

Abstract
Chromosome copy number in cells is controlled so that the frequency of initiation of DNA replication matches that of cell division. In bacteria, this is achieved through regulation of the interaction between the initiator protein DnaA and specific DNA elements arrayed at the origin of replication. DnaA assembles at the origin and promotes DNA unwinding and the assembly of a replication initiation complex. SirA is a DnaA-interacting protein that inhibits initiation of replication in diploid Bacillus subtilis cells committed to the developmental pathway leading to formation of a dormant spore. Here we present the crystal structure of SirA in complex with the N-terminal domain of DnaA revealing a heterodimeric complex. The interacting surfaces of both proteins are α-helical with predominantly apolar side-chains packing in a hydrophobic interface. Site-directed mutagenesis experiments confirm the importance of this interface for the interaction of the two proteins in vitro and in vivo. Localization of GFP–SirA indicates that the protein accumulates at the replisome in sporulating cells, likely through a direct interaction with DnaA. The SirA interacting surface of DnaA corresponds closely to the HobA-interacting surface of DnaA from Helicobacter pylori even though HobA is an activator of DnaA and SirA is an inhibitor.
Introduction
Across the kingdoms of life, DNA replication is tightly regulated to ensure co-ordination with cell growth and development. Failure to maintain and control chromosome copy number is frequently associated with disease or cell death. Regulation of DNA replication is mainly exerted at the initiation step when an initiator protein binds to the origin of replication and promotes the assembly of a nucleoprotein complex from which replication forks diverge.
In prokaryotes, the DNA replication initiator protein is DnaA. In its ATP-bound state, DnaA assembles at the origin of replication, oriC, by binding to a number of 9 bp recognition sequences termed DnaA-boxes (Yoshikawa and Ogasawara, 1991). Recruitment of DnaA to oriC is believed to generate a helical oligomer of DNA-bound DnaA that promotes duplex unwinding at an AT-rich region within the origin termed the DNA unwinding element (Duderstadt et al., 2011). DnaA is a member of the AAA+ (ATPases associated with diverse cellular activities) protein superfamily and is made up of four distinct domains (Kaguni, 2006). Domain I is known to have a number of interaction partners, including replication regulators and DNA helicase (Seitz et al., 2000; Abe et al., 2007). Domain II is thought to be a flexible linker, that may allow nuances in regulatory control (Molt et al., 2009). Domain III binds and hydrolyses ATP, mediates DnaA oligomerization (Erzberger et al., 2006) and binds single stranded DNA thus aiding duplex unwinding (Duderstadt et al., 2011). Domain IV binds double stranded DNA, interacting with the DnaA-box motifs (Fujikawa et al., 2003). The binding of a threshold level of DnaA–ATP at oriC leads to duplex unwinding and the recruitment of other initiation proteins (Leonard and Grimwade, 2011).
Although DnaA is conserved in bacteria, many of the other initiation components are not. This is exemplified by differences in replication initiation observed between E. coli and B. subtilis, organisms which provide our most thorough understanding of the control of replication initiation in Gram-negative and Gram-positive bacteria, respectively. An important early step following duplex unwinding in both organisms, is the recruitment of a DNA helicase (Ec DnaB/Bsu DnaC) to the origin where it is loaded onto the DNA by a helicase loader (Ec DnaC/Bsu DnaI). This is followed by the binding of the primase, DnaG. Curiously, replication initiation in B. subtilis requires two additional proteins, DnaD and DnaB, neither of which is present in E. coli (Ishigo-oka et al., 2001; Rokop et al., 2004) – it should be noted that B. subtilis DnaB is unrelated to E. coli DnaB. DnaD is recruited to the origin through an interaction with DnaA, moreover DnaD binding has been shown to be accompanied by pronounced bending of origin DNA. Unwinding of the duplex appears to be assisted by the formation of DnaD scaffolds, which may provide further anchorage points for DnaA (Zhang et al., 2008). DnaB is subsequently recruited, appearing to play a role in helicase loading along with the helicase loader DnaI (Velten et al., 2003).
During rapid growth, bacteria reinitiate replication before the previous cycle of replication is complete, giving rise to multiple replication forks. Daughter cells thus inherit chromosomes that are already undergoing replication. This emphasizes the need for precise mechanisms to control the frequency of initiation of DNA replication so that it matches the frequency of cell division and nutrient availability. Regulatory mechanisms take the form of proteins and cis-acting DNA elements which typically act on DnaA or oriC. Protein regulators vary between genera. Notably, B. subtilis and E. coli employ a range of replication regulators that lack known homologues in the other species (E. coli Hda, DiaA, SeqA; B. subtilis YabA, Soj, SirA, Spo0A) (Katayama et al., 2010; Briggs et al., 2012), reflecting differences in their mechanisms of regulatory control. For example, in E. coli Hda inactivates DnaA by promoting ATP hydrolysis in DnaA–ATP, whereas its functional homologue in B. subtilis, YabA, acts by both sequestering DnaA at the replication fork, and by inhibiting DnaA oligomerization (Scholefield and Murray, 2013) (Soufo et al., 2008).
An additional specialized DNA replication checkpoint exists in Gram-positive bacteria of the genera Bacilli and Clostridia during sporulation under conditions of nutrient depletion. Sporulation begins with an asymmetric cell division producing genetically identical daughter cells of unequal size. The larger mother cell and the smaller forespore must each inherit a complete copy of the genome in order to drive the developmental program. Therefore, DNA replication is regulated and monitored at the onset of sporulation to ensure that two intact copies of the chromosome are present in the pre-divisional cell. DnaA contributes to this DNA replication check-point through its role as a transcription factor. In B. subtilis, DnaA activates the expression of sda, which encodes an inhibitor of the sporulation sensor kinases, KinA and KinB. Sda thus serves to delay sporulation by limiting the phosphorylation of the master sporulation response regulator, Spo0A. Sda is an intrinsically unstable protein whose levels fluctuate with the cell cycle, reaching a minimum immediately prior to the initiation of a new round of DNA replication (Burkholder et al., 2001; Veening et al., 2009). This creates a small ‘window of opportunity’, for a threshold concentration of Spo0A∼P to be achieved and for sporulation to commence.
SirA, a protein produced under Spo0A∼P regulation, has been identified as an inhibitor of DNA replication that plays a specific role in preventing replication re-initiation in cells committed to sporulation (Rahn-Lee et al., 2009). Although single deletion mutants of sirA, like those of sda, display only mild phenotypes, under conditions of nutrient depletion leading to sporulation sda/sirA double mutants are severely impaired in chromosome copy number control, indicating a shared role in controlling DNA replication (Veening et al., 2009). Artificial induction of expression of sirA in vegetatively growing B. subtilis blocks replication and causes cell death in a DnaA-dependent manner (Wagner et al., 2009). Cells artificially induced to sporulate under conditions of rapid growth undergo a marked decrease in chromosome copy number which is partially relieved by deletion of sirA (Rahn-Lee et al., 2009). These experiments imply that SirA is an inhibitor of DNA replication during sporulation that acts by binding to DnaA. Furthermore, the SirA binding determinants of DnaA have been mapped to its N-terminal domain, DnaADI (Rahn-Lee et al., 2011).
SirA has no significant sequence similarity to other proteins besides orthologues in Bacilli. Here, we have solved the structure of SirA from B. subtilis in complex with DnaADI providing the first structure of a DnaA domain in an inhibitory complex. The structure reveals a heterodimer with an α-helical interface. The importance of this interface for SirA–DnaA interaction in vitro and in vivo has been demonstrated by analysis of a panel of site-directed mutants. Furthermore, localization of GFP–SirA within sporulating cells indicates that the protein accumulates at the replisome, likely through a direct interaction with DnaA. Interestingly, the structure reveals a conserved binding site on DnaADI that is used by DnaA in H. pylori and E. coli to bind the replication activators HobA and DiaA, respectively, implying this surface is functionally important in DNA replication initiation.
Results
Coexpression with DnaADI confers solubility on recombinant SirA
Attempts to produce recombinant SirA in E. coli yielded disappointing results; although SirA could be produced at high levels in a number of E. coli expression strains, the protein always partitioned into the insoluble fraction upon cell lysis. Variations in growth conditions or lysis procedures failed to overcome the insolubility of SirA. Following a report that the determinants of SirA binding to DnaA reside in its N-terminal domain (DnaADI) (Rahn-Lee et al., 2011), we generated a coexpression construct in which sequences encoding DnaADI (fused to an N-terminal cleavable polyhistidine-tag) and SirA were expressed from separate promoters on the same vector. Strikingly, this coexpression strategy led to the appearance of SirA in the soluble fraction following cell lysis, presumably the result of its interaction with DnaADI. Consistent with the notion that recombinant SirA and DnaADI were forming a complex, the two proteins co-purified. SirA was retained on an immobilized nickel affinity column with His-tagged DnaADI and co-eluted with the latter. Moreover, the two proteins eluted together following gel filtration chromatography.
