

Abstract
There are over 30 mouse models with mutations or inactivations in the dystrophin-associated protein complex. This complex is thought to play a crucial role in the functioning of muscle, as both a shock absorber and signalling centre, although its role in the pathogenesis of muscular dystrophy is not fully understood. The first mouse model of muscular dystrophy to be identified with a mutation in a component of the dystrophin-associated complex (dystrophin) was the mdx mouse in 1984. Here, we evaluate the key characteristics of the mdx in comparison with other mouse mutants with inactivations in DAPC components, along with key modifiers of the disease phenotype. By discussing the differences between the individual phenotypes, we show that the functioning of the DAPC and consequently its role in the pathogenesis is more complicated than perhaps currently appreciated.
Keywords: dystroglycan, dystrophin, mdx, mouse models, muscular dystrophy
Introduction
Muscular dystrophy is a family of inherited conditions, with a spectrum of phenotypes, caused by mutations in a number of genes associated with muscle development, structure, maintenance, function or repair. A wide variety and number of different mouse models have been generated (over 55 strains are listed on the Jackson Laboratory web page – http://jaxmice.jax.org/neurobiology/muscular-dystrophy.html) to investigate the phenotype of these human conditions and test potential therapeutic strategies. Consequently, a review of muscular dystrophy mouse models is a huge topic, shown by over 8000 results from a Web of Science search for muscular dystrophy mouse models, so we will restrict ourselves to a particular subset of muscular dystrophy mouse models. It is currently thought in some of these conditions that muscle fibres degenerate because they are unable to withstand the mechanical forces of contraction, due to compromised functioning of the dystrophin-associated protein complex (DAPC). The DAPC (shown as a cartoon schematic in Figure1) is a large multimeric protein complex that links the intracellular actin filaments to the extracellular basement membrane (Michele & Campbell 2003) and is considered to have roles including acting as shock absorber in muscle. Therefore, we have chosen to focus on mouse models where the phenotype is caused by compromised functioning of the DAPC, through mutation and/or inactivation in DAPC components, such as dystrophin, α-dystrobrevin, dystroglycan, the sarcoglycan complex and sarcospan, or through proteins that post-translationally modify DAPC components, such as POMT1, POMT2, POMGnT1, FKRP, fukutin and LARGE, enzymes that are involved in α-dystroglycan glycosylation. At the time of writing, this is still 43 mouse models, although we have excluded a number of mutants – Dag-1 null (Williamson et al. 1997), FKRPKD mouse (Ackroyd et al. 2009), FKRP E310delneo+ (Chan et al. 2010), FKRP P448Lneo+/E310delneo+ compound heterozygotes (Blaeser et al. 2013), fukutin null (Kurahashi et al. 2005), POMT1−/− (Willer et al. 2004) and POMT2 null (Hu et al. 2011) – as they do not exhibit any obvious muscle abnormality because they die either prenatally or in the early postnatal period. We have also chosen to exclude those that do not carry a germline mutation, for example where postnatal knock-down is achieved through shRNA, as well as the fukutin Hp− (Kanagawa et al. 2009), Spsn-deficient (Lebakken et al. 2000), Scge-null (Lancioni et al. 2011) and α-syn−/− mice (Adams et al. 2000), as these do not develop a discernable dystrophic phenotype. Furthermore, we have also chosen to exclude mouse models with mutations or inactivations in neuronal nitric oxide synthase (nNOS) (Huang et al. 1993). The DAPC is thought to fulfil two roles in muscle fibres, a mechanical role, as a shock absorber to minimize contraction-induced damage, and a biochemical role, acting as a signalling centre. All components of the DAPC are involved in both mechanical and biochemical roles, with the exception of nNOS, which is mainly involved with signalling pathways. Whilst these signalling pathways are important for muscle development, maintenance and repair, we feel that encompassing major discussions on these signalling pathways would be outside the scope of this review. Recently, a review has been published on the various signalling roles of the DAPC (Constantin 2014), and we direct the reader to this publication for more details on this topic.
Figure 1.
