

Abstract
Background
The incidence of type 2 diabetes mellitus (T2D) is increasing in youth, yet little is known about the underlying pathophysiology. Decreased insulin suppression of lipolysis and elevated non-esterified free fatty acid (NEFA) concentrations are known to be associated with insulin resistance and T2D in adults, but less is known about the relationship in adolescents.
Objectives
To assess adipose tissue IR (insulin suppression of lipolysis) and its metabolic correlates in lean, obese and T2D adolescents.
Methods
47 lean, obese and T2D youth underwent hyperinsulinemic (80mU*m−2*min−1), euglycemic clamps. NEFAs were measured at baseline and during steady state. Insulin-mediated suppression of lipolysis (%NEFA suppression from baseline) was calculated and metabolic risk factors were assessed by %NEFA suppression tertile.
Results
There was expected variability in %NEFA suppression within obese and T2D youth, but a subset had significantly reduced suppression of lipolysis. NEFA suppression tertile was significantly inversely associated with fasting triglycerides (p=0.0001), log alanine aminotransferase (ALT, p=0.02), and low-density lipoprotein cholesterol (p=0.0002).
Conclusions
Marked adipose tissue IR occurs in some obese and T2D adolescents, which may result in release of triglycerides into the circulation and liver deposition of fatty acids, as evidenced by higher ALT in poor NEFA suppressors.
Keywords: Obesity, adipose tissue insulin resistance, free fatty acids, type 2 diabetes, adolescents, hyperinsulinemic euglycemic clamp
Introduction
The prevalence of obesity is increasing in youth, as are obesity-related diseases, such as type 2 diabetes mellitus (T2D) and fatty liver disease (NAFLD)1, 2. Insulin resistance in skeletal muscle, adipose and hepatic tissue is reported to play a role in developing adult disease and likely contributes to childhood disease. We3 and others4, 5 previously observed impairments in insulin-mediated glucose uptake in obese compared to lean adolescents, but there remains a paucity of data on adipose tissue insulin resistance (i.e., insulin-mediated suppression of lipolysis) in youth6–9.
In adipose, the primary role of insulin is to suppress lipolysis, the release of non-esterified free fatty acids (NEFA) from triglycerides (TG) into the circulation. Adipose tissue lipolysis is normally suppressed at lower doses of insulin (EC50 ~10 μU/mL) than are required to stimulate skeletal muscle glucose uptake (EC50 ~60 μU/mL)10, 11. In obese and T2D adults, adipose often becomes insulin resistant, resulting in chronically elevated NEFAs12. Increased circulating NEFAs are, in turn, associated with increased hepatic13 and intramyocellular14 deposition of triglycerides, thereby exacerbating hepatic and skeletal muscle insulin resistance, respectively. Furthermore, studies evaluating effects of lipid infusion in adults suggest that NEFA elevation induces inflammation, resulting in increased reactive oxygen species and subsequent endothelial dysfunction15, 16. Compared to adults, much less is known about insulin suppression of lipolysis and associated cardiometabolic risk in youth. Thus, our goal was to evaluate the association between markers of cardiometabolic disease and adipose tissue insulin resistance in lean, obese, and T2D adolescents. We hypothesized that impaired insulin suppression of lipolysis is associated with impairments in cardiovascular risk factors (e.g., blood pressure (BP), lipid profile), markers of inflammation (high sensitivity C-reactive protein, hsCRP, interleukin-6, IL-6) and NAFLD (alanine aminotransferase, ALT).
Methods and Procedures
Subjects
We evaluated a cohort of adolescents previously recruited for a study of exercise capacity and insulin-mediated glucose uptake (lean, obese and obese T2D). Characteristics for this cohort were previously reported3. Briefly, entry criteria included Tanner stage >1, age 12–19 years, and sedentary status (<3 hours per week of habitual physical activity). BMI criteria for lean subjects was between the 5th and 85th percentiles, and >95th percentile for obese and T2D subjects. Exclusion criteria included: body weight > 300 pounds, BP >140/90 mmHg, hemoglobin <9 mg/dl, serum creatinine >1.5 mg/dl, hemoglobin A1c (HbA1c) >12%, smoking, medications (other than metformin in T2D subjects) known to affect insulin resistance, insulin, corticosteroids, antihypertensive drugs, pregnancy, or breastfeeding.
A total of 47 adolescents (10 lean, 12 obese and 25 obese T2D) who completed a hyperinsulinemic euglycemic clamp were included (mean age 15±2 years, 63% female). Pubertal staging was performed according to the standards of Tanner and Marshall17, 18. T2D was defined by the American Diabetes Association criteria19, along with negative glutamic acid decarboxylase, islet cell, and insulin antibodies. Diabetes was ruled out in all non-diabetic obese subjects by a 2-hour, 75g oral glucose tolerance test. Average diabetes duration in subjects with T2D was 27 ± 23 months.
