

Abstract
Localized protein synthesis is a fundamental mechanism for creating distinct subcellular environments. Here we developed a generalizable proximity-specific ribosome profiling strategy that enables global analysis of translation in defined subcellular locations. We applied this approach to the endoplasmic reticulum (ER) in yeast and mammals. We observed the large majority of secretory proteins to be cotranslationally translocated, including substrates capable of post-translational insertion in vitro. Distinct translocon complexes engaged nascent chains at different points during synthesis. Whereas most proteins engaged the ER immediately after or even before signal sequence (SS) emergence, a class of Sec66-dependent proteins entered with a looped SS conformation. Finally, we observed rapid ribosome exchange into the cytosol after translation termination. These data provide insights into how distinct translocation mechanisms act in concert to promote efficient cotranslational recruitment.
Eukaryotic cells contain highly specialized subcellular environments including both membrane- and non-membrane-bound compartments. Localized protein synthesis can play a critical role in creating these subcellular structures by allowing protein production at the site of action and in response to local cellular need. Local translation is involved in diverse processes including developmental patterning, cellular motility, synaptic plasticity, and protein trafficking through the secretory pathway (1). Dysfunctional RNA localization is linked to neurodevelopmental and neurodegenerative diseases (2). Numerous microscopy-based studies of individual mRNAs have demonstrated a breadth of subcellular localizations, and recent genome-wide mapping of transcript localization within cells and tissues has further emphasized the widespread spatial control of mRNA (3).
By contrast, global approaches for studying spatial control of protein synthesis are limited to bulk interrogations that cannot uniquely identify proteins – such as the RiboPuroMycylation (4) and FUNCAT (5) methods – or require careful biochemical fractionation of the compartment of interest (6), limiting both the location and resolution of analyses. These considerations motivated us to develop a generalizable strategy for enabling proximity-specific ribosome profiling that preserves in vivo spatiotemporal information about the site of synthesis. We employed a two-step approach wherein we (i) used a spatially-restricted biotin ligase (BirA) to mark ribosomes containing a biotin acceptor peptide (AviTag) in live cells with all membranes and spatial relations intact (7) and (ii) read out the translational activity of purified biotinylated ribosomes with ribosome profiling (the deep sequencing of ribosome protected fragments) (8) that quantitatively reports on genome-wide translation with sub-codon resolution (Fig. 1A).
Fig. 1. A system for in vivo proximity-dependent ribosome biotinylation to monitor local protein synthesis at the ER.

(A) Schematic for proximity-specific ribosome profiling (i) The Escherichia coli biotin ligase BirA is localized to a subcellular site of interest in cells expressing an Avi-tagged ribosomal protein and grown in low-biotin conditions (ii) A biotin pulse is applied resulting in specific biotinylation of ribosomes in close physical proximity to the localized BirA (iii) Ribosome profiling of paired input (gray and red) and isolated biotinylated (red) monosomes reveals codon-resolved translational enrichment specific to the BirA locale. (B) Fractionation of yeast lysates derived from strains containing scarless C-terminal Rps2 or Rpl16a/b hemagglutinin (HA)-TEV-AviTags on 10 to 50% sucrose gradients. Polysome traces demonstrate proper ribosomal assembly and incorporation of tags into polysomes demonstrates their non-perturbative nature. (C) ER localization of BirA fusion proteins used in this study. BirA-mVenus-Ubc6, Sec63-mVenus-BirA and BirA-mVenus-Ssh1 all localize to the perinuclear and cortical ER. (D) Western blot analysis demonstrates that biotinylation of ribosomal AviTags does not occur before the addition of excess biotin or post lysis in our assay. (E) Biotinylation kinetics of 40S and 60S AviTags by BirAs localized to the cytosol or ER (Sec63). Favorable kinetics were achieved independent of localization, and preferential 60S biotinylation demonstrates the specificity of the ER-localized ligase for oriented ER ribosomes. Shaded regions indicate biotinylation times used in subsequent sequencing experiments.
Here we used this proximity-specific ribosome profiling strategy to study protein synthesis at the endoplasmic reticulum (ER), a major site of localized protein synthesis where a diverse set of proteins enter the secretory pathway. Work spanning several decades has revealed multiple routes of targeting nascent proteins to the ER (9). These include the canonical signal recognition particle (SRP)–dependent pathway in which translation is halted upon binding of SRP to hydrophobic sequences, and resumes only when the ribosome engages the translocon. Additionally, there are several SRP-independent pathways, although these are generally considered to mediate posttranslational import (9). Extensive studies have also elucidated the core translocational machinery necessary for protein import across and into the ER membrane, and identified accessory translocon factors in yeast and metazoans thought to increase the efficiency of protein import or assist the translocation of specific proteins (10).
Despite our in-depth mechanistic and structural understanding of these steps, the broader cellular organization of these targeting routes in vivo has remained largely unexplored. Experimental limitations have prevented a systematic characterization of substrate flux through the various ER-targeting pathways in unperturbed cells. Similarly, our understanding of rough ER dynamics remains limited because of the difficulty in precisely measuring both the timing of ribosome-nascent chain (RNC) recruitment to the translocation machinery, as well as RNC fate following translation termination. Here we developed and applied proximity-specific ribosome profiling to address these fundamental questions.