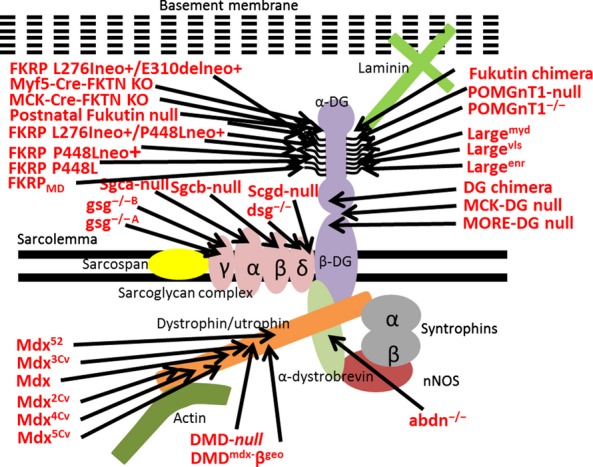
Schematic of dystrophin-associated protein complex (DAPC). Cartoon schematic showing the muscle dystrophin-associated protein complex (DAPC), composed of dystroglycan (both α- and β-subunits are shown), sarcospan, sarcoglycan complex, dystrophin or utrophin, α-dystrobrevin, the syntrophins, actin (which binds to dystrophin) and neuronal nitrogen oxide synthase (nNOS), which binds to α-dystrobrevin and the syntrophins. These different components are labelled in the diagram and are shown in different colours. We will not be describing the structure and function of the different DAPC components, and for more information, the reader is directed to Michele and Campbell (2003). The mouse models mentioned in this review are shown in the figure, with an arrow indicating the affected protein component.
All 32 models included here carrying mutations or inactivations in the components of the DAPC evaluated in the review are listed in Table1. To make the text easier to read, rather than listing the citations after each individual point, the main references for each animal model have been included in Table1. In this review, first we provide a short summary of the most well-known mouse mutants with inactivation in a DAPC component – the dystrophin-deficient mdx models – before comparing these to the other models with mutations/inactivations in the DAPC, exploring what any similarities or differences mean for the functioning of the DAPC. It is important to note here that if no comment is made on a parameter in a certain model, then it simply means that no comment has been made on this parameter in the literature. In this way, we hope to illustrate that the DAPC is an extremely complex structure that is not as well understood as we like to think.
Table 1.
Mouse muscular dystrophy models with inactivations or mutations in DAPC components
| Mouse model name and mutated gene | DAPC Component | Nature of mouse model | References | Model of human disease |
|---|---|---|---|---|
| Mdx Dystrophin | Dystrophin | Spontaneous mutation exon 23 – loss of Dp427 isoform | Bulfield et al. (1984), Barton (2006) | Duchenne muscular dystrophy (DMD) |
| Mdx2Cv Dystrophin | Dystrophin | Mutation intron 42 - loss of Dp427 and Dp260 isoforms | Chapman et al. (1989), Im et al. (1996) | DMD |
| Mdx3Cv Dystrophin | Dystrophin | Mutation exon 65 – loss of Dp427, Dp260, Dp140, Dp116, Dp40 and Dp71 isoforms | Chapman et al. (1989), Cox et al. (1993b) | DMD |
| Mdx4Cv Dystrophin | Dystrophin | Mutation exon 53 – loss of Dp427, Dp260 and Dp140 isoforms | Chapman et al. (1989), Im et al. (1996) | DMD |
| Mdx5Cv Dystrophin | Dystrophin | Mutation exon 10 – loss of Dp427 isoform | Im et al. (1996) | DMD |
| Mdx52 Dystrophin | Dystrophin | Mutation exon 52 – loss of Dp427, Dp260 and Dp140 isoforms | Araki et al. (1997) | DMD |
| DMDmdx−βgeo Dystrophin | Dystrophin | Mutation exon 63 – loss of all dystrophin isoforms | Wertz and Fuchtbauer (1998) | DMD |
| DMD-null Dystrophin | Dystrophin | Global knockout of dystrophin isoforms | Kudoh et al. (2005) | DMD |
| MCK-DG null Dag1 – Dystroglycan | Dystroglycan | Conditional knockout under muscle creatinine kinase (MCK) promoter | Cohn et al. (2002) | N/A |
| MORE-DG null Dag1 | Dystroglycan | Conditional knockout – everywhere except Reichert's membrane | Satz et al. (2008) | N/A |
| DG chimera Dag1 – Dystroglycan1 | Dystroglycan | Dystroglycan chimera | Cote et al. (1999) | N/A |