The study was approved by the Colorado Multiple Institution review board, and appropriate consent and assent were obtained.
Insulin sensitivity
Participants were admitted to the inpatient Clinical Translational Research Center (CTRC) following 3 days of no strenuous exercise and a fixed-macronutrient, weight-maintenance diet, provided by the CTRC metabolic kitchen20. Metformin was discontinued during the 3-day study diet in T2D subjects. All participants fasted overnight and T2D subjects were maintained with an overnight intravenous insulin infusion (average insulin dose = 0.34 U/hr), with a target blood glucose of 100 mg/dl. A 3-hour hyperinsulinemic-euglycemic clamp (80 mU*m−2*min−1 insulin) was performed as previously described20. Of note, this dose is twice as high as routinely used in adult studies because of the known reduction in whole-body insulin sensitivity during puberty21–24. Insulin (radioimmunoassay (RIA), HI-14-K, Millipore) and NEFA (Enzymatic, NEFA-HR2, Wako Chemicals USA) samples were collected at baseline and every 10 minutes during the last 30 minutes of the clamp (steady-state). Steady-state glucose infusion rate (GIR; mg/kg*min−1) was used to measure whole body insulin sensitivity. Percent NEFA suppression was used as a surrogate for adipose tissue insulin sensitivity, calculated as: 1 – (Average NEFA during steady-state/Baseline NEFA).
Additional fasting measures included: direct low-density lipoprotein cholesterol (LDL), high-density lipoprotein cholesterol (HDL), ALT, HbA1c, hsCRP, IL-6, NEFA, insulin, and adiponectin. All assays were performed by standard methods at the CTRC Core Laboratory.
Statistical Analysis
All analyses assumed a two-sided test of hypothesis and an overall significance level of 0.05. Comparisons of outcomes across phenotype groups (lean, obese and T2) were made using one-way ANOVA and linear regression, controlling for Tanner stage and race/ethnicity. Pairwise comparisons were conducted if the overall effect of group was significant. Due to the expected between-group variability in insulin-mediated suppression of lipolysis, subjects were divided into equal tertiles by % suppression of NEFA’s, resulting in low (<73%), moderate (73–90) and high (>90%) suppressors. Multivariate linear regression was performed to assess potential correlates (triglycerides, HDL, LDL, IL-6, hsCRP, GIR, (mg/kg*min−1 and mg/lean kg*min−1), systolic (SBP) and diastolic BP (DBP), ALT, and adiponectin) by NEFA suppression tertile. ALT was log transformed due to skewed distribution. The analyses were repeated to adjust for potential confounders (age, sex, ethnicity, % fat mass, BMI z-score, Tanner stage, diabetes duration, and baseline and end-clamp insulin). Covariates were included in the final model if they resulted in ≥ 10% change in either of the two parameter estimates.
Results
Subject Characteristics
Subject characteristics by group are shown for the 47 participants in Table 1. There was no difference between obese and T2D subjects in percent fat mass, BMI, or BMI z-score by design. There were more females than males, representative of our adolescent obese and T2D population.
Table 1.
Subject characteristics by obesity phenotype
| Characteristic | All N=47 |
Lean N=10 |
Obese N=12 |
T2D N=25 |
p-value |
|---|---|---|---|---|---|
| Male | 17 (36%) | 5 (50%) | 4 (33%) | 8 (32%) | 0.66 |
| Age | 15.3 (2.1) | 14.9 (2.0) | 15.1 (2.0) | 15.6 (2.3) | 0.63 |
| Ethnicity | |||||
| Caucasian | 19 (40%) | 8 (80%) | 6 (50%) | 5 (20%) | |
| Hispanic | 22 (47%) | 1 (10%) | 5 (42%) | 16 (64%) | 0.01 |
| Other | 6 (13%) | 1 (10%) | 1 (8%) | 4 (16%) | |
| Tanner*,† | |||||
| 2 | 1 (2%) | 0 | 0 | 1 (4%) | |
| 3 | 4 (9%) | 1 (10%) | 1 (8.3%) | 2 (8%) | 0.02 |
| 4 | 7 (15%) | 5 (50%) | 1 (8.3%) | 1 (4%) | |
| 5 | 34 (74%) | 4 (40%) | 10 (83.3%) | 20 (83%) | |
| BMI z-score | 1.6 (1.0) | −0.06 (0.71) | 1.9 (0.46)a | 2.1 (0.48)a | <0.0001 |