SirA and DnaADI form a heterodimer
The stoichiometry of the SirA–DnaADI complex was determined using size-exclusion chromatography with multi-angle laser light scattering (SEC-MALLS). In these experiments, samples are fractionated on a gel-filtration column and the absorbance at 280 nm and the refractive index of the eluate are monitored together with the multi-angle laser light scattering of the sample. This enables the weight average molecular weight (Mw) of species in the eluate to be calculated continuously. Samples of DnaADI and SirA–DnaADI were analysed at a series of protein concentrations. As shown in Fig. 1A, DnaADI elutes from the size exclusion column as a single A280 peak at ∼ 27.5 min and has an experimentally determined molecular mass of ∼ 11 kDa. This suggests that DnaADI from B. subtilis is a monomer (calculated molecular mass = 9.7 kDa) in contrast to E. coli DnaADI which is reported to form dimers under similar conditions (Abe et al., 2007).
Figure 1.
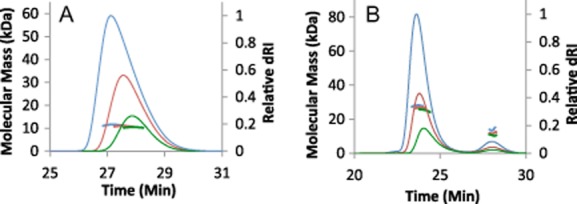
Molecular mass measured from SEC-MALLS analysis. In A and B, the thinner lines trace the differential refractive index of the eluate from a Superdex 10/30 S75 column as a function of time. The thicker lines represent the weight average molecular weight of the species in the eluate, calculated from refractive index and light-scattering measurements.
A. Overlay of chromatograms for DnaADI at 3 concentrations: 1 mg ml−1 (green), 2.5 mg ml−1 (red) and 5 mg ml−1 (blue), revealing species of mass 11 kDa indicating that DnaADI is a monomer.
B. Overlay of chromatograms for SirA–DnaADI at 3 concentrations: 0.5 mg ml−1 (green), 1.0 mg ml−1 (red) and 2.5 mg ml−1 (blue). The derived Mw values for the principal species are 25–28 kDa indicating that the SirA–DnaADI complex is a 1:1 heterodimer. There is evidently, excess/dissociated DnaADI giving rise to the minor peak eluting at ∼28 min.
The SirA–DnaADI elution profile has two peaks: a major peak at ∼ 24 min and a minor peak at ∼ 28 min (Fig. 1B). The minor peak comprises 8–12% of the total protein content and the analysis above suggests this is monomeric DnaADI. The major peak (88–92% of the total protein content), which corresponds to a molecular mass of 25–28 kDa, is consistent with a 1:1 heterodimer of SirA : DnaADI (calculated molecular mass = 28.7 kDa). A discernible shift in the relative sizes of the major and minor peaks occurs as the protein concentration changes in these experiments. There is an increasing area under the minor peak as the protein concentration is lowered, accompanied by a small shift in the elution time associated with the major peak and by a decrease in its associated molecular mass at lower concentrations. This is consistent with increasing complex dissociation at lower protein concentrations. It is clear, however, that under these experimental conditions the SirA–DnaADI heterodimer is the predominant species.
The crystal structure of the SirA–DnaADI complex
The crystal structure of SirA–DnaADI was solved to 1.7 Å resolution using single anomalous dispersion (SAD) phasing techniques (Table 1). Native and selenomethionine-substituted SirA–DnaADI crystals grew under different conditions from PEG 3350 containing solutions (see Experimental procedures). Although both crystals belong to space group P21, the crystals are different (Table 1). SeMet-derivative crystals contain one complex per asymmetric unit (one molecule of SirA, one molecule of DnaADI) while the asymmetric unit of the native crystals contains two complexes. The SeMet structure was solved and partially refined to allow solution of the native structure by molecular replacement. The refined model encompasses residues 2–141 of SirA (Met1 and residues 142–148 being disordered) in both molecules A and C. Residues 1–81 of DnaADI are defined in molecules B and D (the C-terminal Gln82 being disordered). A vestigial N-terminal Gly-Pro-Ala sequence inherited from the DnaADI polyhistidine-tag can be seen in molecule D, and an additional N-terminal Ala is visible in molecule B. SirA–DnaADI is seen as a heterodimer, with molecules A (SirA) and B (DnaADI) forming one heterodimer and molecules C and D forming the other. The electron density maps reveal a 2-mercaptoethanol (BME) molecule linked through a disulphide bond to Cys125 of both SirA chains in the asymmetric unit (Fig. S1). The presence of this adduct explains species observed in the electrospray ionization mass spectrum of SirA of 18 776 Da, 79 Da larger than that of SirA; 18 697 Da. BME was present during the purification steps as it was found to improve the solubility of the DnaADI–SirA complex.
Table 1.
X-ray data collection and refinement statistics
| SirA–DnaADI SeMet | SirA–DnaADI Native I | |
|---|---|---|
| Data collection | ||
| X-ray source | DLS, i24 | DLS, i03 |
| Wavelength (Å) | 0.9789 | 0.9763 |
| Resolution range (Å) | 40.8–2.09 | 62.76–1.65 |
| Space group | P21 | P21 |
| Unit cell parameters | ||
| a, b, c (Å) | 51.35, 35.63, 63.27 | 77.29, 34.69, 84.74 |
| α = γ, β (°) | 90, 92.77 | 90, 102.09 |
| No. of unique reflectionsa | 13 549/989 | 52 893/2598 |
| Completeness (%)a | 98.7/99.1 | 98.9/99.6 |
| Redundancya | 3.2/3.3 | 2.8/2.8 |
| I/σ(I)a | 11.9/1.9 | 12.5/1.9 |
| Rmergeb (%)a | 7.4/79.9 | 3.9/45.8 |
| Refinement and model statistics | ||
| Resolution range (Å) | 62.84–1.65 | |
| R-factorc (Rfreed) | 13.1 (19.7) | |
| Reflections (working/Rfree) | 50172/2706 | |
| Outer-shell/high resolution range | 1.69–1.65 | |
| Outer-shelle/high resolution R-factorc (Rfree)d | 19.0 (27.9) | |
| Outer-shell/high resolution reflections (working/free) | 3677/214 | |
| Molecules per asymmetric unit | 4 | |
| rmsd from ideal geometryf | ||
| Bond lengths (Å) | 0.017 | |
| Bond angles (°) | 1.8 | |
| Average B-factor (Å2) | 27.8 | |
| Ramachandran plotg | 98.16/0.92/0.92 |
The first number refers to the overall data set, the second refers the outer resolution shells; Native: 1.68-1.65 Å; SeMet: 2.15-2.09 Å.
Rmerge = ∑hkl∑i|Ii − < I > |/∑hkl∑i < I > where Ii is the intensity of the ith measurement of a reflection with indexes hkl and < I > is the statistically weighted average reflection intensity.
R-factor = ∑||Fo| − |Fc||/∑|Fo| where Fo and Fc are the observed and calculated structure factor amplitudes respectively.
R-free is the R-factor calculated with 5% of the reflections chosen at random and omitted from refinement.
Outer shell for refinement corresponds to 1.69–1.65 Å.
Root-mean-square deviation of bond lengths and bond angles from ideal geometry.
Percentage of residues in most-favoured/allowed/disallowed regions of the Ramachandran plot.
SirA consists of a single globular domain comprising seven β-strands and five α-helices in the order β1-α1-α2-α3-α4-β2-β3-β4-β5-α5-β6-β7 (Fig. 2A and C). The SirA fold consists of a central seven-stranded twisted β-sheet with strand order β2-β3-β4-β5-β1-β6-β7, flanked on either side by two α-helical regions, one comprising helices α1, α2 and α3 and the other of helices α4 and α5. Comparative analysis of the SirA chain topology using PDBeFold identified the kinase associated domain 1 from the protein KCCP4 (PDB entry 3osm) as the highest Q-scoring hit with 79 Cα atoms overlaying with a positional root mean squared deviation of 2.4 Å. This domain has been identified as a membrane association domain that binds acidic phospholipids (Moravcevic et al., 2010). The region of structural similarity spans residues 2–9 and 53–124 covering the β1-α4-β2-β3-β4-β5-α5 segment. The other highest scoring matches, the core domain of the human ribosomal protein L10 (2pa2) and the yeast mitochondrial protein frataxin (4ec2), exhibit structural similarity to the same region of SirA.
Figure 2.

The structure of the SirA–DnaADI complex.
A and B. Ribbon diagram of the SirA (A) and DnaADI (B) chains from the complex. In each case, the chain is colour-ramped from its N-terminus (red) to the C-terminus (magenta) and the secondary structure elements are labelled. These and subsequent structure figures were produced using the program CCP4mg (McNicholas et al., 2011).
C. The polypeptide chain topology in SirA.
D. Ribbon diagram of the SirA–DnaADI complex with SirA shown in light green and DnaADI shown in blue.
DnaADI from B. subtilis (Fig. 2B) consists of four alpha helices and three beta strands in the order α1-α2-β1-β2-α3-α4-β3 with a β-sheet topology of β1-β2-β3. It has a closely similar topology to the previously determined structures of the corresponding domains of DnaA from E. coli (Abe et al., 2007), M. genitalium (Lowery et al., 2007) and H. pylori (Natrajan et al., 2009). Thus, it shares the K homology domain motif that is widespread in single-stranded nucleic acid binding proteins.
