging” and is believed to be important in the efficacy of IVIG in complement-mediated diseases. Therefore, this mechanism may be especially important in Guillain-Barré syndrome (GBS), since complement activation likely plays a crucial role in its pathogenesis (see section on GBS below). The 2009 study by Kuitwaard et al., which clearly illustrates a correlation between the increments in the serum IgG level achieved after IVIG treatment and the clinical outcome in GBS patients (Fig. 5), is likely an important in vivo example of this phenomenon.
Figure 4.
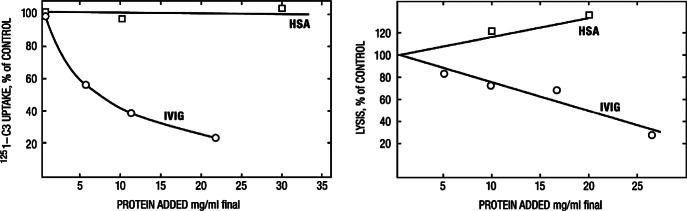
Dose-dependent inhibition by intravenous immunoglobulin (IVIG) of uptake of C3b onto sensitized sheep erythrocytes (left) and also of lysis of the targets (right). Human serum albumin (control) has no effect. Note that a protein concentration in this system of 20 mg/ml is the equivalent of a serum IgG concentration of 2,000 mg/dl, easily achieved during IVIG therapy. From Berger et al. (1985); reproduced with permission from the publisher.
Figure 5.
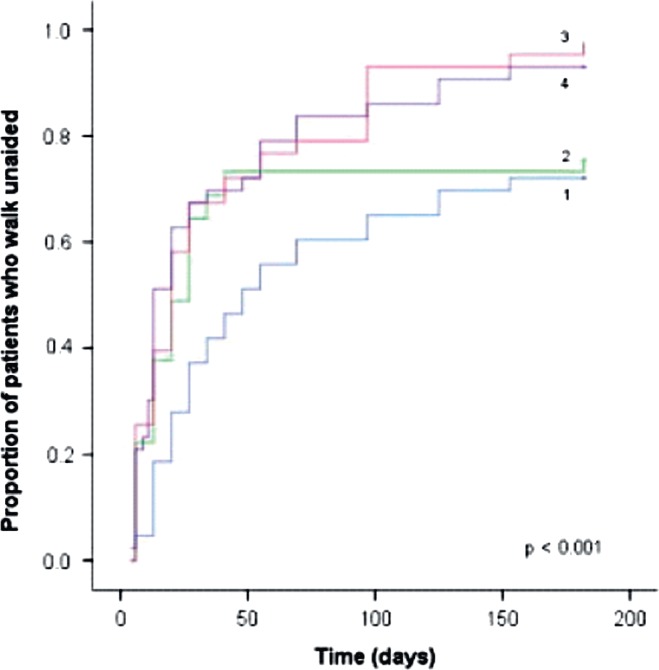
Correlation of clinical outcome with increment in serum IgG after treatment in GBS. Proportion of patients who regained the ability to walk unaided in quartiles based on increase in serum immunoglobulin IgG 2 weeks after treatment with a standard high dose of intravenous immunoglobulin (IVIG). The Kaplan-Meier curves show the cumulative fractions of patients walking unaided along time grouped according to the quartiles (1–4) of increase in serum IgG. Cutoff values of ΔIgG for quartile 1: 3.99 g/l (n = 43); quartile 2: 3.99–7.30 g/l (n = 45); quartile 3: 7.31–10.92 g/l (n = 43); and quartile 4:10.92 g/l (n = 43). p Value is based on the log-rank test for trend. From Kuitwaard et al. (2009); reproduced with permission from the publisher.
Inhibition of C4b and C3b binding also decreases amplification by the complement cascade, decreasing activation of C5 and deposition of the membrane attack complex (MAC). This accounts for the decreased hemolysis of the antibody-coated erythrocytes in Fig. 4 (Berger et al., 1985). Inhibition of formation of the amplification convertases and of C5 cleavage should also reduce generation of the potent chemoattractant C5a and thus decrease infiltration of inflammatory leukocytes. Clinical improvement following IVIG treatment in dermatomyositis has been reported to be accompanied by decreased activation of C3 and decreased deposition of C3b and C5-9 on endomysial capillaries (Basta and Dalakas, 1994; Quick and Tandan, 2011). Basta et al. (2003) also showed that IgG could bind C3a and C5a non-covalently, thereby diminishing their pro-inflammatory effects.
Other Actions of IVIG that Do Not Involve Competition Per Se
Most recent reviews of mechanisms of action of IVIG focus on putative immunomodulatory effects involving networks of T-cells, B-cells, and cytokines (Ballow, 2011; Schwab and Nimmerjahn, 2013). Modulation of macrophages or self-reactive T-cells is more likely to be important when these cells, rather than autoantibodies themselves, are directly responsible for the end-organ pathology. Furthermore, many of these immunomodulatory effects of IVIG have prolonged time courses, and are therefore not likely to be responsible for therapeutic effects that “wear-off” before one half-life of IVIG (Ballow, 2011; Gelfand, 2012; Schwab and Nimmerjahn, 2013). Conversely, highly concentration-dependent and/or short-lived PD effects suggest that the administered IgG is competing with autoantibodies which are directly responsible for the pathology. Symptomatic improvement following IVIG in CIDP, multifocal motor neuropathy (MMN), and MG is transient, and the IVIG must be administered repeatedly at intervals ≤3–4 weeks to maintain its clinical effect (Ruegg et al., 2004; Harbo et al., 2009b; Lin et al., 2011; Pollard and Armati, 2011). Recent reports of individualized regimens show that 28%–60% of CIDP patients require IVIG every 2 weeks or even more frequently for optimal maintenance of strength (Broyles et al., 2013; Kokubun et al., 2013; Kuitwaard et al., 2013; Rajabally et al., 2013). In contrast to transient relief of symptoms, true remission often requires immunomodulators other than IVIG, as shown in Fig. 6A (Pollard and Armati, 2011). Different mechanisms of action of steroids vs. IVIG are also suggested by recent results showing that a higher proportion of CIDP patients treated with high dose methylprednisolone maintained long-term remission than those treated with IVIG (Nobile-Orazio et al., 2012).
Figure 6.

(A) Cyclic gain in foot strength, and wear-off effect, in response to monthly intravenous immunoglobulin (IVIG) in a patient with chronic inflammatory demyelinating polyneuropathy (CIDP), and change in baseline after addition of prednisone and azathioprine at day 79. From Pollard and Armati (2011); reproduced with permission from the publisher. (B) Motor excitability recordings pre-IVIg treatment (open), 1-week (filled), and 2-weeks post-IVIG treatment (hatched) in a single representative patient in strength-duration time-constant (SDTC). (C) Changes in threshold electrotonus waveforms pre (open dots), 1 week post (filled dots) and 2 weeks post (grey dots). (D) SDTC changes before and after individual IVIG infusions longitudinally over 22 months in a single patient. Fig. 6B–D from Lin et al. (2011); reproduced with permission from the publisher.
IVIG can interfere with maturation of dendritic cells in vitro and can inhibit expression of HLA-antigen complexes and co-stimulatory molecules (Bayry et al., 2003a; 2003b; Ballow, 2011). Although IVIG has been reported to decrease the number of antigen-presenting dendritic cells in the CSF of patients with GBS and CIDP, the pathophysiologic role of these cells is not clear, because they do not prominently infiltrate peripheral nerves (Press et al., 2005). Decreasing dendritic cell activity could conceivably limit the duration of GBS, but this seems unlikely to explain the rapid and reversible effects of IVIG in CIDP (Fig. 6A) (Pollard and Armati, 2011).
IVIG can decrease production of the pro-inflammatory cytokines IL-1, IL-12, and IFN-γ; and increase production of regulatory molecules including IL-10 and IL-1 RA (Gupta et al., 2001; Kessel et al., 2007; Ballow, 2011). Altering these cytokines can affect the balance between regulatory (CD25+) and effector (CD8+) T-cells (Kessel et al., 2007), and decrease the inflammatory activity of macrophages. However, the pathology of CIDP, GBS, and MG more prominently involves direct effects of antibodies and/or complement than macrophages or T-cells. It is not clear how modulating T-cells or macrophages could produce the rapid onset and short duration of beneficial effects in several studies of these conditions. One report showed that IVIG decreased the B-cell activating cytokine BAFF, which was elevated in CIDP patients, but the clinical correlation was poor (Bick et al., 2013). Neither the role of BAFF in autoantibody production in vivo, nor the time course of the putative response to decreasing BAFF was described. A recent report that IVIG decreases the number and activity of natural killer cells in CIDP patients more likely represents in vitro blockade of CD16 by immune complexes than genuine physiologic downregulation (Bohn et al., 2011). Furthermore, a role for NK cells in CIDP pathology has not been established.
There has recently been intense interest in the hypothesis that enhanced expression of an inhibitory receptor, FcRIIB, is induced by a small subset of molecules in IVIG. IgG molecules which are fully sialylated are hypothesized to indirectly increase FcRIIB expression, thereby inhibiting inflammatory activity of macrophages (Anthony et al., 2012). This is proposed to account for the need for high dose IVIG to achieve anti-inflammatory effects, since only a small percentage of its molecules are fully sialylated. Although a mouse model showed that 33 mg/kg of a highly sialylated Fc analog could replicate the effects of 1 g/kg of standard IVIG (Kaveri et al., 2008), these results appear to be very strain-specific, with B6 mice requiring 2–2.5 g of IVIG/kg to achieve the same increase in platelet counts in a model of ITP as BALB/c mice given 0.5–1 g/kg (Leontyev et al., 2012a). Thus, the relevance of these mouse studies to diverse human patients is not clear. In a different mouse model of ITP, sialic acid-enriched vs. sialic acid-depleted IVIG preparations did not dramatically differ in efficacy (Leontyev et al., 2012b). Furthermore, “knocking out” FcRIIB in mice does not abrogate the effects of IVIG on ITP (Leontyev et al., 2012a), and knocking out FcRIIB does not cause more severe experimental MG in mice immunized with AChR (Li et al., 2008). Thus, the significance of recent reports of decreased FcRIIB on circulating B lymphocytes in CIDP patients (Tackenberg et al., 2009; Nimmerjahn and Lunemann, 2011), do not shed much light on the role of this receptor in human CIDP, nor how it interacts with IVIG’s effects.
Mechanisms by Which Autoantibodies Can Induce Pathology
To delineate which mechanism(s) of IVIG is/are most important in any given disease, we should first understand as much as possible about its pathogenesis. Therefore, we will review the mechanisms by which autoantibodies induce pathology before we consider how IVIG ameliorates their effects.
Direct effects of Ig binding
Binding of antibody to a self-antigen can have many different consequences, which are not mutually exclusive. First, a bound IgG or IgM molecule may block the interaction of an important cell surface molecule such as a receptor with its ligand or other signaling molecules. Receptor blockade by antibodies is well-recognized in type B insulin resistance (Rodriguez et al., 1992), MG (Drachman et al., 1982) and dysautonomias (Klein et al., 2003). Antibody binding can also cause conformational changes which enhance receptor activity. For example, autoantibodies cause hyperthyroidism in Graves’ disease by binding to and activating the thyroid stimulating hormone receptor (TSHR), and TSHR-antibody complexes closely mimic TSHR-ligand crystals (Nunez Miguel et al., 2009). Receptor-activating autoantibodies are believed to contribute to several other diseases as well (Xia and Kellems, 2011).
Antibody molecules are polyvalent, and can cross-link or aggregate cell surface molecules, bringing them into patches or a “cap” in one area of the membrane (Karnovsky and Unanue, 1978). Such “patching” or “capping” often leads to internalization and removal of the receptors and other proteins from the cell surface, eliminating their function. Antibodies against gangliosides can also cause redistribution of lipids and functionally important proteins into or out of “lipid rafts” (Spiegel et al., 1984; Ueda et al., 2010), thereby disrupting the function of specialized areas of the membrane, such as nodes of Ranvier.
Indirect effects: effector mechanisms activated by the Fc region of Ig
Besides direct effects of IgG or IgM on membrane antigens, the effector mechanisms they recruit also frequently contribute to the pathology (Anthony et al., 2012; Schwab and Nimmerjahn, 2013). When two or more IgG1 or IgG3 molecules bind near each other, or multiple IgM binding sites are engaged, C1q binds and the classical pathway of complement will be activated, culminating in formation of the C5b-9 MAC (Walport, 1969). The MAC is a large, non-selective, protein-lined pore in the cell membrane which allows bidirectional flow of water, ions, and other solutes. A single MAC can lyse an erythrocyte (Walport, 1969), but nucleated cells can protect themselves by shedding or internalizing membrane vesicles (Moskovich and Fishelson, 2007). Although removal of bits of membrane may allow the cell to survive, key receptors and/or other important proteins may be lost, diminishing the cell’s ability to respond to stimuli. This is believed to account for the disappearance of AChRs from the post-synaptic membranes and their so-called “simplification” in MG (Vincent et al., 2006). The presence of a non-selective pore in a cell membrane or axon compromises the electrical potential and ion gradients which are necessary for impulse conduction. Ca2+ influx through MAC pores can activate the intracellular protease calpain, which can then cleave intracellular tails of ion channels and other proteins, disrupting organization of specialized membrane domains such as the node of Ranvier or motor terminal (McGonigal et al., 2010). This is believed to be important in acute motor axonal neuropathy (AMAN), and may be important in other forms of GBS and in CIDP. Plomp and Willison have emphasized that anti-ganglioside antibodies together with complement can have pathological effects at neuromuscular junctions (NMJs) which mimic “demyelination” (Plomp and Willison, 2009).
