

Abstract
Background
Increased hypercoagulability has been reported with low doses of direct thrombin inhibitors but not with direct factor Xa inhibitors.
Objectives
To compare the effects of rivaroxaban with those of melagatran and dabigatran on thrombin generation (TG) and tissue factor-induced hypercoagulability and to explore the possible involvement of the thrombin–thrombomodulin/activated protein C system.
Methods
In normal human plasma and in protein C-deficient plasma, TG was investigated in vitro in the presence and absence of recombinant human soluble thrombomodulin (rhs-TM). TG was determined by calibrated automated thrombography and an ELISA for prothrombin fragments 1+2 (F1+2). In an in vivo rat model, hypercoagulability was induced by tissue factor; levels of thrombin–antithrombin (TAT) and fibrinogen and the platelet count were determined.
Results
Rivaroxaban inhibited TG in a concentration-dependent manner. In the absence of rhs-TM, melagatran and dabigatran also inhibited TG concentration dependently. However, in the presence of rhs-TM, lower concentrations of melagatran (119–474 nmol L–1) and dabigatran (68–545 nmol L−1) enhanced endogenous thrombin potential, peak TG, and F1+2 formation in normal plasma but not in protein C-deficient plasma. In vivo, rivaroxaban dose-dependently inhibited TAT generation, whereas melagatran showed a paradoxical effect, with an increase in TAT and a small decrease in fibrinogen and platelet count at lower doses.
Conclusion
Low concentrations of the direct thrombin inhibitors melagatran and dabigatran enhanced TG and hypercoagulability, possibly via inhibition of the protein C system. In contrast, rivaroxaban reduced TG and hypercoagulability under all conditions studied, suggesting that it does not suppress this negative-feedback system.
Keywords: dabigatran, hypercoagulability, melagatran, rivaroxaban, thrombin
Introduction
Factor Xa (FXa) and thrombin are favorable targets for anticoagulant therapy, and the development of target-specific oral anticoagulants that inhibit either FXa (rivaroxaban, apixaban, edoxaban) or thrombin (dabigatran, melagatran) is rapidly changing clinical practice. The efficacy of these oral anticoagulants has been demonstrated in a variety of clinical studies for the prevention and treatment of thromboembolic disorders 1.
There have been concerns about a trend toward an increase in the incidence of acute coronary events such as myocardial infarction (MI) observed in some trials with the direct thrombin inhibitors (DTIs) ximelagatran and dabigatran. However, in all of these trials, rates of MI/acute coronary syndrome (ACS) were low across treatment groups 2–8, and further analysis of data from the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY) study of dabigatran showed that the increase in the occurrence of MI seen with dabigatran did not reach statistical significance 4.
There is currently no mechanistic explanation for the trend toward an increase in MI/ACS events. One possible explanation is that warfarin may be more efficacious than dabigatran for the prevention of ACS 9, but impairment of the activated protein C system might also be a contributing factor. The phenomenon of a paradoxical activation of coagulation has been observed with low doses of the DTI melagatran in preclinical studies 10–12. In a rat model of disseminated intravascular coagulation (DIC), melagatran aggravated DIC by increasing thrombin generation (TG) at low doses, whereas the FXa inhibitors DX-9065a and edoxaban showed dose-dependent protection against DIC 10,12. In vitro, in human plasma, melagatran increased TG at low concentrations when protein C or thrombomodulin (TM) was present, whereas the FXa inhibitors DX-9065a and edoxaban decreased TG 10. Results from this in vitro study suggest that a DTI such as melagatran may inhibit the activation of protein C through the inhibition of thrombin activity, which may explain the observed increase in TG in the rat model 11,13,14.
Most of the thrombin generated after the activation of coagulation binds to TM present on the surface of endothelial cells, where it rapidly activates protein C. This process is enhanced by the binding of protein C to the endothelial cell protein C receptor (EPCR). When activated, protein C dissociates from EPCR and binds to protein S. The activated protein C–protein S complex then inactivates FVa and FVIIIa, further limiting TG 15. A defect in this feedback pathway has been reported to be associated with microvascular thrombosis―the progression of which could be prevented by the administration of protein C 16.
Rivaroxaban, a direct FXa inhibitor 17, inhibits free and clot-bound FXa 18, as well as prothrombinase activity 17, without affecting the activity of existing thrombin. The aim of this study was to compare the effects of rivaroxaban with those of melagatran and dabigatran on tissue factor (TF)-induced TG in vitro in human plasma and in vivo in a rat model of TF-induced hypercoagulability. The possible involvement of the thrombin–TM/activated protein C system in the enhancement of TG observed with low concentrations of DTIs was also explored.
Methods
Agents
Rivaroxaban, melagatran, and dabigatran were synthesized by Bayer Pharma AG (Wuppertal, Germany). For the in vitro studies, rivaroxaban was dissolved in 100% DMSO and then further diluted with 5% DMSO. Melagatran was dissolved in distilled water. Dabigatran was dissolved in 1 N HCl (1 mg 20 μL−1) and filled up to 1 mL with distilled water. Recombinant human soluble TM (rhs-TM, American Diagnostica, Stamford, CT, USA) was dissolved in 0.9% NaCl. For the in vivo studies, rivaroxaban was dissolved in polyethylene glycol:H2O:glycerol (996:100:60 g), and melagatran was dissolved in 0.9% NaCl. Recombinant TF was obtained from a thromboplastin reagent kit (HemosIL® RecombiPlasTin; Instrumentation Laboratory, Lexington, MA, USA). For the TG assay (calibrated automated thrombogram [CAT] method), platelet-poor plasma (PPP) reagent (5 pmol l–1), thrombin calibrator, and FluCa-Kit (Fluo-buffer and Fluo-substrate) were obtained from Thrombinoscope BV (Maastricht, The Netherlands) and PefablocFG was obtained from Pentapharm (Basel, Switzerland).