The SirA–DnaADI interface
The binding of SirA and DnaADI is mediated by helices α1, α2 and α3 of SirA and α2 and α3 of DnaADI (Figs 2D and 3A). As an extensively α-helical interface the interactions of the two proteins are dominated by side-chain–side-chain contacts (Fig. 3A). Seventeen residues on each chain contribute to the interface which constitutes 12% and 8% of the surface areas of DnaADI and SirA, respectively. In the complex, 1240 Å2 of otherwise accessible surface area is buried in the interface. This buried surface area is at the lower end of the range observed in non-obligate dimeric protein-protein complexes (Janin et al., 2008). In the SirA binding surface of DnaADI, Thr26, Trp27 and Phe49 contribute to the core of the interface with residues Pro22, Ser23, Glu25, Ser30, Pro46, Asn47, Glu48, Asp52, Ser56 and Trp53 prominent in the rim. As shown in Fig. 3C, these residues are very strongly conserved in a set of DnaA orthologues from endospore-forming bacteria. On the corresponding DnaADI binding surface of SirA, Phe14, Tyr18, Gln48 and Ile52 contribute to the core and Glu13, His17, Val24, Leu28, Gln41, Met44, Lys47 and Tyr51 are prominent in the rim. Again core residues are well conserved with some variation observed in the residues constituting the rim (Fig. 3D).
Figure 3.

The SirA–DnaADI interface.
A. Stereoview of the complex between DnaADI (chain D) and SirA (chain C) represented as light blue and light green ribbons, respectively. Side-chains of labelled residues are displayed in cylinder format and coloured by atom type with nitrogen (blue), oxygen (red), sulphur (yellow) and carbons coloured in grey for DnaADI and green for SirA. Water molecules are represented as red spheres, and polar interactions are denoted by dashed lines.
B. Mapping onto the structure of DnaADI the sites corresponding to mutations in dnaA that allow growth of B. subtilis even when sirA is being overexpressed (Rahn-Lee et al., 2011). SirA is rendered as a partially transparent electrostatic surface and DnaADI as a ribbon with the side-chains of residues Asn47, Phe49 and Ala50 in cylinder format.
C and D. Alignment of the sequences of orthologues of DnaADI (C) and SirA (D) from selected Bacillus species; Bsu, B. subtilis; Blic, B. licheniformis; Bant, B. anthracis; Bhal, B. halodurans; Bcla, B. clausii; Gkau, Geobacillus klaustophilus; Oihe, Oceanobacillus iheyensis. Symbols below the alignments indicate interfacial residues in the respective molecules contributing to the core (asterisks) and the rim (triangles). Secondary structure elements and residue numberings are displayed above the alignment. The images were created using ESPript (Gouet, 2003).
The SirA binding determinants of DnaA have previously been explored using genetic approaches. Induction of sirA expression under nutrient rich conditions inhibits growth of B. subtilis. Four strains were identified which harbour mutations in dnaA that were able to suppress this slow growth phenotype accompanying sirA induction (Rahn-Lee et al., 2011). Analysis of the sequence of these dnaA alleles revealed point mutations giving rise to Asn47Asp, Phe49Tyr, Ala50Val and Ala50Thr substitutions (Rahn-Lee et al., 2011). Yeast two hybrid analysis confirmed that mutations at these residues prevent DnaA from interacting with SirA, suggesting they affect SirA–DnaADI complex formation (Rahn-Lee et al., 2011). The SirA–DnaADI structure reveals that Asn47 and Phe49 make direct interactions with SirA (Fig. 3A and B). Asn47 of DnaA forms a pair of hydrogen bonds with Gln48 on helix α3 of SirA, while the side-chain of Phe49 of DnaA projects into a hydrophobic pocket created by helices α1, α2 and α3 of SirA. In contrast, Ala50 does not contact SirA in the complex; instead it is buried within DnaADI in such a way that it determines the structure of the interface (Fig. 3B). It is expected that mutations at this position which introduce bulkier side-chains, such as valine or threonine, will alter the structure of the interaction surface leading to lower affinity binding of SirA. In summary, the structure of SirA–DnaADI confirms previous interpretations of the genetic data.
Substitutions at the SirA–DnaADI interface affect the SirA–DnaA interaction in vitro
To confirm the importance of the protein-protein interface observed in the SirA–DnaADI crystals, we assayed the interactions of site-directed mutants of sirA and dnaADI in vitro. Mixing experiments using the purified proteins are not possible because we are unable to produce soluble SirA in the absence of coexpression with DnaADI. Instead we took advantage of the dependence of SirA solubility on its co-production with, and binding to, DnaADI in developing a qualitative binding assay. We hypothesized that disruption of the interaction between SirA and DnaADI following coexpression would reduce or abolish SirA solubility.
Site-directed mutagenesis was used to introduce alanine substitutions into the pET-YSBLIC3C-DnaADI SirA coexpression vector at codons that specify the SirA–DnaADI interface in the crystal structure. Three alanine substitutions were introduced into SirA: Phe14Ala, Tyr18Ala and Gln48Ala. These residues contribute 45 Å2, 45 Å2 and 80 Å2 of buried surface area respectively to the interface with the phenolic hydroxyl of Tyr18 forming a charge-dipole interaction with Asp52 of DnaA and the side-chain amide of Gln48 forming a pair of hydrogen bonds with the amide of Asn47 of DnaA (Fig. 3A). A further three substitutions were introduced into DnaADI, these being Thr26Ala, Trp27Ala and Phe49Ala. Thr26, Trp27 and Phe49 contribute 75 Å2, 50 Å2 and 135 Å2 of surface area respectively to the interface. After sequencing to confirm the presence of the mutations, the mutated plasmids were introduced into E. coli BL21 and expression experiments were performed. The solubility of the recombinant proteins was compared to that of the wild-type proteins by SDS-PAGE of cell fractions following lysis (Fig. S2).
These experiments show that there are reduced levels of SirA in the soluble lysate fractions (labelled S in Fig. S2) of cells producing SirAF14A and SirAY18A and negligible levels of SirA in these fractions from cells producing DnaADI,W27A and DnaADI,F49A. This suggests weaker binding of SirA by DnaADI. However, interpretation of these experiments is complicated by variability in the levels of DnaADI present in these fractions. Thus, the effect of each mutation on the interaction of SirA with DnaADI was further probed using a pull down assay where the soluble fraction of the cell lysate was loaded onto a Ni-affinity column. The latter was washed extensively with loading buffer and bound proteins were eluted in a buffer containing a high concentration of imidazole. pET-YSBLIC3C-DnaADISirA directs expression of DnaADI with a hexahistidine tag together with untagged SirA. Thus, any retention of SirA is expected to result from its interaction with the histidine-tagged DnaADI. The eluate (E) fractions shown in Fig. S2 were diluted to normalize to an approximately equivalent amount of DnaADI and the samples again resolved by SDS-PAGE with Coomassie staining. As can be seen in Fig. 4A, the DnaADI,W27A and DnaADI,F49A mutations have the most striking effect, with little discernable SirA eluted from the Ni-NTA column in the high imidazole fraction. In marked contrast, DnaADI,T26A supports wild-type levels of SirA recovery after the nickel pull down. For the three SirA mutant proteins, the effects are more modest. Quantification of the DnaADI and SirA band intensities in Fig. 4A using the software ImageJ revealed, relative to the wild type SirA, 1.5-fold lower recovery of SirAF14A and SirAQ48A and a 2.5-fold lower recovery of SirAY18A.
Figure 4.
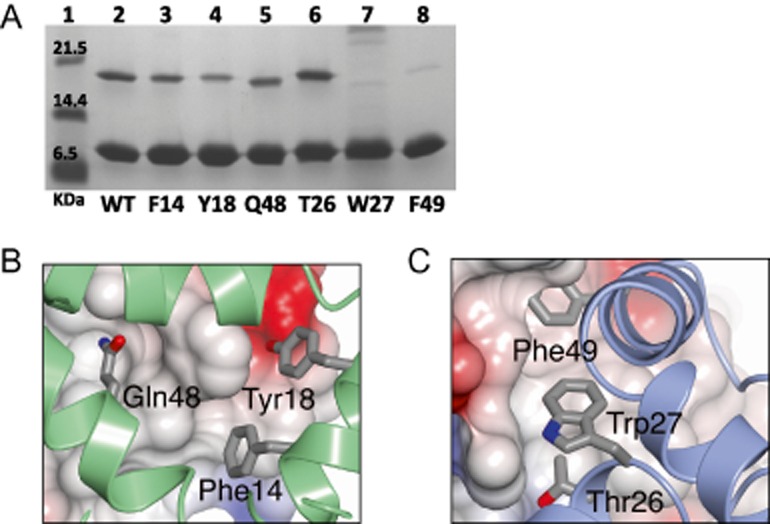
The SirA–DnaADI interface analysed by site-directed mutagenesis.
A. SDS-PAGE. Cultures of cells harbouring plasmids encoding wild type and alanine-substituted variants of His-tagged DnaADI and SirA were grown. Soluble cell lysates were prepared and loaded onto a Ni-NTA column. High imidazole eluate fractions were collected for analysis. Samples of the eluate fractions containing approximately normalized levels of DnaADI were loaded so that the efficiency of SirA pull-down could be compared. Lane 1 contains molecular weight markers. Lane 2: wild type DnaADI–SirA. Lanes 3–5: Native DnaADI and the SirA variants, loaded as follows; Lane 3: SirA(F14A), Lane 4: SirA(Y18A), Lane 5: SirA(Q48A). Lanes 6–8: Samples of native SirA and the DnaADI variants, loaded as follows; Lane 6: DnaADI(T26A), Lane 7: DnaADI(W27A), Lanes 8: DnaADI(F49A).