Interactions of IgG with Fcγ receptors on myeloid cells can activate phagocytosis and cell-mediated cytotoxicity (Anthony et al., 2012; Schwab and Nimmerjahn, 2013). van Sorge et al., (2003) demonstrated that anti-GM1 antibodies from GBS patients induced phagocytosis of GM1-coated beads and leukocyte degranulation. However, the importance of leukocytes, as opposed to complement, in the pathology of GBS in vivo is not clear. Microglia also express FcR, but their function on the microglia is not known (Ulvestad et al., 1994). Inflammatory cells bound to targets by the Fc of IgG can release proteases and highly reactive oxygen radicals. In addition, FcR engagement frequently induces production of pro-inflammatory cytokines and chemoattractants. When autoantibodies recruit inflammatory cells as their principal effectors, the target organ is generally surrounded by or heavily infiltrated with those cells, but that is not a major feature of the diseases discussed here.
Autoantibodies Can Induce Axonal Dysfunction Without Structural Demyelination Per Se
The clinical presentation and course of antibody-mediated peripheral neuropathies likely represent the sum of multiple different effects which may differ in importance as the condition progresses. In MG for example, antibodies alone can block AChR, then subsequent complement damage can lead to loss of post-synaptic folds. In neuropathies, attack on Schwann cells or myelin may spare the axon itself. On the other hand, axon dysfunction can occur with or without significant structural damage, and without demyelination or degeneration per se. Autoantibodies can cause acute ion channel dysfunction, impairing signal propagation and contributing significantly to symptom severity, without structural damage. IgG antibodies with different Fc domains (i.e., different subclasses) have different clinical effects. For example, IgG subclasses 2 and 4 do not strongly activate complement, but may block a functionally important channel without actually inducing membrane damage. On the other hand, IgM and IgG1 and IgG3, which do activate complement, can also initiate inflammation and C5-9 attack on the axolemma, eventually causing it to drop out completely. Delineation of demyelination and remyelination/repair vs. ion channel dysfunction and/or frank axonal injury may hold important keys to understanding the pathophysiology of conduction block and optimizing treatment of peripheral neuropathies.
In early kinetic studies, Saida et al. injected anti-galactocerebroside antibodies into the sciatic nerves of rats. Focal conduction block began within 30–60 min, and progressed to completion within 3–4 h, while demyelination did not begin for several hours and was not complete until 5 days later. This clearly suggested dissociation between antibody-induced conduction failure and observable demyelination (Saida et al., 1980). Spontaneous recovery and remyelination were noted beginning at days 8–9 (Saida et al., 1980). However, a few axons recovered normal latency by 3 days, suggesting a reversible effect independent of demyelination–remyelination per se. They also showed that acute conduction block was associated with a striking reduction in the normally dense staining of Na+ channels in the nodes of Ranvier, at a time when only minimal paranodal demyelination was visible. Thus, the rapid disappearance of the normal architecture in the node, rather than demyelination per se, was considered the morphologic correlate of the loss of axonal excitability (Saida et al., 1984). Yuki drew similar conclusions about axonal damage and degeneration without demyelination from serial studies of patients with AMAN, and from injecting their IgG into rat sciatic nerves (Yuki et al., 2001). GM1 and GD1a are concentrated at nodes of Ranvier, where voltage-gated Na + channels are also concentrated. Dispersing the clustered Na+ channels may be an important means by which anti-ganglioside antibodies disrupt nodal function. Some of these antibodies also activate complement, which could cause detachment of myelin beginning at the paranode. However, Saida’s results suggest that the antibody-mediated dysfunction of ion channels and of the architecture which holds them in place may actually be more important in the overall morbidity than demyelination per se. Certainly, ion channel dysfunction seems more likely to be readily reversible than demyelination per se, and is a more likely explanation for the rapid changes in excitability described by Lin et al. (2011) after vs. before IVIG treatment in vivo. Although some macrophages may be in the proximity of affected nerves, they may be acting more as scavengers of debris and detached myelin than as agents of destruction (Saida et al., 1980; Naba et al., 2000; Kiefer et al., 2001; Yuki and Hartung, 2012).
Deciphering Which Mechanism(s) Are Important In Vivo
A key link in the hypothesis that IVIG acts in direct competition with pathologic autoantibodies is the notion that the autoantibodies are directly involved in the pathophysiology. One of the most compelling criteria for a major role of antibody per se in an autoimmune disease is a response to removal of the antibodies by plasma exchange (PE). PE has been reported to be beneficial in MG, GBS (particularly the acute idiopathic demyelinating polyneuropathy [AIDP] variants), CIDP, and some CNS disorders (Cortese et al., 2011; Lehmann and Hartung, 2011). Unlike several other peripheral neuropathies, MMN does not respond to PE or corticosteroids (Donaghy et al., 1994; Carpo et al., 1998; Lehmann et al., 2008). The reasons for this and a detailed understanding of the pathophysiology of MMN remain elusive (Muley and Parry, 2012). Demonstrating that plasma and/or purified antibodies from patients cause similar symptoms or pathology in animals (i.e., passive transfer using only soluble mediators) or in ex vivo models also strongly supports a major role for antibodies as the effectors. Correlations between antibody titer and symptoms would strengthen the argument that antibodies are directly responsible for neural dysfunction, but the available assays often lack sufficient quantitative sensitivity. Furthermore, in many cases there may be a rapid response to PE even though an antibody is not detectable in vitro. Such observations suggest that an appropriate antigen and/or assay have not yet been identified, or that something other than an antibody (i.e., a soluble toxin or cytokine) has been removed. Ultimately, isolation and molecular characterization of the autoantibodies and their antigen(s) should clarify their role. Obviously, identification of antibody staining to relevant cellular targets in affected patient tissues, when obtainable, would support a role for antibody. However, failure to demonstrate IgG in situ does not rule out internalization, degradation, or binding of the autoantibodies by other proteins. No single one of these criteria is pathognomic for a role of antibodies per se, but the more that are fulfilled, the more likely it is that antibodies are responsible. A few examples of the extent to which these criteria can be applied to different conditions follow.
Myasthenia gravis
MG is one of the best characterized autoantibody-mediated diseases and is thus an important example in which PK-PD data are available and mechanisms of action have been defined. Antibodies against nicotinic AChR or muscle specific tyrosine kinase (MuSK) are detectable in more than 80% of patients with generalized disease, and in 50% of patients with ocular disease (Lindstrom et al., 1976; Vincent et al., 2006; Jacob et al., 2012). In addition to cholinesterase inhibitors, most patients require immunomodulatory therapy. PE and IVIG are considered “rapid immunomodulators,” which have quick onset, relatively short-lived effects, and are particularly useful during myasthenic crises and before thymectomy (Kim et al., 2011; Bird, 2013). Early PE studies demonstrated improvement in strength during the procedure itself in three of five patients, and within hours in the other two (Dau et al., 1977). However, the maximum effect of PE required 24 h or more. These studies lacked sham exchange controls, so placebo effects cannot be ruled out. In another early study, improvements in strength were noted within 2 days of PE in six of seven patients (Newsom-Davis et al., 1978). In both studies, the improvements in strength were associated with decreases in anti-AChR antibodies. Subsequent losses of strength, accompanied by increasing antibody titers, recurred within days in both studies.
In 1982, Drachmann et al. showed that MG patients’ antibodies blocked binding of α-bungarotoxin to the site at which acetylcholine binds to AChR in vitro at 4°C, and also that these antibodies accelerated AChR degradation at 37°C. The different temperatures allow delineation of two different mechanisms: at 4°C, direct blockade of a functionally important site by autoantibodies; vs. at 37°C, cross-linking of AChR leading to internalization and intracellular degradation. Interestingly, there was no correlation between these two different activities in the sera from 44 different patients (Drachman et al., 1982). This is likely due to a spectrum of antibodies against different determinants or subunits of the AChRs in the different patients’ sera (Lindstrom et al., 1976; Herrmann et al., 1985). Subsequently, Jahn et al. demonstrated that IgG from patients with MG caused reversible blockade of AChR channels in vitro within less than 1 min. With prolonged incubation, however, the receptor blockade became irreversible, presumably due to internalization and degradation (Jahn et al., 2000). The ability of the bivalent IgG antibodies to first block and then subsequently to cause cross-linking and internalization of AChR has been emphasized by the observation that cleaving the IgG into monovalent Fab fragments abrogated that effect (Vincent et al., 2006). Furthermore, complement activation by the antibodies contributes to receptor and membrane loss and morphological changes such as the “simplification” of post-synaptic membrane folds seen on microscopic examination (Vincent et al., 2006). Membrane loss can also account for the loss of functionally important ion channels and other proteins besides the AChRs (Vincent et al., 2006; Serra et al., 2012).
Early studies of IVIG treatment in MG used 2 g/kg over 4–5 days. Arsura et al. (1986) reported that 11 of 12 patients responded, beginning at a mean of 3.6 ± 2.7 days. Cosi et al. (1991) reported that 46% of patients responded within 6 days of beginning treatment and 70% responded by 12 days; and Edan and Landgraf (1994) reported that 7 of 10 patients showed definite responses within 7 days. Thus, rapid, if only partial, responses may be seen after a single course of IVIG, but repeated infusions are necessary to maintain the improvement.
Taken together, these observations support the hypotheses that rapidly reversible, functional effects of autoantibodies play a role in the pathogenesis of MG. Competitive binding of anti-ids in the IVIG to the patient’s autoantibodies may be one mechanism of the rapid effects of this therapy, with the response in hours reflecting the time necessary to resynthesize AChR (Newsom-Davis et al., 1978; Vincent et al., 2006). The ability of IVIG to compete for deposition of complement also seems likely to contribute to its therapeutic effects in MG, with the response time again reflecting the time necessary for repairing the membrane (Vincent et al., 2006).
Lambert-Eaton Myasthenic syndrome
Lambert-Eaton Myasthenic syndrome (LEMS) is characterized by proximal muscle weakness and increased fatigability, most often occurring as a paraneoplastic condition in patients with small cell lung cancer (Titulaer et al., 2011; Nobile-Orazio and Gallia, 2013). Antibodies to voltage-gated Ca2+ channels are found in 90% of cases and are believed to have effects on the pre-synaptic membrane analogous to the effects of anti-AChR antibodies on the post-synaptic membrane in MG. In passive transfer experiments, patients’ antibodies caused a reduction in functional Ca2+ channels as well as ultrastructural disorganization of the pre-synaptic active zones (Fukunaga et al., 1983). PE and IVIG are both beneficial. A randomized, crossover, double-blinded placebo controlled study by Bain et al. showed improvements in all measures of strength following IVIG, but there was not a strong correlation with decreased autoantibody titers. Initial attempts to show binding of IVIG to Ca2+ channel antibodies using immunoprecipitation were negative (Bain et al., 1996). Subsequently, however, more sensitive patch-clamp studies of the excitatory potentials induced by release of individual pre-synaptic vesicles clearly showed that IgG from LEMS patients caused profound, concentration-dependent, inhibition within 1 h. Co-incubation of the LEMS IgG with IVIG or monovalent Fab’ fragments reduced or completely blocked this activity (Buchwald et al., 2005). The ability of Fab’ fragments to block the pathologic effect of the LEMS IgG clearly implies anti-id neutralization. The authors concluded that “repeated administration of IVIG keeps the level of inhibitory IgG antibodies high enough to neutralize the pathogenic antibodies and prevent them from binding to … their target antigen,” although they also discussed other possible mechanisms (Buchwald et al., 2005).
Guillain-Barré Syndrome and its Variants
GBS is classically considered a “demyelinating” disease, particularly by non-neurologists, but experts often distinguish variants with predominantly axonal dysfunction/damage (AMAN) from those dominated by demyelination per se (AIDP). AIDP generally predominates, while the prevalence of AMAN varies geographically (Ho et al., 1995; Vucic et al., 2009). Although often described as monophasic and self-limited with a good prognosis, 5%–10% of GBS patients succumb and as many as 20% of survivors have long-term disability (Hughes et al., 2007; Yuki and Hartung, 2012). Thus, there is still room for improvement in recognition and optimal treatment of GBS.
The importance of autoantibodies and the classical pathway of complement in GBS were established in the mid-1980s by studies reporting antibodies to gangliosides in patients’ sera (Ilyas et al., 1988), IgG and C1 bound to peripheral nerve myelin, and soluble C5b-9 complexes in serum (Koski et al., 1986; 1987; van Doorn et al., 1987). Some studies report autoantibodies to gangliosides and/or glycolipids in as many as 60% of AMAN and Miller-Fisher syndrome (MFS) patients (Ariga and Yu, 2005; Chavada and Willison, 2012). Although acute-phase sera patients’ antibodies induced demyelination in animals (Koski et al., 1986), and the titers fell as the patients recovered (Vriesendorp et al., 1991), pathogenic autoantibodies in AIDP variants are not as well defined as in AMAN and acute motor and sensory axonal neuropathy (AMSAN) variants (Kuwabara et al., 1998; Lehmann and Hartung, 2011; Chavada and Willison, 2012). It seems likely that autoantibodies which target Schwann cell antigens are more likely to result in features of AIDP and those which target the axolemma itself more likely cause the AMAN pattern. Nodes of Ranvier, where axonal antigens are exposed but may be surrounded by and held in place by Schwann cell antigens such as the adhesion molecule gliomedin, may be disrupted by either type of antibody, possibly explaining overlapping clinical features (Devaux, 2012; Devaux et al., 2012; Yuki, 2012; Yuki and Hartung, 2012). In vitro studies of antibodies alone vs. antibodies plus complement suggest that functional effects on conduction as well as cytotoxic effects are strongly dependent on complement, with relatively little direct effect of anti-ganglioside and/or other antibodies in the absence of complement (for particularly good examples, see Jacobs et al., 2003; Zhang et al., 2004, respectively).