| % Fat Mass | 36.6 (12.1) | 19.0 (10.6) | 39.6 (6.8)a | 42.4 (6.9)a | <0.0001 |
| GIR (mg/kg*min−1) | 7.6 (5.3) | 15.2 (4.4) | 8.0 (2.8)a | 4.3 (2.4)a,b | <0.0001 |
| Lean GIR (mg/lean kg*min−1) | 12.2 (6.3) | 19.8 (4.3) | 14.0 (4.5)a | 7.9 (3.9)a,b | <0.0001 |
| SBP (mmHg) | 119 (11.0) | 111 (6.2) | 121 (8.9)a | 122 (11.7)a | 0.01 |
| DBP (mmHg) | 71.5 (9.2) | 62.7 (6.4) | 74.6 (7.1)a | 73.5 (9.2)a | 0.002 |
| Triglycerides (mmol/L) | 1.8(1.1) | 1.0 (0.4) | 1.9(1.2) | 2.0(1.2) | 0.07 |
| HDL (mmol/L) | 1.1 (0.3) | 1.2 (0.2) | 1.1 (0.3) | 1.06 (0.3) | 0.49 |
| LDL (mmol/L) | 2.3 (0.6) | 1.8 (0.5) | 2.63 (0.65)a | 2.39 (0.6)a | 0.009 |
| Log (ALT) | 3.6 (0.72) | 3.5 (0.50) | 3.6 (0.41) | 3.7 (0.90) | 0.66 |
| IL6 (pg/ml)* | 3.4 (3.0) | 2.5 (1.1) | 2.4 (0.85) | 4.3 (3.9) | 0.13 |
| hsCRP (nmol/L) | 35.2 (42.9) | 7.5 (9.1) | 10.5(15.2) | 58.1 (47.6)a,b | 0.0002 |
| Adiponectin (μg/ml) | 8.0 (3.8) | 9.5 (3.3) | 10.2 (3.7) | 6.3 (3.2)a,b | 0.002 |
| Baseline Glucose (mmol/L) | 5.6 (1.6) | 4.6 (0.3) | 4.7 (0.4) | 6.5 (1.8)a,b | 0.0001 |
| Baseline NEFA (μmol/L) | 558 (254) | 521 (317) | 583 (155) | 561 (273) | 0.86 |
| NEFA Suppression (%) | 74 (27) | 90 (7) | 68 (36) | 70 (26) | 0.11 |
| Baseline insulin (pmol/L) | 160 (119) | 58 (28) | 113 (49)a | 223 (126)a,b | <0.0001 |
Datarepresent Mean (SD) or N (%). Tanner stage and race/ethnicity were controlled for in the phenotype comparisons of baseline NEFA and NEFA suppression.
significant vs. controls,
significant vs. obese;
one obese subject is missing %fat mass; several subjects are missing IL6, such that lean n=9, obese n=11, T2 n=23; *one T2D subject is missing Tanner stage;
The overall comparison among the groups is tested; pairwise comparisons are not made
Assessment of Insulin Sensitivity
GIR was reduced in obese vs. lean subjects (Table 1), and further reduced in T2D vs. obese, whereas %NEFA suppression tended to be lower in obese and T2D vs. lean subjects (Table 1). There was no difference in steady state insulin between lean (854±254 pmol/L), obese (764±263 pmol/L), and T2D (896±406 pmol/L) groups. However, there was significant variability in %NEFA suppression within the obese and T2D groups. To more directly examine adipose insulin resistance, the entire cohort was evaluated by %NEFA suppression tertiles. After division into tertiles, there was no significant difference in baseline NEFA concentrations by tertile (p=0.16). By design, NEFA suppression was significantly different among the tertiles (p<0.0001). Figure 1 shows distribution of the subject groups among the tertiles. BMI-z tended to be higher in the low (1.99±0.41) vs. moderate (1.57±1.05) and high (1.21±1.28) suppressors (p=0.06). There was also a trend toward higher percent body fat in the low (41.3±8.1%) than moderate (37.0±12.9%) and high (31.5±13.4%) suppressors (p=0.08). There was no significant difference in age, gender, BMI z-score, percent fat mass, or overnight insulin dose (T2D subjects) by tertile (data not shown).
Figure 1.
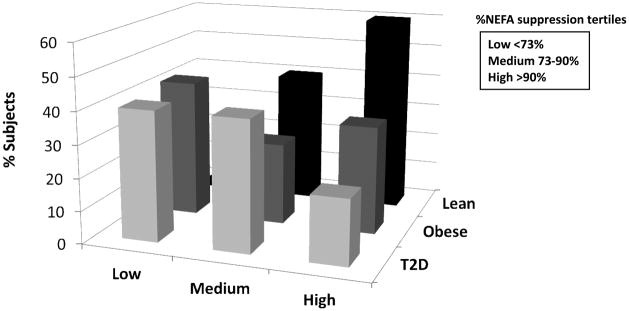
Participant groups (Lean, Obese, T2D) broken down by NEFA suppression tertiles. The distribution of obese and T2D subjects across the suppressor tertiles indicates the high variability in adipose tissue insulin resistance in the obese and T2D groups.
Variables associated with NEFA suppression in regression analysis included: triglycerides, GIR, log ALT, LDL, and SBP. After controlling for confounders, GIR and SBP were no longer significantly different across NEFA suppression tertiles (p=0.28 and p=0.50, respectively). However, a significant association remained for LDL (p=0.0002), triglycerides (p=0.0001) and log ALT (p=0.02) (Table 2). Post-hoc analyses were performed to further evaluate tertile differences in triglycerides, LDL and log ALT, again controlling for potential confounders (Table 2).