B and C. Core residues from the DnaA DI-interacting surface of SirA (B) and the SirA interacting surface of DnaADI (C) which were sites of alanine substitution. (B) DnaADI is shown as an electrostatic surface with SirA represented as a green ribbon with the side-chains of F14, Y18 and Q48 displayed as cylinders. (C) SirA is shown as an electrostatic surface with DnaADI represented as a blue ribbon with the side-chains of T26, W27 and F49 displayed as cylinders.
Collectively, these results correlate with the SirA–DnaA interface in the crystal structure. Residues Phe14, Trp18 and Gln48 of SirA form contacts with DnaADI which would be weakened upon truncation of these side-chains to alanine (Fig. 4B). The side-chains of residues Trp27 and Phe49 in DnaADI project away from the surface of the protein into a hydrophobic groove on the SirA surface, forming extensive van der Waals contacts across the SirA–DnaA interface (Fig. 4C). The results indicate that truncation of either of these large hydrophobic residues strongly affects the SirA–DnaA interaction due to the loss of these contacts. By contrast, Thr26 of DnaADI binds at the edge of a hydrophobic groove and although it is largely buried, its hydroxyl is able to form a hydrogen bond to a recessed water molecule on the protein surface. Moreover, substitution of threonine with alanine is a less drastic change, evidently allowing DnaADI, T26A to maintain its interaction with SirA.
GFP–SirA colocalizes with the replisome in sporulating cells
The biochemical analysis of SirA–DnaADI complex formation provides strong evidence that the interaction observed in the crystal structure is also formed between the two proteins in solution. To study the physiological relevance of the proposed SirA–DnaA interface, SirA activity was examined in vivo. Visualization of SirA was achieved by replacing the endogenous gene with gfp–sirA (expressed from its native transcriptional and translational regulatory sequences; Fig. 5A), inducing cells to sporulate by nutrient deprivation, and detecting the localization of GFP–SirA using epifluorescence microscopy (Fig. 5B). A time-course experiment showed that GFP–SirA foci began to appear approximately 90 min after cells were resuspended in starvation medium. By 150 min, the number of cells containing a GFP–SirA focus reached its maximum (∼ 20%; Fig. 5C). In the majority of cases (> 80%) GFP–SirA foci were located near mid-cell. Previous studies suggested that SirA inhibits new rounds of DNA replication by inhibiting the binding of DnaA to the origin of replication (Wagner et al., 2009). However, the mid-cell localization of GFP–SirA foci is contrary to origin positioning in sporulating cells where the two origins are positioned towards the cell poles, suggesting that SirA was not accumulating at oriC.
Figure 5.

Localization of GFP–SirA in vivo.
A. Schematic diagram showing the modified sirA locus used for localization studies. Chromosomal sirA was replaced with gfp–sirA under the control of its native expression system.
B. GFP–SirA localization during sporulation of B. subtilis 150 min post resuspension in starvation media. Membrane dye FM5-95 was used to highlight the outlines of the cells. Scale bar = 3 μm. gfp–sirA (NR3).
C. Temporal analysis of GFP–SirA foci formation during sporulation. At least 500 cells were analysed at each time-point.
D. Colocalization of GFP–SirA with oriC during sporulation (150 min post resuspension in starvation media). More than 200 cells were analysed and a representative image is shown. Scale bar = 3 μm. gfp–sirA oriCtetO/TetR−mCherry (NR164).
E. Colocalization of GFP–SirA with the replisome during sporulation (150 min post resuspension in starvation media). More than 100 cells were analysed and a representative image is shown. Scale bar = 3 μm. gfp–sirA dnaN–mCherry (NR168).
In order to further investigate GFP–SirA localization we constructed a strain that allowed visualization of both origin regions and SirA. An array of tet operators was inserted near oriC and the Tet repressor was fused to a red fluorescent protein (TetR-mCherry); interaction of TetR-mCherry with the tetO array produces a fluorescent focus near each origin of replication. Cells were induced to sporulate by nutrient deprivation and the localization of GFP–SirA was determined in respect to the origin regions. In the majority of cells with a GFP–SirA focus there was no colocalization of SirA with oriC (78% with non-overlapping signals; Fig. 5D). Only 8% of the GFP–SirA foci appeared to colocalize with the origin regions, with the remaining 14% of cells containing a GFP–SirA focus that partially overlapped with the origin marker (Fig. 5D). These results show that during sporulation GFP–SirA mainly accumulates away from the replication origin.
A previous study in B. subtilis reported that DnaA colocalizes with the replisome at mid-cell via the YabA–DnaN complex during DNA replication (Soufo et al., 2008). We hypothesized that SirA might be interacting with DnaA when it is bound to the replication machinery. To begin testing this model we examined GFP–SirA localization in cells that contained a fusion of a red fluorescent protein to the sliding clamp of the replisome (DnaN–mCherry). Cells were induced to sporulate by nutrient deprivation and the localization of GFP–SirA was determined in respect to the replisome. Strikingly, in cells containing a GFP–SirA focus the vast majority colocalized with DnaN–mCherry (88%; Fig. 5E). This result indicates that GFP–SirA accumulates at the replisome.
Substitutions at the SirA–DnaADI interface affect GFP–SirA localization in vivo
To investigate whether the localization of GFP–SirA at the replisome was dependent upon an interaction with DnaA, first the wild-type sirA (from the gfp–sirA chimera) was replaced with sirA mutants altering the residues identified in the SirA–DnaADI structure implicated in complex formation (gfp–sirAF14A, gfp–sirAY18A or gfp–sirAQ48A) and the localization GFP–SirA proteins was determined during sporulation. All of the sirA mutants caused a significant decrease in the number of cells containing a fluorescent focus (Fig. 6A). Both gfp–sirAF14A and gfp–sirAY18A mutants reduced the number of GFP foci to background levels (i.e. – in the absence of a GFP fusion), while the gfp–sirAQ48A mutant decreased foci formation 2.5-fold. These results indicate that the amino acid residues in SirA identified in the structure at the interface with DnaADI are required for GFP–SirA localization at the replisome.
Figure 6.
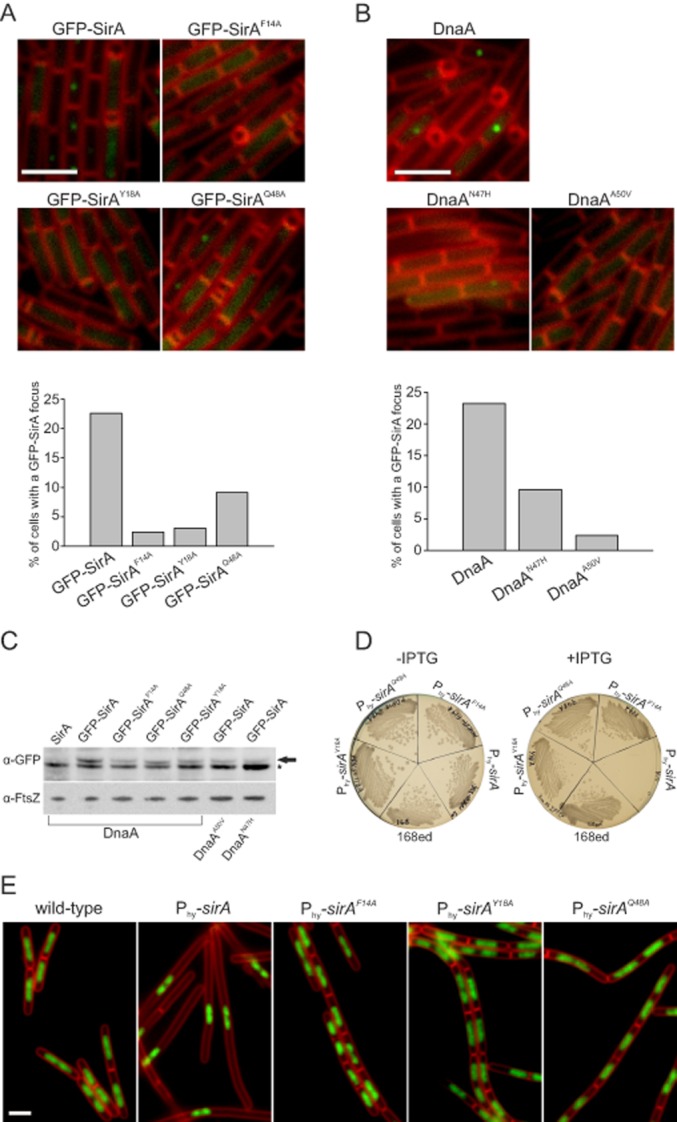
Examination of the SirA–DnaA interface in vivo.
A. Amino acid substitutions in SirA inhibit GFP–SirA foci formation. For each strain over 700 cells were analysed and the experiment was repeated at least three times. Quantification of a representative dataset is shown below. Scale bar = 3 μm. gfp–sirA (NR3); gfp–sirAF14A (NR130); gfp–sirAY18A (NR156); gfp–sirAQ48A (NR131).