A general approach for subcellular ribosome profiling: Development and application to the ER
To establish the proximity-specific ribosome profiling method, we implemented the following five steps: (i) introduction of a non-perturbing ribosome tag consisting of a tobacco etch virus (TEV) protease-cleavable AviTag; (ii) genetic targeting of BirA to a subcellular location of interest; (iii) temporal control of ribosome biotinylation in vivo; (iv) inhibition of post-lysis biotinylation; and (v) selective isolation of biotinylated ribosomes and specific elution via TEV cleavage (Fig. 1A). We developed and validated these steps in the budding yeast Saccharomyces cerevisiae as well as in the human embryonic kidney–293 (HEK-293) cell line.
Informed by a recent structure of the yeast 80S ribosome (11), we expressed Avi-tagged versions of several candidate ribosomal proteins with surface-accessible termini. We identified multiple subunits that when tagged and expressed from their endogenous loci, including the natural 3′ untranslated region (UTR), were incorporated into ribosomes and covered growth defects seen in deletion mutants. These included C-terminally tagged RPL16 and RPS2 [also called uL13 and uS5 (12)], which were used for subsequent experiments (Fig. 1B). N-terminally tagged RPL10a [uL1] was used for mammalian studies (13) (fig. S1A).
For our yeast studies, we constructed three different ER-localized BirA fusion proteins, as well as cytosolic and mitochondrial controls (Fig. 1C). To broadly capture the translational activity of all ER-associated ribosomes, we localized BirA to the ER using the C-terminal tail-anchor (TA) from UBC6 (14). To more specifically examine translation at two known translocation entry points to the ER, we fused BirA to SEC63, a member of the SEC complex that specifically associates with the Sec61 translocon (15), and to SSH1, a paralog of the canonical Sec61 translocon that interacts with SRP but not the SEC complex (16). For the mitochondrial studies, we used a BirA-fusion to OM45, a major constituent of the mitochondrial outer membrane (MOM). Finally, the mammalian studies used a BirA fusion to Sec61β that uniformly labeled the ER (17). In all cases, the BirA fusion proteins showed the expected localization (Fig. 1C and fig. S1B).
Because of the potential cycling of ribosomes between different cellular locations, especially following translation termination, it was critical to be able to induce rapid ribosome biotinylation while also suppressing any constitutive background BirA activity. Although biotin is an essential cofactor for both yeast and mammalian cells, titrating biotin levels in the growth media (7) suppressed BirA activity to undetectable levels without affecting cell growth (fig. S2). Brief biotin pulses were sufficient to give a robust biotinylation signal in live cells (Fig. 1E) and it was possible to prevent post-lysis biotinylation by depleting lysates of biotin and adenosine 5′-triphosphate (ATP) (Fig. 1D and fig. S2). This procedure allowed us to achieve rapid (on the time scale of polypeptide synthesis) and efficient biotinylation of both our 40S and 60S Avi-tagged ribosomes using a cytosolic BirA. In marked contrast, ER-localized BirAs failed to label the 40S Avi-tagged ribosomal subunit but retained the ability to robustly label the 60S Avi-tagged subunit (Fig. 1E). Based on the length of our BirA tether, which is too short to allow biotinylation of the 40S subunit of a docked, translocating ribosome, this result demonstrates the specific biotinylation of oriented translocating ribosomes over those that passively encounter the ER membrane. Finally, we optimized the purification of biotinylated, ribonuclease-digested monosomes (fig. S3) (18).
Validation of proximity-specific ribosome profiling
We performed proximity-specific ribosome profiling in S. cerevisiae using the three different ER-localized BirA constructs as well as the cytosolic and mitochondrially-localized controls (Fig. 2A), and in mammalian HEK-293 cells using an ER-localized BirA fusion protein. For each experiment, brief treatment with the translation elongation inhibitor cycloheximide (CHX), which preserves the ribosome position along an mRNA, was followed by a biotin pulse. Subsequent to processing and sequencing, we determined an enrichment value for each gene by taking the log2 ratio of ribosome footprint densities in the matched streptavidin-pulldown versus input whole-cell ribosome profiling samples. Enrichment metrics obtained from the same BirA were highly reproducible between replicates (Fig. 2B; Ssh1 Pearson r = 0.97; Sec63 Pearson r = 0.98) and robust across expression levels (fig. S4)
Fig. 2. Specificity of proximity-dependent ribosome profiling across multiple systems.

(A) Boxplots of the log2 enrichment distributions for secretome (blue), curated mitochondrial (red), and all other (gray) gene categories obtained from proximity-specific ribosome profiling experiments in yeast using different BirA fusions. Biotinylation was carried out in the presence of CHX for 2 min (cytosolic, mitochondria) or 7 min (ER). Enrichments were computed for each reliably expressed gene as the log2 ratio of biotinylated footprint density (RPM) over the corresponding density from the matched input whole-cell ribosome profiling experiment. Where possible, lines connect the same gene across experiments. (B) Enrichments shown for representative proximity-specific ribosome profiling replicates using the BirA-Ssh1 fusion protein. Colors match those in (A). (C) Histograms of log2 enrichments for Sec63-BirA in yeast. Enrichment thresholds were determined by receiver operating characteristic (ROC) analysis (fig. S5). Shown below are the corresponding enrichment analyses of GO-slim cellular components for robustly enriched genes versus expressed genes. Colors match those in (A). (D) As in (C) for BirA-Sec61β in HEK293T cells. Additionally, GO-term analysis of dis-enriched secretome genes versus expressed secretome genes is shown. (E) Gene enrichments obtained with the general BirA-Ubc6 ER marker in yeast, for well-expressed CHX-independent peroxisomal genes. SS and TMD annotations were predicted by SignalP and TMHMM, respectively. * denotes necessarily post-translational tail-anchor TMDs [see (18)].