| POMGnT1-null POMGnT1 – Protein-O-mannose β-1,2-N-acetylglucosaminyltransferase | Dystroglycan glycosylation | Global knockout of POMGnT1 | Liu et al. (2006) | Walker–Warburg syndrome (WWS), muscle–eye–brain (MEB) disease, limb girdle muscular dystrophy type 2M (LGMD2O) |
| POMGnT1−/− POMGnT1 | Dystroglycan glycosylation | Global knockout POMGnT1 | Miyagoe-Suzuki et al. (2009) | WWS, MEB disease, LGMD2O |
| FKRPMD Fkrp – fukutin-related protein | Dystroglycan glycosylation | Knock-down in all cells except those expressing Sox1 | Whitmore et al. (2014) | WWS, MEB disease, congenital muscular dystrophy type 1C (MDC1C), LGMD 2I |
| FKRP P448Lneo+ Fkrp | Dystroglycan glycosylation | Global knock-down in expression | Chan et al. (2010) | WWS, MEB disease, MDC1C, LGMD2I |
| FKRP P448L Fkrp | Dystroglycan glycosylation | Global point mutation | Blaeser et al. (2013) | WWS, MEB disease, MDC1C, LGMD2I |
| FKRP L276Ineo+/P448Lneo+ Fkrp – fukutin-related protein | Dystroglycan glycosylation | Global point mutation + reduction in expression | Blaeser et al. (2013) | WWS, MEB disease, MDC1C, LGMD2I |
| FKRP L276Ineo+/E310delneo+Fkrp | Dystroglycan glycosylation | Global point mutation + reduction in expression | Blaeser et al. (2013) | WWS, MEB disease, MDC1C, LGMD2I |
| Largemyd Large – Like-acetylglucosaminyltransferase | Dystroglycan glycosylation | Global knockout Large | Lane et al. (1976), Mathews et al. (1995) Grewal et al. (2005) | WWS, MEB disease, MDC1D |
| Largeenr Large | Dystroglycan glycosylation | Global knockout Large | Kelly et al. (1994) Levedakou et al. (2005) | WWS, MEB disease, MDC1D |
| Largevls Large | Dystroglycan glycosylation | Global knockout Large | Lee et al. (2005) | WWS, MEB disease, MDC1D |
| Myf5-Cre-FKTN-KO Fktn – fukutin | Dystroglycan glycosylation | Conditional knockout under Myf5 promoter | Beedle et al. (2012) | WWS, MEB disease, FCMD, LGMD2M |
| MCK-Cre-FKTN-KO Fktn | Dystroglycan glycosylation | Conditional knockout of under MCK promoter | Beedle et al. (2012) | WWS, MEB disease, FCMD, LGMD2M |
| Postnatal fukutin null Fktn – fukutin | Dystroglycan glycosylation | Global knockout of fukutin from 6 weeks | Beedle et al. (2012) | WWS, MEB disease, FCMD, LGMD2M |
| Fukutin chimera Fktn – fukutin | Dystroglycan glycosylation | Fukutin chimera | Takeda et al. (2003) | WWS, MEB disease, Fukuyama congenital muscular dystrophy (FCMD), LGMD2M |
| Sgca-null Sgca – α-sarcoglycan | α-Sarcoglycan | Global knockout | Duclos et al. (1998), Consolino et al. (2005), Jakubiec-Puka et al. (2005), Patel et al. (2003) | LGMD2D |
| Sgcb-null Sgcb – β-sarcoglycan | β-Sarcoglycan | Global knockout | Durbeej et al. (2000), Andersson et al. (2012) | LGMD2E |
| gsg−/−A Sgcg – γ-sarcoglycan | γ-Sarcoglycan | Global knockout | Hack et al. (1998, 1999), Barton (2006) | LGMD2C |
| gsg−/−B Sgcg | γ-Sarcoglycan | Global knockout | Sasaoka et al. (2003) | LGMD2C |
| dsg−/− Sgcd – δ-sarcoglycan | δ-Sarcoglycan | Global knockout | Hack et al. (2000) | LGMD2F |
| Scgd-null Sgcd | δ-Sarcoglycan | Global knockout | Coral-Vazquez et al. (1999), Wansapura et al. (2011) | LGMD2F |
| abdn−/− abdn – α-dystrobrevin | α-Dystrobrevin | Global knockout of α-dystrobrevin splice isoforms with exon 3 | Grady et al. (1999), Bunnell et al. (2008) | Left ventricular non-compaction 1 (LVNC1) |
This table lists all the mouse models mentioned in this review by name, identifying which part of the DAPC is affected and describing the nature of the model. For ease of reading, we have left the frequently cited references out of the text, so this table also contains those references used to gather the majority of the information about each mouse model.