Table 2.
Cardiometabolic risk markers and peripheral insulin sensitivity by NEFA suppression tertile
| Outcome | Low Suppression (n=15) | Moderate Suppression (n=17) | High Suppression (n=15) | p-value |
|---|---|---|---|---|
| Triglycerides(mmol/L) | 2.40 (0.26)a,b | 1.52 (0.24)b | 0.79 (0.25) | 0.0001 |
| HDL (mmol/L) | 1.18 (0.12) | 1.06 (0.11) | 1.22 (0.12) | 0.35 |
| LDL (mmol/L) | 2.86 (0.14)a,b | 2.07 (0.13) | 2.09 (0.14) | 0.0002 |
| IL6 (pg/ml) | 4.09 (0.75) | 3.39 (0.71) | 2.69 (0.79) | 0.45 |
| hsCRP (nmol/L) | 71.5 (13.2) | 78.0 (12.2) | 83.2 (13.6) | 0.71 |
| GIR (mg/kg*m−1) | 7.46 (1.10) | 7.94 (1.00) | 8.79 (1.07) | 0.50 |
| Lean GIR (mg/lean kg*min−1) | 11.58 (1.59) | 12.05 (1.45) | 12.86 (1.62) | 0.74 |
| SBP (mmHg) | 124.8 (3.78) | 119.2 (3.51) | 118.8 (3.91) | 0.28 |
| DBP (mmHg) | 73.3 (1.97) | 69.7 (1.85) | 70.5 (1.99) | 0.40 |
| Log(ALT) | 4.07 (0.17)a,b | 3.46 (0.16) | 3.40 (0.17) | 0.02 |
| Adiponectin (μ/ml) | 7.35 (1.44) | 7.55 (1.19) | 7.49 (1.27) | 0.98 |
Data represent estimated Means (SD) from the regression analyses, controlling for covariates as described in the methods
Significant vs. moderate suppression;
Significant vs. high suppression
Discussion
This study presents novel information regarding adipose tissue insulin resistance in obese adolescents with and without diabetes. Typically, insulin is a potent suppressor of lipolysis at much lower concentrations than are needed to stimulate glucose uptake. Data presented here demonstrate that adipose tissue insulin sensitivity (%NEFA suppression) is much more variable within obese and T2D youth than between these groups. Indeed, a subset of our obese and T2D youth appeared to have severe adipose insulin resistance, as demonstrated by negligible NEFA suppression even after high-dose insulin exposure (twice as high as typically used in adult hyperinsulinemic clamps). Because of this variability we evaluated cardiometabolic risk factors by %NEFA suppression tertiles and found that adipose tissue insulin resistance was associated with elevated triglycerides, LDL, and ALT, possibly indicative of early changes in fat storage. In particular, higher triglycerides and ALT may be clinically relevant markers of adipose tissue insulin resistance, in addition to their known associations with peripheral insulin resistance and NAFLD, respectively.
Obese adults with and without T2D are reported to have both higher fasting NEFA and reduced suppression of lipolysis relative to lean controls25–27. In contrast, we did not observe significant group differences in our adolescent subjects. The lack of a statistical difference between obese and lean may be due to small sample size, as there was a trend toward greater NEFA suppression in lean vs. obese and T2D subjects (p=0.11). However, declines in adipose tissue insulin sensitivity may also be a physiologic adaptation that takes time to manifest. Metabolic changes during puberty6, 7 may also impact lipolysis and fat oxidation. Thus, our group variation in pubertal development may have contributed to our lack of difference in NEFA suppression, in addition to possible racial/ethnic and sex differences in fatty acid metabolism. However, after adjusting for Tanner stage, sex, and race/ethnicity, our results remain unchanged. All groups, including lean, were sedentary, so our lean subjects may also have greater adipose insulin resistance than a lean, active population. Finally, studies in BMI-matched obese adults with and without T2D suggest the degree of adipose tissue insulin resistance does not differ between these groups28, 29.