Targeting of BirA to the cytosol yielded a narrow range of enrichment values (90% of genes fell within -0.2 to +0.2 log2 enrichment units) demonstrating that our protocol for isolating biotinylated monosomes introduced minimal bias. We detected a modest but significant (P < 1 × 10-15, Kolmogorov-Smirnov test) depletion of secreted genes, consistent with the expected lower accessibility of ER-docked ribosomes. By contrast, BirA targeted to mitochondria produced a clear bimodal distribution, enriching for genes annotated to localize to this cellular compartment (19). An in-depth analysis of translation at the mitochondrial outer membrane is presented in an accompanying manuscript (20). Targeting of BirA to the ER membrane inverted the mitochondrial enrichment pattern, cleanly separating secreted proteins from those synthesized in the cytosol or targeted to mitochondria. Ssh1, Sec63, and Ubc6 ER-localized BirA fusion constructs labeled ribosomes translating similar sets of secretome genes [defined in (21)], though we observed pronounced differences in the point during translation at which RNCs interact with these BirA fusions (explored below). To determine whether other gene categories were significantly overrepresented in the enriched populations of our ER data sets, we performed gene ontology (GO-) term analysis on gene categories in yeast and mammalian cells that were enriched above a threshold derived from a receiver operator characteristic analysis (Fig. 2, C and D, and fig. S5). In both yeast and HEK-293 cells, enriched gene sets were exclusively from the secretome (18). However, a substantial number of mammalian secretome transcripts predicted by Phobius (22) to encode secretory proteins were not enriched in our assay. This set of genes was enriched in GO-terms for nucleus and cytosol when compared to all secretome genes, arguing that these proteins represent potential false positives in the computationally-predicted secretory gene set. This discrepancy serves to highlight both the sensitivity and utility of our approach for experimentally defining proteins that are targeted to specific cellular compartments.
We noted that peroxisomal proteins exhibited heterogeneous ER translational enrichment. The peroxisome is a highly conserved organelle responsible for lipid catabolism whose mechanism of biogenesis has been controversial. There is evidence for both de novo peroxisome generation from ER-derived vesicles, as well as for derivation from pre-existing peroxisomes through growth and fission (23). Our data reveal that 16 of 54 yeast peroxisomal proteins showed clear co-translational enrichment. Consistent with previous targeted studies in yeast (24), a unifying determinant for this ER targeting is the presence of one or more transmembrane domains (TMDs)—we found no evidence for the enrichment of peroxisomal matrix proteins (Fig. 2E). Notably, this partitioning was also seen in mammalian cells (fig. S6). Thus peroxisomes appear to obtain transmembrane proteins from the ER and matrix proteins from the cytosol.
Cotranslational targeting of SRP-dependent and –independent substrates in vivo
Whereas a subset of proteins are strictly reliant on SRP for ER targeting, a process that is thought to be obligatorily cotranslational, import of other proteins occurs efficiently without SRP when measured both in vitro and in vivo (21, 25). A recent in silico analysis revealed that ∼40% of yeast secretome substrates use the less-studied SRP-independent pathway (21). SRP-independent translocation depends on translocon accessory factors as well as the luminal chaperone Kar2/BiP and in vitro can occur efficiently after translation (26, 27) (Fig. 3A).
Fig. 3. Global characterization of co- versus posttranslational translocation in vivo.

(A) Overview of current models for SRP-dependent and -independent targeting to and translocation into the ER. Predictions for the proportion of substrates that partition between pathways are taken from (21). (B) Cumulative distribution of the Sec63-BirA log2 enrichments for SRP-dependent (blue), SRP independent (red), and nonsecreted (gray) genes with or without CHX. Biochemically validated genes (dashed lines) were consolidated from (21) and (25). (C) Venn diagram summarizing the SVM classifications for CHX dependence in the context of the Phobius-predicted secretome. The Sec63-BirA +CHX enrichment profile was fit as a mixture of two normal distributions, and all genes enriched above the 99th percentile of the dis-enriched distribution were classified by the SVM. (D) Number of codons downstream of the first hydrophobic domain of Phobius-secretome genes versus the position of this domain relative to overall gene length, plotted for genes in different SVM-classified enrichment categories. Contour lines are added for specific gene sets for visual clarity and represent Gaussian density fits of the corresponding points in that set. Colors match those in (C) with the tail-anchored genes (dark-blue) overlaid as open circles. (E) Proportion of genes for which a hydrophobic feature was predicted by either TMHMM or SignalP, for different gene categories. Colors and gene sets match those in (C) with the addition of nonsecretome genes (gray), as predicted by Phobius.
Notably, we found that the vast majority of secretory proteins undergo cotranslational targeting in vivo, irrespective of their dependence on SRP (Fig. 3B). This pattern held across all BirA fusions (fig. S7) for 162 genes experimentally validated as SRP-dependent or –independent (21, 25), as well as for an additional 756 genes whose SRP dependence was predicted using a hydropathy-based analysis (21).
It was a formal possibility that the apparent co-translational ER enrichment of these translating messages was a result of brief treatment with the translation elongation inhibitor (CHX) prior to biotinylation, because this provides extra time for the RNC complex to engage the translocon. We evaluated this possibility by omitting translation inhibitors and labeling with biotin for 1 min, a time scale comparable to that of a single round of polypeptide synthesis. For the large majority of SRP-dependent and -independent substrates, levels of translational enrichment were not dependent on CHX (Fig. 3B). Intriguingly, ribosomes translating a small minority of the SRP-independent proteins lost their enrichment, suggesting that in an unperturbed setting these proteins translocate posttranslationally.