In vitro studies
Plasma preparation
Human blood was obtained from healthy subjects who had not received medication during the 10 days before the study. Blood was collected via venipuncture and was allowed to drip freely into plastic tubes containing 1/10 volume of 3.12% trisodium citrate. PPP was obtained by immediate centrifugation at 1000× g for 20 minutes at room temperature.
Thrombin generation assay in human plasma using the CAT method
TG was determined by the CAT method (Thrombinoscope, Maastricht, The Netherlands) in accordance with the manufacturer's instructions with some modifications. PPP (76 μL) from individual donors was spiked with 2 μL of increasing concentrations of rivaroxaban (n = 9), melagatran (n = 8), dabigatran (n = 12), or the appropriate vehicles in the presence or absence of 2 μL rhs-TM (final concentration 10 nmol L–1). Diluted PPP reagent (20 μL, diluted with Fluo-buffer) was added to the plasma to achieve a final TF concentration of 1.67 pmol L–1 and phospholipid concentration of 1.33 μmol L–1. Incubation was carried out at 37 °C for 5 minutes, and the reaction was then started by adding 20 μL FluCa (HEPES, calcium chloride, and fluorogenic substrate). Fluorescence was read in a fluorometer (Fluoroskan Ascent®; Thermo Scientific, Waltham, MA, USA). In addition, thrombin calibration curves were performed for each individual plasma sample spiked with the appropriate solvent. TG curves were calculated using the Thrombinoscope software (Thrombinoscope BV). The following parameters were determined: peak TG (Cmax) and endogenous thrombin potential (ETP). TG in protein C-deficient plasma (lyophilized protein C-deficient human plasma; Kordia, Leiden, The Netherlands) was determined as described for normal PPP.
Determination of prothrombin fragment 1+2 in human plasma
To circumvent the small paradoxical stimulation of TG observed at low concentrations of a DTI in some experiments using the CAT assay 19, prothrombin fragment 1+2 (F1+2) was measured as an additional marker for TG. Briefly, human PPP (76 μL) was spiked with 2 μL of increasing concentrations of rivaroxaban (n = 3), melagatran (n = 3), dabigatran (n = 4–8), or the appropriate vehicles in the presence or absence of 2 μL rhs-TM (final concentration 10 nmol L−1). TG (generation of F1+2) was induced according to the CAT method with the following modifications. PPP was defibrinated by the addition of PefablocFG (2 μL; final concentration of 6 mg mL−1). TG was started by the addition of Fluo-buffer (HEPES, calcium chloride, and 5% DMSO [instead of Fluo-Substrate]). The reaction was stopped by adding 2 μL PPACK (Calbiochem®, Darmstadt, Germany; final concentration 10 μmol L−1) after 12 minutes. Determination of F1+2 in protein C-deficient plasma was performed as described earlier. Samples (120 μL) were then transferred to microtiter plates containing EDTA (final concentration 100 mmol L−1) at 4 °C and then stored at −20 °C. F1+2 was determined by an ELISA (Enzygnost® F1+2 monoclonal; Dade Behring, Marburg, Germany) in accordance with the manufacturer's instructions.
In vivo study―tissue factor-induced hypercoagulability in rats
RecombiPlasTin (8 mg) was reconstituted in RecombiPlasTin Diluent (0.5 mL) and further diluted with 0.5 mL 0.9% NaCl. Male Wistar rats, weighing 227–273 g, were fasted overnight and anesthetized by intraperitoneal injection of pentobarbital-Na (Narcoren® 80–100 mg kg−1; 5 mL kg−1; Merial GmbH, Hallbergmoos, Germany). The animals were randomized (n = 10) to receive intravenous rivaroxaban (0.001–0.3 mg kg−1), melagatran (0.001–0.3 mg kg−1), or the appropriate vehicle (1 mL kg−1). After 5 minutes, RecombiPlasTin (8 mg kg−1) or its vehicle (sham control groups) was administered slowly as an intravenous bolus. Blood was obtained via puncture of the abdominal aorta 10 minutes after the administration of RecombiPlasTin or vehicle using 3.12% trisodium citrate (for measurements of clotting times) or EDTA 1.8 mg ml−1 (for measurements of thrombin–antithrombin [TAT], fibrinogen, platelet count) as an anticoagulant. PPP was obtained as described for the in vitro studies and stored at −20 °C. All animal procedures were conducted in accordance with the German Animal Protection Act (Deutsches Tierschutzgesetz).
TAT levels
Plasma TAT complex levels were measured using a commercially available ELISA kit (Enzygnost®; Dade Behring) in accordance with the manufacturer's instructions. The TAT concentrations of the sham groups were used as baseline values to calculate fold increases.
Fibrinogen levels
Plasma fibrinogen was measured using a commercially available ELISA kit (Rat Fibrinogen ELISA, Immunology Consultants Laboratory, Newberg, OR, USA) in accordance with the manufacturer's instructions.
Measurement of platelet count
Platelet count was determined in whole blood in a Coulter counter (Beckman Coulter, Krefeld, Germany).
Clotting time measurements
Prothrombin time (PT; Neoplastin® Plus; Diagnostica Stago, Asnières-sur-Seine, France) was measured in the rivaroxaban study and activated partial thromboplastin time (APTT; STA APTT; Diagnostica Stago) was determined in the melagatran study using a ball coagulometer KC10A (Amelung, Lemgo, Germany), in accordance with the manufacturer's instructions.
