

Abstract
BACKGROUND
Under conditions promoting early afterdepolarizations (EADs), ventricular tissue can become bi-excitable, that is, capable of wave propagation mediated by either the Na current (INa) or the L-type calcium current (ICa,L), raising the possibility that ICa,L-mediated reentry may contribute to polymorphic ventricular tachycardia (PVT) and torsades de pointes. ATP-sensitive K current (IKATP) activation suppresses EADs, but the effects on ICa,L-mediated reentry are unknown.
OBJECTIVE
To investigate the effects of IKATP activation on ICa,L-mediated reentry.
METHODS
We performed optical voltage mapping in cultured neonatal rat ventricular myocyte monolayers exposed to BayK8644 and isoproterenol. The effects of pharmacologically activating IKATP with pinacidil were analyzed.
RESULTS
In 13 monolayers with anatomic ICa,L-mediated reentry around a central obstacle, pinacidil (50 μM) converted ICa,L-mediated reentry to INa-mediated reentry. In 33 monolayers with functional ICa,L-mediated reentry (spiral waves), pinacidil terminated reentry in 17, converted reentry into more complex INa-mediated reentry resembling fibrillation in 12, and had no effect in 4. In simulated 2-dimensional bi-excitable tissue in which ICa,L- and INa-mediated wave fronts coexisted, slow IKATP activation (over minutes) reliably terminated rotors but rapid IKATP activation (over seconds) often converted ICa,L-mediated reentry to INa-mediated reentry resembling fibrillation.
CONCLUSIONS
IKATP activation can have proarrhythmic effects on EAD-mediated arrhythmias if ICa,L-mediated reentry is present.
Keywords: Early afterdepolarizations, Reentry, Bi-excitability, Torsades de pointes, ATP-sensitive K channels, Pinacidil
Introduction
In normal ventricular and atrial tissue, wave-front propagation is driven by activation of the Na current (INa). Recently, we showed that under conditions promoting early afterdepolarizations (EADs), ventricular tissue can become bi-excitable, that is, capable of wave propagation mediated by either the INa or the L-type calcium current (ICa,L), separately or simultaneously, in the same tissue.1 This raises the possibility that in addition to focal activations, ICa,L-mediated reentry may contribute to EAD-mediated arrhythmias such as polymorphic ventricular tachycardia (PVT) and torsades de pointes (TdP). Consistent with this possibility, ICa,L-mediated rotors exhibit a much longer cycle length than INa-mediated rotors, more compatible with the typical rates of PVT and TdP, and their core meandering reproduces characteristic electrocardiographic features of PVT and TdP.1
Many experimental and clinical studies2–9 have shown that pharmacological activation of sarcolemmal ATP-sensitive K (KATP) channels shortens action potential (AP) duration and abolishes EADs, preventing EAD-mediated triggered activity and suppressing PVT and TdP.2–9 Since hypotension is a common consequence of PVT and TdP, the resulting ischemia-related KATP channel activation may be a factor contributing to spontaneous termination of PVT and TdP and restoration of sinus rhythm by abolishing EAD-mediated focal excitations. Occasionally, however, TdP or PVT degenerate into ventricular fibrillation (VF), causing sudden cardiac death.10–12 Why most episodes of TdP or PVT terminate spontaneously, but some degenerate into VF, is poorly understood. To explore these issues, we investigated the effects of KATP channel activation on ICa,L-mediated reentry, given its potential relevance to PVT and TdP. By using optical voltage mapping in neonatal rat ventricular myocyte (NRVM) monolayers, combined with computer simulations, we find that the activation of KATP channels often terminates ICa,L-mediated reentry but also has a significant probability of converting ICa,L-mediated reentry into INa-mediated reentry, which can then degenerate into multiwavelet or mother rotor VF, especially if ATP-sensitive K current (IKATP) activation occurs rapidly.
Methods
All protocols used conform to the standard set forth by the National Institutes of Health in the Guide for the Care and Use of Animals (NIH Publication No 85-23, revised 1996).
Monolayer preparation
We created monolayers of NRVMs by plating 1 × 106 cells on 21-mm fibronectin-coated plastic coverslips, as previously described.13 Briefly, the hearts harvested from 2- to 3-day-old neonatal Sprague-Dawley rats were digested with collagenase (0.02; Worthington Biochemical Corp, Lakewood, NJ) and pancreatin (0.06; Sigma-Aldrich, St Louis, MO). Myocytes were isolated with the use of a Percoll (Pharmacia Biotech AB, Uppsala, Sweden) gradient and plated at a density of 106 cells/mm3 per coverslip.
Optical mapping
Arrhythmias were imaged by optical mapping performed after 11–14 days in culture. Coverslips were visually inspected under a microscope, and monolayers with obvious gaps in confluence were discarded. Acceptable coverslips were then stained with the voltage-sensitive dye di-4-ANEPPS (5 μmol/L for 5 minutes), after which they were continuously superfused with warm (36 °C) oxygenated (normal) Tyrode’s solution containing 135 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 0.33 mM NaH2PO4, 5 mM HEPES, and 5 mM glucose.
APs were recorded by using a charge-coupled device-based optical imaging system (Photometrics Cascade 128+;128 × 128 pixels). Voltage signals were acquired continuously over 10–180 seconds at 0.6–5 ms/frame. Signals were digitized with 16 bits of precision and processed offline, as described previously.14 Data were stored, displayed, and analyzed by using custom software written in Visual C++ (Microsoft) and MATLAB (MathWorks).