Although direct comparisons of the adult and adolescent literature are difficult due to methodological reasons, reports in obese adults generally show better suppression of lipolysis at much lower doses of insulin than we observed in our adolescents. The average steady state insulin concentration in our obese and T2D subjects was 882 pmol/L. Yet, at that insulin concentration, 24% of obese and T2D subjects had <50% NEFA suppression and 8% had <15% NEFA suppression. Such severely impaired NEFA suppression has not previously been described in any age group. One adult study of lean and obese subjects showed some suppression of palmitate flux even at a serum insulin concentration of 69.4 pmol/L, and all subjects had > 50% suppression of palmitate flux with high doses of insulin, resulting in insulin concentrations of 694 pmol/L27. In another study of obese women, 80% NEFA suppression was achieved with relatively low insulin doses, with serum insulin concentrations averaging 201 pmol/L30. Adults with T2D also show greater insulin suppression of lipolysis than our adolescents. For example, in one study evaluating effects of low-dose insulin infusion on relatively lean adults with T2D, approximately 30% NEFA suppression was achieved at serum insulin concentrations of 69–104 pmol/L31. In a second study of 13 adults with T2D (average BMI 28.7 kg/m2), 70–85% NEFA suppression was seen with serum insulin concentrations of 451 pmol/L32. Two small pediatric studies including lean and obese (but not T2D) adolescents showed a shift in the dose-response curve in insulin-suppression of lipolysis in obese versus lean adolescents6, 7. One of these pediatric studies included 13 obese and 13 lean adolescents and showed 58% NEFA suppression in the obese and 75% NEFA suppression in the lean youth at half the dose of insulin used for our current study (40 mU*m−2*min−1, with insulin concentrations of 552 pmol/L in obese and 378 pmol/L in lean adolescents)6. Of note, 6 obese adults were also included in this study and had less impaired NEFA suppression (68%), when compared with lean adults (79%) at the same insulin dose as the adolescents. The second pediatric study, also using 40 mU*m−2*min−1 insulin, had similar findings in 7 obese and 7 lean adolescents (60% and 71% NEFA suppression, respectively)7. An additional pediatric study evaluated NEFA appearance using a palmitate isotope with 40 mU*m−2*min−1 insulin in 18 obese subjects with and without NAFLD. Results showed NEFA suppression of 81% and 86% in obese subjects with and without NAFLD, respectively33. In summary, our results are consistent with previous studies in adults and adolescents, but are the first to include T2D adolescents. In addition, by using a higher dose of insulin, we are the first to demonstrate such a high degree of adipose tissue IR in obese youth.
It is important to note that insulin-suppression of NEFA occurs along a continuum. Furthermore, there are no established clinically relevant cutoffs for failure of NEFA suppression. However, it does appear that a subset of our cohort of adolescents has significantly impaired ability to suppress lipolysis. Thus, some adolescents with early-onset obesity and diabetes may have much more significant adipose tissue insulin resistance than similar adults with longer diabetes duration.
In terms of markers of adipose tissue insulin resistance in adolescents, the strongest independent association observed in our cohort was between triglycerides and NEFA suppression tertile; those in the low suppression tertile had the highest triglycerides, a finding not confounded by age, sex, %fat mass, BMI z-score or Tanner stage. As was observed with NEFA suppression, triglycerides were highly variable within the obese non-diabetic and obese T2D groups. Fasting triglycerides are an accepted marker of insulin resistance in adults34 and in youth35. The fact that triglycerides are significantly different among the NEFA suppression tertiles, but not different among the lean, obese and T2D groups, suggests that fasting triglycerides may be more specifically an indication of adipocyte dysfunction in youth. This adipocyte insulin resistance may, in turn, contribute to ectopic lipid deposition, as suggested by the higher ALT seen in the moderate and low suppressors. Previous pediatric studies have shown an association between peripheral insulin resistance and hepatic fat fraction as measured by MRI36, 37. MRI imaging studies in these youth are now underway to further evaluate fat compartmentalization, including hepatic, visceral, subcutaneous, and intra- and extramyocellular lipid.
Interestingly, low NEFA suppression was also associated with higher LDL. While dietary fatty acids were previously shown to increase LDL in humans38, 39, the extent to which elevated endogenous NEFAs impact LDL is not known. It is speculated that elevated NEFA may cause down-regulation of hepatic LDL-receptor activity, thereby decreasing hepatic clearance of LDL40. This concept is supported by several in vitro studies41–43. Although our studies were not designed test this hypothesis specifically, the fact that LDL concentrations were higher in the low suppressors remains intriguing.
There are several limitations to our study. First, a single dose insulin clamp is not the gold standard measure of adipose tissue insulin resistance. Typically a dose-response to both lower and higher insulin concentrations is used to construct a suppression curve or the insulin concentration needed to half maximally suppress NEFA or glycerol (EC50) is used to estimate adipose insulin sensitivity. Since we only had NEFA concentrations in the basal state and during high dose insulin infusion, we estimated lipolysis from NEFA suppression. However, the fact that a subset of adolescents had minimal suppression of lipolysis despite such a high dose is remarkable and different from what is reported in adults. The lack of isotopes and calorimetry in our current methodology does not account for intracellular recycling of NEFA, differences in NEFA clearance from the circulation, or differences in fatty acid oxidation. For these reasons, additional studies are now underway using staged insulin dosing, glycerol tracers, and indirect calorimetry in a larger sample size. A second limitation is that an overnight insulin infusion was required to avoid baseline hyperglycemia in T2D subjects, which may have resulted in some suppression of fasting NEFAs and, thus, masked differences in fasting NEFAs. However, there was no significant difference among the T2D subjects in each tertile suppression group in overnight insulin dose, so the overnight insulin is unlikely to have had a significant impact.