Thus, SRP independence is not synonymous with posttranslational translocation; import concurrent with protein synthesis is the principal route into the ER in vivo. By effectively coupling translation and translocation for the large majority of proteins entering the secretory pathway, the cell minimizes the dangers associated with having a cytosolic cohort of un-translocated, aggregation-prone proteins (28). Understanding how the cell achieves co-translational translocation of SRP- independent messages remains unclear.
Comprehensive analysis of co- versus post-translational translocation in vivo
Having uncoupled SRP independence from posttranslational translocation, we sought to better understand the determinants for partitioning between the co- and posttranslational import pathways. To classify genes on the basis of their ER translational enrichment, we systematically identified genes whose enrichments were dependent on CHX using a support vector machine (SVM) classifier trained to distinguish between ∼140 proteins characterized empirically as being CHX-dependent or -independent (18). This SVM analysis enabled us to systematically characterize the import of proteins as being either cotranslational (CHX-independent), cotranslational translocation that is dependent upon (or enhanced by) treatment with a translation inhibitor (CHX-dependent), or obligatorily posttranslational (dis-enriched) (Fig. 3C). The enrichment of CHX-dependent proteins was greatest for Sec63, consistent with its role in co- and posttranslational translocation (fig. S8). These differences demonstrate the specificity of ribosome labeling by BirA fusion proteins and suggest that cotranslational insertion (CHX-independent) typically occurs through both translocons.
The SVM analysis indicated that the large majority (681 of 837) of Phobius-predicted secretory genes were translated at the ER independent of CHX. Of the remaining predicted secretory proteins, 63 were dependent on CHX for enrichment, whereas 93 were not enriched under any condition tested. The latter dis-enriched group contained nearly all of the roughly 50 annotated TA proteins whose C-terminal TMDs preclude cotranslational recognition and that are known to be targeted to the ER posttranslationally through the guided entry of TA proteins (GET) pathway (29, 30).
What then accounts for the remaining proteins whose translocation is not strictly cotranslational? The position of an ER-targeting signal within a protein imposes restrictions on when during synthesis targeting may occur, and thus might be an important determinant. Indeed, robustly enriched CHX-independent secreted genes and dis-enriched TA genes fall on the opposite sides of this spectrum, with extreme N- and C-terminal ER-targeting signals, respectively (Fig. 3D). The targeting signals of proteins dependent on CHX for cotranslational targeting to the ER fall in-between these two extremes, often present far downstream in relation to their overall gene length. Following co-translational targeting to the ER, translation of long downstream regions leads to prolonged mRNA retention on the ER surface, which would be mediated by multiple translocating RNCs. In contrast, short downstream regions are unlikely to stably tether the mRNA to the ER following targeting (fig. S9). The CHX dependence of genes with short downstream regions suggests that ER retention mediated by polysomes promotes efficient translocon targeting.
Our SVM classification also revealed co-translational, CHX-independent ER enrichment of 70 genes for which no hydrophobic domains were detected by Phobius (Fig. 3C). However, most of these were predicted to contain a hydrophobic domain by alternate hydrophobic prediction algorithms (TMHMM or SignalP) (31, 32) (Fig. 3E). These genes thus likely represent genuine secretory proteins that were missed by Phobius, highlighting the value of our studies as an experimental complement to computational algorithms for globally identifying secreted and transmembrane proteins.
Timing and specificity of cotranslational targeting to the ER
We next asked when during translation RNCs are recruited to the ER, which is expected to depend upon the mechanism of recruitment. For example, SRP binds preferentially to short nascent chains containing cytosolically accessible hydrophobic sequences (33) and halts translation elongation until the RNC reaches the ER. By contrast, SRP-independent transport through the SEC complex relies on a poorly understood network of cytosolic chaperones, none of which are known to arrest translation.
In yeast, translocation occurs through two paralogous channels, Sec61 and Ssh1. The essential Sec61 translocon associates with several accessory factors, including both essential (Sec63 and Sec62) and nonessential (Sec66 and Sec72) peripheral components to form the SEC complex, or separately with the SRP receptor (SR). By contrast, the nonessential Ssh1 is a simpler translocon thought to interact peripherally only with SR (16). We reasoned that fusing BirA to specific complexes would allow us to globally monitor the timing and specificity of translocation of substrates through these distinct translocons (Fig. 4A).
Fig. 4. Timing and specificity of cotranslational targeting to the ER.