GBS is considered the archetypical example of the molecular mimicry theory of autoimmune disease, because the carbohydrate moieties of gangliosides such as GM1 are found both in the lipooligosaccharide (LOS) of C. jejuni and in human peripheral nerves. Most experts now consider GBS a spectrum of diseases whose predominant clinical features are determined by the specificities of the autoantibodies produced by particular patients in response to different specific pathogens (Arcila-Londono and Lewis, 2012; Yuki, 2012; Yuki and Hartung, 2012). The role of antecedent C. jejuni infection was postulated in the early 1980s based on epidemiologic and serologic studies (Rhodes and Tattersfield, 1982; Kaldor and Speed, 1984). The association was further strengthened by Yuki et al. (1990) and Rees et al. (1995a; 1995b). A recent survey from France showed that 43.8% of GBS patients had serologic evidence of recent infection: 24.6% with C. jejuni; 12.4% with cytomegalovirus; 3.2% with mycoplasma and 1.3% with Epstein-Barr virus (Caudie et al., 2011). Of those with serologic evidence of recent infection, 61% had anti-ganglioside antibodies (Caudie et al., 2011). MFS patients also frequently recount infection with C. jejuni or Haemophilus influenzae (Koga et al., 2005; Yuki and Hartung, 2012). Differences in incidence and clinical presentations in different regions of the world may be due to infection with pathogens that bear LOS with different structures, leading to formation of different spectrums of autoantibodies (Arcila-Londono and Lewis, 2012; Yuki, 2012).
An important role of carbohydrate antigens in GBS was confirmed by inducing axonal injury in animals with sera from AMAN patients which contained antibody to GD1a (Goodfellow et al., 2005). Furthermore, attempted therapeutic administration of bovine ganglioside mixtures to humans induced formation of anti-ganglioside antibodies and neuropathic symptoms (Yuki, 2012). Subsequently, these gangliosides, which were withdrawn from human use, were shown to induce anti-ganglioside antibodies and an AMAN-like syndrome in rabbits (Yuki et al., 2001; Yuki, 2012). In animal models, anti-GM1 and anti-GD1a can induce complement-mediated disruption of nodal architecture, resulting in conduction block (Susuki et al., 2012). Antibodies to GM1 and GD1a have been identified in AMAN patients and are more prevalent in those with antecedent diarrhea (Capasso et al., 2003; Sekiguchi et al., 2012). Kuwabara et al. (1998) found statistically significant correlations between anti-GM1 titer and electrophysiologic diagnoses in GBS. In GM1-antibody positive patients, conduction block resolved rapidly as the antibody titers fell. Recovery was accompanied by rapid increases in amplitude of distal compound muscle action potentials, rather than prolonged duration or polyphasic action potentials, which would be more typical of remyelination per se. They therefore concluded that anti-GM1 antibodies caused reversible physiologic impairment of nodal function by effects on the axolemma. Ito et al. reported that 65% of acute ataxic GBS patients had anti-GQ1b (Ito et al., 2011). Of 31 patients with C. jejuni infection preceding pharyngeal-cervical-brachial weakness, 51% had anti-GT1a and 39% had anti-GQ1b (Nagashima et al., 2007). Anti-GQ1b antibodies were reported in 68% of patients with Bickerstaff’s brainstem encephalitis (Shahrizaila and Yuki, 2013) and anti-GQ1b antibodies which bind to human oculomotor nerves are prevalent in MFS patients with ataxia, areflexia, and ophthalmoplegia (Chiba et al., 1993). Thus, the different clinical presentations likely reflect the exact distribution of gangliosides in different nerves and the spectrum of anti-ganglioside antibodies produced by any particular patient. A recent study using sensitive assays with tissue culture cells transfected with individual nodal proteins reported that IgG from ≈25% of patients with CIDP and the AMAN and AIDP forms of GBS bound to nodal proteins such as neurofascin (NF186), gliomedin, and/or neuronal cell adhesion molecule (Devaux et al., 2012). Interestingly, 25% of AMAN patients’ sera contained antibodies to GM1 or GD1b as well as nodal proteins (Devaux et al., 2012). Some of these additional antibodies may be formed against antigens released following initial nerve injury by a narrower spectrum of self-reactive antibodies (i.e., against the gangliosides alone), but further work is necessary to test that possibility.
PE was first reported to be effective in GBS in 1978 (Brettle et al., 1978) and was well established by progressively larger case series through the 1980s (Zerbi et al., 1981; Tharakan et al., 1989), and a large randomized controlled trial (RCT) at Johns Hopkins (McKhann et al., 1988). A French RCT showed that at least two PEs of 50 ml/kg were effective, but four or five treatments were used in subsequent studies (French Cooperative Group on Plasma Exchange in Guillain-Barre Syndrome, 1987; 1997). Yuki et al. (1998) reported that a single exchange of 40 ml of plasma/kg reduced IgG anti-ganglioside antibody titers fourfold. A further twofold reduction could be achieved by a second exchange, but subsequent exchanges yielded little further reduction (Yuki et al., 1998). PE is now established as a first-line treatment for GBS (Cortese et al., 2011), strongly suggesting that decreasing the titer of autoantibodies is beneficial. IVIG became available in the early to mid-1980s and reports of its use in GBS soon followed. A randomized, multicenter study of 150 GBS patients in Holland showed that IVIG was more often successful than PE and led to faster recovery amongst responders (van der Meche and Schmitz, 1988). Since then, multiple studies have shown that IVIG is at least as effective as PE, often with fewer adverse effects (Cortese et al., 2011; Hughes et al., 2012; Patwa et al., 2012; Vriesendorp, 2013).
The similarity in results with IVIG or PE is consistent with the hypothesis that both act by reducing the amounts of pathologic autoantibodies. It seems clear that at least one mechanism of action of PE is removing anti-ganglioside antibodies. Several studies suggest that anti-id blocking is a major mechanism by which the IVIG reduces autoantibodies. van Doorn et al. demonstrated that antibodies in the sera of GBS patients bound to neuroblastoma cells, and that this binding could be blocked by F(ab’)2 of IVIG, or F(ab’)2 of IgG from patients who had recovered (van Doorn et al., 1988; 1990; Lundkvist et al., 1993). Subsequently, IVIG and its F(ab’)2 fragments were reported to block binding of GBS patients’ antibodies to GM1 in an ELISA (Malik et al., 1996; Yuki and Miyagi, 1996). Lopez et al. showed that F(ab’)2 fragments of normal IgG inhibited binding of GBS patients’ IgG to GM1 in a dose-dependent fashion. The normal F(ab’)2 fragments could not bind GM1 themselves, suggesting that they block antigen-binding by the pathologic antibodies (Lopez et al., 2000). Jacobs et al. (2003) reported that IVIG caused dose-dependent inhibition of binding of patient’s antibodies to GQ1b in an ELISA, using sera from 12 patients with ophthalmoplegia due to GBS or MFS. They also went a step further, using an ex vivo mouse diaphragm NMJ model (Jacobs et al., 2002). The patients’ sera had complement-dependent “α-latrotoxin-like effects” on the NMJs, including increased miniature endplate potential frequency and visible twitching. This was prevented if IVIG was present during the incubation of the patients’ sera with the NMJ preparation; but not if the NMJs were pre-incubated with IVIG before the patients’ sera. If the IVIG was added after the patient’s IgG, it did not inhibit the activity of the patients’ antibodies. This suggests the IVIG was blocking binding of the patient’s antibodies to the antigen, not the subsequent complement activation. Of course, since it is the pathologic antibody which initiates the complement activation, inhibiting its binding will decrease the complement-mediated damage as well. Importantly, IVIG was effective in vitro at concentrations readily achievable during high dose therapy: 5–25 mg/ml. Finally, it was shown that patient sera obtained 2 and 4 weeks after IVIG treatment in vivo lost the α-latrotoxin-like effects which were present before treatment. Zhang et al. (2004) showed that anti-GD1a antibodies in GBS sera are particularly effective inducers of complement-mediated cytotoxicity of neuronal cell lines and that IVIG inhibits this by blocking both the antibody binding and complement deposition stages. van Doorn’s observation that F(ab’)2 from convalescent sera blocked the binding of acute-phase GBS IgG to neuroblastoma cells (van Doorn et al., 1990) supports the hypothesis that an anti-id response is important in limiting the cross-reacting anti-self antibody production in vivo.
These observations which support anti-id binding do not exclude competition of IVIG for complement and/or its ability to increase catabolism of the pathologic IgG by saturating FcRn. Better understanding of the contribution of each mechanism should allow improvements in therapy. The PK of IVIG in GBS patients varies widely, but the report by Kuitwaard et al. (2009) that greater increases in serum IgG concentrations 2 weeks following IVIG treatment correlated with better outcomes certainly supports a competitive mechanism of action. Press et al. (2001) reported that in a majority of GBS patients, initially elevated titers of IgG anti-GM1 and/or anti-GD1a fell rapidly after IVIG treatment. Together, these observations support a model in which a monophasic antibody response to an acute but limited infection induces production of a single wave of self-reactive antibodies. IVIG and/or PE are most likely successful when they can reduce the titers of these antibodies before irreversible damage to nerves occurs. In that case, a single course of therapy may be sufficient, especially if the patient’s own anti-id response and/or other regulatory mechanisms “kick-in” in time. However, if the titers and/or affinities of the autoantibodies exceed the capacity of PE or IVIG to remove or neutralize them, and if autoantibody production continues, the patient may have continuing pathology and significant long-term disability. Additional studies looking not only at levels of total IgG after IVIG treatment, but also concomitantly at anti-ganglioside antibody levels are likely to shed important light on the most important mechanism(s) of action of IVIG, and how treatment can be optimized to improve outcomes.
Chronic Inflammatory Demyelinating Polyneuropathy
Unlike GBS, CIDP patients do not usually recall recent infections, and no causative or triggering agent has been identified. Nevertheless, an autoimmune etiology does not seem to be in doubt. Participation of humoral as well as cellular immune mechanisms in CIDP has been postulated, but their respective contributions have not been delineated (Lehmann and Hartung, 2011). “Onion bulbs” may be seen in CIDP, and are usually attributed to repeated cycles of injury and repair, but it is important to remember that macrophages may be removing debris, rather than damaging Schwann cells or axons per se (Saida et al., 1980; Naba et al., 2000; Yuki and Hartung, 2012). One difficulty defining the immunopathogenesis of CIDP is the failure to identify a major putative antigen(s). However, sensitive assays using animal models and nodal proteins (Devaux et al., 2012), and the efficacy of PE, strongly support an autoantibody-mediated pathogenesis. Autoantibodies against gangliosides as well as proteins have been reported, but there is no agreement about any individual predominant antigen or autoantibody (Peltier and Donofrio, 2012). Nerve excitability studies (Kiernan et al., 2000) suggest that pathologic autoantibodies cause a hyperpolarized state by disrupting clustering of Na+ channels in the nodes and altering the activity of Na+/K+ ATPase (Cappelen-Smith et al., 2000; Sung et al., 2004; Lin et al., 2011). These findings have been replicated in laboratory models with anti-ganglioside IgG and complement (McGonigal et al., 2010). However, immunization of animals with gliomedin, an adhesion molecule that maintains the localization of Na+ channels within the node, also induces a CIDP-like syndrome (Devaux, 2012).
The effects of PE in CIDP may be fairly rapid, with symptomatic improvement within days (Dyck et al., 1986; Hahn et al., 1996a). A single series of PE treatments rarely leads to remission, however, and multiple courses are usually necessary (Choudhary and Hughes, 1995). This is consistent with the conclusion that PE removes autoantibodies but does not stop their production. This may be analogous to the major action of IVIG as well. IVIG and steroids are considered first-line treatments for mixed sensory and motor CIDP, and IVIG is considered first-line treatment for pure motor CIDP (Joint Task Force of the EFNS and the PNS, 2010; Van den Bergh et al., 2010). In randomized clinical trials, IVIG has been reported to be effective in 54%–76% of patients (Hahn et al., 1996b; Mendell et al., 2001; Hughes et al., 2008). Electrophysiologic results suggesting that IVIG treatment in CIDP modulates axonal excitability, stabilizes membrane potential, and promotes functional recovery also show that these effects wane within weeks (Boerio et al., 2010; Lin et al., 2011). Transient blocking of the effects of autoantibodies on axonal excitability by IVIG could explain anecdotal reports showing increasing strength in affected muscles within days after IVIG treatment but recurrent weakness after only a week or two at the plateau. This type of response is illustrated in Fig. 6A (Pollard and Armati, 2011). Nobile-Orazio and Terenghi (2005) have also emphasized the rapid but transient character of the response to IVIG in CIDP, and the prolonged need for periodic infusions. In the randomized, double-blinded “ICE” trial of IVIG in CIDP, improvements in grip strength and INCAT score correlated with improvements in compound muscle action potential measurements and were seen at the first post-IVIG determination, 16 days after treatment. Further improvement was recorded at 3 and 6 weeks, but repeated measurements were not taken before and after subsequent doses of IVIG (Bril et al., 2010; Latov et al., 2010; Vanhoutte et al., 2013). “Wearing off” of the effect of each dose of IVIG may be one reason that as many as 30% of CIDP patients received IVIG at intervals ≤15 days in a recent US survey (Broyles et al., 2013), and 27% received IVIG at intervals of ≤21 days in a UK study aimed at minimizing the dose of IVIG (Rajabally et al., 2006). Kuitwaard et al. (2013) recently reported that use of a protocol for optimizing IVIG treatment resulted in 60% of the patients receiving IVIG at intervals ≤2 weeks, and a study using frequent hand grip strength measurements to assess clinical responses in two CIDP patients found that weekly IVIG treatment and serum IgG levels >1,750 mg/dl were required to maintain optimal strength (Kokubun et al., 2013). There are several anecdotal reports of patients asking for “booster” doses of IVIG before their next monthly dose is due. Harbo et al. reported that 6 of 11 CIDP patients on individualized regimens began to lose strength within a few days when their IVIG was delayed beyond the usual interval. Conversely, they began to regain strength in ≤5 days after an IVIG dose, although a plateau was not achieved for 15 days (Harbo et al., 2009b). Further studies should compare IgG levels with strength measurements at multiple time intervals during each IVIG treatment cycle, as illustrated by Kokubun et al. (2013). Ultimately, daily measurements with hand grip dynamometers or other devices the patient can use at home should help identify the best treatment regimens. Anecdotal results suggest that weekly dosing, which results in steady-state serum IgG levels, leads to better continuous maintenance of motor performance (Kokubun et al., 2013) but larger and longer-term studies are needed. Transient responses seem more compatible with mechanisms of action of IVIG involving a competitive balance between pathologic and therapeutic IgG, or competition for complement binding, than with the time course of remyelination and demyelination. Similarly, the prolonged need for periodic treatment seems more compatible with a reversible competitive effect, as contrasted with longer-lasting effects on the underlying autoimmune response. The latter seems more likely to explain the long-term remissions achieved with steroids (Nobile-Orazio et al., 2012).