Experimental protocols
Reentrant arrhythmias were initiated by either rapid pacing or incubating monolayers in the presence of the L-type Ca channel agonist BayK8644 (BayK, 2.5 μM) and the beta-adrenergic receptor agonist isoproterenol (Iso; 1 μM) for > 2 hours. The dependence of wave propagation on INa vs ICa,L was tested in the subset of monolayers exhibiting reentry (n = 10) by using the Na channel blocker tetrodotoxin (TTX; 50 μM) or the Ca channel blocker nitrendipine (5 μM). KATP channels were activated by adding pinacidil (100 μM) to the superfusate.
Data analysis
The baseline drift due to photobleaching of potentiometric dyes was reduced by subtracting a third-order polynomial best fit curve of the optical signals. To reduce noise in the optical signals, a 7-point median filter was applied to the detrended data. Movies of electrical propagation were generated from signals that were low-pass filtered between 0 and 100 Hz. The activation time was defined as the instant of maximum positive slope. For each data set, the mean and accompanying 95 confidence intervals are reported. The conventional percentile bootstrap-resampling approach with 10,000 replications was used for estimating 95 confidence interval.15 A P value of <.05 was considered statistically significant. Conduction velocity was estimated as described previously.1,14
Computer simulations
Computer simulations used the ventricular AP model by Mahajan et al,16 modified to produce EADs and bi-excitability1. IKATP, based on the formulation by Ferrero et al,17 was added to the myocyte model with a density of 3.8 channels/μm2. To simulate the effects of pinacidil, we increased the fraction of open KATP channels (fATP) from 0 to 0.0025 (over time periods of 50 ms, 10 seconds, or 5 minutes). All simulations were performed in a monodomain 2-dimensional (2D) tissue of 300 × 300 cells (4.5 cm × 4.5 cm), as described previously.1 A linear gradient in the maximum conductance of IKs from 0.512 to 2.048 mS/cm2 was imposed from center to periphery in the tissue to facilitate the joint appearance of both INa- and ICa,L-mediated wave fronts as described previously to mimic the electrocardiographic appearance of TdP.1 Simulations were performed on NVIDA Tesla C2050 General Purpose Graphics Processing Units.
Results
Effects of pinacidil on ICa,L-mediated reentry in NRVM monolayers
To generate ICa,L-mediated wave propagation and reentry, we used our previously characterized model of bi-excitability by superfusing NRVM monolayers with BayK + Iso.1,18 When exposed to BayK + Iso, monolayers developed bursts of EAD-mediated focal activity, typically arising from multiple sites, resulting in a complex mixture of focal activity and reentry, as illustrated in Figure 1. The accompanying optical voltage trace recorded from a representative site in the monolayer during the initiation of a burst of focal activity illustrates the incomplete repolarization between beats, which is consistent with EAD-mediated triggered activity as described previously under these conditions.1
Figure 1.
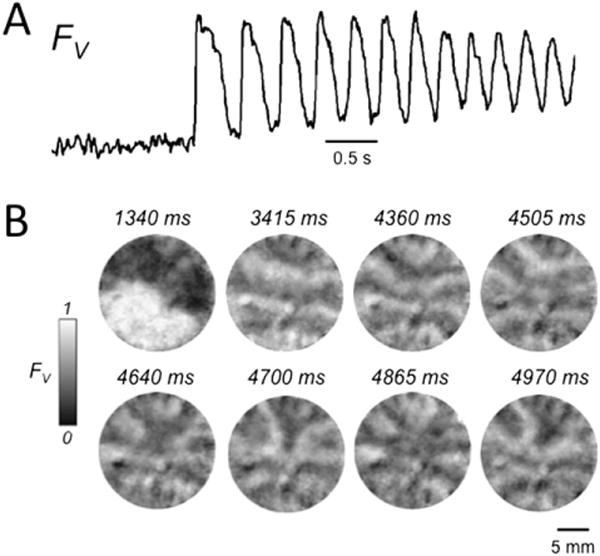
Early afterdepolarization (EAD)-related arrhythmias induced by BayK4688 + isoproterenol in neonatal rat ventricular myocyte monolayers. A: Optical voltage trace (FV) illustrating a burst of EAD-mediated triggered activity following a paced beat. Note the incomplete repolarization between beats. B: Snapshots of voltage fluorescence (FV) at the times indicated following the paced beat at 1340 ms, revealing a complex pattern of wave fronts due to a mixture of focal activity and reentry. FV is shown on a gray scale, with 0 = maximally repolarized (<−70 mV) and 1 = maximally depolarized (>0 mV), on the basis of our previously reported patch electrode recording estimates.19
Since reentry during BayK + Iso-induced arrhythmias in NRVM monolayers tended to be complex and intermittent due to meandering and frequent interruption by focal activations, we prefabricated monolayers with a hole in the center, which facilitated stable ICa,L-mediated reentry by anchoring reentry and also preventing spontaneous termination due to the core meandering and colliding with a tissue border. In these monolayers, incubation with BayK + Iso frequently resulted in sustained arrhythmias owing to either sustained reentry around the central obstacle (Figures 2 and 3) or more complex patterns in which rotors due to functional reentry were present (Figures 4 and 5). Both anchored and functional reentry in the presence of BayK + Iso were dependent on ICa,L-mediated wave propagation since reentry was consistently abolished by the L-type Ca channel blocker nitrendipine (n = 10) but not by the Na channel blocker TTX (n = 13) as in the examples of reentry around the central obstacle in Figures 2 and 3. Although TTX did not abolish reentry, it made conduction velocity more uniform in some monolayers by slowing rapid propagation in some regions (Figure 3). This indicates that although reentry was dependent on ICa,L-mediated wave propagation, INa contributed to more rapid propagation in some regions of the monolayer. Thus, BayK + Iso-treated monolayers contained a mixture of ICa,L-and INa-mediated wave propagation, even though reentry was dependent on ICa,L rather than INa.
Figure 2.