Plasma concentrations of rivaroxaban and melagatran
Plasma concentrations of rivaroxaban and melagatran were determined by liquid chromatography–tandem mass spectrometry using a stable isotope-labeled internal standard. The applied sample preparation procedure involved protein precipitation followed by HPLC separation and tandem mass spectrometric detection. The HPLC system was coupled online to a tandem mass spectrometer (Biosystems/MDS Sciex API 4000) via a TurboIonSpray® interface.
Statistics
Results are shown as mean ± standard error of the mean. IC50 values were calculated using GraphPad Prism 6.0 (GraphPad Software, San Diego, CA, USA). One-way analysis of variance (anova) followed by Tukey's multiple comparison test was used for statistical analysis or unpaired parametric t test to compare two groups, with a P-value < 0.05 considered to be statistically significant.
Results
Effect of rivaroxaban, melagatran, or dabigatran on thrombin generation in normal and protein C-deficient human plasma (CAT method)
In control PPP, the levels of TG (ETP and Cmax) were similar across all studies investigating rivaroxaban, melagatran, or dabigatran. The addition of 10 nmol L−1 rhs-TM significantly decreased TG in the control plasma, as shown by the reduction in Cmax (by 63 ± 4%, 63 ± 5%, and 69 ± 4% in the rivaroxaban, melagatran, and dabigatran groups, respectively) and the ETP (by 72 ± 3%, 71 ± 3%, and 76 ± 3% in the rivaroxaban, melagatran, and dabigatran groups, respectively) (Table1).
Table 1.
Thrombin generation in control samples of normal human plasma and protein C-deficient plasma with and without thrombomodulin comparing the rivaroxaban, melagatran, and dabigatran study
| Rivaroxaban | Melagatran | Dabigatran | ||||
|---|---|---|---|---|---|---|
| −TM | +TM | −TM | +TM | −TM | +TM | |
| Normal PPP | ||||||
| Cmax (nmol L−1 thrombin) | 273 ± 9 | 100 ± 11*** | 276 ± 14 | 101 ± 13*** | 261 ± 12 | 81 ± 11*** |
| ETP (nmol L−1 thrombin × min) | 2070 ± 74 | 586 ± 61*** | 2019 ± 90 | 583 ± 67*** | 1970 ± 84 | 472 ± 61*** |
| Protein C-deficient plasma | ||||||
| Cmax (nmol L−1 thrombin) | 287 ± 15 | 290 ± 19 | 269 ± 45 | 294 ± 22 | 271 ± 45 | 298 ± 21 |
| ETP (nmol L−1 thrombin × min) | 1997 ± 69 | 2022 ± 46 | 1913 ± 182 | 1993 ± 62 | 1856 ± 186 | 1980 ± 73 |
Data are control values and are shown as mean ± standard error of the mean (n = 8–12 for rivaroxaban, melagatran, and dabigatran in normal plasma from healthy volunteers; n = 3 in protein C-deficient plasma).
P < 0.001 vs. –TM.
Cmax, peak thrombin generation; ETP, endogenous thrombin potential; PPP, platelet-poor plasma; TM, thrombomodulin.
Rivaroxaban significantly inhibited TG in a concentration-dependent manner in normal human PPP (Figs1A, 2A, and 3A,B), resulting in IC50 for Cmax of 44 ± 6 and 66 ± 5 nmol L−1 in the presence and absence of rhs-TM, respectively, and for ETP of 57 ± 8 and 697 ± 106 nmol L−1 in the presence and absence of rhs-TM, respectively (Table2).
Fig 1.

Representative thrombograms without and with rivaroxaban (3–1090 nmol L−1), melagatran (3–948 nmol L−1), or dabigatran (3–1090 nmol L−1) in normal human platelet-poor plasma (A, C, E) and in protein C-deficient plasma (B, D, F) in the absence of recombinant human soluble thrombomodulin (rhs-TM).
Fig 2.

Representative thrombograms without and with rivaroxaban (3–1090 nmol L−1), melagatran (3–948 nmol L−1), or dabigatran (3–1090 nmol L−1) in normal human platelet-poor plasma (A, C, E) and in protein C-deficient plasma (B, D, F) in the presence of recombinant human soluble thrombomodulin (rhs-TM; 10 nmol L−1).
Fig 3.

Peak thrombin generation (Cmax) and endogenous thrombin potential (ETP) in normal human platelet-poor plasma spiked with rivaroxaban (n = 9), melagatran (n = 8), or dabigatran (n = 12) in the absence and presence of recombinant human soluble thrombomodulin (rhs-TM; 10 nmol L−1). Results are shown as mean ± standard error of the mean. *P < 0.05, **P < 0.01, ***P < 0.001 vs. respective controls.
Table 2.
Thrombin generation in human platelet-poor plasma spiked with rivaroxaban, melagatran, or dabigatran without and with thrombomodulin
| Parameter | Rivaroxaban | Melagatran | Dabigatran | |||
|---|---|---|---|---|---|---|
| −TM | +TM | −TM | +TM | −TM | +TM | |
| Cmax IC50 (nmol L−1) | 66 ± 5 | 44 ± 6 | 940 ± 79 | >950 | 976 ± 71 | >1090 |
| ETP IC50 (nmol L−1) | 697 ± 106 | 57 ± 8 | 732 ± 43 | >950 | 722 ± 44 | >1090 |
Results are shown as mean ± standard error of the mean; n = 8–12 in normal plasma from healthy volunteers.
Cmax, peak thrombin generation; ETP, endogenous thrombin potential; IC50, maximal drug concentration to cause 50% inhibition; TM, thrombomodulin.