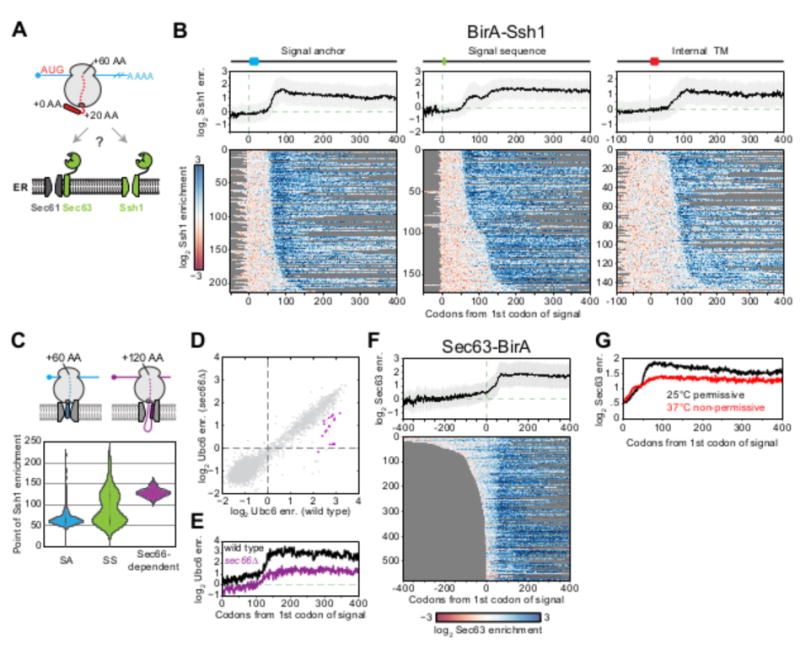
(A) Schematic of the yeast translocon-specific BirAs used to examine ribosome accessibility at two translocational entry points into the ER. (B) Metagene plots of log2 BirA-Ssh1 enrichment per codon (mean ± SD) as a function of ribosome position relative to the first codon of the first Phobius-predicted hydrophobic element for the indicated signal class. Heat maps below represent single-gene positional enrichments used to derive the corresponding averaged metagene plot, sorted by increasing distance to the point at which enrichment occurs. (C) Violin plot showing the distribution of the point of enrichment for BirA-Ssh1 relative to the first hydrophobic element, for different types of hydrophobic features and Sec66-dependent genes as defined in (D). Shown above are two RNC conformations consistent with nascent chain lengths. (D) Gene enrichments obtained with the general BirA-Ubc6 ER marker in yeast in wild-type versus sec66Δ backgrounds. Sec66-dependent genes are defined in fig. S11. (E) Metagene plot as in (B) of log2 BirA-Ubc6 enrichments for Sec66-dependent genes in wild-type (black) and sec66Δ (purple) backgrounds. (F) Metagene plot as in (B) of log2 Sec63-BirA enrichments. (G) Meta-gene plot of log2 enrichments as in (F) in a sec65-1 SRP temperature sensitive background at the permissive (25°C, black) and non-permissive (37°C, red) temperatures.
Ssh1
Ssh1 is expected to receive RNCs from SRP and should therefore interact with the ribosomes after the hydrophobic sequence emerges from the ribosome peptide exit tunnel. Consistent with this model, for all secreted and TMD proteins regardless of the location of their targeting sequence, BirA-Ssh1 enrichment began only after the hydrophobic domain was fully accessible (∼60 amino acids from the start of targeting sequence) (34) (Fig. 4B and fig. S10A).
Type II signal anchors (SAs) and cleavable signal sequences (SSs) are oriented in a looped conformation with their N termini facing the cytosol (Fig. 4C). Models of how SSs and SAs achieve this topology within the translocon differ in the efficiency of RNC recruitment; i.e. the head-first model stipulates early RNC binding and subsequent signal inversion, whereas the looped-insertion model requires delayed RNC binding in which the nascent chain is correctly oriented before binding (Fig. 4C). Studies of model substrates are consistent with the head-first model (35, 36). Efficient targeting to Ssh1 immediately after translation of the hydrophobic sequence is also consistent with such a mechanism. Here, the timing of engagement was bimodally distributed: Most of the secretome was efficiently recruited to Ssh1 immediately after exit of the hydrophobic sequence from the ribosome, however, a prominent subset of proteins with cleavable SSs engaged only after enough synthesis (∼120 amino acids) to allow the nascent chain to acquire a looped topology (Fig. 4, B and C). Thus both head-first and looped-insertion can occur in vivo, depending on the protein. Deletion of SEC66, a nonessential component of the SEC complex that mediates translocation of SRP-independent substrates, exclusively affected translocation of the looped-insertion substrates (Fig. 4, C to E and fig. S11). Thus proximity-specific ribosome profiling can decipher how distinct translocon components enable the efficient handling of diverse targeting sequences and topologies (35, 37, 38).
Sec63
Sec63 mediates both SRP-independent and -dependent translocation. Without a strict requirement for SRP-induced translational pausing in the cytosol, Sec63 translocation of SRP-independent substrates naïvely might have been expected to result in a delayed and more broadly distributed timing of ER targeting. Surprisingly, the opposite was observed; Sec63 began to interact with RNCs translating secretory proteins well before the emergence of the hydrophobic sequence from the ribosome (Fig. 4F), and maximal engagement occurred shortly after this element was fully solvent accessible. Thus Sec63 interacts with ribosomes through two distinct modes: One depends on the presence of an accessible hydrophobic element, and the second reflects an interaction with ribosomes while the targeting sequence is in the exit tunnel. Consistent with this interpretation, acute loss of SRP function using a temperature-sensitive SRP allele (fig. S12) did not affect the early engagement, but did compromise the late (exposed targeting sequence) enrichment (Fig. 4G).
Dynamics of ER-associated ribosomes
Upon translation termination, ER-associated ribosomes can either immediately dissociate and return to the pool of cytosolic ribosomes or preferentially undergo multiple rounds of translation on ER-associated mRNAs. To investigate these dynamics in the context of living cells, we harvested samples for proximity-specific profiling after increasing lengths of biotinylation time in the absence of CHX. Enrichment for secretory messages is expected to decrease at a rate proportional to the time scale at which biotinylated ribosomes, originating from the ER, exchange into the cytosol and begin translating cytosolic messages (Fig. 5A). We observed rapid collapse of our bimodal enrichment distribution into a single population on the order of minutes, although secreted messages remain on the enriched side of the distribution at all time points tested as expected from continual biotinylation of ER ribosomes (Fig. 5B). Based on a median gene length of ∼425 codons and a translation rate of ∼5.5 codons per second (39), translation of a single secretory protein is expected to take ∼77 seconds. Thus ribosomes at the yeast ER are highly dynamic, freely exchanging into the cytosol within at most a few rounds of translation.