There are several advantages to our study. Most importantly, it was a well-controlled study using a hyperinsulinemic euglycemic clamp, the reference method for measuring insulin-mediated glucose uptake. We also used a controlled study diet and limited exercise in the 3 days prior to the clamp, observed the subjects overnight to ensure fasting status, and had a fixed period of fasting, as intake and length of fasting highly affects NEFA levels and insulin sensitivity. The obese and T2D groups were similar for mean BMI, fat mass, and pubertal stage, and all 3 groups were similar for age and physical activity level. In addition, we evaluated a wide variety of cardiometabolic risk factors known to correlate with whole body insulin resistance.
In summary, we present the novel finding that a subset of obese and T2D adolescents have markedly decreased ability of insulin to suppress lipolysis. This early manifestation of adipose tissue insulin resistance in some children and adolescents with obesity and T2D suggests a rapidly progressing pathology or transient changes in adipocyte biology that could lead to early complications, and which will likely have a significant impact on health over time. These data emphasize a need to follow obese and T2D adolescents as they become adults to determine hepatic and cardiovascular disease outcomes. They also suggest that ALT, LDL, and triglyceride elevations may be specifically relevant clinical markers of adipose tissue insulin resistance in youth. More importantly, these data further support a need to increase obesity prevention efforts in youth.
What is already known about this subject
Type 2 diabetes is increasing in youth, but little is known about underlying pathophysiology
Elevated fasting free fatty acids and failure of insulin to suppress lipolysis is well described in adults
Small studies suggest that obese youth with and without type 2 diabetes have impaired suppression of lipolysis, but there are little data on associated factors
What this study adds
Assessment of insulin suppression of lipolysis in a larger cohort of adolescents using hyperinsulinemic euglycemic clamp technique at higher insulin doses than have been previously reported
Novel finding that a some obese youth with and without type 2 diabetes have impaired suppression of lipolysis even at very high doses of insulin
Factors associated with impaired insulin suppression of lipolysis include: high triglycerides, high alanine aminotransferase, and high low-density lipoprotein cholesterol
Acknowledgments
The authors would like to acknowledge funding from the following sources:
NIH Building Interdisciplinary Careers in Women’s Health Award 5K12HD057022-04
NIH/NCRR Colorado CTSI Grant Number UL1 RR025780
University of Colorado Center for Women’s Health Research
Juvenile Diabetes Research Foundation JDRF 11-2010-343
NIH/NIDDK 1R56DK088971-01
NIH/NCRR K23 RR020038-05
Juvenile Diabetes Research Foundation JDRF5-2008-291
Footnotes
Disclosures:
Megan Kelsey, MD is receiving research funding for clinical trials sponsored by Daiichi-Sankyo, Merck and Bristol Myers Squibb. These are not related to the data presented in this manuscript.
References
- 1.Pinhas-Hamiel O, Zeitler P. Acute and chronic complications of type 2 diabetes mellitus in children and adolescents. Lancet. 2007;369:1823–31. doi: 10.1016/S0140-6736(07)60821-6. [DOI] [PubMed] [Google Scholar]
- 2.Schwimmer JB, Deutsch R, Kahen T, et al. Prevalence of fatty liver in children and adolescents. Pediatrics. 2006;118:1388–93. doi: 10.1542/peds.2006-1212. [DOI] [PubMed] [Google Scholar]
- 3.Nadeau KJ, Ehlers L, Draznin B, et al. Exercise Capacity is Abnormal in Youth with Type 2 Diabetes. Diabetes. 2007;56(Suppl 1):A278. [Google Scholar]
- 4.Rossetti L, Smith D, Shulman GI, et al. Correction of hyperglycemia with phlorizin normalizes tissue sensitivity to insulin in diabetic rats. The Journal of clinical investigation. 1987;79:1510–5. doi: 10.1172/JCI112981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tessari P, Nosadini R, Trevisan R, et al. Defective suppression by insulin of leucine-carbon appearance and oxidation in type 1, insulin-dependent diabetes mellitus. Evidence for insulin resistance involving glucose and amino acid metabolism. The Journal of clinical investigation. 1986;77:1797–804. doi: 10.1172/JCI112504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caprio S, Bronson M, Sherwin RS, et al. Co-existence of severe insulin resistance and hyperinsulinaemia in pre-adolescent obese children. Diabetologia. 1996;39:1489–97. doi: 10.1007/s001250050603. [DOI] [PubMed] [Google Scholar]