Melagatran and dabigatran inhibited TG in a concentration-dependent manner in the absence of rhs-TM (Figs1C,E and 3C–F). However, there was a slight, but not significant, increase in Cmax (maximal increase of 8% and 11% for melagatran and dabigatran, respectively) and ETP (maximal increase of 6% and 5% for melagatran and dabigatran, respectively) over baseline at the lower concentration range measured. In the presence of rhs-TM, melagatran and dabigatran exhibited a paradoxical effect on Cmax and ETP. There was a concentration-dependent increase over control of Cmax and ETP at concentrations ≥ 10 nmol L−1, reaching a maximum at approximately 200–300 nmol L−1. The ETP and Cmax were significantly increased by 1.9–2.2-fold and by 2.2–2.7-fold, respectively, with melagatran at 119–474 nmol L−1 vs. control (Fig.3C,D). Similarly, in the presence of dabigatran, the ETP was significantly increased at 136–545 nmol L−1 by 2.2–2.6-fold, and the Cmax was significantly enhanced at 68–545 nmol L−1 by 2.0–3.3-fold, respectively (Fig.3E,F). Furthermore, even at the highest concentration of melagatran and dabigatran tested (∼1000 nmol L−1), TG was not reduced to values below those measured in the controls (Fig.3C–F). Thus, in the presence of rhs-TM, IC50 for Cmax and ETP increased from 940 to 732 nmol L−1, respectively, for melagatran and from 976 to 722 nmol L−1, respectively, for dabigatran, to values above the highest concentrations measured (Table2).
In protein C-deficient plasma, rhs-TM had no influence on TG or the inhibition of TG by rivaroxaban, melagatran, or dabigatran (Figs1B,D,F and 2B,D,F). No increase in ETP or Cmax was observed with melagatran or dabigatran in the absence or presence of rhs-TM in protein C-deficient plasma.
F1+2 generation
To confirm that the results obtained with the CAT method were not biased by the methodology, TG was measured under similar conditions but using the measurement of F1+2 as a read-out system. Similar results were obtained by measuring F1+2 generation (Fig.4). In control plasma, the amount of F1+2 generated was similar across all studies. The addition of rhs-TM decreased the amount of F1+2 (Fig.4B). Rivaroxaban concentration-dependently inhibited F1+2 generation in the absence or presence of rhs-TM, with IC50 values of 12 ± 7 and 14 ± 7 nmol L−1, respectively (Fig.4A,B).
Fig 4.
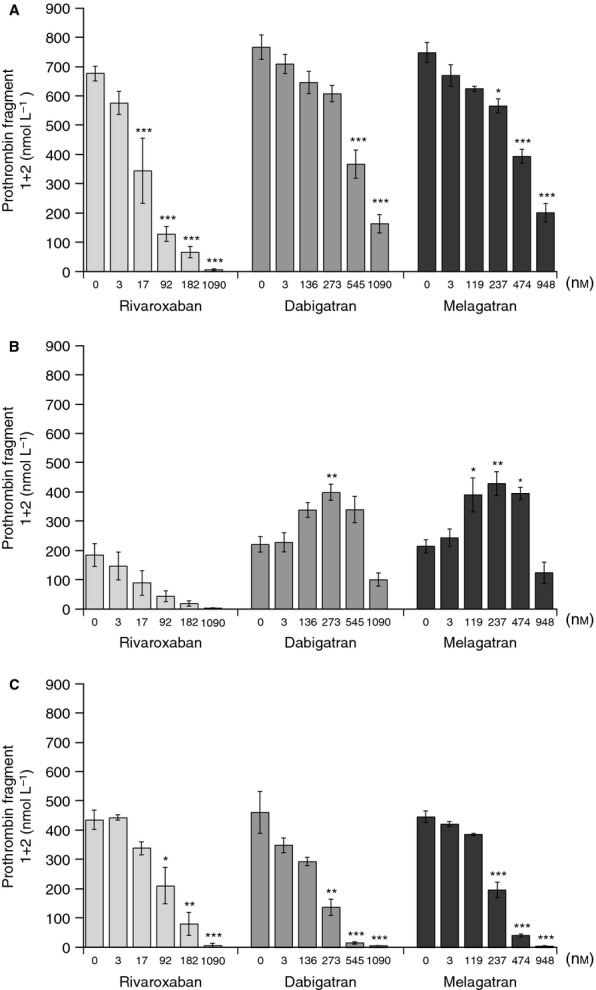
Effects of rivaroxaban (3–1090 nmol L−1), melagatran (3–948 nmol L−1), and dabigatran (3–1090 nmol L−1) on prothrombin fragment 1+2 generation in the absence (A) and presence (B) of recombinant human soluble thrombomodulin (rhs-TM; 10 nmol L−1) in normal human platelet-poor plasma and in protein C-deficient human plasma in the presence of rhs-TM (C). *P < 0.05, **P < 0.01, ***P < 0.001 vs. respective controls. rhs-TM, recombinant human soluble thrombomodulin.
In the absence of TM, melagatran and dabigatran inhibited F1+2 generation in a concentration-dependent manner (Fig.4A). The IC50 values were 368 ± 40 nmol L−1 for melagatran and 387 ± 63 nmol L−1 for dabigatran. However, in the presence of rhs-TM, both melagatran and dabigatran increased F1+2 generation. F1+2 was significantly increased within the same concentration range as observed by the CAT method, by 1.8–2.0-fold and 1.6–1.9-fold with melagatran and dabigatran, respectively (Fig.4B).
In protein C-deficient plasma, rivaroxaban, as well as melagatran and dabigatran, concentration-dependently inhibited the generation of F1+2 in the presence of TM. Neither dabigatran nor melagatran enhanced F1+2 generation (Fig.4C).