Fig. 5. Dynamics of ER-associated ribosomes in vivo.

(A) Overview of the pulse labeling experiment to assay the kinetics of ribosome exchange from the ER in vivo. (B) Histograms of log2 Sec63-BirA enrichment values for well-expressed secretome (blue) and all other (gray) genes over the exchange time course. Times represent the total time of ribosome biotinylation in the absence of CHX. (C) Working model consistent with the positional enrichments observed for the translocon-specific BirAs and ribosome recycling. (1) Initial recruitment to the ER depends on a fully accessible signal sequence. (2) Ribosomes translating ER-tethered mRNAs can interact with SEC early. (3) Upon termination, ribosomes recycle into the cytosolic pool.
Discussion
Here we present a proximity-based ribosome profiling strategy that can monitor translation for any location at which it is possible to target a BirA fusion protein. We applied this strategy to analyze modes of cotranslational translocation into the ER. Nearly one quarter of the proteome is imported into the ER; accordingly, this process has been the focus of intense research. Much of this previous work, however, has explored the behavior of a small group of model substrates often outside of a cellular context. Proximity-specific ribosome profiling allowed us to simultaneously probe the ER engagement of nascent chains across the full proteome in vivo, in the context of competing and redundant targeting pathways. This comprehensive characterization revealed several principles of how cells integrate distinct targeting pathways with the translocation machinery to allow for unexpectedly robust co-translational ER import of a diverse set of substrates.
Foremost is the critical role of the timing of translation of the ER targeting sequence relative to translation termination for determining the propensity of a protein to undergo import cotranslationally. This stands in contrast to the view that cotranslational import is dictated by the factors that mediate targeting (e.g., SRP). It had previously been appreciated that TA proteins must insert posttranslationally because the targeting sequence is obscured prior to translation termination. However, these represent a single point on a broader spectrum of signal positions. Proteins with targeting domains near the C-terminus typically were targeted as RNCs only when the kinetics of translation were crippled. By contrast, the predominantly SRP-independent (21) set of substrates containing N-terminal SSs are robustly cotranslationally targeted. Indeed, the full range of co-translational substrates could engage both the essential SEC and “alternate” Ssh1 translocons. Moreover, RNCs were able to interact with the SEC complex prior to exposure of a hydrophobic sequence, even though there are no known mechanisms for coordinating translation and recruitment to SEC.
The above findings suggest a model wherein a pioneering round of translation is responsible for recruiting the RNC to the ER surface, after which the message remains tethered to the ER by ongoing translation by downstream ribosomes (Fig. 5C). As evidenced by the lack of enrichment for proteins with low (i.e., length-limited) ribosome occupancy downstream of the hydrophobic targeting sequence, the tethered state appears to be crucial for efficient cotranslational engagement.
Consistent with our understanding of SRP function, SRP likely plays a critical role in establishing specificity and ensuring translocation competency through its ability to halt translation. Subsequent rounds of ribosome initiation in the context of this ER-tethered mRNA would obviate the need for SRP to survey every translation event, particularly for messages with extensive downstream regions that can accommodate multiple ribosomes. Such a mechanism is consistent with the observed 1:50 stoichiometry of SRP to the ribosome (40), and would simplify the problem of cellular protein sorting while minimizing the toxicity associated with solvent-exposed hydrophobic domains. Although initial recruitment may direct an RNC to a specific translocon, once tethered to the ER the high effective concentration would enable upstream RNCs to engage any translocon. Alternatively, the apparent lack of translocon substrate specificity could be due to ribosome biotinylation in trans, though the observed differences in position and CHX-dependent enrichments argues against this. In either case, inhibition of translocation is known to induce a massive cytoplasmic stress response (28) underscoring the danger of having ER-targeted proteins in the cytosol even when they can be post-translationally translocated.
Our studies also revealed a class of SSs that emphasize an intimate connection between the timing of import and protein topology, mediated by translocon accessory factors. The bimodal timing of targeting to Ssh1 suggests that insertion can occur in either a head-first or looped orientation. Our results implicate Sec66 in mediating the import of those proteins that undergo looped insertion. This functionality may be necessary for certain substrates whose insertion kinetics would preclude re-orientation within the translocon. A clear future application of our method is probing the elusive roles of other translocon accessory factors, such as the translocating chain-associated membrane protein (TRAM) and translocon-associated protein (TRAP) in mammals (37, 38).
A final principle that emerged from our studies is the dynamic nature of ER-associated ribosomes in yeast, which cycle readily between cellular compartments. This is in contrast to evidence from in vitro exchange experiments that showed stable association of the 60S ribosome subunit with the mammalian ER (41). It will be interesting to explore whether such a pattern holds in more specialized secretory cells, such as plasma cells, which rely on efficient translation at the ER. Indeed, electron micrographs have revealed the presence of circular polysomes (42) in these cells, consistent with a “closed-loop model” of translation (43) that is presumed to promote efficient translation reinitiation.
The principles uncovered here highlight the ability of proximity-specific ribosome profiling to synergize with prior mechanistic studies of ER targeting pathways. Diverse biological systems localize mRNAs to generate cellular structure and function, yet compared to the ER, much less is known about how cotranslational protein targeting contributes to asymmetry at these sites. More generally, this approach enables the profiling of subpools of ribosomes that interact, even transiently, with cellular proteins of interest, e.g. those involved in protein folding, quality control, targeting, and posttranslational modification. As a flexible, precise, and global method, proximity-specific ribosome profiling provides a tool for exploring the interface between translation and cell biology.