Of course, any given molecule of pathologic IgG bound to a protein or ganglioside at the node of Ranvier can not only alter nodal function, but may also cause detachment of myelin and/or recruit complement and other effector mechanisms. Repeated cycles of increased effector recruitment could eventually lead to irreversible axon damage/destruction, and to long-term disability, as shown in Fig. 6D. Viewed in that context, monthly recurrences, and potentially negative effects of lengthening the intervals between infusions (Mata et al., 2006) may portend a less than optimal long-term outcome. Should we shorten the interval between IVIG doses or strive to maintain high steady-state IgG levels to prevent this?
Although IVIG reduces symptoms in a high percentage of CIDP patients, optimal doses, and infusion intervals have yet to be clearly established, and individualization of treatment has been recommended (Joint Task Force of the EFNS and the PNS, 2005; Hughes et al., 2006). Dutch investigators and the report of Kokubun suggest that higher therapeutic IgG levels correlate with improved efficacy (van Doorn et al., 2011). Achieving and maintaining adequate increases in serum IgG may be critical for optimizing symptom improvement and minimizing long-term disability, but the necessary IgG level likely varies in different patients. An increased dosing frequency might be beneficial, and a current clinical trial is aimed at maintaining high steady-state IgG concentrations by the use of weekly subcutaneous IgG (SCIG) infusions (NTC # 01545076).
Multifocal Motor Neuropathy
Multifocal conduction blocks with eventual axon loss and weakness are characteristic of MMN (Vlam et al., 2012). The putative target antigen(s) in MMN and the possible contributions of anti-ganglioside or other autoantibodies to disease pathology are not clear. Approximately half of MMN patients have anti-GM1 antibodies, primarily of the IgM isotype, which correlate with more severe weakness, disability, and axon loss as compared with antibody-negative patients (Cats et al., 2010a). MMN sera containing IgM anti-GM1 led to complement deposition in vitro (Piepers et al., 2010; Yuki et al., 2011). Thus, analogously to GBS, autoantibody-induced complement attack on ganglioside-rich membrane domains may be important in MMN. Interestingly, while IVIG is widely considered to be effective in MMN (Cats et al., 2010b), neither PE nor prednisone is very effective (Azulay et al., 1994; Van den Berg et al., 1995; Federico et al., 2000; Leger et al., 2001). It is possible that in MMN the IVIG is mainly inhibiting complement, since IgM is a very potent activator of that system. Reports of reductions in serum C1q and C4 levels in MMN patients support the latter possibility (Piepers et al., 2010). Because of the multivalency of IgM, PE may be less effective in removing high-affinity IgM than it would be for IgG. Since IgM is not recycled by the FcRn (Yu and Lennon, 1999), and has a short half-life (Fahey and Sell, 1965), it is unlikely that IVIG promotes catabolism of IgM autoantibodies. In most situations, however, IgM is efficiently removed by PE because, unlike IgG, it is mostly intravascular. One hypothesis which could account for efficacy of IVIG but not PE is that the pathology involves the alternative pathway of complement, perhaps facilitated by abnormalities in nerve membranes, and that IVIG is acting mainly by inhibiting complement activation initiated by a small amount of high-affinity IgM. Precedents for alternative pathway-mediated pathology are provided by paroxysmal nocturnal hemoglobinuria and atypical hemolytic-uremic syndrome, both of which are effectively treated by the anti-C5 monoclonal antibody eculizumab (Holers, 2008).
Despite the lack of efficacy of PE (Piepers et al., 2010; Yuki et al., 2011), it is still possible that anti-id antibodies contribute to the efficacy of IVIG in MMN. A fascinating single case study strongly suggests that IVIG acts by a competitive mechanism. A patient with “antibody-negative” MMN, who was nevertheless responsive to IVIG, was requiring increasingly frequent infusions to maintain strength. He was then treated with the anti-B cell monoclonal antibody rituximab, which presumably reduced autoantibody production, resulting in near-doubling of the interval at which IVIG was required (Ruegg et al., 2004). IVIG has been shown to block binding of anti-ganglioside antibodies from MMN patients in an ELISA (Yuki et al., 2011), but IVIG can also have anti-id effects on specific T-cell receptors (Lacroix-Desmazes et al., 1996). As occurs in CIDP, a decrease in strength-duration time-constant for CMAP follows IVIG infusions in MMN, suggesting that decreased axonal excitability contributes to the pathology (Boerio et al., 2010). Axonal excitability improves shortly after IVIG infusions, but these effects wane in subsequent weeks, before the next infusion is due (Priori et al., 2002; Van den Berg-Vos et al., 2002; Terenghi et al., 2004). Weekly subcutaneous IgG has shown efficacy in MMN (Eftimov et al., 2009; Harbo et al., 2009a; Misbah et al., 2011). Frequent IgG dosing may better prevent reversion of axonal electrophysiology to the pre-administration state and thus maintain a continuous level of symptom relief, but this has so far been reported only anecdotally (Dacci et al., 2010).
Conclusions and Future Research Questions
IVIG is remarkably effective in a wide variety of autoimmune neuromuscular disorders, despite our incomplete understanding of its mechanisms of action or the immunopathogenesis of these disorders. Anti-ganglioside antibodies are believed to play major roles in the pathophysiology of GBS, and perhaps CIDP, but have not been as clearly implicated in MMN (Willison and Yuki, 2002; Yuki and Hartung, 2012). In contrast, protein antigens are the likely targets in MG and LEMS (Vincent et al., 2006). In MG and likely in GBS, the major effect of IVIG may be to protect the targets of the autoantibodies by competing for complement binding. PD studies in CIDP and MMN show that the effects of IVIG are transient and reversible. Thus, the therapeutic IVIG may more likely be interfering with the effector mechanisms actually producing the patients’ symptoms than by inducing more profound and longer-lasting immunomodulatory effects. This seems particularly likely when the benefits of IVIG peak and wear-off rapidly, consistent with direct competition between the therapeutic antibodies and pathologic autoantibodies which interfere with nerve function. Functional effects of autoantibodies (and complement) on the nodes of Ranvier may be important in CIDP, and perhaps MMN and GBS as well. Additional nerve excitability studies which attempt to correlate the results with total IgG as well as autoantibody levels should improve our understanding of the pathologic processes contributing to patients’ symptoms, as well as the mechanism(s) by which IVIG ameliorates them. Adopting the working hypothesis that IVIG competes with pathogenic autoantibodies could lead to optimizing therapy by determining and maintaining the level of IgG necessary to block or reduce the titers of the autoantibodies and the symptoms they induce. Individualization of IgG doses and treatment intervals is gaining increasingly wide acceptance in primary antibody deficiency diseases, and is becoming adopted in neuromuscular disorders as well (Joint Task Force of the EFNS and the PNS, 2005; Hughes et al., 2006). Recent results showing that higher serum IgG levels and shorter dosing intervals are associated with better outcomes (Kuitwaard et al., 2009; van Doorn et al., 2011; Kokubun et al., 2013) suggest that individualization of therapy may be preferable to the use of empiric “one-size-fits-all” regimens. In particular, maintaining consistently high steady-state serum IgG levels with weekly or even more frequent IgG infusions may lead to more stable symptom control and better long-term outcomes in many neuropathy patients.
References
- Anthony RM, Wermeling F, Ravetch JV. Novel roles for the IgG Fc glycan. Ann N Y Acad Sci. 2012;1253:170–180. doi: 10.1111/j.1749-6632.2011.06305.x. [DOI] [PubMed] [Google Scholar]
- Arcila-Londono X, Lewis RA. Guillain-Barre syndrome. Semin Neurol. 2012;32:179–186. doi: 10.1055/s-0032-1329196. [DOI] [PubMed] [Google Scholar]
- Ariga T, Yu RK. Antiglycolipid antibodies in Guillain-Barre syndrome and related diseases: review of clinical features and antibody specificities. J Neurosci Res. 2005;80:1–17. doi: 10.1002/jnr.20395. [DOI] [PubMed] [Google Scholar]
- Arsura EL, Bick A, Brunner NG, Namba T, Grob D. High-dose intravenous immunoglobulin in the management of myasthenia gravis. Arch Intern Med. 1986;146:1365–1368. [PubMed] [Google Scholar]
- Azulay JP, Blin O, Pouget J, Boucraut J, Bille-Turc F, Carles G, Serratrice G. Intravenous immunoglobulin treatment in patients with motor neuron syndromes associated with anti-GM1 antibodies: a double-blind, placebo-controlled study. Neurology. 1994;44:429–432. doi: 10.1212/wnl.44.3_part_1.429. [DOI] [PubMed] [Google Scholar]
- Bain PG, Motomura M, Newsom-Davis J, Misbah SA, Chapel HM, Lee ML, Vincent A, Lang B. Effects of intravenous immunoglobulin on muscle weakness and calcium-channel autoantibodies in the Lambert-Eaton myasthenic syndrome. Neurology. 1996;47:678–683. doi: 10.1212/wnl.47.3.678. [DOI] [PubMed] [Google Scholar]
- Ballow M. The IgG molecule as a biological immune response modifier: mechanisms of action of intravenous immune serum globulin in autoimmune and inflammatory disorders. J Allergy Clin Immunol. 2011;127:315–323. doi: 10.1016/j.jaci.2010.10.030. [DOI] [PubMed] [Google Scholar]
- Basta M, Dalakas MC. High-dose intravenous immunoglobulin exerts its beneficial effect in patients with dermatomyositis by blocking endomysial deposition of activated complement fragments. J Clin Invest. 1994;94:1729–1735. doi: 10.1172/JCI117520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basta M, Van Goor F, Luccioli S, Billings EM, Vortmeyer AO, Baranyi L, Szebeni J, Alving CR, Carroll MC, Berkower I, Stojilkovic SS, Metcalfe DD. F(ab)’2-mediated neutralization of C3a and C5a anaphylatoxins: a novel effector function of immunoglobulins. Nat Med. 2003;9:431–438. doi: 10.1038/nm836. [DOI] [PubMed] [Google Scholar]
- Bayry J, Lacroix-Desmazes S, Carbonneil C, Misra N, Donkova V, Pashov A, Chevailler A, Mouthon L, Weill B, Bruneval P, Kazatchkine MD, Kaveri SV. Inhibition of maturation and function of dendritic cells by intravenous immunoglobulin. Blood. 2003a;101:758–765. doi: 10.1182/blood-2002-05-1447. [DOI] [PubMed] [Google Scholar]
- Bayry J, Lacroix-Desmazes S, Delignat S, Mouthon L, Weill B, Kazatchkine MD, Kaveri SV. Intravenous immunoglobulin abrogates dendritic cell differentiation induced by interferon-alpha present in serum from patients with systemic lupus erythematosus. Arthritis Rheum. 2003b;48:3497–3502. doi: 10.1002/art.11346. [DOI] [PubMed] [Google Scholar]
- Berger M, Rosenkranz P, Brown CY. Intravenous and standard immune serum globulin preparations interfere with uptake of 125I-C3 onto sensitized erythrocytes and inhibit hemolytic complement activity. Clin Immunol Immunopathol. 1985;34:227–236. doi: 10.1016/0090-1229(85)90027-3. [DOI] [PubMed] [Google Scholar]
- Bick S, Tschernatsch M, Karg A, Fuehlhuber V, Trenczek TE, Faltermeier K, Hackstein H, Kaps M, Blaes F. Intravenous immunoglobulin inhibits BAFF production in chronic inflammatory demyelinating polyneuropathy - a new mechanism of action? J Neuroimmunol. 2013;256:84–90. doi: 10.1016/j.jneuroim.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Bird SJ. 2013. Treatment of Myasthenia gravis. Wolters Kluwer Health. Available at: http://www.uptodate.com/contents/treatment-of-myasthenia-gravis. Accessed May 13, 2013.