Early afterdepolarization-related reentry around a central obstacle in a neonatal rat ventricular myocyte monolayer. A: Snapshot (left) and trace (right) of voltage fluorescence (FV), illustrating that reentry around the central obstacle was unaffected by tetrodotoxin (TTX) (50 μM). B: In contrast, nitrendipine (5 μM) terminated reentry. C: Bar graph summarizing incidence of termination of reentry by tetrodotoxin (TTX) vs nitrendipine in 23 monolayers. FV gray scale as described in Figure 1 legend. BayK + Iso = BayK4688 + isoproterenol.
Figure 3.
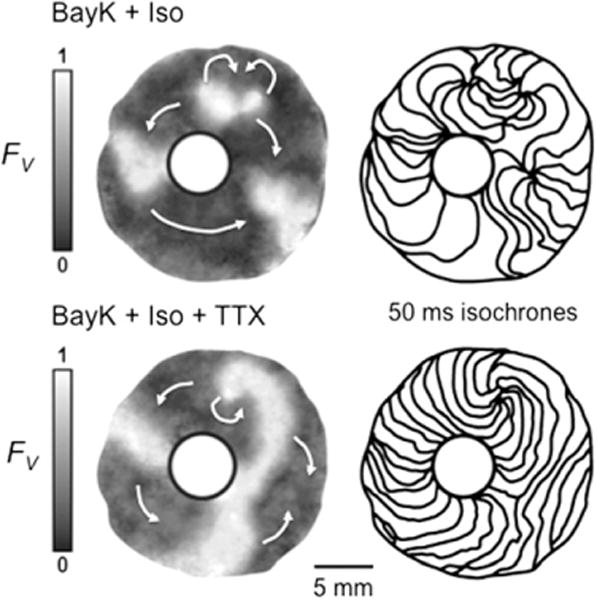
Effect of tetrodotoxin (TTX) on early afterdepolarization (EAD)-related reentry around a central obstacle in a neonatal rat ventricular myocyte monolayer. Top panels: Sustained reentry was driven by a figure-eight rotor (curved arrows at 1 o’clock). At right, isochrone maps (50 ms between isochrones) show slow propagation (closely spaced isochrones) clockwise from 9 to 6 o’clock, consistent with L-type calcium current (ICa,L)-mediated propagation, but much faster (widely spaced isochrones) from 6 to 9 o’clock, consistent with a significant contribution of Na current (INa)-mediated propagation. Bottom panels: After TTX (50 μM), a single rotor with the same cycle length (~500 ms) continues, but conduction velocity is now uniformly slow, consistent with ICa,L-mediated rotor and wave propagation throughout the tissue. FV gray scale as described in Figure 1 legend. BayK + Iso = BayK4688 + isoproterenol.
Figure 4.
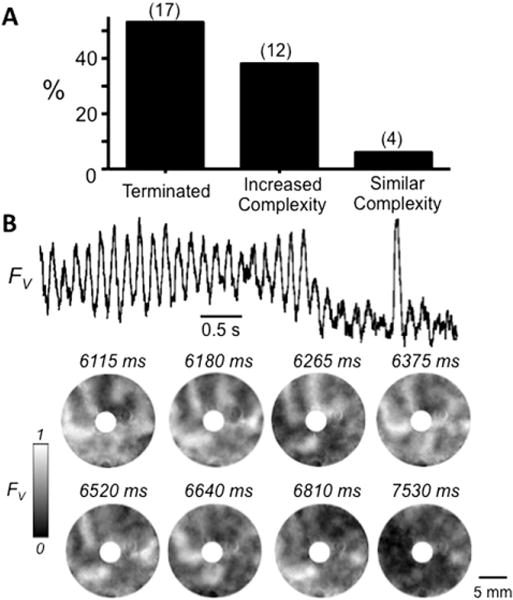
Effect of the ATP-sensitive K channel agonist pinacidil in neonatal rat ventricular myocyte (NRVM) monolayers with complex sustained early afterdepolarization (EAD)-mediated arrhythmias. A: Histogram illustrating the outcomes of pinacidil (100 μM) treatment on complex EAD-mediated arrhythmias induced by BayK4688 + isoproterenol in 33 NRVM monolayers with a central obstacle. B: Representative optical trace (top) and snapshots (below) of voltage fluorescence (FV) at the times indicated, illustrating termination of a complex EAD-mediated arrhythmia by pinacidil. In this example, the arrhythmia was driven primarily by a functional rotor (spiral wave) at 1 o’clock. FV gray scale as described in Figure 1 legend.
Figure 5.
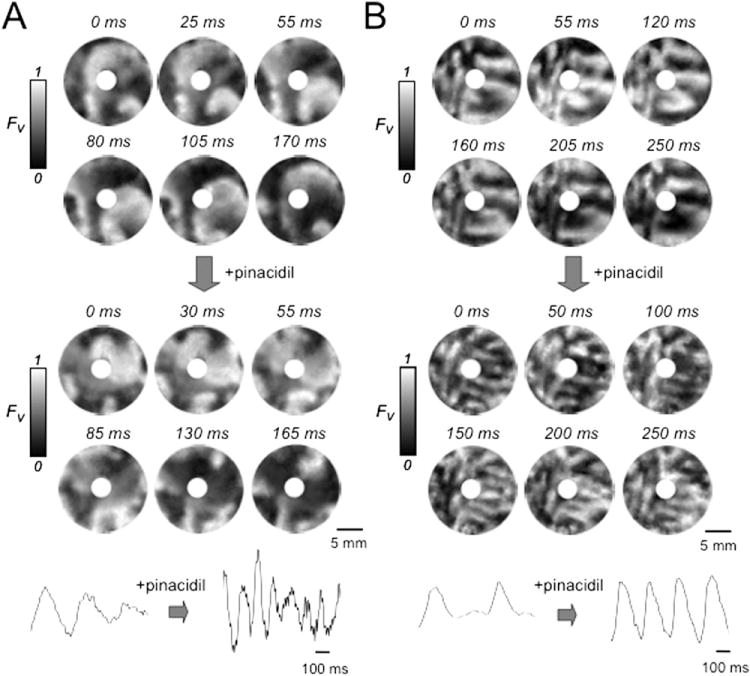
Effect of the ATP-sensitive K channel agonist pinacidil in neonatal rat ventricular myocyte monolayers with complex sustained early afterdepolarization-mediated arrhythmias. A: Snapshots of voltage fluorescence (FV) at the times indicated in a monolayer with a central obstacle in which BayK4688 + isoproterenol induced an arrhythmia driven primarily by functional rotors (spiral waves) at 6 and 8 o’clock (left panels). Pinacidil (100 μM, right panels) accelerated but did not significantly affect the complexity of the arrhythmia. B: Voltage snapshots from another monolayer with a central obstacle, in which pinacidil (right panels) increased the complexity of the BayK4688 + isoproterenol-induced arrhythmia, as evident from the greater number of wave fronts. FV gray scale as described in Figure 1 legend.