B. Amino acid substitutions in DnaA inhibit GFP–SirA foci formation. For each strain over 700 cells were analysed and the experiment was repeated at least three times. Quantification of a representative dataset is shown below. Scale bar = 3 μm. gfp–sirA (NR3); gfp–sirA dnaAA50V (NR5); gfp–sirA dnaAN47H (NR154).
C. Immunoblot analysis showing levels of GFP-tagged SirA proteins. Cell samples were taken 150 min post resuspension in starvation media. The arrow points to GFP–SirA, the star highlights a contaminating band. Immunoblot of FtsZ was utilized to standardize the amount of protein from different samples. Wild-type (168ed); gfp–sirA (NR3); gfp–sirAF14A (NR130); gfp–sirAY18A (NR156); gfp–sirAQ48A (NR131); gfp–sirA dnaAA50V (NR5); gfp–sirA dnaAN47H (NR154).
D. Wild-type and mutant sirA were placed under control of an IPTG-inducible promoter and streaked on nutrient agar plates in the presence and absence of IPTG (3 mM). Wild-type (168ed); Phyperspank-sirA (NR171); Phyperspank-sirAF14A (NR172); Phyperspank-sirAQ48A (NR173); Phyperspank-sirAY18A (NR174).
E. The effects of overexpressing various SirA proteins at the single cell level were studied by growing cells in liquid CH medium. The images were taken 180 min after induction of gene expression with IPTG (3 mM). Membrane dye FM5-95 was used to highlight the outlines of the cells, DAPI was used to stain the DNA. Scale bar = 3 μm. Wild-type (168ed); Phyperspank-sirA (NR171); Phyperspank-sirAF14A (NR172); Phyperspank-sirAQ48A (NR173); Phyperspank-sirAY18A (NR174).
Next we attempted to replace dnaA with dnaAT26A, dnaAW27A and dnaAF49A; however, we were unable to isolate any of these mutants (see Discussion). In contrast, mutations in dnaA at locations that were previously shown to inhibit SirA activity in vivo (dnaAA50V and dnaAN47H) could be readily generated; therefore, GFP–SirA localization was determined using these dnaA alleles. Figure 6B shows that both DnaA variants inhibited GFP–SirA foci formation, with DnaAA50V reducing foci formation to background levels and DnaAN47H decreasing foci formation 2.4-fold. Immunoblot analysis of GFP-SirA proteins showed similar levels in all mutants tested, indicating that the absence of foci formation was not due to altered protein expression (Fig. 6C). Taken together with the analysis of the sirA mutants, these results show that the SirA–DnaADI interface identified in the crystal structure is critical for GFP–SirA localization in vivo and they suggest that GFP–SirA localization is mediated through a direct interaction with replisome-bound DnaA.
Substitutions at SirA–DnaADI interface render cells resistant to lethal effects of SirA overexpression in vegetatively growing cells
We attempted to determine whether GFP–DnaA colocalized with the replisome during sporulation, but unfortunately the previously published gfp–dnaA reporter strain displayed a severe sporulation defect (Soufo et al., 2008). Therefore, to test whether SirA variants that displayed decreased foci formation were also defective in DnaA regulation, SirA proteins were overexpressed. Wild-type and mutant sirA genes were placed under the control of an IPTG-inducible promoter (Phyperspank) integrated at an ectopic locus. Induction of wild-type SirA inhibited cell growth on solid media, in contrast to the SirA variants (SirAF14A, SirAY18A, SirAQ48A; Fig. 6D). Induction of wild-type SirA during vegetative growth in liquid media inhibited DNA replication, producing elongated cells that contained a single nucleoid (Fig. 6E). Induction of SirA variants did not affect DNA distribution or cell morphology, and these cells were indistinguishable from a control strain lacking the ectopic sirA construct (Fig. 6E). These results show that amino acid residue substitutions in SirA that impair protein localization also affect the ability of SirA to inhibit DnaA activity.
A conserved binding site on DnaADI
The structure of the SirA–DnaADI complex and that formed between DnaADI from H. pylori and HobA (Natrajan et al., 2009), a regulator of DNA replication in this pathogen, were compared with each other (Fig. 7). It is apparent that HobA and SirA bind to the same structural site on DnaADI, burying equivalent surface residues (Fig. 7A and B). This is surprising given the divergent effects on DnaA exerted by SirA and HobA. HobA is an essential stimulator of replication initiation in H. pylori, in contrast to SirA which is a replication inhibitor. Thus, HobA and SirA achieve opposing regulatory functions by binding to the same structural site on DnaADI. Despite their close tertiary structural correspondence, residues on the regulatory protein binding site on DnaADI are poorly conserved between H. pylori and B. subtilis, perhaps reflecting a divergence in their respective regulatory mechanisms (Fig. 7C). Nevertheless, this indicates an important structural site on DnaADI for the regulation of replication initiation.
Figure 7.
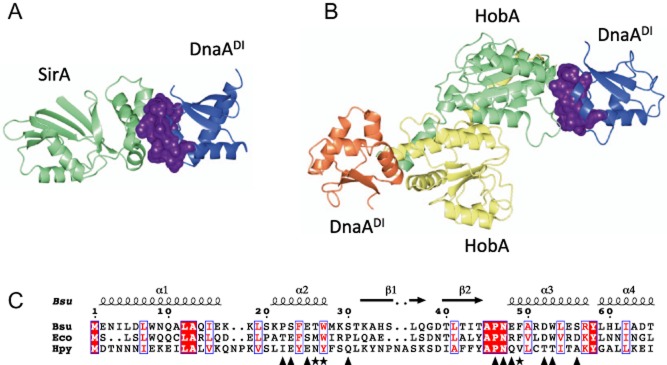
Comparison of B. subtilis SirA–DnaADI with H. pylori HobA-DnaADI.
A. Ribbon diagram of the B. subtilis SirA–DnaADI complex with SirA shown in light green and DnaADI shown in blue.
B. Ribbon diagram of the H. pylori HobA-DnaADI complex (PDB id code: 2wp0) with two molecules of HobA shown in yellow and light green, and two molecules of DnaADI shown in blue and coral. The SirA and HobA binding surfaces on DnaADI in (A) and (B) respectively are shown in purple.
C. Alignment of DnaADI from B. subtilis, E. coli and H. pylori. Symbols below the alignments indicate interfacial residues on DnaADI in the SirA–DnaADI structure contributing to the core (asterisks) and the rim (triangles). Secondary structure elements and residue numberings are displayed above the alignment. The images were created using ESPript (Gouet, 2003).
Discussion
The initiator of bacterial DNA replication, DnaA, is stringently regulated so that DNA replication is co-ordinated with cell growth and differentiation. Five negative regulators of DNA replication have been identified in B. subtilis: SirA (Rahn-Lee et al., 2009), YabA (Noirot-Gros et al., 2006), Spo0A (Castilla-Llorente et al., 2006) Soj (Murray and Errington, 2008) and DnaD (Bonilla and Grossman, 2012). YabA, Soj and DnaD bind to domain III of DnaA and are thought to block the assembly of helical DnaA filaments at oriC (Cho et al., 2008; Scholefield et al., 2012; Scholefield and Murray, 2013). Phosphorylated Spo0A binds to a set of Spo0A-boxes at oriC which overlap with DnaA-boxes, suggesting that Spo0A∼P occludes DnaA from the replication origin (Boonstra et al., 2013). SirA is distinct and represents the first, and so far only, B. subtilis regulator that interacts with domain I of DnaA (Rahn-Lee et al., 2011). In other organisms however, regulators have been identified which interact with DnaADI namely, E. coli DiaA (Keyamura et al., 2009) and Hda (Su'etsugu et al., 2013), and H. pylori HobA (Natrajan et al., 2009). For E. coli, DnaADI has also been shown to interact with the DNA helicase, DnaB (Sutton et al., 1998; Seitz et al., 2000), and to play a role in the oligomeriation of DnaA (Weigel et al., 1999; Felczak et al., 2005), forming dimers in vitro (Abe et al., 2007). Here we have elucidated the structure of the SirA–DnaADI from B. subtilis revealing DnaADI bound in an inhibitory complex. This structure complements that of HobA-DnaADI from H. pylori in which DnaADI is bound in a complex that leads to activation (Natrajan et al., 2009).
As previously inferred (Rahn-Lee et al., 2011), SirA binds to a site on DnaADI that corresponds closely to that bound by the regulators HobA from H. pylori (Natrajan et al., 2009) and DiaA from E. coli (Keyamura et al., 2009). HobA and DiaA are structural homologues which form tetramers that promote DnaA oligomerization and activate the initiation of DNA replication (Zawilak-Pawlik et al., 2011). Each HobA/DiaA tetramer binds to four DnaADI molecules in a way that is thought to facilitate DnaA-binding to the array of DnaA-boxes distributed at oriC (Natrajan et al., 2009). In marked contrast, SirA binds a single molecule of DnaADI and inhibits DNA replication initiation. Although SirA and HobA/DiaA have quite different three dimensional structures, each buries a structurally equivalent site on DnaADI. It is intriguing therefore that this elicits different regulatory outcomes.