Both γ-sarcoglycan mouse mutants are referred to in their publications as gsg−/−, so the letters A and B refer to the models so the two models can be differentiated in the text. Additionally, in the sections on mdx mouse models, we have specified which have lost the Dp40 isoform based on information from Tozawa et al. (2012).
Dystrophin-deficient models
The original dystrophin-deficient model, the mdx mouse, was discovered by chance in 1984 as it had increased levels of muscle creatinine kinase (MCK) and pyruvate kinase in the serum, along with histological indicators of skeletal muscle degeneration and regeneration (Carnwath & Shotton 1987; Coulton et al. 1988). It was then found to lack the protein dystrophin that is missing in human Duchenne muscular dystrophy patients (Hoffman et al. 1987) and to have a point mutation in exon 23 of the dystrophin gene (Sicinski et al. 1989). In the 30 years since its discovery, the mdx mouse has been used as a preclinical model of dystrophin restoration mediated by means such as stem or precursor cells (Partridge et al. 1989), plasmids, viral vectors [reviewed (Konieczny et al. 2013)] stop codon read-through and antisense oligonucleotide-mediated exon skipping. Several other mouse models for dystrophin deficiency exist (Table1), which have mutations in different parts of dystrophin – the mutation in mdx2cv is in intron 42, mdx4cv in exon 53, mdx5cv in exon 10, mdx3cv in exon 65, mdx52 in exon 52 and DMDmdx-βgeo in exon 63. However, with the exception of the DMDmdx-βgeo mouse, all of these models express shorter dystrophin isoforms (Chamberlain et al. 1993) [reviewed (Willmann et al. 2009)] and have naturally occurring revertant (dystrophin-expressing) fibres (Hoffman et al. 1990; Lu et al. 2000), although mdx4cv, mdx5cv and mdx52 have fewer reverent fibres than mdx mice (Danko et al. 1992; Echigoya et al. 2013). As all of these dystrophin-deficient mouse models have a similar, although not always identical, pathology (Beastrom et al. 2011), we will discuss, unless otherwise indicated, the most widely used model, the original mdx.
Age of onset
These mouse models of muscular dystrophy have a range in the reported age of onset, varying from birth for the MORE-DG null to 10 days for mdx mice (Torres & Duchen 1987); 2 weeks for the Largemyd, Scgb-null, Scga-null, Largeenr and Sgcd-null models; 12 weeks in the MCK-Cre-FKTN-KO; and finally 14 weeks in the postnatal fukutin null, although the most common age of onset for these models is 4–8 weeks of age (fukutin chimera, Myf5-Cre-FKTN-KO, FKRP P448L-neo+, FKRPMD, Largeenr, Largevls, MCK-DG null, POMGnT1-null, dsg−/−, gsg−/−A, abdn−/−). This wide range in the age of onset can most likely be attributed to a combination of two reasons – first the presence of central nervous system defects in some of these models, which may exacerbate the general phenotype or make it more pronounced, so symptoms are evident at an earlier age. Many studies have shown that the DAPC is involved in cortical development and function [reviewed in (Waite et al. 2009)], and this can be seen in these animal models with inactivations in the DAPC as many display central nervous system defects, including the MORE-DG null, Largemyd, Largevls, Largeenr and the mdx mouse. However, the different components of the DAPC have varying roles in the central nervous system (Waite et al. 2009) with α-dystroglycan shown to be important for neuronal migration and dystrophin crucial for postsynaptic clustering of GABAergic receptors (Brunig et al. 2002) and functioning of cholinergic synapses (Parames et al. 2014). This is reflected in the different cortical phenotypes of the mouse models, with alterations in α-dystroglycan expression or structure (MORE-DG, Largemyd) more severe than those seen in the mdx. Second, some models included in this review have tissue-specific or induced gene loss (to overcome the lethality of a complete null phenotype), for example the postnatal fukutin null, which in some cases results in the later onset of the phenotype.
In combination, these data suggest that the age of onset does not vary depending on which specific component of the DAPC is lost, but that the characteristics of each individual mouse model, for example global knockout or tissue-specific knockout, determine the age of symptom onset.