- Boerio D, Creange A, Hogrel JY, Gueguen A, Bertrand D, Lefaucheur JP. Nerve excitability changes after intravenous immunoglobulin infusions in multifocal motor neuropathy and chronic inflammatory demyelinating neuropathy. J Neurol Sci. 2010;292:63–71. doi: 10.1016/j.jns.2010.02.002. [DOI] [PubMed] [Google Scholar]
- Bohn AB, Nederby L, Harbo T, Skovbo A, Vorup-Jensen T, Krog J, Jakobsen J, Hokland ME. The effect of IgG levels on the number of natural killer cells and their Fc receptors in chronic inflammatory demyelinating polyradiculoneuropathy. Eur J Neurol. 2011;18:919–924. doi: 10.1111/j.1468-1331.2010.03333.x. [DOI] [PubMed] [Google Scholar]
- Bonagura VR, Marchlewski R, Cox A, Rosenthal DW. Biologic IgG level in primary immunodeficiency disease: the IgG level that protects against recurrent infection. J Allergy Clin Immunol. 2008;122:210–212. doi: 10.1016/j.jaci.2008.04.044. [DOI] [PubMed] [Google Scholar]
- Brettle RP, Gross M, Legg NJ, Lockwood M, Pallis C. Treatment of acute polyneuropathy by plasma exchange. Lancet. 1978;2:1100. doi: 10.1016/s0140-6736(78)91837-8. [DOI] [PubMed] [Google Scholar]
- Bril V, Banach M, Dalakas MC, Deng C, Donofrio P, Hanna K, Hartung HP, Hughes RA, Katzberg H, Latov N, Merkies IS, Van Doorn PA ICE Study Group. Electrophysiologic correlations with clinical outcomes in CIDP. Muscle Nerve. 2010;42:492–497. doi: 10.1002/mus.21733. [DOI] [PubMed] [Google Scholar]
- Brown EJ, Berger M, Joiner KA, Frank MM. Classical complement pathway activation by antipneumococcal antibodies leads to covalent binding of C3b to antibody molecules. Infect Immun. 1983;42:594–598. doi: 10.1128/iai.42.2.594-598.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broyles R, Rodden L, Riley P, Berger M. Variability in intravenous immunoglobulin G regimens for autoimmune neuromuscular disorders. Postgrad Med. 2013;125:65–72. doi: 10.3810/pgm.2013.03.2619. [DOI] [PubMed] [Google Scholar]
- Buchwald B, Ahangari R, Weishaupt A, Toyka KV. Presynaptic effects of immunoglobulin G from patients with Lambert-Eaton myasthenic syndrome: their neutralization by intravenous immunoglobulins. Muscle Nerve. 2005;31:487–494. doi: 10.1002/mus.20269. [DOI] [PubMed] [Google Scholar]
- Capasso M, Caporale CM, Pomilio F, Gandolfi P, Lugaresi A, Uncini A. Acute motor conduction block neuropathy Another Guillain-Barre syndrome variant. Neurology. 2003;61:617–622. doi: 10.1212/wnl.61.5.617. [DOI] [PubMed] [Google Scholar]
- Cappelen-Smith C, Kuwabara S, Lin CS, Mogyoros I, Burke D. Activity-dependent hyperpolarization and conduction block in chronic inflammatory demyelinating polyneuropathy. Ann Neurol. 2000;48:826–832. [PubMed] [Google Scholar]
- Carpo M, Cappellari A, Mora G, Pedotti R, Barbieri S, Scarlato G, Nobile-Orazio E. Deterioration of multifocal motor neuropathy after plasma exchange. Neurology. 1998;50:1480–1482. doi: 10.1212/wnl.50.5.1480. [DOI] [PubMed] [Google Scholar]
- Cats EA, Jacobs BC, Yuki N, Tio-Gillen AP, Piepers S, Franssen H, van Asseldonk JT, van den Berg LH, van der Pol WL. Multifocal motor neuropathy: association of anti-GM1 IgM antibodies with clinical features. Neurology. 2010a;75:1961–1967. doi: 10.1212/WNL.0b013e3181ff94c2. [DOI] [PubMed] [Google Scholar]
- Cats EA, van der Pol WL, Piepers S, Franssen H, Jacobs BC, van den Berg-Vos RM, Kuks JB, van Doorn PA, van Engelen BG, Verschuuren JJ, Wokke JH, Veldink JH, van den Berg LH. Correlates of outcome and response to IVIg in 88 patients with multifocal motor neuropathy. Neurology. 2010b;75:818–825. doi: 10.1212/WNL.0b013e3181f0738e. [DOI] [PubMed] [Google Scholar]
- Cavill D, Waterman SA, Gordon TP. Anti-idiotypic antibodies neutralize autoantibodies that inhibit cholinergic neurotransmission. Arthritis Rheum. 2003;48:3597–3602. doi: 10.1002/art.11343. [DOI] [PubMed] [Google Scholar]
- Caudie C, Quittard Pinon A, Taravel D, Sivadon-Tardy V, Orlikowski D, Rozenberg F, Sharshar T, Raphael JC, Gaillard JL. Preceding infections and anti-ganglioside antibody profiles assessed by a dot immunoassay in 306 French Guillain-Barre syndrome patients. J Neurol. 2011;258:1958–1964. doi: 10.1007/s00415-011-6042-9. [DOI] [PubMed] [Google Scholar]
- Chavada G, Willison HJ. Autoantibodies in immune-mediated neuropathies. Curr Opin Neurol. 2012;25:550–555. doi: 10.1097/WCO.0b013e328357a77f. [DOI] [PubMed] [Google Scholar]
- Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I. Serum anti-GQ1b IgG antibody is associated with ophthalmoplegia in Miller Fisher syndrome and Guillain-Barre syndrome: clinical and immunohistochemical studies. Neurology. 1993;43:1911–1917. doi: 10.1212/wnl.43.10.1911. [DOI] [PubMed] [Google Scholar]
- Choudhary PP, Hughes RA. Long-term treatment of chronic inflammatory demyelinating polyradiculoneuropathy with plasma exchange or intravenous immunoglobulin. QJM. 1995;88:493–502. [PubMed] [Google Scholar]
- Cortese I, Chaudhry V, So YT, Cantor F, Cornblath DR, Rae-Grant A. Evidence-based guideline update: Plasmapheresis in neurologic disorders: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2011;76:294–300. doi: 10.1212/WNL.0b013e318207b1f6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosi V, Lombardi M, Piccolo G, Erbetta A. Treatment of myasthenia gravis with high-dose intravenous immunoglobulin. Acta Neurol Scand. 1991;84:81–84. doi: 10.1111/j.1600-0404.1991.tb04912.x. [DOI] [PubMed] [Google Scholar]
- Dacci P, Riva N, Scarlato M, Andresen I, Schmidt D, Comi G, Fazio R. Subcutaneous immunoglobulin therapy for the treatment of multifocal motorneuropathy: a case report. Neurol Sci. 2010;31:829–831. doi: 10.1007/s10072-010-0352-z. [DOI] [PubMed] [Google Scholar]
- Dalakas MC, Fujii M, Li M, Lutfi B, Kyhos J, McElroy B. High-dose intravenous immune globulin for stiff-person syndrome. N Engl J Med. 2001;345:1870–1876. doi: 10.1056/NEJMoa01167. [DOI] [PubMed] [Google Scholar]
- Dau PC, Lindstrom JM, Cassel CK, Denys EH, Shev EE, Spitler LE. Plasmapheresis and immunosuppressive drug therapy in myasthenia gravis. N Engl J Med. 1977;297:1134–1140. doi: 10.1056/NEJM197711242972102. [DOI] [PubMed] [Google Scholar]
- Devaux JJ. Antibodies to gliomedin cause peripheral demyelinating neuropathy and the dismantling of the nodes of Ranvier. Am J Pathol. 2012;181:1402–1413. doi: 10.1016/j.ajpath.2012.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux JJ, Odaka M, Yuki N. Nodal proteins are target antigens in Guillain-Barre syndrome. J Peripher Nerv Syst. 2012;17:62–71. doi: 10.1111/j.1529-8027.2012.00372.x. [DOI] [PubMed] [Google Scholar]
- Dietrich G, Kazatchkine MD. Normal immunoglobulin G (IgG) for therapeutic use (intravenous Ig) contain antiidiotypic specificities against an immunodominant, disease-associated, cross-reactive idiotype of human anti-thyroglobulin autoantibodies. J Clin Invest. 1990;85:620–625. doi: 10.1172/JCI114483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaghy M, Mills KR, Boniface SJ, Simmons J, Wright I, Gregson N, Jacobs J. Pure motor demyelinating neuropathy: deterioration after steroid treatment and improvement with intravenous immunoglobulin. J Neurol Neurosurg Psychiatry. 1994;57:778–783. doi: 10.1136/jnnp.57.7.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drachman DB, Adams RN, Josifek LF, Self SG. Functional activities of autoantibodies to acetylcholine receptors and the clinical severity of myasthenia gravis. N Engl J Med. 1982;307:769–775. doi: 10.1056/NEJM198209233071301. [DOI] [PubMed] [Google Scholar]
- Dyck PJ, Daube J, O’Brien P, Pineda A, Low PA, Windebank AJ, Swanson C. Plasma exchange in chronic inflammatory demyelinating polyradiculoneuropathy. N Engl J Med. 1986;314:461–465. doi: 10.1056/NEJM198602203140801. [DOI] [PubMed] [Google Scholar]
- Edan G, Landgraf F. Experience with intravenous immunoglobulin in myasthenia gravis: a review. J Neurol Neurosurg Psychiatry. 1994;57(Suppl):55–56. doi: 10.1136/jnnp.57.suppl.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eftimov F, Vermeulen M, de Haan RJ, van den Berg LH, van Schaik IN. Subcutaneous immunoglobulin therapy for multifocal motor neuropathy. J Peripher Nerv Syst. 2009;14:93–100. doi: 10.1111/j.1529-8027.2009.00218.x. [DOI] [PubMed] [Google Scholar]
- Evans MJ, Suenaga R, Abdou NI. Detection and purification of antiidiotypic antibody against anti-DNA in intravenous immune globulin. J Clin Immunol. 1991;11:291–295. doi: 10.1007/BF00918187. [DOI] [PubMed] [Google Scholar]
- Fahey JL, Sell S. The immunoglobulins of mice. V. The metabolic (catabolic) properties of five immunoglobulin classes. J Exp Med. 1965;122:41–58. doi: 10.1084/jem.122.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federico P, Zochodne DW, Hahn AF, Brown WF, Feasby TE. Multifocal motor neuropathy improved by IVIg: randomized, double-blind, placebo-controlled study. Neurology. 2000;55:1256–1262. doi: 10.1212/wnl.55.9.1256. [DOI] [PubMed] [Google Scholar]
- French Cooperative Group on Plasma Exchange in Guillain-Barre Syndrome. Efficiency of plasma exchange in Guillain-Barre syndrome: role of replacement fluids. French Cooperative Group on Plasma Exchange in Guillain-Barre syndrome. Ann Neurol. 1987;22:753–761. doi: 10.1002/ana.410220612. [DOI] [PubMed] [Google Scholar]
- French Cooperative Group on Plasma Exchange in Guillain-Barre Syndrome. Appropriate number of plasma exchanges in Guillain-Barre syndrome. The French Cooperative Group on Plasma Exchange in Guillain-Barre Syndrome. Ann Neurol. 1997;41:298–306. doi: 10.1002/ana.410410304. [DOI] [PubMed] [Google Scholar]
- Fukunaga H, Engel AG, Lang B, Newsom-Davis J, Vincent A. Passive transfer of Lambert-Eaton myasthenic syndrome with IgG from man to mouse depletes the presynaptic membrane active zones. Proc Natl Acad Sci U S A. 1983;80:7636–7640. doi: 10.1073/pnas.80.24.7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfand EW. Intravenous immune globulin in autoimmune and inflammatory diseases. N Engl J Med. 2012;367:2015–2025. doi: 10.1056/NEJMra1009433. [DOI] [PubMed] [Google Scholar]
- Goodfellow JA, Bowes T, Sheikh K, Odaka M, Halstead SK, Humphreys PD, Wagner ER, Yuki N, Furukawa K, Plomp JJ, Willison HJ. Overexpression of GD1a ganglioside sensitizes motor nerve terminals to anti-GD1a antibody-mediated injury in a model of acute motor axonal neuropathy. J Neurosci. 2005;25:1620–1628. doi: 10.1523/JNEUROSCI.4279-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M, Noel GJ, Schaefer M, Friedman D, Bussel J, Johann-Liang R. Cytokine modulation with immune gamma-globulin in peripheral blood of normal children and its implications in Kawasaki disease treatment. J Clin Immunol. 2001;21:193–199. doi: 10.1023/a:1011039216251. [DOI] [PubMed] [Google Scholar]
- Hahn AF, Bolton CF, Pillay N, Chalk C, Benstead T, Bril V, Shumak K, Vandervoort MK, Feasby TE. Plasma-exchange therapy in chronic inflammatory demyelinating polyneuropathy. A double-blind, sham-controlled, cross-over study. Brain. 1996a;119:1055–1066. doi: 10.1093/brain/119.4.1055. [DOI] [PubMed] [Google Scholar]
- Hahn AF, Bolton CF, Zochodne D, Feasby TE. Intravenous immunoglobulin treatment in chronic inflammatory demyelinating polyneuropathy. A double-blind, placebo-controlled, cross-over study. Brain. 1996b;119:1067–1077. doi: 10.1093/brain/119.4.1067. [DOI] [PubMed] [Google Scholar]
- Hansen RJ, Balthasar JP. Pharmacokinetic/pharmacodynamic modeling of the effects of intravenous immunoglobulin on the disposition of antiplatelet antibodies in a rat model of immune thrombocytopenia. J Pharm Sci. 2003;92::1206–1215. doi: 10.1002/jps.10364. [DOI] [PubMed] [Google Scholar]