We then examined the effects of KATP channel activation with 5 μM pinacidil in monolayers with sustained anatomic or functional ICa,L-mediated reentry for > 10 minutes. In 13 monolayers with sustained anatomic reentry around the central obstacle, as in Figures 2 and 3, pinacidil did not terminate reentry but changed its pharmacological profile. Although reentry was consistently terminated by nitrendipine (Figure 2B) but not by TTX before pinacidil (Figures 2A and 3), the converse was true after pinacidil; that is, reentry was terminated by TTX in all monolayers tested, whereas nitrendipine terminated reentry in only 2 of 10 monolayers tested (20) (Figure 6). This result indicates that pinacidil converted ICa,L-mediated reentry to INa-mediated reentry. Consistent with this interpretation, conduction velocity after pinacidil increased by an average of 89 ± 82% (95% confidence interval 46–141) (Figure 7A). This is illustrated by the increased spacing of the isochrone lines after pinacidil in the example shown in Figure 7B.
Figure 6.
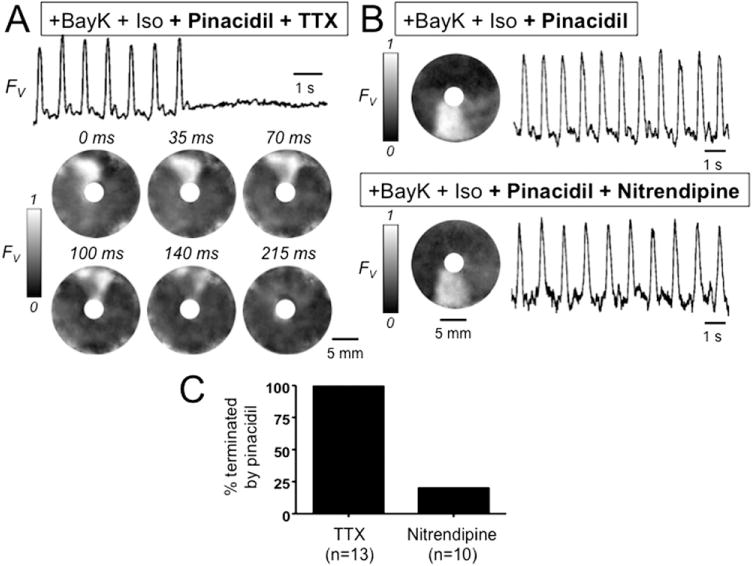
Effect of pinacidil on early afterdepolarization-mediated reentry around a central obstacle in a neonatal rat ventricular myocyte monolayer. A: Optical trace (above) and snapshots (below) of voltage fluorescence (FV) in a monolayer in which BayK4688 + isoproterenol induced sustained reentry around a central obstacle. After pinacidil treatment, the addition of tetrodotoxin (TTX) (50 μM) terminated reentry. B: Same, but with nitrendipine (5 μM) added instead of TTX, showing that reentry persists. C: Bar graph summarizing the incidence of termination of reentry by TTX vs nitrendipine in 23 monolayers exposed to BayK4688 + isoproterenol + pinacidil. FV gray scale as described in Figure 1 legend. BayK + Iso = BayK4688 + isoproterenol.
Figure 7.
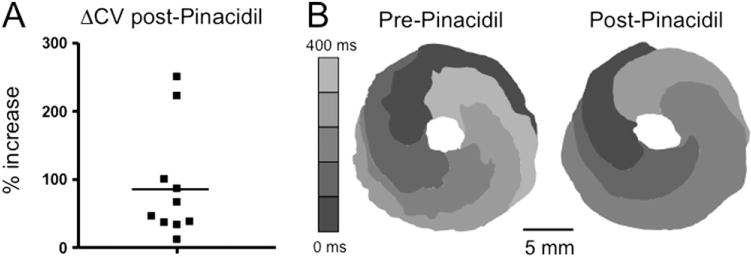
Effect of pinacidil on conduction velocity of early afterdepolarization-mediated reentry around a central obstacle in neonatal rat ventricular myocyte monolayers. A: The percent increase in conduction velocity after pinacidil in 10 monolayers (solid squares) in which BayK4688 + isoproterenol induced sustained reentry around a central obstacle. Mean increase is indicated by the horizontal bar. B: Isochrome map during reentry in a representative monolayer, pre-, and post-pinacidil treatment. Increased spacing between 80-ms isochrome lines after pinacidil reflects increased conduction velocity. FV gray scale as described in Figure 1 legend.