TF-induced hypercoagulability in vivo in rats
TF significantly increased TAT levels by 13-fold from 9 ± 5 to 116 ± 11 μg L−1 and from 7 ± 2 to 88 ± 10 μg L−1 in the control animals of the rivaroxaban and melagatran groups, respectively, within 10 minutes after injection, without affecting fibrinogen concentration or platelet count (Fig.5). There was a slight increase in APTT but not PT in the control animals (Table3).
Fig 5.

Effects of rivaroxaban and melagatran (0.001–0.3 mg kg−1 i.v.) on thrombin–antithrombin (TAT) complex (A, B), fibrinogen levels (C, D), and platelet counts (E, F) 10 minutes after injection of tissue factor (8 mg kg−1) in male rats. Results are mean ± standard error of the mean (n = 10). **P < 0.01, ***P < 0.001 vs. tissue factor control group; †P < 0.05, ††P < 0.01, †††P < 0.001 vs. sham control group. i.v., intravenous.
Table 3.
Prothrombin time (PT) and activated partial thromboplastin time (APTT) prolongation and plasma levels of rivaroxaban and melagatran 15 minutes after intravenous drug administration and 10 minutes after injection of tissue factor (TF)
| Drug dose (mg kg−1) | Rivaroxaban | Melagatran | ||
|---|---|---|---|---|
| Plasma concentration (μg L−1) | PT prolongation (x-fold over baseline) | Plasma concentration (μg L−1) | APTT prolongation (x-fold over baseline) | |
| Sham | – | 1.00 ± 0.03 | – | 1.00 ± 0.05 |
| TF control | – | 1.01 ± 0.03 | – | 1.23 ± 0.04 |
| 0.001 | 2.9 ± 0.4 | 1.00 ± 0.04 | <2.9† | 1.26 ± 0.05 |
| 0.003 | 4.8 ± 0.9 | 1.02 ± 0.02 | 6.3 ± 0.3 | 1.34 ± 0.05 |
| 0.01 | 15.4 ± 2.6 | 0.96 ± 0.02 | 19.2 ± 0.9 | 1.68 ± 0.03 |
| 0.03 | 60.7 ± 9.1 | 1.03 ± 0.02 | 45.9 ± 5.7 | 2.17 ± 0.23* |
| 0.1 | 276.8 ± 35.4 | 1.26 ± 0.06*** | 199.8 ± 25.8 | 2.08 ± 0.18** |
| 0.3 | 765.9 ± 68.0 | 1.92 ± 0.05*** | 524.7 ± 26.1 | 3.11 ± 0.27*** |
Results are shown as mean ± standard error of the mean.
P < 0.05
P < 0.01
P < 0.001 vs. sham control.
†Value below the detection limit of the assay.
Rivaroxaban dose-dependently inhibited the TF-induced increase in TAT levels over a broad dose range (0.001–0.3 mg kg−1); the effect was highly significant at doses ≥ 0.03 mg kg−1 (P < 0.001), with complete inhibition at 0.3 mg kg−1 rivaroxaban (Fig.5A). This dose range corresponded to plasma levels of rivaroxaban of 3–766 μg L−1 and prolongation of PT of 1.0–1.9-fold over baseline (Table3). Platelet count and fibrinogen concentration were not affected over the whole dose range tested (Fig.5C,E). Melagatran also reduced TF-induced TAT generation at the highest dose (0.3 mg kg−1; corresponding to a plasma level of 525 μg L−1 and APTT prolongation of 3.1-fold) (Table3). However, melagatran potentiated TF-induced hypercoagulability at lower doses (0.01–0.03 mg kg−1; corresponding to plasma levels of 19–46 μg L−1). At a melagatran dose of 0.01 mg kg−1 (corresponding to a plasma level of 19 μg L−1 and APTT prolongation of 1.7-fold), TAT increased by 1.6-fold vs. TF control (P < 0.01; Fig.5B). Furthermore, fibrinogen levels decreased significantly from 2.34 ± 0.08 g L−1 (TF control) to 1.41 ± 0.20 g L−1 at 0.03 mg kg−1 melagatran (P < 0.001) (Fig.5D), and platelet count decreased from 644 × 103 μL−1 (TF control) to 559 × 103 μL−1 at 0.01 mg kg−1 melagatran (Fig.5F).
Discussion
In this study, we investigated the phenomenon of a paradoxical activation of coagulation at low concentrations of DTIs, as reported by Furugohri et al. 10–12, and compared the effects of dabigatran, melagatran, and rivaroxaban in vitro and melagatran and rivaroxaban in vivo. In contrast to the in vivo studies by Furugohri et al., we used less severe hypercoagulable conditions, which avoided the development of DIC in the control animals. In these studies, the increase in TAT was higher (519 μg L−1 10 and 385 μg L−1 12), which resulted in a strong decrease in platelet count (27% and 42%, respectively), compared with TAT levels of 88 μg L−1 (melagatran study) and 116 μg L−1 (rivaroxaban study) in our study without a reduction in platelet count or fibrinogen levels in the control groups. Furthermore, in the study by Furugohri et al. 12, there was a dose-dependent or time-dependent high mortality rate (up to 67% and 88% mortality, respectively) in the ximelagatran-treated animals. We used this modified model of TF-induced intravascular coagulation to reflect more closely the clinical situation of atherothrombosis. Our study showed that rivaroxaban protected against TF-induced hypercoagulation over a broad dose range. Higher doses of melagatran also exhibited a protective effect; however, at low doses/plasma concentrations, direct inhibition of thrombin with melagatran increased hypercoagulability, as shown by an increase in TAT and a decrease in fibrinogen levels, and a mild decrease in platelet count. These results are consistent with the previous studies showing a paradoxical activation of coagulation with these DTIs, in contrast to FXa inhibitors 10,12.