- Harbo T, Andersen H, Hess A, Hansen K, Sindrup SH, Jakobsen J. Subcutaneous versus intravenous immunoglobulin in multifocal motor neuropathy: a randomized, single-blinded cross-over trial. Eur J Neurol. 2009a;16:631–638. doi: 10.1111/j.1468-1331.2009.02568.x. [DOI] [PubMed] [Google Scholar]
- Harbo T, Andersen H, Jakobsen J. Acute motor response following a single IVIG treatment course in chronic inflammatory demyelinating polyneuropathy. Muscle Nerve. 2009b;39:439–447. doi: 10.1002/mus.21305. [DOI] [PubMed] [Google Scholar]
- Herrmann C, Jr, Lindstrom JM, Keesey JC, Mulder DG. Myasthenia gravis – current concepts. West J Med. 1985;142:797–809. [PMC free article] [PubMed] [Google Scholar]
- Ho TW, Mishu B, Li CY, Gao CY, Cornblath DR, Griffin JW, Asbury AK, Blaser MJ, McKhann GM. Guillain-Barre syndrome in northern China. Relationship to Campylobacter jejuni infection and anti-glycolipid antibodies. Brain. 1995;118:597–605. doi: 10.1093/brain/118.3.597. [DOI] [PubMed] [Google Scholar]
- Holers VM. The spectrum of complement alternative pathway-mediated diseases. Immunol Rev. 2008;223:300–316. doi: 10.1111/j.1600-065X.2008.00641.x. [DOI] [PubMed] [Google Scholar]
- Hughes RA, Bouche P, Cornblath DR, Evers E, Hadden RD, Hahn A, Illa I, Koski CL, Leger JM, Nobile-Orazio E, Pollard J, Sommer C, Van den Bergh P, van Doorn PA, van Schaik IN. European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society. Eur J Neurol. 2006;13:326–332. doi: 10.1111/j.1468-1331.2006.01278.x. [DOI] [PubMed] [Google Scholar]
- Hughes RA, Swan AV, Raphael JC, Annane D, van Koningsveld R, van Doorn PA. Immunotherapy for Guillain-Barre syndrome: a systematic review. Brain. 2007;130:2245–2257. doi: 10.1093/brain/awm004. [DOI] [PubMed] [Google Scholar]
- Hughes RA, Donofrio P, Bril V, Dalakas MC, Deng C, Hanna K, Hartung HP, Latov N, Merkies IS, van Doorn PA. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): a randomised placebo-controlled trial. Lancet Neurol. 2008;7:136–144. doi: 10.1016/S1474-4422(07)70329-0. [DOI] [PubMed] [Google Scholar]
- Hughes RA, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barre syndrome. Cochrane Database Syst Rev. 2012;7:CD002063. doi: 10.1002/14651858.CD002063.pub5. [DOI] [PubMed] [Google Scholar]
- Ilyas AA, Willison HJ, Quarles RH, Jungalwala FB, Cornblath DR, Trapp BD, Griffin DE, Griffin JW, McKhann GM. Serum antibodies to gangliosides in Guillain-Barre syndrome. Ann Neurol. 1988;23:440–447. doi: 10.1002/ana.410230503. [DOI] [PubMed] [Google Scholar]
- Imbach P, Barandun S, d’Apuzzo V, Baumgartner C, Hirt A, Morell A, Rossi E, Schöni M, Vest M, Wagner HP. High-dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood. Lancet. 1981;1:1228–1231. doi: 10.1016/s0140-6736(81)92400-4. [DOI] [PubMed] [Google Scholar]
- Bonilla FA. Pharmacokinetics of immunoglobulin administered via intravenous or subcutaneous routes. Immunol Allergy Clin North Am. 2008;28:803–819. doi: 10.1016/j.iac.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Ito M, Matsuno K, Sakumoto Y, Hirata K, Yuki N. Ataxic Guillain-Barre syndrome and acute sensory ataxic neuropathy form a continuous spectrum. J Neurol Neurosurg Psychiatry. 2011;82:294–299. doi: 10.1136/jnnp.2010.222836. [DOI] [PubMed] [Google Scholar]
- Jacob S, Viegas S, Leite MI, Webster R, Cossins J, Kennett R, Hilton-Jones D, Morgan BP, Vincent A. Presence and pathogenic relevance of antibodies to clustered acetylcholine receptor in ocular and generalized myasthenia gravis. Arch Neurol. 2012;69:994–1001. doi: 10.1001/archneurol.2012.437. [DOI] [PubMed] [Google Scholar]
- Jacobs BC, Bullens RW, O’Hanlon GM, Ang CW, Willison HJ, Plomp JJ. Detection and prevalence of alpha-latrotoxin-like effects of serum from patients with Guillain-Barre syndrome. Muscle Nerve. 2002;25:549–558. doi: 10.1002/mus.10060. [DOI] [PubMed] [Google Scholar]
- Jacobs BC, O’Hanlon GM, Bullens RW, Veitch J, Plomp JJ, Willison HJ. Immunoglobulins inhibit pathophysiological effects of anti-GQ1b-positive sera at motor nerve terminals through inhibition of antibody binding. Brain. 2003;126:2220–2234. doi: 10.1093/brain/awg235. [DOI] [PubMed] [Google Scholar]
- Jahn K, Franke C, Bufler J. Mechanism of block of nicotinic acetylcholine receptor channels by purified IgG from seropositive patients with myasthenia gravis. Neurology. 2000;54:474–479. doi: 10.1212/wnl.54.2.474. [DOI] [PubMed] [Google Scholar]
- Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society. J Peripher Nerv Syst. 2005;10:220–228. doi: 10.1111/j.1085-9489.2005.10302.x. [DOI] [PubMed] [Google Scholar]
- Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society – First Revision. J Peripher Nerv Syst. 2010;15:1–9. doi: 10.1111/j.1529-8027.2010.00245.x. [DOI] [PubMed] [Google Scholar]
- Junghans RP, Anderson CL. The protection receptor for IgG catabolism is the beta2-microglobulin-containing neonatal intestinal transport receptor. Proc Natl Acad Sci U S A. 1996;93:5512–5516. doi: 10.1073/pnas.93.11.5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaldor J, Speed BR. Guillain-Barre syndrome and Campylobacter jejuni: a serological study. Br Med J (Clin Res Ed) 1984;288:1867–1870. doi: 10.1136/bmj.288.6434.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnovsky MJ, Unanue ER. Cell surface changes in capping studied by correlated fluorescence and scanning electron microscopy. Lab Invest. 1978;39:554–564. [PubMed] [Google Scholar]
- Kaveri SV, Lacroix-Desmazes S, Bayry J. The antiinflammatory IgG. N Engl J Med. 2008;359:307–309. doi: 10.1056/NEJMcibr0803649. [DOI] [PubMed] [Google Scholar]
- Kessel A, Ammuri H, Peri R, Pavlotzky ER, Blank M, Shoenfeld Y, Toubi E. Intravenous immunoglobulin therapy affects T regulatory cells by increasing their suppressive function. J Immunol. 2007;179:5571–5575. doi: 10.4049/jimmunol.179.8.5571. [DOI] [PubMed] [Google Scholar]
- Kiefer R, Kieseier BC, Stoll G, Hartung HP. The role of macrophages in immune-mediated damage to the peripheral nervous system. Prog Neurobiol. 2001;64:109–127. doi: 10.1016/s0301-0082(00)00060-5. [DOI] [PubMed] [Google Scholar]
- Kiernan MC, Burke D, Andersen KV, Bostock H. Multiple measures of axonal excitability: a new approach in clinical testing. Muscle Nerve. 2000;23:399–409. doi: 10.1002/(sici)1097-4598(200003)23:3<399::aid-mus12>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Kim JY, Park KD, Richman DP. Treatment of myasthenia gravis based on its immunopathogenesis. J Clin Neurol. 2011;7:173–183. doi: 10.3988/jcn.2011.7.4.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein CM, Vernino S, Lennon VA, Sandroni P, Fealey RD, Benrud-Larson L, Sletten D, Low PA. The spectrum of autoimmune autonomic neuropathies. Ann Neurol. 2003;53:752–758. doi: 10.1002/ana.10556. [DOI] [PubMed] [Google Scholar]
- Koga M, Gilbert M, Li J, Koike S, Takahashi M, Furukawa K, Hirata K, Yuki N. Antecedent infections in Fisher syndrome: a common pathogenesis of molecular mimicry. Neurology. 2005;64:1605–1611. doi: 10.1212/01.WNL.0000160399.08456.7C. [DOI] [PubMed] [Google Scholar]
- Kokubun N, Sada T, Yuki N, Okabe M, Hirata K. Optimization of intravenous immunoglobulibn in chronic inflammatory demyelinating polyneuropathy evaluated by grip strength measurement. Eur Neurol. 2013;70:65–69. doi: 10.1159/000350287. [DOI] [PubMed] [Google Scholar]
- Koski CL, Gratz E, Sutherland J, Mayer RF. Clinical correlation with anti-peripheral-nerve myelin antibodies in Guillain-Barre syndrome. Ann Neurol. 1986;19:573–577. doi: 10.1002/ana.410190609. [DOI] [PubMed] [Google Scholar]
- Koski CL, Sanders ME, Swoveland PT, Lawley TJ, Shin ML, Frank MM, Joiner KA. Activation of terminal components of complement in patients with Guillain-Barre syndrome and other demyelinating neuropathies. J Clin Invest. 1987;80:1492–1497. doi: 10.1172/JCI113231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuitwaard K, de Gelder J, Tio-Gillen AP, Hop WC, van Gelder T, van Toorenenbergen AW, van Doorn PA, Jacobs BC. Pharmacokinetics of intravenous immunoglobulin and outcome in Guillain-Barre syndrome. Ann Neurol. 2009;66:597–603. doi: 10.1002/ana.21737. [DOI] [PubMed] [Google Scholar]
- Kuitwaard K, van Doorn PA, Vermeulen M. Serum IgG levels in IV immunoglobulin treated chronic inflammatory demyelinating polyneuropathy. J Neurol Neurosurg Psychiatry. 2013;84:859–861. doi: 10.1136/jnnp-2012-304670. [DOI] [PubMed] [Google Scholar]
- Kuwabara S, Yuki N, Koga M, Hattori T, Matsuura D, Miyake M, Noda M. IgG anti-GM1 antibody is associated with reversible conduction failure and axonal degeneration in Guillain-Barre syndrome. Ann Neurol. 1998;44:202–208. doi: 10.1002/ana.410440210. [DOI] [PubMed] [Google Scholar]
- Lacroix-Desmazes S, Mouthon L, Spalter SH, Kaveri S, Kazatchkine MD. Immunoglobulins and the regulation of autoimmunity through the immune network. Clin Exp Rheumatol. 1996;14(Suppl 15):S9–S15. [PubMed] [Google Scholar]
- Latov N, Deng C, Dalakas MC, Bril V, Donofrio P, Hanna K, Hartung HP, Hughes RA, Merkies IS, van Doorn PA. Timing and course of clinical response to intravenous immunoglobulin in chronic inflammatory demyelinating polyradiculoneuropathy. Arch Neurol. 2010;67:802–807. doi: 10.1001/archneurol.2010.105. [DOI] [PubMed] [Google Scholar]
- Leger JM, Chassande B, Musset L, Meininger V, Bouche P, Baumann N. Intravenous immunoglobulin therapy in multifocal motor neuropathy: a double-blind, placebo-controlled study. Brain. 2001;124:145–153. doi: 10.1093/brain/124.1.145. [DOI] [PubMed] [Google Scholar]
- Lehmann HC, Hartung HP. Plasma exchange and intravenous immunoglobulins: mechanism of action in immune-mediated neuropathies. J Neuroimmunol. 2011;231:61–69. doi: 10.1016/j.jneuroim.2010.09.015. [DOI] [PubMed] [Google Scholar]
- Lehmann HC, Hoffmann FR, Fusshoeller A, Meyer zu Hörste G, Hetzel R, Hartung HP, Schroeter M, Kieseier BC. The clinical value of therapeutic plasma exchange in multifocal motor neuropathy. J Neurol Sci. 2008;271:34–39. doi: 10.1016/j.jns.2008.02.022. [DOI] [PubMed] [Google Scholar]
- Leontyev D, Katsman Y, Branch DR. Mouse background and IVIG dosage are critical in establishing the role of inhibitory Fc g receptor for the amelioration of experimental ITP. Blood. 2012a;119:5261–5264. doi: 10.1182/blood-2012-03-415695. [DOI] [PubMed] [Google Scholar]
- Leontyev D, Katsman Y, Ma XZ, Miescher S, Kasermann F, Branch DR. Sialylation-independent mechanism involved in the amelioration of murine immune thrombocytopenia using intravenous gammaglobulin. Transfusion (Paris) 2012b;52:1799–1805. doi: 10.1111/j.1537-2995.2011.03517.x. [DOI] [PubMed] [Google Scholar]
- Li N, Zhao M, Hilario-Vargas J, Prisayanh P, Warren S, Diaz LA, Roopenian DC, Liu Z. Complete FcRn dependence for intravenous Ig therapy in autoimmune skin blistering diseases. Clin Invest. 2005;115:3440–3450. doi: 10.1172/JCI24394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Tüzün E, Wu XR, Qi HB, Allman W, Saini SS, Christadoss P. Inhibitory IgG receptor FcgammaRIIB fails to inhibit experimental autoimmune myasthenia gravis pathogenesis. J Neuroimmunol. 2008;194:44–53. doi: 10.1016/j.jneuroim.2007.11.005. [DOI] [PubMed] [Google Scholar]
- Liblau R, Gajdos P, Bustarret FA, el Habib R, Bach JF, Morel E. Intravenous gamma-globulin in myasthenia gravis: interaction with anti-acetylcholine receptor autoantibodies. J Clin Immunol. 1991;11:128–131. doi: 10.1007/BF00918680. [DOI] [PubMed] [Google Scholar]