In contrast, among 33 monolayers with more complex arrhythmia patterns involving functional ICa,L-mediated rotors as in Figures 4 and 5, the administration of pinacidil spontaneously terminated the arrhythmia in 17 (52%) (Figure 4). In the remaining 16 monolayers, complex reentry persisted after pinacidil, but with a significantly shorter average cycle length (Figure 5). In 4 monolayers (12%), the complexity of reentry was not significantly changed (Figure 5 A), but in 12 (36%), the complexity increased with the appearance of many additional and more frequent wave fronts (Figure 5B).
Computer simulations in 2D tissue
To ions by using the Mahajan et al16 ventricular AP model, with parameters modified to produce EADs with bi-excitable wave propagation in tissue, as described previously. In homogeneous 2D tissue, either INa- or ICa,L-mediated reentry could be induced, depending on the stimulation protocol used to initiate reentry, indicative of bi-excitability. To study the effects of IKATP activation on functional reentry driven by an ICa,L-mediated rotor in which INa-mediated wave fronts were also intermittently present (as previously documented in bi-excitable monolayers1), we created a heterogeneous 2D tissue with a linearly increasing gradient in IKs from center to periphery, such that the center of the tissue had a longer AP duration than did the periphery. When an ICa,L-mediated rotor was induced, the rotor meandered throughout the central region, with the arm of the rotor in the outer tissue containing a mixture of ICa,L- and INa-mediated wave propagation whose relative proportions varied in time (Figure 8).
Figure 8.
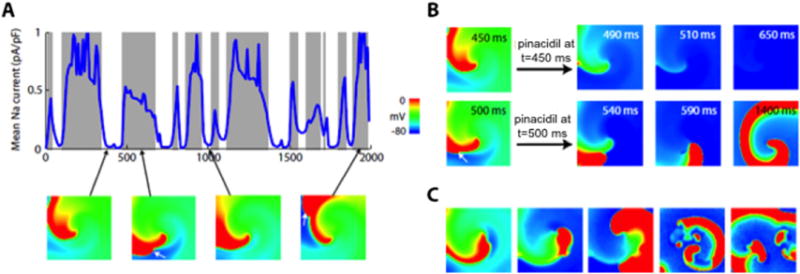
Effect of ATP-sensitive K current (IKATP) activation on L-type calcium current (ICa,L)-mediated reentry in simulated 2-dimensional heterogeneous cardiac tissue (300 × 300 myocytes). A: Outcomes of rapidly activating IKATP at various time points during reentry driven by an ICa,L-mediated rotor in the center of the tissue. Gray zones indicate that ICa,L-mediated reentry converted to Na current (INa)-mediated reentry after IKATP activation; white zones indicate that reentry terminated. The blue line shows INa amplitude averaged over all cells in the tissue over time. IKATP activation terminated reentry only if no significant INa-mediated wave fronts were present. Voltage snapshots (below) show the ICa,L-mediated rotor in the center of the tissue, with INa-mediated wave fronts intermittently present along the spiral arm (2nd and 4th panels; white arrows). B: Voltage snapshots showing termination of reentry by IKATP activation at t = 450 ms (top row) but conversion to INa-mediated reentry at t = 500 ms (bottom row). C: When the number of KATP channels was randomly varied from myocyte to myocyte (using a Gaussian distribution with mean 3.8 channels/μm2 and standard deviation 2.0), IKATP activation caused the stable ICa,L-mediated rotor to convert to an INa-mediated rotor, which then broke up into multiwavelet VF. FV scale as described in Figure 1 legend.
To simulate the effects of pinacidil, we added a formulation of IKATP17 whose conductance throughout the tissue increased linearly to a maximum over a specified time period. Under these conditions, the outcome of IKATP activation during the arrhythmia was probabilistic in nature, sensitive to both the timing and the speed of IKATP activation. When IKATP was activated rapidly over 50 ms, reentry terminated and the tissue repolarized to a quiescent state in 41 of 201 trials. In the remaining 59 trials, the ICa,L-mediated rotor was converted to an INa-mediated rotor (Figure 8A). The specific outcome depended on whether INa-mediated wave fronts were present in the tissue when IKATP was activated. If IKATP was activated at a time when no INa wave fronts happened to be present in the arm of the rotor, reentry terminated (Figure 8A, unshaded areas) because no INa wave fronts were present to form a new rotor after the ICa,L-mediated rotor terminated. In contrast, if IKATP was activated at a time when INa wave fronts were present in the arm of the rotor (Figure 8A, shaded areas), these INa wave fronts were unaffected by IKATP activation. Transformation from an ICa,L-mediated to an INa-mediated rotor was achieved by the INa-mediated wave fronts whipping around the front end of the vanishing ICa,L-mediated rotor, reactivating the fully repolarized tissue previously occupied by the ICa,L-mediated wave front (Figure 8B).
In this scenario, the speed of IKATP activation also played a critical role. When IKATP was activated slowly—over 10 seconds or 5 minutes—the incidence of termination increased to 54% (101 trials) and 100% (21 trials), respectively. This is because when IKATP reached a sufficient amplitude to terminate all ICa,L-mediated wave fronts, the AP duration and wavelength were still too long to allow an INa-mediated rotor to become sustained. That is, if an INa-mediated rotor formed at this point, it meandered to the tissue border and self-terminated (Movie 1, online supplementary material). Only if IKATP continued to activate rapidly did the AP duration and wavelength shorten sufficiently to allow the INa-mediated rotor to stabilize before colliding with a tissue border and self-terminating (Movie 2, online supplementary material). If the tissue size was increased to prolong the lifetime of the INa-mediated rotor when the ICa,L-mediated rotor terminated, the percentage of trials that resulted in termination for the same time course of IKATP activation decreased.