It has been previously proposed that SirA inhibits DnaA binding to oriC, based on the observations that SirA disrupts DnaA–GFP localization at oriC during vegetative growth, and that there is a SirA-dependent decrease in the amount of DnaA at oriC following artificial induction of sporulation (Wagner et al., 2009; Rahn-Lee et al., 2011). At first glance, SirA may achieve this by inhibiting DnaA-oligomerization at oriC, since domain I fragments of E. coli DnaA form dimers in vitro, and the dimerization surface has been identified (Felczak et al., 2005). However, the corresponding surface in B. subtilis DnaADI is located on the opposite side of DnaADI to the SirA binding surface so that SirA binding would not be expected to prevent dimer formation. Furthermore, we did not observe dimers or oligomers of B. subtilis DnaADI in vitro. Thus, it seems unlikely that SirA influences DnaA assembly simply by inhibiting DnaADI-DnaADI interactions.
Our localization studies indicate that SirA accumulates away from oriC and with the replisome near mid-cell during sporulation. We hypothesize that SirA could interact with replisome-bound DnaA to stabilize replisome–DnaA complexes, thereby inhibiting DnaA rebinding at the origin. This is reminiscent of a DnaA-tethering model proposed for YabA (Soufo et al., 2008). Alternatively, our finding that mutations directing alanine substitutions of three residues on the SirA binding surface of DnaA could not be introduced into dnaA suggests that SirA could act by inhibiting a critical interaction of DnaA with other components of the initiation complex. In E. coli, DnaADI is implicated directly in the recruitment of the helicase to the nascent replicative complex (Abe et al., 2007). There is no evidence for a DnaA-helicase interaction in B. subtilis however, in which helicase recruitment involves additional DNA remodelling proteins (Zhang et al., 2005). DnaD forms multimeric scaffolds on the DNA and recruits DnaB, which in turn is thought to bridge an interaction with the helicase–helicase loader (Zhang et al., 2008; Smits et al., 2010). Thus, we speculate that SirA may inhibit a DnaA–DnaD interaction arresting assembly of the initiation complex.
In summary, this work has defined the interaction surfaces of SirA and DnaADI and the stoichiometry of their complex. Moreover, we have shown that their interaction is required for GFP-SirA foci formation at the replisome in sporulating B. subtilis. These observations will help elucidate the mechanism of action of SirA, the understanding of which is currently limited by imprecise knowledge of the function of domain I of DnaA in this organism.
Experimental procedures
Cloning
DNA fragments encoding SirA and domain I of DnaA (DnaADI) were amplified from B. subtilis genomic DNA by the polymerase chain reaction (PCR) and inserted into the expression vector pET-YSBLIC3C (Fogg and Wilkinson, 2008) using a ligation independent cloning method. Two constructs were created: one encoding SirA with an N-terminal, 3C protease cleavable His-tag (pET-YSBL3C-SirA) and the other a duet construct containing fragments encoding DnaADI and SirA cloned upstream and downstream respectively of a LIC Duet Minimal Adaptor (Novagen). The recombinant plasmid (pET-YSBLIC3C-DnaADISirA) directs the expression of DnaADI fused to a 3C protease cleavable N-terminal His-tag (His–DnaADI) and SirA from separate bacteriophage T7 promoters. Alanine substitution mutations were introduced into pET-YSBLIC3C-DnaADISirA by site-directed mutagenesis. The sequences of oligonucleotides used for the cloning and subsequent site-directed mutagenesis of pET-YSBLIC3C-DnaADISirA are listed in Tables S1 and S2 respectively.
Expression
The plasmid pET-YSBLIC3C-DnaADISirA was introduced into E. coli BL21 (DE3) cells for the co-overproduction of SirA and His-tagged DnaADI. Overnight cell cultures were used to inoculate 500 ml of Luria–Bertani (LB) media supplemented with 30 μg ml−1 kanamycin. Cultures were grown to an OD600 of 0.6–0.9 at 37°C with shaking at 180 rpm before protein production was induced with 1 mM isopropyl-β-d-1-thiogalactoside (IPTG). Following induction, cultures were grown at 37°C (180 rpm shaking) for a further 4 h before cells were harvested by centrifugation. SirA/DnaADI proteins harbouring site-directed mutations were produced in an analogous manner.
For SeMet substituted protein production, overnight cultures of E. coli BL21 (DE3) harbouring pET-YSBLIC3C-DnaADISirA were used to inoculate 500 ml minimal media supplemented with 30 μg ml−1 kanamycin. Cultures were grown to an OD600 of 0.6–0.8 at 37°C (180 rpm shaking) prior to the addition of an amino acid mixture (50 mg lysine, 50 mg phenylalanine, 50 mg threonine, 25 mg isoleucine, 25 mg leucine, 25 mg valine) to suppress methionine production (Doublié, 1997), and 30 mg selenomethionine. Cultures were grown at 37°C (180 rpm shaking) for a further 15 min prior to induction of recombinant protein production with 1 mM IPTG. Cultures were subsequently grown at 30°C (180 rpm shaking) overnight (16–20 h) before cells were harvested by centrifugation.
DnaADI–SirA purification
The protein purification procedure was identical for native and SeMet substituted proteins. Harvested cells were resuspended in 50 mM Tris pH 8.5, 200 mM KCl, 20 mM imidazole and 10 mM BME, and an EDTA-free protease inhibitor cocktail tablet (Roche) was added. Resuspended cells were lysed by sonication and the lysate clarified by centrifugation. The cell lysate was loaded on to a His Trap FF crude Ni-affinity column (GE Healthcare) and bound protein eluted with an increasing imidazole concentration gradient (20–500 mM). This step was followed by size-exclusion chromatography on a HiLoad 16/60 Superdex S75 column (GE Healthcare) equilibrated with 50 mM Tris pH 8.5, 200 mM KCl, 10 mM BME. The chromatogram exhibited two protein peaks. SDS-PAGE analysis of the peak fractions showed the earlier eluting peak corresponded to the SirA : His–DnaADI complex with the later eluting peak containing His–DnaADI which is produced in excess in the duet expression system. The protein complex and DnaA domain I fractions were combined separately and the N-terminal histidine tag was removed from DnaADI in both cases by incubation with 3C protease overnight (protease : protein ratio of 1:50). Passage through a second Ni-affinity column to remove the histidine tag and protease yielded pure protein in a buffer of 50 mM Tris pH 8.5, 200 mM KCl, 10 mM BME. The proteins were judged to be pure according to Coomassie staining of SDS-polyacrylamide gels.
Crystallization and structure solution
Native crystals of SirA–DnaADI were grown in hanging-drops containing a 1:1 ratio of concentrated protein solution and reservoir solution. Native crystals suitable for data collection were obtained following mixing of a protein solution of 6.4 mg ml−1 and a reservoir solution of 100 mM HEPES pH 7.5, 200 mM NH4 acetate, 25% (w/v) PEG 3350, 1% (v/v) DMF. Crystals were transferred to a cryoprotectant solution consisting of the reservoir solution containing 20% (v/v) glycerol before being cryocooled in liquid nitrogen. X-ray diffraction data were collected to 1.7 Å resolution on beamline I03 at the Diamond Light Source (DLS), Harwell. The crystal belongs to space group P21 with unit cell dimensions a = 77.3 Å, b = 34.7 Å, c = 84.7 Å and α = γ = 90°, β = 102.1°. SeMet crystals were grown in hanging-drops containing a 2:1 ratio of concentrated protein solution : reservoir solution. SeMet-substituted crystals suitable for data collection were obtained using a protein concentration of 1.9 mg ml−1 and a reservoir solution of 100 mM MMT pH 6.0 (dl-malic acid, MES and Tris buffers in a molar ratio of 1:2:2), 20% (w/v) PEG 3350, 2% (v/v) DMF. Crystals were soaked in a cryoprotectant solution consisting of the reservoir solution containing 20% (v/v) glycerol before being cryocooled in liquid nitrogen. X-ray diffraction data were collected to 2.1 Å resolution on beamline I24 at DLS. The crystal belongs to space group P21 with cell dimensions a = 51.4 Å, b = 35.6 Å, c = 63.3 Å and α = γ = 90°, β = 92.8°.
Diffraction datasets obtained from the SeMet derivative and native crystals were processed using the automated data processing pipeline Xia2 (Winter, 2009) with options that run XDS (Kabsch, 2010). Data were merged using AIMLESS (Evans, 2006). The structure of SirA–DnaADI was solved by single-wavelength anomalous dispersion (SAD) phasing. Heavy atom substructure determination, density modification and model building were carried out using the CRANK (Pannu et al., 2011) pipeline available within the Collaborative Computational Project No. 4 (CCP4) program suite (Winn et al., 2001). Nine selenium atom sites were identified using SHELX C/D (Sheldrick, 2008) and their positions refined using BP3. The correct hand for the phases was identified using SOLOMON (Abrahams and Leslie, 1996) and density modification carried out in PARROT (Cowtan, 2010) before atomic model building in BUCCANEER (Cowtan, 2006). The SeMet–SirA–DnaADI model was partially refined using maximum-likelihood methods in REFMAC (Murshudov et al., 1997) and manual model building in COOT (Emsley and Cowtan, 2004). The partially refined SeMet–SirA–DnaADI model was used as a model for the solution of native SirA–DnaADI by molecular replacement with MOLREP (Vagin and Teplyakov, 1997), the search for SirA molecules preceding that for DnaADI domains. The native SirA–DnaADI model was refined through iterative cycles of refinement in REFMAC and manual model building in COOT to an R-factor of 13.1 (Rfree = 19.7). Refinement statistics are shown in Table 1.