- Lin CS, Krishnan AV, Park SB, Kiernan MC. Modulatory effects on axonal function after intravenous immunoglobulin therapy in chronic inflammatory demyelinating polyneuropathy. Arch Neurol. 2011;68:862–869. doi: 10.1001/archneurol.2011.137. [DOI] [PubMed] [Google Scholar]
- Lindstrom JM, Seybold ME, Lennon VA, Whittingham S, Duane DD. Antibody to acetylcholine receptor in myasthenia gravis. Prevalence, clinical correlates, and diagnostic value. Neurology. 1976;26:1054–1059. doi: 10.1212/wnl.26.11.1054. [DOI] [PubMed] [Google Scholar]
- Lopez PH, Irazoqui FJ, Nores GA. Normal human plasma contains antibodies that specifically block neuropathy-associated human anti-GM1 IgG-antibodies. J Neuroimmunol. 2000;105:179–183. doi: 10.1016/s0165-5728(99)00276-3. [DOI] [PubMed] [Google Scholar]
- Lucas M, Lee M, Lortan J, Lopez-Granados E, Misbah S, Chapel H. Infection outcomes in patients with common variable immunodeficiency disorders: relationship to immunoglobulin therapy over 22 years. J Allergy Clin Immunol. 2010;125:1354–1360. doi: 10.1016/j.jaci.2010.02.040. [DOI] [PubMed] [Google Scholar]
- Lundkvist I, van Doorn PA, Vermeulen M, Brand A. Spontaneous recovery from the Guillain-Barre syndrome is associated with anti-idiotypic antibodies recognizing a cross-reactive idiotype on anti-neuroblastoma cell line antibodies. Clin Immunol Immunopathol. 1993;67:192–198. doi: 10.1006/clin.1993.1064. [DOI] [PubMed] [Google Scholar]
- Malik U, Oleksowicz L, Latov N, Cardo LJ. Intravenous gamma-globulin inhibits binding of anti-GM1 to its target antigen. Ann Neurol. 1996;39:136–139. doi: 10.1002/ana.410390121. [DOI] [PubMed] [Google Scholar]
- Mata S, Borsini W, Caldini A, De Scisciolo G, Piacentini S, Taiuti R. Long-term evolution of anti-ganglioside antibody levels in patient with chronic dysimmune neuropathy under IVIg therapy. J Neuroimmunol. 2006;181:141–144. doi: 10.1016/j.jneuroim.2006.08.010. [DOI] [PubMed] [Google Scholar]
- McGonigal R, Rowan EG, Greenshields KN, Halstead SK, Humphreys PD, Rother RP, Furukawa K, Willison HJ. Anti-GD1a antibodies activate complement and calpain to injure distal motor nodes of Ranvier in mice. Brain. 2010;133:1944–1960. doi: 10.1093/brain/awq119. [DOI] [PubMed] [Google Scholar]
- McKhann GM, Griffin JW, Cornblath DR, Quaskey SA, Mellits ED. Role of therapeutic plasmapheresis in the acute Guillain-Barré syndrome. J Neuroimmunol. 1988;20:297–300. doi: 10.1016/0165-5728(88)90177-4. [DOI] [PubMed] [Google Scholar]
- Mendell JR, Barohn RJ, Freimer ML, Kissel JT, King W, Nagaraja HN, Rice R, Campbell WW, Donofrio PD, Jackson CE, Lewis RA, Shy M, Simpson DM, Parry GJ, Rivner MH, Thornton CA, Bromberg MB, Tandan R, Harati Y, Giuliani MJ Working Group on Peripheral N. Randomized controlled trial of IVIg in untreated chronic inflammatory demyelinating polyradiculoneuropathy. Neurology. 2001;56:445–449. doi: 10.1212/wnl.56.4.445. [DOI] [PubMed] [Google Scholar]
- Misbah SA, Baumann A, Fazio R, Dacci P, Schmidt DS, Burton J, Sturzenegger M. A smooth transition protocol for patients with multifocal motor neuropathy going from intravenous to subcutaneous immunoglobulin therapy: an open-label proof-of-concept study. J Peripher Nerv Syst. 2011;16:92–97. doi: 10.1111/j.1529-8027.2011.00330.x. [DOI] [PubMed] [Google Scholar]
- Moskovich O, Fishelson Z. Live cell imaging of outward and inward vesiculation induced by the complement c5b-9 complex. J Biol Chem. 2007;282:29977–29986. doi: 10.1074/jbc.M703742200. [DOI] [PubMed] [Google Scholar]
- Muley SA, Parry GJ. Multifocal motor neuropathy. J Clin Neurosci. 2012;19:1201–1209. doi: 10.1016/j.jocn.2012.02.011. [DOI] [PubMed] [Google Scholar]
- Naba I, Yoshikawa H, Sakoda S, Itabe H, Suzuki H, Kodama T, Yanagihara T. Onion-bulb formation after a single compression injury in the macrophage scavenger receptor knockout mice. Exp Neurol. 2000;166:83–89. doi: 10.1006/exnr.2000.7495. [DOI] [PubMed] [Google Scholar]
- Nagashima T, Koga M, Odaka M, Hirata K, Yuki N. Continuous spectrum of pharyngeal-cervical-brachial variant of Guillain-Barre syndrome. Arch Neurol. 2007;64:1519–1523. doi: 10.1001/archneur.64.10.1519. [DOI] [PubMed] [Google Scholar]
- Newsom-Davis J, Pinching AJ, Vincent A, Wilson SG. Function of circulating antibody to acetylcholine receptor in myasthenia gravis: investigation by plasma exchange. Neurology. 1978;28:266–272. doi: 10.1212/wnl.28.3.266. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn F, Lunemann JD. Expression and function of the inhibitory Fcgamma-receptor in CIDP. J Peripher Nerv Syst. 2011;16(Suppl 1):41–44. doi: 10.1111/j.1529-8027.2011.00305.x. [DOI] [PubMed] [Google Scholar]
- Nobile-Orazio E, Gallia F. Multifocal motor neuropathy: current therapies and novel strategies. Drugs. 2013;73:397–406. doi: 10.1007/s40265-013-0029-z. [DOI] [PubMed] [Google Scholar]
- Nobile-Orazio E, Terenghi F. IVIg in idiopathic autoimmune neuropathies: analysis in the light of the latest results. J Neurol. 2005;252(Suppl 1):I7–I13. doi: 10.1007/s00415-005-1103-6. [DOI] [PubMed] [Google Scholar]
- Nobile-Orazio E, Cocito D, Jann S, Uncini A, Beghi E, Messina P, Antonini G, Fazio R, Gallia F, Schenone A, Francia A, Pareyson D, Santoro L, Tamburin S, Macchia R, Cavaletti G, Giannini F, Sabatelli M Group IMCT. Intravenous immunoglobulin versus intravenous methylprednisolone for chronic inflammatory demyelinating polyradiculoneuropathy: a randomised controlled trial. Lancet Neurol. 2012;11:493–502. doi: 10.1016/S1474-4422(12)70093-5. [DOI] [PubMed] [Google Scholar]
- Nunez Miguel R, Sanders J, Chirgadze DY, Furmaniak J, Rees Smith B. Thyroid stimulating autoantibody M22 mimics TSH binding to the TSH receptor leucine rich domain: a comparative structural study of protein-protein interactions. J Mol Endocrinol. 2009;42:381–395. doi: 10.1677/JME-08-0152. [DOI] [PubMed] [Google Scholar]
- Oak S, Gilliam LK, Landin-Olsson M, Torn C, Kockum I, Pennington CR, Rowley MJ, Christie MR, Banga JP, Hampe CS. The lack of anti-idiotypic antibodies, not the presence of the corresponding autoantibodies to glutamate decarboxylase, defines type 1 diabetes. Proc Natl Acad Sci U S A. 2008;105:5471–5476. doi: 10.1073/pnas.0800578105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwa HS, Chaudhry V, Katzberg H, Rae-Grant AD, So YT. Evidence-based guideline: intravenous immunoglobulin in the treatment of neuromuscular disorders: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2012;78:1009–1015. doi: 10.1212/WNL.0b013e31824de293. [DOI] [PubMed] [Google Scholar]
- Peltier AC, Donofrio PD. Chronic inflammatory demyelinating polyradiculoneuropathy: from bench to bedside. Semin Neurol. 2012;32:187–195. doi: 10.1055/s-0032-1329194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piepers S, Jansen MD, Cats EA, van Sorge NM, van den Berg LH, van der Pol WL. IVIg inhibits classical pathway activity and anti-GM1 IgM-mediated complement deposition in MMN. J Neuroimmunol. 2010;229:256–262. doi: 10.1016/j.jneuroim.2010.08.023. [DOI] [PubMed] [Google Scholar]
- Pirofsky B. Intravenous immune globulin therapy in hypogammaglobulinemia. A review. Am J Med. 1984;76:53–60. doi: 10.1016/0002-9343(84)90320-6. [DOI] [PubMed] [Google Scholar]
- Plomp JJ, Willison HJ. Pathophysiological actions of neuropathy-related anti-ganglioside antibodies at the neuromuscular junction. J Physiol. 2009;587:3979–3999. doi: 10.1113/jphysiol.2009.171702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard JD, Armati PJ. CIDP - the relevance of recent advances in Schwann cell/axonal neurobiology. J Peripher Nerv Syst. 2011;16:15–23. doi: 10.1111/j.1529-8027.2011.00323.x. [DOI] [PubMed] [Google Scholar]
- Press R, Mata S, Lolli F, Zhu J, Andersson T, Link H. Temporal profile of anti-ganglioside antibodies and their relation to clinical parameters and treatment in Guillain-Barre syndrome. J Neurol Sci. 2001;190:41–47. doi: 10.1016/s0022-510x(01)00580-9. [DOI] [PubMed] [Google Scholar]
- Press R, Nennesmo I, Kouwenhoven M, Huang YM, Link H, Pashenkov M. Dendritic cells in the cerebrospinal fluid and peripheral nerves in Guillain-Barre syndrome and chronic inflammatory demyelinating polyradiculoneuropathy. J Neuroimmunol. 2005;159:165–176. doi: 10.1016/j.jneuroim.2004.09.020. [DOI] [PubMed] [Google Scholar]
- Priori A, Cinnante C, Pesenti A, Carpo M, Cappellari A, Nobile-Orazio E, Scarlato G, Barbieri S. Distinctive abnormalities of motor axonal strength-duration properties in multifocal motor neuropathy and in motor neurone disease. Brain. 2002;125:2481–2490. doi: 10.1093/brain/awf255. [DOI] [PubMed] [Google Scholar]
- Quick A, Tandan R. Mechanisms of action of intravenous immunoglobulin in inflammatory muscle disease. Curr Rheumatol Rep. 2011;13:192–198. doi: 10.1007/s11926-011-0171-0. [DOI] [PubMed] [Google Scholar]
- Rajabally YA, Jacob S, Abbott RJ. Clinical heterogeneity in mild chronic inflammatory demyelinating polyneuropathy. Eur J Neurol. 2006;13:958–962. doi: 10.1111/j.1468-1331.2006.01403.x. [DOI] [PubMed] [Google Scholar]
- Rajabally YA, Wong SL, Kearney DA. Immunoglobulin G level variations in treated chronic inflammatory demyelinating polyneuropathy: clues for future treatment regimens? J Neurol. 2013;260:2052–2056. doi: 10.1007/s00415-013-6938-7. [DOI] [PubMed] [Google Scholar]
- Rees JH, Gregson NA, Hughes RA. Anti-ganglioside GM1 antibodies in Guillain-Barre syndrome and their relationship to Campylobacter jejuni infection. Ann Neurol. 1995a;38:809–816. doi: 10.1002/ana.410380516. [DOI] [PubMed] [Google Scholar]
- Rees JH, Soudain SE, Gregson NA, Hughes RA. Campylobacter jejuni infection and Guillain-Barre syndrome. N Engl J Med. 1995b;333:1374–1379. doi: 10.1056/NEJM199511233332102. [DOI] [PubMed] [Google Scholar]
- Reinhart WH, Berchtold PE. Effect of high-dose intravenous immunoglobulin therapy on blood rheology. Lancet. 1992;339:662–664. doi: 10.1016/0140-6736(92)90806-e. [DOI] [PubMed] [Google Scholar]
- Rhodes KM, Tattersfield AE. Guillain-Barre syndrome associated with Campylobacter infection. Br Med J (Clin Res Ed) 1982;285:173–174. doi: 10.1136/bmj.285.6336.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez O, Collier E, Arakaki R, Gorden P. Characterization of purified autoantibodies to the insulin receptor from six patients with type B insulin resistance. Metabolism. 1992;41:325–331. doi: 10.1016/0026-0495(92)90279-j. [DOI] [PubMed] [Google Scholar]
- Rossi F, Sultan Y, Kazatchkine MD. Anti-idiotypes against autoantibodies and alloantibodies to VIII:C (anti-haemophilic factor) are present in therapeutic polyspecific normal immunoglobulins. Clin Exp Immunol. 1988;74:311–316. [PMC free article] [PubMed] [Google Scholar]
- Ruegg SJ, Fuhr P, Steck AJ. Rituximab stabilizes multifocal motor neuropathy increasingly less responsive to IVIg. Neurology. 2004;63:2178–2179. doi: 10.1212/01.wnl.0000145706.04340.25. [DOI] [PubMed] [Google Scholar]
- Saida K, Sumner AJ, Saida T, Brown MJ, Silberberg DH. Antiserum-mediated demyelination: relationship between remyelination and functional recovery. Ann Neurol. 1980;8:12–24. doi: 10.1002/ana.410080103. [DOI] [PubMed] [Google Scholar]
- Saida K, Saida T, Kayama H, Nishitani H. Rapid alterations of the axon membrane in antibody-mediated demyelination. Ann Neurol. 1984;15:581–589. doi: 10.1002/ana.410150611. [DOI] [PubMed] [Google Scholar]
- Schroeder HW, Jr, Cavacini L. Structure and function of immunoglobulins. J Allergy Clin Immunol. 2010;125:S41–S52. doi: 10.1016/j.jaci.2009.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol. 2013;13:176–189. doi: 10.1038/nri3401. [DOI] [PubMed] [Google Scholar]