If random cell-to-cell variability in the number of KATP channels was introduced into the tissue model to simulate a heterogeneous distribution of KATP channels among cardiac myocytes, then for the cases in which INa-mediated rotors did not terminate during IKATP activation, the increased dispersion of repolarization led to unidirectional conduction block, causing the INa-mediated rotor to break up into multiwavelet VF (Figure 8C). This was similar to the finding in monolayers in which pinacidil increased arrhythmia complexity (Figure 5B).
In summary, these simulations show that IKATP activation can either terminate ICa,L-mediated reentry or convert it to more complex INa-mediated reentry in heterogeneous tissue, depending on the timing and speed of IKATP activation, with slower IKATP activation favoring termination.
Discussion
Activation of sarcolemmal KATP channels shortens the cardiac AP duration and generally suppresses EADs by increasing repolarization reserve, as demonstrated previously in many studies.2–9 Therefore, when ventricular arrhythmias such as PVT and TdP are caused by EAD-mediated triggered activity arising from one or more focal sites, IKATP activation tends to be antiarrhythmic by suppressing EAD formation. However, the extent to which EAD-mediated arrhythmias are focal or reentrant remains controversial.20,21 We recently demonstrated that under EAD-promoting conditions, cardiac tissue can become bi-excitable, that is, capable of supporting both INa- and ICa,L-mediated wave fronts.1 The theory of bi-excitability posits that myocytes can develop 2 quasi-stable resting membrane potentials under appropriate conditions in which repolarization reserve is reduced (eg, by BayK + Iso)—a fully repolarized state from which INa-mediated wave fronts emanate and a partially depolarized state at which INa is inactivated but ICa,L can still mediate propagating wave fronts. We previously demonstrated this phenomenon in BayK + Iso-treated monolayers without a central obstacle in which nonsustained bursting behavior emanates from a partially depolarized membrane potential, which repolarizes back to the fully repolarized resting membrane potential when the burst terminates.18 Here, we have shown the analogous phenomenon for ICa,L-mediated reentry in bi-excitable tissue. In the optical trace in Figure 2B, termination of ICa,L-mediated reentry by nitrendipine allowed membrane potential to fully repolarize. In Figure 4B, termination of ICa,L-mediated by pinacidil had a similar effect, with full repolarization and a subsequent full amplitude AP. It is possible that by increasing repolarization reserve, pinacidil hyperpolarized the fully repolarized resting potential to some extent. However, the similarity between the effects of nitrendipine and pinacidil implies that full repolarization after pinacidil, such as nitrendipine, is primarily due to the destabilization of the partially depolarized resting state made possible by BayK + Iso, rather by additional hyperpolarization of the fully repolarized resting membrane potential by IKATP activation.
ICa,L-mediated rotors that can form under bi-excitable conditions exhibit electrocardiographic features and cycle lengths resembling PVT and TdP.1 Given the possibility that ICa,L-mediated reentry may be involved in the pathogenesis of PVT or TdP, the question arises as to whether IKATP activation has exclusively antiarrhythmic effects on this form of reentry (as it does on EAD-mediated triggered activity) or whether it can be proarrhythmic as well. This is an important question, because IKATP activation agonists have been proposed as therapeutic agents for TdP and also because hypotension associated with prolonged episodes of PVT and TdP may result in IKATP activation physiologically. We hoped that the investigation of this issue might provide insight into why PVT and TdP episodes, which usually self-terminate, occasionally degenerate to VF to cause sudden cardiac death.10–12
By using pinacidil to activate IKATP in NRVM monolayers, we found IKATP activation often converted ICa,L-mediated anatomic or functional reentry to INa-mediated reentry with faster conduction velocity, accelerating the rate of the tachycardia. These findings imply that if ICa,L-mediated reentry is present during PVT or TdP, activation of IKATP by drugs or ischemia might accelerate the tachycardia, potentially resulting in wavebreak and precipitating multiwavelet or mother rotor VF. We consistently observed that anatomic ICa,L-mediated reentry around a central obstacle was converted to anatomic INa-mediated reentry. For functional ICa,L-mediated rotors in which ICa,L- and INa-mediated wave fronts coexisted, however, the effects of IKATP activation were probabilistic. As elucidated in computer simulations, if IKATP was activated during an epoch when no INamediated wave fronts happened to be present in the tissue, reentry terminated. However, if IKATP was activated when both ICa,L- and INa-mediated wave fronts were present, the ICa,L-mediated wave fronts were eliminated but the INa-mediated wave fronts were unaffected, allowing INa-mediated rotors to form and potentially break up into multiwavelet or mother rotor VF (Figure 8). An important factor influencing whether functional ICa,L-mediated rotors terminated or were converted to INa-mediated reentry and VF was the rapidity with which IKATP was activated. As IKATP was activated more slowly, the probability of termination progressively increased and the probability of degeneration to VF concomitantly decreased. This was because the tissue size was not large enough to support a stable INa-mediated rotor when IKATP first became large enough to eliminate the ICa,L-mediated rotor. Only if subsequent IKATP activation was rapid did the AP duration and wavelength subsequently shorten quickly enough to stabilize the INa-mediated rotor before it meandered to a tissue border and self-terminated. This implies that tissue size, relative to wavelength, is also an important determinant of whether functional ICa,L-mediated rotors terminate or are converted to sustained INa-mediated reentry, with termination being favored by smaller tissue size. However, even for the large tissue size (4.5 cm × 4.5 cm) that we simulated, the incidence of termination was still 41% even with very rapid IKATP activation over 50 ms and 100% for slow activation over 5 minutes. Given that spontaneous IKATP activation due to systemic hypotension during PVT or TdP occurs gradually over minutes, our findings suggest that hypotension in this setting is more likely to favor termination of these arrhythmias than promote degeneration to VF but the latter still has a finite probability. In addition, there are many other reasons why episodes of PVT or TdP due to ICa,L-mediated reentry might spontaneously terminate before IKATP activation becomes significant, including heterogeneity-induced spiral drifting, wave collision, and dynamical instabilities.22–25 Also, under conditions in which PVT or TdP are maintained primarily by focal activation, rather than ICa,L-mediated reentry, IKATP activation would be expected to suppress the arrhythmia, whether activated rapidly or slowly. However, our findings raise the possibility that an acute intravenous administration of an IKATP agonist to terminate PVT or TdP in the clinical setting could run a significant risk of converting the arrhythmia to VF if the arrhythmia involved ICa,L-mediated reentry and the drug was administered rapidly.