Supplementary Material
Acknowledgments
We thank members of the Weissman and P. Walter labs, as well as A. Ting, for discussion; E. Costa, M. Kampmann, P. Kimmig, K. Kostova, and M. Smith for critical reading of the manuscript. Technical assistance was provided by the UCSF CAT, NIC and the CFR. This research was supported by the Center for RNA Systems Biology and the Howard Hughes Medical Institute. C.H.J. is the Rebecca Ridley Kry Fellow of the Damon Runyon Cancer Research Foundation (DRG-2085-11). C.C.W. is an NSF GRFP. Data are deposited in GEO under accession numbers GSE61011and GSE61012.
References and Notes
- 1.St Johnston D. Moving messages: the intracellular localization of mRNAs. Nat Rev Mol Cell Biol. 2005;6:363–375. doi: 10.1038/nrm1643. [DOI] [PubMed] [Google Scholar]
- 2.Jung H, Gkogkas CG, Sonenberg N, Holt CE. Remote Control of Gene Function by Local Translation. Cell. 2014;157:26–40. doi: 10.1016/j.cell.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee JH, et al. Highly Multiplexed Subcellular RNA Sequencing in Situ. Science. 2014;343:1360–1363. doi: 10.1126/science.1250212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.David A, et al. Nuclear translation visualized by ribosome-bound nascent chain puromycylation. J Cell Biol. 2012;197:45–57. doi: 10.1083/jcb.201112145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dieterich DC, et al. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat Neurosci. 2010;13:897–905. doi: 10.1038/nn.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stephens SB, Nicchitta CV. In vitro and tissue culture methods for analysis of translation initiation on the endoplasmic reticulum. Methods Enzymol. 2007;431:47–60. doi: 10.1016/S0076-6879(07)31004-5. [DOI] [PubMed] [Google Scholar]
- 7.Fernández-Suárez M, Chen TS, Ting AY. Protein−Protein Interaction Detection in Vitro and in Cells by Proximity Biotinylation. J Am Chem Soc. 2008;130:9251–9253. doi: 10.1021/ja801445p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS. Genome-Wide Analysis in Vivo of Translation with Nucleotide Resolution Using Ribosome Profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ast T, Schuldiner M. All roads lead to Rome (but some may be harder to travel): SRP-independent translocation into the endoplasmic reticulum. Crit Rev Biochem Mol Biol. 2013;48:273–288. doi: 10.3109/10409238.2013.782999. [DOI] [PubMed] [Google Scholar]
- 10.Mandon EC, Trueman SF, Gilmore R. Protein translocation across the rough endoplasmic reticulum. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a013342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ben-Shem A, et al. The Structure of the Eukaryotic Ribosome at 3.0 A Resolution. Science. 2011;334:1524–1529. doi: 10.1126/science.1212642. [DOI] [PubMed] [Google Scholar]
- 12.Ban N, et al. A new system for naming ribosomal proteins. Curr Opin Struct Biol. 2014;24:165–169. doi: 10.1016/j.sbi.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heiman M, et al. A Translational Profiling Approach for the Molecular Characterization of CNS Cell Types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kornmann B, et al. An ER-Mitochondria Tethering Complex Revealed by a Synthetic Biology Screen. Science. 2009;325:477–481. doi: 10.1126/science.1175088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finke K, et al. A second trimeric complex containing homologs of the Sec61p complex functions in protein transport across the ER membrane of S. cerevisiae. EMBO J. 1996;15:1482–1494. [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang Y, Cheng Z, Mandon EC, Gilmore R. An interaction between the SRP receptor and the translocon is critical during cotranslational protein translocation. J Cell Biol. 2008;180:1149–1161. doi: 10.1083/jcb.200707196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shibata Y, et al. The Reticulon and Dp1/Yop1p Proteins Form Immobile Oligomers in the Tubular Endoplasmic Reticulum. J Biol Chem. 2008;283:18892–18904. doi: 10.1074/jbc.M800986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Materials and methods are available as supplementary material on Science Online.