- Sekiguchi Y, Uncini A, Yuki N, Misawa S, Notturno F, Nasu S, Kanai K, Noto Y, Fujimaki Y, Shibuya K, Ohmori S, Sato Y, Kuwabara S. Antiganglioside antibodies are associated with axonal Guillain-Barre syndrome: a Japanese-Italian collaborative study. J Neurol Neurosurg Psychiatry. 2012;83:23–28. doi: 10.1136/jnnp-2011-300309. [DOI] [PubMed] [Google Scholar]
- Serra A, Ruff RL, Leigh RJ. Neuromuscular transmission failure in myasthenia gravis: decrement of safety factor and susceptibility of extraocular muscles. Ann N Y Acad Sci. 2012;1275:129–135. doi: 10.1111/j.1749-6632.2012.06841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahrizaila N, Yuki N. Bickerstaff brainstem encephalitis and Fisher syndrome: anti-GQ1b antibody syndrome. J Neurol Neurosurg Psychiatry. 2013;84:576–583. doi: 10.1136/jnnp-2012-302824. [DOI] [PubMed] [Google Scholar]
- Shiraishi H, Motomura M, Iwanaga H, Tsujino A, Nishiura Y, Shirabe S, Nakamura T, Yoshimura T. Successful treatment in a patient with a focal form of stiff-person syndrome using plasma exchange and intravenous immunoglobulin therapy. Rinsho Shinkeigaku. 2002;42:766–770. [PubMed] [Google Scholar]
- Smith AJ, Jackson MW, Wang F, Cavill D, Rischmueller M, Gordon TP. Neutralization of muscarinic receptor autoantibodies by intravenous immunoglobulin in Sjögren syndrome. Hum Immunol. 2005;66:411–416. doi: 10.1016/j.humimm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- Spiegel S, Kassis S, Wilchek M, Fishman PH. Direct visualization of redistribution and capping of fluorescent gangliosides on lymphocytes. J Cell Biol. 1984;99:1575–1581. doi: 10.1083/jcb.99.5.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung JY, Kuwabara S, Kaji R, Ogawara K, Mori M, Kanai K, Nodera H, Hattori T, Bostock H. Threshold electrotonus in chronic inflammatory demyelinating polyneuropathy: correlation with clinical profiles. Muscle Nerve. 2004;29:28–37. doi: 10.1002/mus.10516. [DOI] [PubMed] [Google Scholar]
- Susuki K, Yuki N, Schafer DP, Hirata K, Zhang G, Funakoshi K, Rasband MN. Dysfunction of nodes of Ranvier: a mechanism for anti-ganglioside antibody-mediated neuropathies. Exp Neurol. 2012;233:534–542. doi: 10.1016/j.expneurol.2011.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tackenberg B, Jelcic I, Baerenwaldt A, Oertel WH, Sommer N, Nimmerjahn F, Lunemann JD. Impaired inhibitory Fcgamma receptor IIB expression on B cells in chronic inflammatory demyelinating polyneuropathy. Proc Natl Acad Sci U S A. 2009;106:4788–4792. doi: 10.1073/pnas.0807319106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon N, Jayne DR, McGregor AM, Weetman AP. Analysis of anti-idiotypic antibodies against anti-microsomal antibodies in patients with thyroid autoimmunity. J Autoimmun. 1992;5:557–570. doi: 10.1016/0896-8411(92)90153-h. [DOI] [PubMed] [Google Scholar]
- Terenghi F, Cappellari A, Bersano A, Carpo M, Barbieri S, Nobile-Orazio E. How long is IVIg effective in multifocal motor neuropathy? Neurology. 2004;62:666–668. doi: 10.1212/01.wnl.0000110185.23464.a1. [DOI] [PubMed] [Google Scholar]
- Tharakan J, Ferner RE, Hughes RA, Winer J, Barnett M, Brown ER, Smith G. Plasma exchange for Guillain-Barre syndrome. J R Soc Med. 1989;82:458–461. doi: 10.1177/014107688908200805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 2011;10:1098–1107. doi: 10.1016/S1474-4422(11)70245-9. [DOI] [PubMed] [Google Scholar]
- Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302:575–581. doi: 10.1038/302575a0. [DOI] [PubMed] [Google Scholar]
- Ueda A, Shima S, Miyashita T, Ito S, Ueda M, Kusunoki S, Asakura K, Mutoh T. Anti-GM1 antibodies affect the integrity of lipid rafts. Mol Cell Neurosci. 2010;45:355–362. doi: 10.1016/j.mcn.2010.07.008. [DOI] [PubMed] [Google Scholar]
- Ulvestad E, Williams K, Bjerkvig R, Tiekotter K, Antel J, Matre R. Human microglial cells have phenotypic and functional characteristics in common with both macrophages and dendritic antigen-presenting cells. J Leukoc Biol. 1994;56:732–740. doi: 10.1002/jlb.56.6.732. [DOI] [PubMed] [Google Scholar]
- Van den Berg LH, Kerkhoff H, Oey PL, Franssen H, Mollee I, Vermeulen M, Jennekens FG, Wokke JH. Treatment of multifocal motor neuropathy with high dose intravenous immunoglobulins: a double blind, placebo controlled study. J Neurol Neurosurg Psychiatry. 1995;59:248–252. doi: 10.1136/jnnp.59.3.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bergh PY, Hadden RD, Bouche P, Cornblath DR, Hahn A, Illa I, Koski CL, Leger JM, Nobile-Orazio E, Pollard J, Sommer C, van Doorn PA, van Schaik IN. European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society - first revision. Eur J Neurol. 2010;17:356–363. doi: 10.1111/j.1468-1331.2009.02930.x. [DOI] [PubMed] [Google Scholar]
- Van den Berg-Vos RM, Franssen H, Wokke JH, Van den Berg LH. Multifocal motor neuropathy: long-term clinical and electrophysiological assessment of intravenous immunoglobulin maintenance treatment. Brain. 2002;125:1875–1886. doi: 10.1093/brain/awf193. [DOI] [PubMed] [Google Scholar]
- Van der Meche FG, Schmitz PI. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barre syndrome. Dutch Guillain-Barre Study Group. N Engl J Med. 1992;326:1123–1129. doi: 10.1056/NEJM199204233261705. [DOI] [PubMed] [Google Scholar]
- Van Doorn PA, Brand A, Vermeulen M. Clinical significance of antibodies against peripheral nerve tissue in inflammatory polyneuropathy. Neurology. 1987;37:1798–1802. doi: 10.1212/wnl.37.11.1798. [DOI] [PubMed] [Google Scholar]
- Van Doorn PA, Brand A, Vermeulen M. Anti-neuroblastoma cell line antibodies in inflammatory demyelinating polyneuropathy: inhibition in vitro and in vivo by IV immunoglobulin. Neurology. 1988;38:1592–1595. doi: 10.1212/wnl.38.10.1592. [DOI] [PubMed] [Google Scholar]
- Van Doorn PA, Rossi F, Brand A, van Lint M, Vermeulen M, Kazatchkine MD. On the mechanism of high-dose intravenous immunoglobulin treatment of patients with chronic inflammatory demyelinating polyneuropathy. J Neuroimmunol. 1990;29:57–64. doi: 10.1016/0165-5728(90)90147-f. [DOI] [PubMed] [Google Scholar]
- Van Doorn PA, Kuitwaard K, Jacobs BC. Serum IgG levels as biomarkers for optimizing IVIg therapy in CIDP. J Peripher Nerv Syst. 2011;16(Suppl 1):38–40. doi: 10.1111/j.1529-8027.2011.00304.x. [DOI] [PubMed] [Google Scholar]
- Van Sorge NM, Van den Berg LH, Geleijns K, Van Strijp JA, Jacobs BC, Van Doorn PA, Wokke JH, Van de Winkel JG, Leusen JH, Van der Pol WL. Anti-GM1 IgG antibodies induce leukocyte effector functions via Fcgamma receptors. Ann Neurol. 2003;53:570–579. doi: 10.1002/ana.10503. [DOI] [PubMed] [Google Scholar]
- Vanhoutte EK, Latov N, Deng C, Hanna K, Hughes RA, Bril V, Dalakas MC, Donofrio P, van Doorn PA, Hartung HP, Merkies IS. Vigorimeter grip strength in CIDP: a responsive tool that rapidly measures the effect of IVIG – the ICE study. Eur J Neurol. 2013;20:748–755. doi: 10.1111/j.1468-1331.2012.03851.x. [DOI] [PubMed] [Google Scholar]
- Vincent A, Whiting PJ, Heidenreich F, Roberts A. Monoclonal anti-acetylcholine receptor antibodies as probes for human acetylcholine-receptor in myasthenia gravis. J Recept Res. 1988;8:143–159. doi: 10.3109/10799898809048984. [DOI] [PubMed] [Google Scholar]
- Vincent A, Lang B, Kleopa KA. Autoimmune channelopathies and related neurological disorders. Neuron. 2006;52:123–138. doi: 10.1016/j.neuron.2006.09.024. [DOI] [PubMed] [Google Scholar]
- Vlam L, van der Pol WL, Cats EA, Straver DC, Piepers S, Franssen H, van den Berg LH. Multifocal motor neuropathy: diagnosis, pathogenesis and treatment strategies. Nat Rev Neurol. 2012;8:48–58. doi: 10.1038/nrneurol.2011.175. [DOI] [PubMed] [Google Scholar]
- Vriesendorp FJ. 2013. Pathogenesis of Guillain-Barre syndrome in adults. Available at: http://www.uptodate.com/contents/pathogenesis-of-guillain-barre-syndrome-in-adults. Accessed May 13, 2013.
- Vriesendorp FJ, Mayer RF, Koski CL. Kinetics of anti-peripheral nerve myelin antibody in patients with Guillain-Barre syndrome treated and not treated with plasmapheresis. Arch Neurol. 1991;48:858–861. doi: 10.1001/archneur.1991.00530200100027. [DOI] [PubMed] [Google Scholar]
- Vucic S, Kiernan MC, Cornblath DR. Guillain-Barre syndrome: an update. J Clin Neurosci. 2009;16:733–741. doi: 10.1016/j.jocn.2008.08.033. [DOI] [PubMed] [Google Scholar]
- Waldmann TA, Strober W. Metabolism of immunoglobulins. Prog Allergy. 1969;13:1–110. doi: 10.1159/000385919. [DOI] [PubMed] [Google Scholar]
- Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- Wani MA, Haynes LD, Kim J, Bronson CL, Chaudhury C, Mohanty S, Waldmann TA, Robinson JM, Anderson CL. Familial hypercatabolic hypoproteinemia caused by deficiency of the neonatal Fc receptor, FcRn, due to a mutant beta2-microglobulin gene. Proc Natl Acad Sci USA. 2006;103:5084–5089. doi: 10.1073/pnas.0600548103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willison HJ, Yuki N. Peripheral neuropathies and anti-glycolipid antibodies. Brain. 2002;125:2591–2625. doi: 10.1093/brain/awf272. [DOI] [PubMed] [Google Scholar]
- Xia Y, Kellems RE. Receptor-activating autoantibodies and disease: preeclampsia and beyond. Expert Rev Clin Immunol. 2011;7:659–674. doi: 10.1586/eci.11.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Lennon VA. Mechanism of intravenous immune globulin therapy in antibody-mediated autoimmune diseases. N Engl J Med. 1999;340:227–228. doi: 10.1056/NEJM199901213400311. [DOI] [PubMed] [Google Scholar]
- Yuki N. Guillain-Barre syndrome and anti-ganglioside antibodies: a clinician-scientist’s journey. Proc Jpn Acad Ser B Phys Biol Sci. 88:299–326. doi: 10.2183/pjab.88.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuki N, Hartung HP. Guillain-Barre syndrome. N Engl J Med. 2012;366:2294–2304. doi: 10.1056/NEJMra1114525. [DOI] [PubMed] [Google Scholar]
- Yuki N, Miyagi F. Possible mechanism of intravenous immunoglobulin treatment on anti-GM1 antibody-mediated neuropathies. J Neurol Sci. 1996;139:160–162. [PubMed] [Google Scholar]
- Yuki N, Yoshino H, Sato S, Miyatake T. Acute axonal polyneuropathy associated with anti-GM1 antibodies following Campylobacter enteritis. Neurology. 1990;40:1900–1902. doi: 10.1212/wnl.40.12.1900. [DOI] [PubMed] [Google Scholar]
- Yuki N, Tagawa Y, Hirata K. Minimal number of plasma exchanges needed to reduce immunoglobulin in Guillain-Barre syndrome. Neurology. 1998;51:875–877. doi: 10.1212/wnl.51.3.875. [DOI] [PubMed] [Google Scholar]
- Yuki N, Yamada M, Koga M, Odaka M, Susuki K, Tagawa Y, Ueda S, Kasama T, Ohnishi A, Hayashi S, Takahashi H, Kamijo M, Hirata K. Animal model of axonal Guillain-Barre syndrome induced by sensitization with GM1 ganglioside. Ann Neurol. 2001;49:712–720. [PubMed] [Google Scholar]
- Yuki N, Watanabe H, Nakajima T, Spath PJ. IVIG blocks complement deposition mediated by anti-GM1 antibodies in multifocal motor neuropathy. J Neurol Neurosurg Psychiatry. 2011;82:87–91. doi: 10.1136/jnnp.2010.205856. [DOI] [PubMed] [Google Scholar]
- Zerbi D, Celano I, Forlani G, Garelli S, Mosconi L, Valbonesi M. Plasmapheresis in the treatment of four cases of Guillain-Barre syndrome (acute form) Ital J Neurol Sci. 1981;2:331–336. [PubMed] [Google Scholar]
- Zhang G, Lopez PH, Li CY, Mehta NR, Griffin JW, Schnaar RL, Sheikh KA. Anti-ganglioside antibody-mediated neuronal cytotoxicity and its protection by intravenous immunoglobulin: implications for immune neuropathies. Brain. 2004;127:1085–1100. doi: 10.1093/brain/awh127. [DOI] [PubMed] [Google Scholar]