In extrapolating to the clinical relevance of the present findings, a number of caveats and limitations should be recognized. ICa,L-mediated reentry was induced by using BayK + Iso in NRVM monolayers, which, despite the advantages for optical mapping, have important electrophysiological differences with human myocardium. It is also not possible to calibrate membrane potential absolutely by using optical mapping, although in a previous study,19 we documented a resting membrane potential < −70 mV, as confirmed in other studies,26,27 but values from −65 to −70 mV have also been reported,19 at which INa may be significantly reduced. BayK + Iso is an artificial means of inducing EADs, with arguably limited direct physiological relevance, although Iso alone has been shown to generate EAD bursting in this preparation and is well known to be a potentiating arrhythmogenic factor in long QT syndromes.28,29 Among many other effects, BayK and Iso both accentuate mode 2 gating of L-type Ca channels, which is believed to contribute to the genetic defect in LQT8.30 In addition, ICa,L-mediated rotors due to bi-excitability have yet to be documented experimentally in intact native animal or human cardiac tissues subjected to more physiologically relevant stressors causing EADs. The experimental model (NRVM) and computer AP model (rabbit ventricle) also represented different species. On the other hand, the observation that different experimental and computer models yielded consistent results increases our confidence that these findings are generalizable and may have relevance to clinical EAD-mediated arrhythmias such as TdP and PVT.
Conclusions
IKATP activation can have pro-arrhythmic or anti-arrhythmic effects on ICa,L-induced reentry in the setting of EAD-mediated arrhythmias such as TdP and PVT.
Supplementary Material
Acknowledgments
This study was supported by National Institutes of Health (NIH) grants P01 HL078931 (to Dr Weiss and Dr Qu), R01 HL103622 (to Dr Weiss and Dr Qu), MSTP T32GM008042, and MCIP T32GM065823; a fellowship award for advanced researchers from the Swiss Foundation for Grants in Biology and Medicine (Dr de Lange); and the Laubisch and Kawata Endowments (to Dr Weiss).
ABBREVIATIONS
- 2D
2-dimensional
- AP
action potential
- EAD
early afterdepolarization
- ICa,L
L-type calcium current
- IKATP
ATP-sensitive K current
- INa
Na current
- Iso
isoproterenol
- KATP
ATP-sensitive K
- NRVM
neonatal rat ventricular myocyte
- PVT
polymorphic ventricular tachycardia
- TdP
torsades de pointes
- TTX
tetrodotoxin
- VF
ventricular fibrillation
Appendix: Supplementary data
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.hrthm.2012.12.017.
References
- 1.Chang MG, Sato D, de Lange E, et al. Bi-stable wave propagation and early afterdepolarization-mediated cardiac arrhythmias. Heart Rhythm. 2012;9:115–122. doi: 10.1016/j.hrthm.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fish FA, Prakash C, Roden DM. Suppression of repolarization-related arrhythmias in vitro and in vivo by low-dose potassium channel activators. Circ Res. 1990;82:1362–1369. doi: 10.1161/01.cir.82.4.1362. [DOI] [PubMed] [Google Scholar]
- 3.Spinelli W, Sorota S, Siegal M, et al. Antiarrhythmic actions of the ATP-regulated K current activated by pinacidil. Circ Res. 1991;68:1127–1137. doi: 10.1161/01.res.68.4.1127. [DOI] [PubMed] [Google Scholar]
- 4.Carlsson L, Abrahamsson C, Drews L, et al. Antiarrhythmic effects of potassium channel openers in rhythm abnormalities related to delayed repolarization. Circulation. 1992;85:1491–1500. doi: 10.1161/01.cir.85.4.1491. [DOI] [PubMed] [Google Scholar]
- 5.Sato T, Hata Y, Yamamoto M, Morita H, et al. Early afterdepolarization abolished by potassium channel opener in a patient with idiopathic long QT syndrome. J Cardiovasc Electrophysiol. 1995;6:279–282. doi: 10.1111/j.1540-8167.1995.tb00400.x. [DOI] [PubMed] [Google Scholar]
- 6.Riccioppo Neto F, Mesquita Junior O, Olivera GB. Antiarrhythmic and electrophysiological effects of the novel KATP channel opener, rilmakalim, in rabbit cardiac cells. Gen Pharmacol. 1997;29:201–205. doi: 10.1016/s0306-3623(96)00403-x. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe O, Okumura T, Takeda H, et al. Nicorandil, a potassium channel opener, abolished torsades de pointes in a patient with complete atrioventricular block. Pacing Clin Electrophysiol. 1999;22:686–688. doi: 10.1111/j.1540-8159.1999.tb00516.x. [DOI] [PubMed] [Google Scholar]
- 8.Shimizu W, Antzelevitch C. Effects of a K channel opener to reduce transmural dispersion of repolarization and prevent torsade de pointes in LQT1, LQT2, and LQT3 models of the long-QT syndrome. Circulation. 2000;102:706–712. doi: 10.1161/01.cir.102.6.706. [DOI] [PubMed] [Google Scholar]
- 9.Chinushi M, Kasai H, Tagawa M, et al. Triggers of ventricular tachyarrhythmias and therapeutic effects of nicorandil in canine models of LQT2 and LQT3 syndromes. J Am Coll Cardiol. 2002;40:555–562. doi: 10.1016/s0735-1097(02)01975-7. [DOI] [PubMed] [Google Scholar]