The atomic co-ordinates and crystallographic structure factors have been deposited in the Protein Data Bank with the Accession Code 4TPS.
SEC-MALLS
SEC-MALLS analysis of DnaADI and SirA–DnaADI was carried out at a range of protein concentrations: DnaADI was analysed at 1.0 mg ml−1, 2.5 mg ml−1 and 5.0 mg ml−1 and SirA–DnaADI was analysed at 0.5 mg ml−1, 1.0 mg ml−1 and 2.5 mg ml−1. For each run, 100 μl of sample was loaded onto a Superdex 75 HR 10/30 size-exclusion column equilibrated with 50 mM Tris pH 8.5, 200 mM KCl at a flow rate of 0.5 ml min−1. The eluate was analysed successively by a SPD20A UV/Vis detector, a Wyatt Dawn HELEOS-II 18-angle light scattering detector and a Wyatt Optilab rEX refractive index monitor as described previously (Colledge et al., 2011). Data were analysed with Astra software (Wyatt).
Solubility assay
E. coli BL21 (DE3), harbouring wild type and mutated pET-YSBLIC3C-DnaADISirA plasmids were grown in 200 ml LB-kanamycin until the A600 reached ∼ 0.6. A portion of cells (uninduced) was set aside and grown separately while IPTG was added to the remaining cells. After a further 4 h growth, aliquots of the uninduced (U) and induced (I) cells were taken and used to prepare total cell samples. The remaining cells from the induced culture were harvested by centrifugation and the cell pellets were re-suspended in 20 ml of 50 mM Tris pH 8.5, 200 mM KCl, 20 mM imidazole, 10 mM BME. Cells were lysed by sonication and the lysate clarified by centrifugation. An aliquot of this soluble fraction (S) was retained. The remaining lysate was loaded onto a 1 ml HisTrap FF crude Ni-affinity column (GE Healthcare), washed with 6 ml re-suspension buffer, and bound protein eluted with 4 ml of 50 mM Tris pH 8.5, 200 ml KCl, 500 mM imidazole, 10 mM BME and the eluate (E) was collected. For each of the wild type and alanine variants, samples of the total fractions from uninduced (U) and induced (I) cells together with the soluble lysis (S) and high imidazole column eluate (E) fractions were analysed by SDS-PAGE followed by Coomassie blue staining.
B. subtilis strains, media and growth conditions
Strains used in this study are listed in Table S3. Nutrient agar (Oxoid) was used as the solid media for growth of B. subtilis. LB medium was used for growing cells to extract genomic DNA. Antibiotics were added to the growth media as required: chloramphenicol (5 μg ml−1), spectinomycin (50 μg ml−1). To induce sporulation Bacillus subtilis cells grown in hydrolysed casein media at 37°C were induced to sporulate according to the resuspension method of Sterlini and Mandelstam (1969) as modified by Partridge and Errington (1993).
Microscopy
After induction of sporulation, samples were taken every 30 min and visualized using fluorescence microscopy. Microscopy was performed using glass slides covered with a ∼ 1.5% agarose pad containing sporulation media. A glass coverslip (0.17 mm VWR) covered cells immobilized on the agarose pad. The dye FM5-95 was added to the agar pads to visualize the membrane (2.9 μg ml−1 final). To visualize the nucleoid the DNA was stained with 4′-6-diamidino-2-phenylindole (DAPI 2.5 μg ml−1 final). Microscopy was performed on an inverted epifluorescence microscope (Zeiss Axiovert 200M) fitted with a Plan-Neofluar objective (Zeiss 100×/1.30 Oil Ph3). Light was transmitted from a 300 Watt xenon arc-lamp through a liquid light guide (Sutter Instruments) and images were collected using a Sony CoolSnap HQ cooled CCD camera (Roper Scientific). All filters were Modified Magnetron ET Sets from Chroma and details are available upon request. Digital images were acquired and analysed using METAMORPH software (version V.6.2r6).
Western blot analysis
Proteins were separated by electrophoresis using a NuPAGE 4–12% Bis-Tris gradient gel run in MES buffer (Life Technologies) and transferred to a Hybond-P PVDF membrane (GE Healthcare) using a semi-dry apparatus (Hoefer Scientific Instruments). Proteins of interest were probed with polyclonal primary antibodies and then detected with an anti-rabbit horseradish peroxidase-linked secondary antibody using an ImageQuant LAS 4000 mini digital imaging system (GE Healthcare).
Acknowledgments
This work was supported by the EU-funded BaSysBio project LSHG-CT-2006-037469, by BBSRC studentships awarded to K.J. at York and N.R. at Newcastle and by a Royal Society University Research Fellowship to H.M. A.G. was funded by an ERASMUS programme. We would like to thank the DIAMOND Light Source (Harwell) and the Biology Technology Facility at York for access to equipment and experimental advice. We also wish to thank Sheila Taylor and Sam Hart for technical assistance, Jean Whittingham for advice and Alan Koh for providing a plasmid.
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher's web-site.
Supporting information
Supporting information
Supporting information
References
- Abe Y, Jo T, Matsuda Y, Matsunaga C, Katayama T. Ueda T. Structure and function of DnaA N-terminal domains: specific sites and mechanisms in inter-DnaA interaction and in DnaB helicase loading on oriC. J Biol Chem. 2007;282:17816–17827. doi: 10.1074/jbc.M701841200. [DOI] [PubMed] [Google Scholar]
- Abrahams JP. Leslie AG. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr D Biol Crystallogr. 1996;52:30–42. doi: 10.1107/S0907444995008754. [DOI] [PubMed] [Google Scholar]
- Bonilla CY. Grossman AD. The primosomal protein DnaD inhibits cooperative DNA binding by the replication initiator DnaA in Bacillus subtilis. J Bacteriol. 2012;194:5110–5117. doi: 10.1128/JB.00958-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonstra M, de Jong IG, Scholefield G, Murray H, Kuipers OP. Veening J-W. Spo0A regulates chromosome copy number during sporulation by directly binding to the origin of replication in Bacillus subtilis. Mol Microbiol. 2013;87:925–938. doi: 10.1111/mmi.12141. [DOI] [PubMed] [Google Scholar]
- Briggs GS, Smits WK. Soultanas P. Chromosomal replication initiation machinery of low-G + C-content Firmicutes. J Bacteriol. 2012;194:5162–5170. doi: 10.1128/JB.00865-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkholder WF, Kurtser I. Grossman AD. Replication initiation proteins regulate a developmental checkpoint in Bacillus subtilis. Cell. 2001;104:269–279. doi: 10.1016/s0092-8674(01)00211-2. [DOI] [PubMed] [Google Scholar]
- Castilla-Llorente V, Muñoz-Espín D, Villar L, Salas M. Meijer WJJ. Spo0A, the key transcriptional regulator for entrance into sporulation, is an inhibitor of DNA replication. EMBO J. 2006;25:3890–3899. doi: 10.1038/sj.emboj.7601266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho E, Ogasawara N. Ishikawa S. The functional analysis of YabA, which interacts with DnaA and regulates initiation of chromosome replication in Bacillus subtils. Genes Genet Syst. 2008;83:111–125. doi: 10.1266/ggs.83.111. [DOI] [PubMed] [Google Scholar]
- Colledge VL, Fogg MJ, Levdikov VM, Leech A, Dodson EJ. Wilkinson AJ. Structure and organisation of SinR, the master regulator of biofilm formation in Bacillus subtilis. J Mol Biol. 2011;411:597–613. doi: 10.1016/j.jmb.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- Cowtan K. Recent developments in classical density modification. Acta Crystallogr D Biol Crystallogr. 2010;66:470–478. doi: 10.1107/S090744490903947X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doublié S. Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 1997;276:523–530. [PubMed] [Google Scholar]
- Duderstadt KE, Chuang K. Berger JM. DNA stretching by bacterial initiators promotes replication origin opening. Nature. 2011;478:209–213. doi: 10.1038/nature10455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P. Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Erzberger JP, Mott ML. Berger JM. Structural basis for ATP-dependent DnaA assembly and replication-origin remodeling. Nat Struct Mol Biol. 2006;13:676–683. doi: 10.1038/nsmb1115. [DOI] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Felczak MM, Simmons LA. Kaguni JM. An essential tryptophan of Escherichia coli DnaA protein functions in oligomerization at the E. coli replication origin. J Biol Chem. 2005;280:24627–24633. doi: 10.1074/jbc.M503684200. [DOI] [PubMed] [Google Scholar]
- Fogg MJ. Wilkinson AJ. Higher-throughput approaches to crystallization and crystal structure determination. Biochem Soc Trans. 2008;36:771–775. doi: 10.1042/BST0360771. [DOI] [PubMed] [Google Scholar]