Pathology
Histological evidence of skeletal muscle degeneration and regeneration, typified by centrally nucleated muscle fibres and a variation in fibre size, was identified in all models of muscular dystrophy discussed here, with the exception of POMGnT1−/− where neither feature was identified. Similarly to the mdx, inflammatory infiltrates were identified in the DG chimera, adbn−/−, gsg−B, Largemyd, FKRPMD and fukutin chimera, but not the Largeenr, POMGnT1−/−, postnatal fukutin null, Myf5-Cre-Fktn-KO or MCK-Cre-Fktn-KO. Although these inflammatory infiltrates are generally considered to be macrophages, which are important for clearing debris from necrotic or degenerating muscle fibres, other cell types including T cells, mast cells, eosinophils and neutrophils have been identified in dystrophic muscle (Nahirney et al. 1997; Cai et al. 2000; Whitehead et al. 2006). These cells directly contribute to muscle pathology, and in the mdx, it has been shown that altering the behaviour of these cells influences muscle pathology; for example, modulating macrophage or T cell populations reduces pathological features such as fibrosis (Morrison et al. 2000; Farini et al. 2007; Villalta et al. 2009; reviewed in (Evans et al. 2009; Mann et al. 2011)). Therefore, a lack of inflammatory infiltrate could be interpreted as evidence of muscle pathology of reduced severity. This does not seem to segregate with components of the DAPC – although a loss of dystrophin results in inflammatory infiltrates, a loss of α-dystroglycan glycosylation induces inflammatory infiltrates in some models (FKRPMD) but not others (POMGnT1−/−). Even mutations in the same gene do not necessarily result in the same pathology, with inflammatory infiltrates documented in the Largemyd but not the Largeenr. However, all mouse models, with the exception of POMGnT1−/−, have centrally nucleated muscle fibres, suggesting that the absence of an inflammatory infiltrate does not prevent degeneration (and regeneration) of muscle fibres.
Mdx mice have a florid phase of skeletal muscle degeneration and regeneration between 3 and 5 weeks of age (Carnwath & Shotton 1987; Coulton et al. 1988), with muscle fibre necrosis and regeneration continuing, although less conspicuously, throughout life (Pagel & Partridge 1999; Spurney et al. 2009). Similarly, gsg−/−B and FKRPMD mice also have a crisis period, albeit at slightly later onset (7–10 and 12 weeks of age, respectively), when histopathological features such as Evans blue dye uptake and muscle fibre degeneration/regeneration were worsened. In the other models, either no crisis period has been reported, or it is thought that they have ongoing fibre necrosis and degeneration with age, for example Sgca-null and abdn−/− mice. Thus, there does not seem to be segregation of the occurrence of a crisis phase with components of the DAPC, with a loss of a sarcoglycan, albeit different ones, inducing a crisis phase in one model (gsg−/−B) and not another (Sgca-null). However, it should be noted that with the exception of the FKRPMD, all other models with loss of α-dystroglycan glycosylation do not report a crisis phase. This could be due to differences in the nature of the different dystroglycan models – the FKRPMD has a loss of Fkrp expression in all tissues apart from those expressing Sox1, that is, the central nervous system. This suggests that potentially other cell types or factors could be involved. Despite this, an important point to consider is that a lack of reporting on a crisis phase of disease pathology does not mean that it does not exist; it may be that it has not been explored or identified in these models. For example, if the pathology of the mouse model was only evaluated at one age, then it would be difficult to tell whether a crisis period was present in that mutant at a different time. This would also affect the identification of inflammatory infiltrates, as if these models did have a crisis period, and histological evaluation did not occur during this period, then it might be expected that inflammatory infiltrates would be absent from the muscle.