- 10.Krikler DM, Curry PV. Torsade de pointes, an atypical ventricular tachycardia. Br Heart J. 1976;38:117–120. doi: 10.1136/hrt.38.2.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.El-Sherif N, Turitto G. Torsade de pointes. Curr Opin Cardiol. 2003;18:6–13. doi: 10.1097/00001573-200301000-00002. [DOI] [PubMed] [Google Scholar]
- 12.Drew BJ, Ackerman MJ, Funk M, et al. Prevention of torsade de pointes in hospital settings: a scientific statement from the American Heart association and the American College of Cardiology Foundation. Circulation. 2010;121:1047–1060. doi: 10.1161/CIRCULATIONAHA.109.192704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abraham MR, Henrikson CA, Tung L, et al. Antiarrhythmic engineering of skeletal myoblasts for cardiac transplantation. Circ Res. 2005;97:159–167. doi: 10.1161/01.RES.0000174794.22491.a0. [DOI] [PubMed] [Google Scholar]
- 14.de Diego C, Pai RK, Dave AS, et al. Spatially discordant alternans in cardiomyocyte monolayers. Am J Physiol Heart Circ Physiol. 2008;294:H1417–H1425. doi: 10.1152/ajpheart.01233.2007. [DOI] [PubMed] [Google Scholar]
- 15.Calmettes G, Drummond GB, Vowler SL. Making do with what we have: use your bootstraps. J Physiol. 2012;590:3403–3406. doi: 10.1113/jphysiol.2012.239376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahajan A, Shiferaw Y, Sato D, et al. A rabbit ventricular action potential model replicating cardiac dynamics at rapid heart rates. Biophys J. 2008;94:392–410. doi: 10.1529/biophysj.106.98160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferrero JM, Jr, Saiz J, Ferrero JM, et al. Simulation of action potentials from metabolically impaired cardiac myocytes: role of ATP-sensitive K current. Circ Res. 1996;79:208–221. doi: 10.1161/01.res.79.2.208. [DOI] [PubMed] [Google Scholar]
- 18.Chang MG, Chang CY, de Lange E, et al. Dynamics of early afterdepolarization-mediated triggered activity in cardiac monolayers. Biophys J. 2012;102:2706–2714. doi: 10.1016/j.bpj.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang MG, Zhang Y, Chang CY, et al. Spiral waves and reentry dynamics in an in vitro model of the healed infarct border zone. Circ Res. 2009;105:1062–1071. doi: 10.1161/CIRCRESAHA.108.176248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Asano Y, Davidenko JM, Baxter WT, et al. Optical mapping of drug-induced polymorphic arrhythmias and torsade de pointes in the isolated rabbit heart. J Am Coll Cardiol. 1997;29:831–842. doi: 10.1016/s0735-1097(96)00588-8. [DOI] [PubMed] [Google Scholar]
- 21.Choi BR, Burton F, Salama G. Cytosolic Ca triggers early afterdepolarizations and torsade de pointes in rabbit hearts with type 2 long QT syndrome. J Physiol. 2002;543:615–631. doi: 10.1113/jphysiol.2002.024570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nattel S, Kneller J, Zou R, et al. Mechanisms of termination of atrial fibrillation by class I antiarrhythmic drugs: Evidence from clinical, experimental, and mathematical modeling studies. J Cardiovasc Electrophysiol. 2003;14:S133–S139. doi: 10.1046/j.1540.8167.90302.x. [DOI] [PubMed] [Google Scholar]
- 23.Qu Z, Weiss JN. Effects of Na and K channel blockade on vulnerability to and termination of fibrillation in simulated normal cardiac tissue. Am J Physiol Heart Circ Physiol. 2005;289:H1692–H1701. doi: 10.1152/ajpheart.00241.2005. [DOI] [PubMed] [Google Scholar]
- 24.Qu Z. Critical mass hypothesis revisited: Role of dynamical wave stability in spontaneous termination of cardiac fibrillation. Am J Physiol. 2006;290:H255–H263. doi: 10.1152/ajpheart.00668.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamazaki M, Honjo H, Nakagawa H, et al. Mechanisms of destabilization and early termination of spiral wave reentry in the ventricle by a class III antiarrhythmic agent, nifekalant. Am J Physiol Heart Circ Physiol. 2007;292:H539–H548. doi: 10.1152/ajpheart.00640.2006. [DOI] [PubMed] [Google Scholar]
- 26.Sekar RB, Kizana E, Cho HC, et al. IK1 heterogeneity affects genesis and stability of spiral waves in cardiac myocyte monolayers. Circ Res. 2009;104:355–364. doi: 10.1161/CIRCRESAHA.108.178335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pedrotty DM, Klinger RY, Kirkton RD, et al. Cardiac fibroblast paracrine factors alter impulse conduction and ion channel expression of neonatal rat cardiomyocytes. Cardiovasc Res. 2009;83:688–697. doi: 10.1093/cvr/cvp164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kossmann CE. The long QT interval and syndromes. Adv Intern Med. 1987;32:87–110. [PubMed] [Google Scholar]
- 29.Vermeulen JT. Mechanisms of arrhythmias in heart failure. J Cardiovasc Electrophysiol. 1998;9:208–221. doi: 10.1111/j.1540-8167.1998.tb00902.x. [DOI] [PubMed] [Google Scholar]
- 30.Splawski I, Timothy KW, Decher N, et al. Inaugural article: Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci. 2005;102:8089–8096. doi: 10.1073/pnas.0502506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.