In normal human PPP, rivaroxaban concentration-dependently inhibited TG in the absence and presence of rhs-TM. Melagatran and dabigatran also inhibited TG in a concentration-dependent manner in the absence of TM. The slight, but not significant, increase in Cmax and ETP over baseline may be due to an increase in the activity of α2-macroglobulin-thrombin (α2-M-thrombin) in the presence of low concentrations of a DTI, but this transient increase is not subtracted by the mathematical algorithm used by the CAT technique to correct the α2-M-thrombin activity. Thus, the increase might not be attributed to thrombin itself 19. However, in the presence of TM, only high concentrations of melagatran and dabigatran reduced ETP and Cmax, whereas at lower concentrations both DTIs significantly enhanced peak levels of TG; this effect was not observed in protein C-deficient plasma. This is consistent with previous findings that DTIs, such as melagatran, increased TG at low concentrations in the presence of protein C 10 or TM 11. To exclude the possibility that the observed increase in TG in the presence of TM was due to the readout system of the CAT method, TG was also determined by measuring the concentration of F1+2. The results showed that low concentrations of melagatran (119–474 nmol L−1) and dabigatran (136–545 nmol L−1) enhanced F1+2 generation in the presence but not in the absence of rhs-TM, confirming the results obtained with the CAT method.
In the control samples of normal plasma, the concentrations of F1+2 were 1.5–1.7-fold higher compared with those in protein C-deficient plasma. In contrast, ETP was similar in normal and in protein C-deficient plasma, as measured by using the CAT method. These differences may be at least partly due to the different batches of protein C-deficient plasma used in both methods. We also excluded the possibility that contact activation at low TF concentrations may have influenced the TG profile. The results were similar in plasma obtained from blood withdrawn in the presence of corn trypsin inhibitor (data not shown).
In normal human PPP, the addition of 10 nmol L−1 rhs-TM suppressed the amount of TG by approximately 60%, as a result of the activation of protein C and the negative-feedback system. The increase in TG at low concentrations of melagatran and dabigatran in the presence of TM and protein C suggests that these DTIs may inhibit the activation of protein C through the inhibition of TM-bound thrombin, which subsequently reduces the TM/activated protein C negative-feedback system. This may be due in part to an increase in the affinity of the thrombin inhibitor for the thrombin bound to TM, which may reduce the concentrations of the active TM–thrombin complex below a critical level. A twofold increase in this affinity was shown for melagatran and inogatran 13. Melagatran, inogatran, and dabigatran are derivatives of the peptide-like, benzamidine-based thrombin inhibitors and show structural similarities. It should be noted that the paradoxical activation may not occur with DTIs in general. Structural changes of the inhibitors leading to different binding kinetics and thermodynamics can have significant effects on the interaction of the inhibitor with its different target enzyme complexes 20.
Low thrombin inhibitor concentrations might insufficiently block the feedback activation of FXI, FV, and FVIII by thrombin; in this situation, the protein C pathway may have a dominant role in the regulation of TG. Higher inhibitor concentrations are sufficient to inhibit thrombin activity and TG because of the inhibition of the feedback activation. Of note, in the presence of TM, Cmax and ETP reached basal levels only at the highest DTI concentration tested, indicating that higher concentrations are necessary to inhibit TG effectively in the presence of TM. In contrast, in the presence of a FXa inhibitor there remains enough active TM–thrombin complex for the regulation of TG. It is noteworthy that, in the presence of TM, the IC50 value for ETP inhibition by rivaroxaban was 12-fold lower compared with the IC50 in the absence of TM; this can be explained by the reduction in TG in the presence of TM. Recently, it was suggested that the addition of TM to a TG assay might reflect more closely a physiological situation and, furthermore, this assay might be used to detect thrombophilic phenotypes 21,22.
The increase in TG may be responsible for the increased hypercoagulation observed with low doses of melagatran in vivo. It remains to be investigated whether there is a link between hypercoagulability and the interaction with the thrombin–TM/activated protein C system observed in preclinical studies and the reported higher risk of ACS (than the comparators) in some clinical studies of dabigatran and melagatran. The precise mechanisms are not known, but potential explanations have been discussed. It was suggested recently that, owing to the massive release of FV when platelets are involved and the subsequent formation of the prothrombinase complex, there may be too much thrombin produced to be inhibited by a thrombin inhibitor. In contrast, a direct FXa inhibitor inhibits prothrombinase activity and thereby reduces the amount of thrombin 23. Furthermore, a recent analysis of markers of inflammation using blood samples from a phase II study of the first oral DTI ximelagatran (Efficacy and Safety of the oral Thrombin inhibitor ximelagatran in combination with aspirin, in patiEnts with rEcent Myocardial damage [ESTEEM]) showed that long-term treatment with ximelagatran increased the levels of several markers of inflammation 24. In contrast, rivaroxaban was shown to suppress thrombin-induced proinflammatory gene expression in human umbilical vein endothelial cells; in contrast, in the same study, low-dose dabigatran exerted a proinflammatory effect 25. Given the role of inflammation in the pathogenesis of ACS, further insights may be provided by clinical studies investigating the effect of long-term treatment with the direct FXa inhibitors and the DTIs on the thrombin–TM/activated protein C system, inflammation and endothelial function.
Our studies have some limitations. The ‘low concentrations’ used in the in vitro studies are close to the reported Ctrough and Cmax levels after therapeutic doses of dabigatran and ximelagatran 26,27; however, data obtained under in vitro experimental conditions may not directly mirror ex vivo drug levels in patients.