- 7.Robinson C, Tamborlane WV, Maggs DG, et al. Effect of insulin on glycerol production in obese adolescents. Am J Physiol. 1998;274:E737–43. doi: 10.1152/ajpendo.1998.274.4.E737. [DOI] [PubMed] [Google Scholar]
- 8.Danadian K, Lewy V, Janosky JJ, et al. Lipolysis in African-American children: is it a metabolic risk factor predisposing to obesity? The Journal of clinical endocrinology and metabolism. 2001;86:3022–6. doi: 10.1210/jcem.86.7.7626. [DOI] [PubMed] [Google Scholar]
- 9.Weiss R, Taksali SE, Dufour S, et al. The “obese insulin-sensitive” adolescent: importance of adiponectin and lipid partitioning. The Journal of clinical endocrinology and metabolism. 2005;90:3731–7. doi: 10.1210/jc.2004-2305. [DOI] [PubMed] [Google Scholar]
- 10.Rossetti L, Lauglin MR. Correction of chronic hyperglycemia with vanadate, but not with phlorizin, normalizes in vivo glycogen repletion and in vitro glycogen synthase activity in diabetic skeletal muscle. The Journal of clinical investigation. 1989;84:892–9. doi: 10.1172/JCI114250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Del Prato S, Castellino P, Simonson DC, et al. Hyperglucagonemia and insulin-mediated glucose metabolism. The Journal of clinical investigation. 1987;79:547–56. doi: 10.1172/JCI112846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heilbronn L, Smith SR, Ravussin E. Failure of fat cell proliferation, mitochondrial function and fat oxidation results in ectopic fat storage, insulin resistance and type II diabetes mellitus. Int J Obes Relat Metab Disord. 2004;28 (Suppl 4):S12–S21. doi: 10.1038/sj.ijo.0802853. [DOI] [PubMed] [Google Scholar]
- 13.Seppala-Lindroos A, Vehkavaara S, Hakkinen AM, et al. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. The Journal of clinical endocrinology and metabolism. 2002;87:3023–8. doi: 10.1210/jcem.87.7.8638. [DOI] [PubMed] [Google Scholar]
- 14.Kelley DE, Williams KV, Price JC, et al. Plasma fatty acids, adiposity, and variance of skeletal muscle insulin resistance in type 2 diabetes mellitus. The Journal of clinical endocrinology and metabolism. 2001;86:5412–9. doi: 10.1210/jcem.86.11.8027. [DOI] [PubMed] [Google Scholar]
- 15.Tripathy D, Mohanty P, Dhindsa S, et al. Elevation of free fatty acids induces inflammation and impairs vascular reactivity in healthy subjects. Diabetes. 2003;52:2882–7. doi: 10.2337/diabetes.52.12.2882. [DOI] [PubMed] [Google Scholar]
- 16.Steinberg HO, Tarshoby M, Monestel R, et al. Elevated circulating free fatty acid levels impair endothelium-dependent vasodilation. The Journal of clinical investigation. 1997;100:1230–9. doi: 10.1172/JCI119636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marshall WA, Tanner JM. Variations in pattern of pubertal changes in girls. Arch Dis Child. 1969;44:291–303. doi: 10.1136/adc.44.235.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marshall WA, Tanner JM. Variations in the pattern of pubertal changes in boys. Arch Dis Child. 1970;45:13–23. doi: 10.1136/adc.45.239.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.American Diabetes A. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2012;35 (Suppl 1):S64–71. doi: 10.2337/dc12-s064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nadeau KJ, Zeitler PS, Bauer TA, et al. Insulin resistance in adolescents with type 2 diabetes is associated with impaired exercise capacity. Journal of Clinical Endocrinology Metabolism. 2009;94:3687–95. doi: 10.1210/jc.2008-2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ball GD, Huang TT, Gower BA, et al. Longitudinal changes in insulin sensitivity, insulin secretion, and beta-cell function during puberty.[see comment] Journal of Pediatrics. 2006;148(1):16–22. doi: 10.1016/j.jpeds.2005.08.059. [DOI] [PubMed] [Google Scholar]
- 22.Amiel SA, Sherwin RS, Simonson DC, et al. Impaired insulin action in puberty. A contributing factor to poor glycemic control in adolescents with diabetes. New England Journal of Medicine. 1986;315(4):215–9. doi: 10.1056/NEJM198607243150402. [DOI] [PubMed] [Google Scholar]
- 23.Moran A, Jacobs DR, Jr, Steinberger J, et al. Insulin resistance during puberty: results from clamp studies in 357 children. Diabetes. 1999;48(10):2039–44. doi: 10.2337/diabetes.48.10.2039. [DOI] [PubMed] [Google Scholar]
- 24.Travers SH, Labarta JI, Gargosky SE, et al. Insulin-like growth factor binding protein-I levels are strongly associated with insulin sensitivity and obesity in early pubertal children. Journal of Clinical Endocrinology & Metabolism. 1998;83(6):1935–9. doi: 10.1210/jcem.83.6.4857. [DOI] [PubMed] [Google Scholar]