- 19.Elstner M, Andreoli C, Klopstock T, Meitinger T, Prokisch H. The mitochondrial proteome database: MitoP2. Methods Enzymol. 2009;457:3–20. doi: 10.1016/S0076-6879(09)05001-0. [DOI] [PubMed] [Google Scholar]
- 20.Williams CC, Jan CH, Weissman JS. Targeting and Plasticity of Mitochondrial Proteins Revealed by Proximity-Specific Ribosome Profiling. Science (submitted) 2014 doi: 10.1126/science.1257522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ast T, Cohen G, Schuldiner M. A Network of Cytosolic Factors Targets SRP-Independent Proteins to the Endoplasmic Reticulum. Cell. 2013;152:1134–1145. doi: 10.1016/j.cell.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Käll L, Krogh A, Sonnhammer EL. A Combined Transmembrane Topology and Signal Peptide Prediction Method. J Mol Biol. 2004;338:1027–1036. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 23.Smith JJ, Aitchison JD. Peroxisomes take shape. Nat Rev Mol Cell Biol. 2013;14:803–817. doi: 10.1038/nrm3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van der Zand A, Braakman I, Tabak HF. Peroxisomal Membrane Proteins Insert into the Endoplasmic Reticulum. Mol Biol Cell. 2010;21:2057–2065. doi: 10.1091/mbc.E10-02-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ng DT, Brown JD, Walter P. Signal sequences specify the targeting route to the endoplasmic reticulum membrane. J Cell Biol. 1996;134:269–278. doi: 10.1083/jcb.134.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Young BP. Sec63p and Kar2p are required for the translocation of SRP-dependent precursors into the yeast endoplasmic reticulum in vivo. EMBO J. 2001;20:262–271. doi: 10.1093/emboj/20.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Panzner S, Dreier L, Hartmann E, Kostka S, Rapoport TA. Posttranslational protein transport in yeast reconstituted with a purified complex of Sec proteins and Kar2p. Cell. 1995;81:561–570. doi: 10.1016/0092-8674(95)90077-2. [DOI] [PubMed] [Google Scholar]
- 28.Brandman O, et al. A Ribosome-Bound Quality Control Complex Triggers Degradation of Nascent Peptides and Signals Translation Stress. Cell. 2012;151:1042–1054. doi: 10.1016/j.cell.2012.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schuldiner M, et al. The GET complex mediates insertion of tail-anchored proteins into the ER membrane. Cell. 2008;134:634–645. doi: 10.1016/j.cell.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stefanovic S, Hegde RS. Identification of a Targeting Factor for Posttranslational Membrane Protein Insertion into the ER. Cell. 2007;128:1147–1159. doi: 10.1016/j.cell.2007.01.036. [DOI] [PubMed] [Google Scholar]
- 31.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 32.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 33.Siegel V, Walter P. The affinity of signal recognition particle for presecretory proteins is dependent on nascent chain length. EMBO J. 1988;7:1769–1775. doi: 10.1002/j.1460-2075.1988.tb03007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kowarik M, Küng S, Martoglio B, Helenius A. Protein folding during cotranslational translocation in the endoplasmic reticulum. Mol Cell. 2002;10:769–778. doi: 10.1016/s1097-2765(02)00685-8. [DOI] [PubMed] [Google Scholar]
- 35.Devaraneni PK, et al. Stepwise Insertion and Inversion of a Type II Signal Anchor Sequence in the Ribosome-Sec61 Translocon Complex. Cell. 2011;146:134–147. doi: 10.1016/j.cell.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goder V. Molecular mechanism of signal sequence orientation in the endoplasmic reticulum. EMBO J. 2003;22:3645–3653. doi: 10.1093/emboj/cdg361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Görlich D, Hartmann E, Prehn S, Rapoport TA. A protein of the endoplasmic reticulum involved early in polypeptide translocation. Nature. 1992;357:47–52. doi: 10.1038/357047a0. [DOI] [PubMed] [Google Scholar]
- 38.Fons RD. Substrate-specific function of the translocon-associated protein complex during translocation across the ER membrane. J Cell Biol. 2003;160:529–539. doi: 10.1083/jcb.200210095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bonven B, Gulløv K. Peptide chain elongation rate and ribosomal activity in Saccharomyces cerevisiae as a function of the growth rate. Mol Gen Genet MGG. 1979;170:225–230. doi: 10.1007/BF00337800. [DOI] [PubMed] [Google Scholar]
- 40.Ogg SC, Walter P. SRP samples nascent chains for the presence of signal sequences by interacting with ribosomes at a discrete step during translation elongation. Cell. 1995;81:1075–1084. doi: 10.1016/s0092-8674(05)80012-1. [DOI] [PubMed] [Google Scholar]
- 41.Borgese D, Blobel G, Sabatini DD. In vitro exchange of ribosomal subunits between free and membrane-bound ribosomes. J Mol Biol. 1973;74:415–438. doi: 10.1016/0022-2836(73)90037-5. [DOI] [PubMed] [Google Scholar]
- 42.Christensen AK, Kahn LE, Bourne CM. Circular polysomes predominate on the rough endoplasmic reticulum of somatotropes and mammotropes in the rat anterior pituitary. Am J Anat. 1987;178:1–10. doi: 10.1002/aja.1001780102. [DOI] [PubMed] [Google Scholar]
- 43.Wells SE, Hillner PE, Vale RD, Sachs AB. Circularization of mRNA by eukaryotic translation initiation factors. Mol Cell. 1998;2:135–140. doi: 10.1016/s1097-2765(00)80122-7. [DOI] [PubMed] [Google Scholar]
- 44.Rothstein R. Targeting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Methods Enzymol. 1991;194:281–301. doi: 10.1016/0076-6879(91)94022-5. [DOI] [PubMed] [Google Scholar]
- 45.Meinema AC, et al. Long Unfolded Linkers Facilitate Membrane Protein Import Through the Nuclear Pore Complex. Science. 2011;333:90–93. doi: 10.1126/science.1205741. [DOI] [PubMed] [Google Scholar]
- 46.Longtine MS, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast Chichester Engl. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 47.Meerbrey KL, et al. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc Natl Acad Sci. 2011 doi: 10.1073/pnas.1019736108. 201019736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stirling CJ, Rothblatt J, Hosobuchi M, Deshaies R, Schekman R. Protein translocation mutants defective in the insertion of integral membrane proteins into the endoplasmic reticulum. Mol Biol Cell. 1992;3:129–142. doi: 10.1091/mbc.3.2.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Algire MA, et al. Development and characterization of a reconstituted yeast translation initiation system. RNA N Y N. 2002;8:382–397. doi: 10.1017/s1355838202029527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brar GA, et al. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science. 2012;335:552–557. doi: 10.1126/science.1215110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tabas-Madrid D, Nogales-Cadenas R, Pascual-Montano A. GeneCodis3: a non-redundant and modular enrichment analysis tool for functional genomics. Nucleic Acids Res. 2012;40:W478–483. doi: 10.1093/nar/gks402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pagliarini DJ, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arava Y, et al. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc Natl Acad Sci. 2003;100:3889–3894. doi: 10.1073/pnas.0635171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.