In the in vitro study, we used rhs-TM, but TM is physiologically active as a transmembrane thrombin receptor and this may affect the binding of the thrombin inhibitor to the TM–thrombin complex. Furthermore, the EPCR augments thrombin–TM-dependent protein C activation in vivo 28. Thus, the presence of endothelial cells might affect the results, and similar investigations in the presence of endothelial cells are warranted to confirm these findings. The data from our in vivo study cannot be compared directly with the underlying pathophysiology of atherothrombosis. Therefore, these results generate a hypothesis for a biochemical mechanism; biomarkers from clinical studies that demonstrate an increase in thrombin activation or a reduction of activated protein C are necessary to support the hypothesis.
In conclusion, the present study indicates a paradoxical activation of coagulation at low plasma concentrations of melagatran and dabigatran, which may be caused by suppression of the thrombin–TM/activated protein C feedback system. In contrast, rivaroxaban did not exhibit such an effect, suggesting that direct FXa inhibition may not be associated with a paradoxically increased prothrombotic state. However, the clinical relevance remains to be evaluated in clinical studies measuring prothrombotic markers.
Addendum
Study concept and design: E. Perzborn and S. Heitmeier; analysis and interpretation of data: E. Perzborn, S. Heitmeier, U. Buetehorn, and V. Laux; drafting, revisions of the manuscript for important intellectual content, and approval of the submission draft: E. Perzborn, S. Heitmeier, U. Buetehorn, and V. Laux.
Acknowledgments
This study was funded by Bayer Pharma AG. The authors would like to acknowledge Elke Fischer, Michaela Harwardt, Uwe Lange, and Axel Trabandt for their technical support and Yong-Ling Liu, who provided editorial support with funding from Bayer HealthCare Pharmaceuticals and Janssen Scientific Affairs, LLC.
Disclosure of Conflict of Interests
E. Perzborn is a former employee of Bayer HealthCare Pharmaceuticals. S. Heitmeier, U. Buetehorn, and V. Laux are employees of Bayer HealthCare Pharmaceuticals.
References
- 1.Eriksson BI, Quinlan DJ, Eikelboom JW. Novel oral Factor Xa and thrombin inhibitors in the management of thromboembolism. Annu Rev Med. 2011;62:41–57. doi: 10.1146/annurev-med-062209-095159. [DOI] [PubMed] [Google Scholar]
- 2.Executive Steering Committee of behalf of the SPORTIF III Investigators. Stroke prevention with the oral direct thrombin inhibitor ximelagatran compared with warfarin in patients with non-valvular atrial fibrillation (SPORTIF III): randomised controlled trial. Lancet. 2003;362:1691–8. doi: 10.1016/s0140-6736(03)14841-6. [DOI] [PubMed] [Google Scholar]
- 3.Uchino K, Hernandez AV. Dabigatran association with higher risk of acute coronary events: meta-analysis of noninferiority randomized controlled trials. Arch Intern Med. 2012;172:397–402. doi: 10.1001/archinternmed.2011.1666. [DOI] [PubMed] [Google Scholar]
- 4.Hohnloser SH, Oldgren J, Yang S, Wallentin L, Ezekowitz M, Reilly P, Eikelboom J, Brueckmann M, Yusuf S, Connolly SJ. Myocardial ischemic events in patients with atrial fibrillation treated with dabigatran or warfarin in the RE-LY (Randomized Evaluation of Long-Term Anticoagulation Therapy) trial. Circulation. 2012;125:669–76. doi: 10.1161/CIRCULATIONAHA.111.055970. [DOI] [PubMed] [Google Scholar]
- 5.Schulman S, Kearon C, Kakkar AK, Schellong S, Eriksson H, Baanstra D, Kvamme AM, Friedman J, Mismetti P, Goldhaber SZ. Extended use of dabigatran, warfarin, or placebo in venous thromboembolism. N Engl J Med. 2013;368:709–18. doi: 10.1056/NEJMoa1113697. [DOI] [PubMed] [Google Scholar]
- 6.Mak KH. Coronary and mortality risk of novel oral antithrombotic agents: a meta-analysis of large randomised trials. BMJ Open. 2012;2:e001592. doi: 10.1136/bmjopen-2012-001592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fiessinger JN, Huisman MV, Davidson BL, Bounameaux H, Francis CW, Eriksson H, Lundström T, Berkowitz SD, Nyström P, Thorsén M, Ginsberg JS. THRIVE Treatment Study Investigators. Ximelagatran vs low-molecular-weight heparin and warfarin for the treatment of deep vein thrombosis: a randomized trial. JAMA. 2005;293:681–9. doi: 10.1001/jama.293.6.681. [DOI] [PubMed] [Google Scholar]
- 8.Ruyi H. Integrated executive summary of FDA review for NDA 21-686 Exanta (ximelagatran) 2004. http://www.fda.gov/ohrms/dockets/ac/04/briefing/2004-4069B1_03_FDA-Backgrounder-Execsummaryredacted.pdf. Accessed 24 March 2014.