- 25.Reaven GM, Hollenbeck C, Jeng CY, et al. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes. 1988;37:1020–4. doi: 10.2337/diab.37.8.1020. [DOI] [PubMed] [Google Scholar]
- 26.Jensen MD, Haymond MW, Rizza RA, et al. Influence of body fat distribution on free fatty acid metabolism in obesity. The Journal of clinical investigation. 1989;83:1168–73. doi: 10.1172/JCI113997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jensen MD, Nielsen S. Insulin dose response analysis of free fatty acid kinetics. Metabolism. 2007;56:68–76. doi: 10.1016/j.metabol.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 28.Basu A, Basu R, Shah P, et al. Systemic and regional free fatty acid metabolism in type 2 diabetes. American journal of physiology Endocrinology and metabolism. 2001;280:E1000–6. doi: 10.1152/ajpendo.2001.280.6.E1000. [DOI] [PubMed] [Google Scholar]
- 29.Riemens SC, Sluiter WJ, Dullaart RP. Enhanced escape of non-esterified fatty acids from tissue uptake: its role in impaired insulin-induced lowering of total rate of appearance in obesity and Type II diabetes mellitus. Diabetologia. 2000;43:416–26. doi: 10.1007/s001250051324. [DOI] [PubMed] [Google Scholar]
- 30.Hickner RC, Racette SB, Binder EF, et al. Suppression of whole body and regional lipolysis by insulin: effects of obesity and exercise. Journal of Clinical Endocrinology Metabolism. 1999;84:3886–95. doi: 10.1210/jcem.84.11.6137. [DOI] [PubMed] [Google Scholar]
- 31.Singh BM, Krentz AJ, Nattrass M. Insulin resistance in the regulation of lipolysis and ketone body metabolism in non-insulin dependent diabetes is apparent at very low insulin concentrations. Diabetes Res Clin Pract. 1993;20:55–62. doi: 10.1016/0168-8227(93)90023-x. [DOI] [PubMed] [Google Scholar]
- 32.Webber J, Whitelaw D, Smith JM, et al. Glucose and fatty acid metabolism in type 2 diabetes mellitus: an assessment using low-dose insulin infusion and the hyperinsulinaemic euglycaemic clamp. Diabetes Obes Metab. 1999;1:173–78. doi: 10.1046/j.1463-1326.1999.00011.x. [DOI] [PubMed] [Google Scholar]
- 33.Fabbrini E, deHaseth D, Deivanayagam S, et al. Alterations in fatty acid kinetics in obese adolescents with increased intrahepatic triglyceride content. Obesity (Silver Spring) 2009;17:25–9. doi: 10.1038/oby.2008.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.German JP, Wisse BE, Thaler JP, et al. Leptin deficiency causes insulin resistance induced by uncontrolled diabetes. Diabetes. 2010;59:1626–34. doi: 10.2337/db09-1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schauer IE, Snell-Bergeon JK, Bergman BC, et al. Insulin resistance, defective insulin-mediated fatty acid suppression, and coronary artery calcification in subjects with and without type 1 diabetes: The CACTI study. Diabetes. 2011;60:306–14. doi: 10.2337/db10-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D’Adamo E, Cali AM, Weiss R, et al. Central role of fatty liver in the pathogenesis of insulin resistance in obese adolescents. Diabetes Care. 2010;33:1817–22. doi: 10.2337/dc10-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim JS, Le KA, Mahurkar S, et al. Influence of elevated liver fat on circulating adipocytokines and insulin resistance in obese Hispanic adolescents. Pediatric obesity. 2012;7:158–64. doi: 10.1111/j.2047-6310.2011.00014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grundy SM, Denke MA. Dietary influences on serum lipids and lipoproteins. Journal of lipid research. 1990;31:1149–72. [PubMed] [Google Scholar]
- 39.Mensink RP, Katan MB. Effect of dietary fatty acids on serum lipids and lipoproteins. A meta-analysis of 27 trials. Arteriosclerosis and thrombosis: a journal of vascular biology/American Heart Association. 1992;12:911–9. doi: 10.1161/01.atv.12.8.911. [DOI] [PubMed] [Google Scholar]
- 40.Hernandez TL, Sutherland JP, Wolfe P, et al. Lack of suppression of circulating free fatty acids and hypercholesterolemia during weight loss on a high-fat, low-carbohydrate diet. Am J Clin Nutr. 2010;91:578–85. doi: 10.3945/ajcn.2009.27909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–40. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 42.Duncan EA, Brown MS, Goldstein JL, et al. Cleavage site for sterol-regulated protease localized to a leu-Ser bond in the lumenal loop of sterol regulatory element-binding protein-2. The Journal of biological chemistry. 1997;272:12778–85. doi: 10.1074/jbc.272.19.12778. [DOI] [PubMed] [Google Scholar]
- 43.Sato R, Yang J, Wang X, et al. Assignment of the membrane attachment, DNA binding, and transcriptional activation domains of sterol regulatory element-binding protein-1 (SREBP-1) The Journal of biological chemistry. 1994;269:17267–73. [PubMed] [Google Scholar]