- Fujikawa N, Kurumizaka H, Nureki O. Terada T. Structural basis of replication origin recognition by the DnaA protein. Nucleic Acids Res. 2003;31:2077–2086. doi: 10.1093/nar/gkg309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouet P. ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003;31:3320–3323. doi: 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishigo-oka D, Ogasawara N. Moriya S. DnaD protein of Bacillus subtilis interacts with DnaA, the initiator protein of replication. J Bacteriol. 2001;183:1–4. doi: 10.1128/JB.183.6.2148-2150.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janin J, Bahadur RP. Chakrabarti P. Protein-protein interaction and quaternary structure. Q Rev Biophys. 2008;41:133–180. doi: 10.1017/S0033583508004708. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaguni JM. DnaA: controlling the initiation of bacterial DNA replication and more. Annu Rev Microbiol. 2006;60:351–375. doi: 10.1146/annurev.micro.60.080805.142111. [DOI] [PubMed] [Google Scholar]
- Katayama T, Ozaki S, Keyamura K. Fujimitsu K. Regulation of the replication cycle: conserved and diverse regulatory systems for DnaA and oriC. Nat Rev Microbiol. 2010;8:163–170. doi: 10.1038/nrmicro2314. [DOI] [PubMed] [Google Scholar]
- Keyamura K, Abe Y, Higashi M, Ueda T. Katayama T. DiaA dynamics are coupled with changes in initial origin complexes leading to helicase loading. J Biol Chem. 2009;284:25038–25050. doi: 10.1074/jbc.M109.002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AC. Grimwade JE. Regulation of DnaA assembly and activity: taking directions from the genome. Annu Rev Microbiol. 2011;65:19–35. doi: 10.1146/annurev-micro-090110-102934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery TJ, Pelton JG, Chandonia J-M, Kim R, Yokota H. Wemmer DE. NMR structure of the N-terminal domain of the replication initiator protein DnaA. J Struct Funct Genomics. 2007;8:11–17. doi: 10.1007/s10969-007-9022-7. [DOI] [PubMed] [Google Scholar]
- McNicholas S, Potterton E, Wilson KS. Noble MEM. Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr D Biol Crystallogr. 2011;67:386–394. doi: 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molt KL, Sutera VA, Moore KK. Lovett ST. A role for nonessential domain II of initiator protein, DnaA, in replication control. Genetics. 2009;183:39–49. doi: 10.1534/genetics.109.104760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moravcevic K, Mendrola J. Schmitz K. Kinase associated-1 (KA1) domains drive MARK/PAR1 kinases to membrane targets by binding acidic phospholipids. Cell. 2010;143:966–977. doi: 10.1016/j.cell.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray H. Errington J. Dynamic control of the DNA replication initiation protein DnaA by Soj/ParA. Cell. 2008;135:74–84. doi: 10.1016/j.cell.2008.07.044. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA. Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Natrajan G, Noirot-Gros M-F, Zawilak-Pawlik A, Kapp U. Terradot L. The structure of a DnaA/HobA complex from Helicobacter pylori provides insight into regulation of DNA replication in bacteria. Proc Natl Acad Sci USA. 2009;106:21115–21120. doi: 10.1073/pnas.0908966106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noirot-Gros M-F, Velten M, Yoshimura M, McGovern S, Morimoto T, Ehrlich SD, et al. Functional dissection of YabA, a negative regulator of DNA replication initiation in Bacillus subtilis. Proc Natl Acad Sci USA. 2006;103:2368–2373. doi: 10.1073/pnas.0506914103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannu NS, Waterreus WJ, Skubák P, Sikharulidze I, Abrahams JP. de Graaff RAG. Recent advances in the CRANK software suite for experimental phasing. Acta Crystallogr D Biol Crystallogr. 2011;67:331–337. doi: 10.1107/S0907444910052224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge SR. Errington J. The importance of morphological events and intercellular interactions in the regulation of prespore-specific gene expression during sporulation in Bacillus subtilis. Mol Microbiol. 1993;8:945–955. doi: 10.1111/j.1365-2958.1993.tb01639.x. [DOI] [PubMed] [Google Scholar]
- Rahn-Lee L, Gorbatyuk B, Skovgaard O. Losick R. The conserved sporulation protein YneE inhibits DNA replication in Bacillus subtilis. J Bacteriol. 2009;191:3736–3739. doi: 10.1128/JB.00216-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahn-Lee L, Merrikh H, Grossman AD. Losick R. The sporulation protein SirA inhibits the binding of DnaA to the origin of replication by contacting a patch of clustered amino acids. J Bacteriol. 2011;193:1302–1307. doi: 10.1128/JB.01390-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokop ME, Auchtung JM. Grossman AD. Control of DNA replication initiation by recruitment of an essential initiation protein to the membrane of Bacillus subtilis. Mol Microbiol. 2004;52:1757–1767. doi: 10.1111/j.1365-2958.2004.04091.x. [DOI] [PubMed] [Google Scholar]
- Scholefield G. Murray H. YabA and DnaD inhibit helix assembly of the DNA replication initiation protein DnaA. Mol Microbiol. 2013;90:147–159. doi: 10.1111/mmi.12353. [DOI] [PubMed] [Google Scholar]
- Scholefield G, Errington J. Murray H. Soj/ParA stalls DNA replication by inhibiting helix formation of the initiator protein DnaA. EMBO J. 2012;31:1542–1555. doi: 10.1038/emboj.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz H, Weigel C. Messer W. The interaction domains of the DnaA and DnaB replication proteins of Escherichia coli. Mol Microbiol. 2000;37:1270–1279. doi: 10.1046/j.1365-2958.2000.02096.x. [DOI] [PubMed] [Google Scholar]
- Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Smits WK, Goranov AI. Grossman AD. Ordered association of helicase loader proteins with the Bacillus subtilis origin of replication in vivo. Mol Microbiol. 2010;75:452–461. doi: 10.1111/j.1365-2958.2009.06999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufo CD, Soufo HJD, Noirot-Gros M-F, Steindorf A, Noirot P. Graumann PL. Cell-cycle-dependent spatial sequestration of the DnaA replication initiator protein in Bacillus subtilis. Dev Cell. 2008;15:935–941. doi: 10.1016/j.devcel.2008.09.010. [DOI] [PubMed] [Google Scholar]
- Sterlini JM. Mandelstam J. Commitment to sporulation in Bacillus subtilis and its relationship to development of actinomycin resistance. Biochem J. 1969;113:29–37. doi: 10.1042/bj1130029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su'etsugu M, Harada Y, Keyamura K, Matsunaga C, Kasho K, Abe Y, et al. The DnaA N-terminal domain interacts with Hda to facilitate replicase clamp-mediated inactivation of DnaA. Environ Microbiol. 2013;15:3183–3195. doi: 10.1111/1462-2920.12147. [DOI] [PubMed] [Google Scholar]
- Sutton MD, Carr KM, Vicente M. Kaguni JM. Escherichia coli DnaA protein. The N-terminal domain and loading of DnaB helicase at the E. coli chromosomal origin. J Biol Chem. 1998;273:34255–34262. doi: 10.1074/jbc.273.51.34255. [DOI] [PubMed] [Google Scholar]
- Vagin A. Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- Veening J-W, Murray H. Errington J. A mechanism for cell cycle regulation of sporulation initiation in Bacillus subtilis. Genes Dev. 2009;23:1959–1970. doi: 10.1101/gad.528209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velten M, McGovern S, Marsin S, Ehrlich SD, Noirot P. Polard P. A two-protein strategy for the functional loading of a cellular replicative DNA helicase. Mol Cell. 2003;11:1009–1020. doi: 10.1016/s1097-2765(03)00130-8. [DOI] [PubMed] [Google Scholar]
- Wagner JK, Marquis KA. Rudner DZ. SirA enforces diploidy by inhibiting the replication initiator DnaA during spore formation in Bacillus subtilis. Mol Microbiol. 2009;73:963–974. doi: 10.1111/j.1365-2958.2009.06825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel C, Schmidt A, Seitz H, Tüngler D, Welzeck M. Messer W. The N-terminus promotes oligomerization of the Escherichia coli initiator protein DnaA. Mol Microbiol. 1999;34:53–66. doi: 10.1046/j.1365-2958.1999.01568.x. [DOI] [PubMed] [Google Scholar]
- Winn M, Ballard C, Cowtan K, Dodson E, Emsley P, Evans P, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2001;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G. Xia2: an expert system for macromolecular crystallography data reduction. J Appl Crystallogr. 2009;43:186–190. [Google Scholar]
- Yoshikawa H. Ogasawara N. Structure and function of DnaA and the DnaA-box in eubacteria: evolutionary relationships of bacterial replication origins. Mol Microbiol. 1991;5:2589–2597. doi: 10.1111/j.1365-2958.1991.tb01967.x. [DOI] [PubMed] [Google Scholar]
- Zawilak-Pawlik A, Donczew R, Szafrański S, Mackiewicz P, Terradot L. Zakrzewska-Czerwińska J. DiaA/HobA and DnaA: a pair of proteins co-evolved to cooperate during bacterial orisome assembly. J Mol Biol. 2011;408:238–251. doi: 10.1016/j.jmb.2011.02.045. [DOI] [PubMed] [Google Scholar]
- Zhang W, Carneiro MJVM, Turner IJ, Allen S, Roberts CJ. Soultanas P. The Bacillus subtilis DnaD and DnaB proteins exhibit different DNA remodelling activities. J Mol Biol. 2005;351:66–75. doi: 10.1016/j.jmb.2005.05.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Machón C, Orta A, Phillips N, Roberts CJ, Allen S. Soultanas P. Single-molecule atomic force spectroscopy reveals that DnaD forms scaffolds and enhances duplex melting. J Mol Biol. 2008;377:706–714. doi: 10.1016/j.jmb.2008.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Supporting information