- 9.Eikelboom JW, Weitz JI. Anticoagulation therapy. Dabigatran and risk of myocardial infarction. Nat Rev Cardiol. 2012;9:260–2. doi: 10.1038/nrcardio.2012.34. [DOI] [PubMed] [Google Scholar]
- 10.Furugohri T, Shiozaki Y, Muramatsu S, Honda Y, Matsumoto C, Isobe K, Sugiyama N. Different antithrombotic properties of Factor Xa inhibitor and thrombin inhibitor in rat thrombosis models. Eur J Pharmacol. 2005;514:35–42. doi: 10.1016/j.ejphar.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 11.Furugohri T, Sugiyama N, Morishima Y, Shibano T. Antithrombin-independent thrombin inhibitors, but not direct Factor Xa inhibitors, enhance thrombin generation in plasma through inhibition of thrombin-thrombomodulin-protein C system. Thromb Haemost. 2011;106:1076–83. doi: 10.1160/TH11-06-0382. [DOI] [PubMed] [Google Scholar]
- 12.Furugohri T, Fukuda T, Tsuji N, Kita A, Morishima Y, Shibano T. Melagatran, a direct thrombin inhibitor, but not edoxaban, a direct Factor Xa inhibitor, nor heparin aggravates tissue factor-induced hypercoagulation in rats. Eur J Pharmacol. 2012;686:74–80. doi: 10.1016/j.ejphar.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 13.Mattsson C, Menschik-Lundin A, Nylander S, Gyzander E, Deinum J. Effect of different types of thrombin inhibitors on thrombin/thrombomodulin modulated activation of protein C in vitro. Thromb Res. 2001;104:475–86. doi: 10.1016/s0049-3848(01)00392-9. [DOI] [PubMed] [Google Scholar]
- 14.Linder R, Frebelius S, Grip L, Swedenborg J. The influence of direct and antithrombin-dependent thrombin inhibitors on the procoagulant and anticoagulant effects of thrombin. Thromb Res. 2003;110:221–6. doi: 10.1016/s0049-3848(03)00344-x. [DOI] [PubMed] [Google Scholar]
- 15.Esmon CT. The protein C pathway. Chest. 2003;124:26S–32S. doi: 10.1378/chest.124.3_suppl.26s. [DOI] [PubMed] [Google Scholar]
- 16.Dreyfus M, Magny JF, Bridey F, Schwarz HP, Planche C, Dehan M, Tchernia G. Treatment of homozygous protein C deficiency and neonatal purpura fulminans with a purified protein C concentrate. N Engl J Med. 1991;325:1565–8. doi: 10.1056/NEJM199111283252207. [DOI] [PubMed] [Google Scholar]
- 17.Perzborn E, Strassburger J, Wilmen A, Pohlmann J, Roehrig S, Schlemmer KH, Straub A. In vitro and in vivo studies of the novel antithrombotic agent BAY 59-7939 − an oral, direct Factor Xa inhibitor. J Thromb Haemost. 2005;3:514–21. doi: 10.1111/j.1538-7836.2005.01166.x. [DOI] [PubMed] [Google Scholar]
- 18.Depasse F, Busson J, Mnich J, Le Flem L, Gerotziafas GT, Samama MM. Effect of BAY 59-7939 − a novel, oral, direct Factor Xa inhibitor − on clot-bound Factor Xa activity in vitro. J Thromb Haemost. 2005;3(Suppl 1) Abstract P1104. [Google Scholar]
- 19.Wagenvoord RJ, Deinum J, Elg M, Hemker HC. The paradoxical stimulation by a reversible thrombin inhibitor of thrombin generation in plasma measured with thrombinography is caused by alpha-macroglobulin-thrombin. J Thromb Haemost. 2010;8:1281–9. doi: 10.1111/j.1538-7836.2010.03822.x. [DOI] [PubMed] [Google Scholar]
- 20.Winquist J, Geschwindner S, Xue Y, Gustavsson L, Musil D, Deinum J, Danielson UH. Identification of structural-kinetic and structural-thermodynamic relationships for thrombin inhibitors. Biochemistry. 2013;52:613–26. doi: 10.1021/bi301333z. [DOI] [PubMed] [Google Scholar]
- 21.Dargaud Y, Trzeciak MC, Bordet JC, Ninet J, Negrier C. Use of calibrated automated thrombinography +/− thrombomodulin to recognise the prothrombotic phenotype. Thromb Haemost. 2006;96:562–7. [PubMed] [Google Scholar]
- 22.Hemker HC, Giesen P, al Dieri R, Regnault V, de Smedt E, Wagenvoord R, Lecompte T, Beguin S. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb. 2003;33:4–15. doi: 10.1159/000071636. [DOI] [PubMed] [Google Scholar]
- 23.Esmon CT. What did we learn from new oral anticoagulant treatment? Thromb Res. 2012;130(Suppl 1):S41–3. doi: 10.1016/j.thromres.2012.08.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christersson C, Oldgren J, Wallentin L, Siegbahn A. Treatment with an oral direct thrombin inhibitor decreases platelet activity but increases markers of inflammation in patients with myocardial infarction. J Intern Med. 2011;270:215–23. doi: 10.1111/j.1365-2796.2011.02354.x. [DOI] [PubMed] [Google Scholar]
- 25.Ellinghaus P, Laux V, Perzborn E. Effect of rivaroxaban on thrombin-induced pro-inflammatory gene expression in human umbilical vein endothelial cells. J Thromb Haemost. 2011;9:491. Abstract P-TU-577. [Google Scholar]
- 26.Cullberg M, Eriksson UG, Wåhlander K, Eriksson H, Schulman S, Karlsson MO. Pharmacokinetics of ximelagatran and relationship to clinical response in acute deep vein thrombosis. Clin Pharmacol Ther. 2005;77:279–90. doi: 10.1016/j.clpt.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 27.Hawes EM, Deal AM, Funk-Adcock D, Gosselin R, Jeanneret C, Cook AM, Taylor JM, Whinna HC, Winkler AM, Moll S. Performance of coagulation tests in patients on therapeutic doses of dabigatran: a cross-sectional pharmacodynamic study based on peak and trough plasma levels. J Thromb Haemost. 2013;11:1493–502. doi: 10.1111/jth.12308. [DOI] [PubMed] [Google Scholar]
- 28.Taylor FB, Jr, Peer GT, Lockhart MS, Ferrell G, Esmon CT. Endothelial cell protein C receptor plays an important role in protein C activation in vivo. Blood. 2001;97:1685–8. doi: 10.1182/blood.v97.6.1685. [DOI] [PubMed] [Google Scholar]