Furthermore, there is some variation in the occurrence of other histological indicators of muscular dystrophy between the different models, particularly replacement of muscle fibres by adipocytes or fibrotic tissue. Pronounced fat infiltration is observed in Scga-null, Scgb-null, Scgd-null, POMGnT1-null, Largemyd, dsg−/−, Largeenr, gsg−/−A, Largevls, gsg−/−B, dystroglycan chimera and Myf5-Cre-FKTN-null models, but not the mdx, FKRPMD, fukutin chimera, abdn−/−, postnatal fukutin null, MCK-Cre-FKTN-KO or the MCK-DG null dystroglycan knockout. Fibrosis was not reported in the FKRPMD, POMGnT1-null, POMGnT1−/−, abdn−/−, postnatal fukutin null or MCK-DG null, but it was observed in other models – mdx, Largemyd, gsg−/−A, Largevls, Largeenr, gsg−/−B, fukutin chimera, dystroglycan chimera Scgb-null, FKRP P448Lneo+, Sgca-null, FKRP L276Ineo+/P448Lneo+, Sgcd-null and FKRP L276Ineo+/E310delneo+. Here, there are differences between the different DAPC components – models carrying mutations or inactivations in one of the sarcoglycans have replacement of muscle fibres with fibroadipogenic tissue, but those with perturbed α-dystrobrevin expression do not. The mdx, with loss of dystrophin, has fibrosis, but not replacement of muscle fibres with adipocytes. Dystroglycan is more heterogeneous – loss of dystroglycan results in replacement of muscle tissue with fibroadipogenic tissue, but loss of dystroglycan glycosylation is more variable, some models (Largemyd) have replacement by fibrosis and adipocytes, whereas others (FKRPMD) do not.
In combination, these data show that the histopathological features of muscular dystrophy pathology (degree of muscle fibre degeneration, loss and replacement by fibrotic tissue or adipocytes) vary between the different mouse models. These data also show, as in the POMGnT1−/−, that if there is not a great deal of muscle fibre loss induced by the mutation, then consequently inflammation and replacement of muscle fibres by adipocytes or fibrotic tissue will not be as prevalent. This variability suggests that identification of the pathological features present in the muscle depends on the disease process and implies that certain components of the DAPC are more essential than others for the maintenance of muscle structure. However, it should also be noted that the nature of the gene inactivation in the mouse model affects the severity of the pathology – FKRP L276Ineo+/P448Lneo+, with its global knock-down in Fkrp, has replacement of muscle fibres with fibroadipogenic tissue, suggesting that it has a more severe pathology than FKRPMD, which has Fkrp expression in the CNS and no obvious loss of muscle fibres.
In human patients, a number of muscular dystrophies have a characteristic pattern of muscle involvement, with calf hypertrophy associated with a number of neuromuscular conditions (Reimers et al. 1996) and further more specific patterns associated with certain conditions; for example, in the limb girdle muscular dystrophies, the most severely affected muscles are those of the upper and lower limb girdles. A specific pattern of muscle involvement has also been reported in the mdx mouse – although dystrophic pathology has been documented in various muscles throughout the animal, the extraocular (Porter et al. 1998), toe (Dowling et al. 2002) and intrinsic laryngeal muscles (Marques et al. 2007) seem to be relatively spared by the disease process, whereas contrastingly the diaphragm displays comparatively worsened histopathology. This suggests that the presence of dystrophin at the DAPC is not uniformly important for the integrity of each muscle in the body and these three muscles are most likely spared because they are not load-bearing, so consequently not subjected to marked contractile stresses. For most of the other models described in this review, a detailed description of the muscle involvement pattern was not provided in the publications, with the exception of the Largemyd, where all muscle groups were reported to be equally affected, with the exception of the tongue, which was spared, and the FKRPMD where the soleus was reported to be less affected than the gastrocnemius and diaphragm. So, in contrast to dystrophin deficiency, the Largemyd suggests that correctly glycosylated α-dystroglycan is uniformly important for all muscles. As this glycosylation arrangement on α-dystroglycan is crucial for binding to the basement membrane, it is most likely that this phenotype is associated with a loss of signalling at the DAPC, along with a reduction to contraction-induced injury. However, this is somewhat contrasted by the FKRPMD mouse reporting reduced pathology in the soleus, although this can most likely be attributed to the soleus, a postural muscle, being composed of mostly slow type I muscle fibres, which are more resistant to contraction-induced injury (Macpherson et al. 1996).
Interestingly, the phenomenon of worsened histopathology in the diaphragm is seen in a number of other mouse muscular dystrophy models – FKRP L276Ineo+/P448Lneo+, FKRP L276Ineo+/E310delneo+, FKRP P448Lneo+ and abdn−/− mice, but not the FKRPMD mouse. It is thought to occur because the diaphragm is the mostly used muscle in laboratory mice, therefore suffering from the most contraction-induced damage. Unfortunately, the pathology of the diaphragm compared to other hindlimb muscles has not been reported for all the models in this review. These data would be very interesting, as they would elucidate the importance of the different DAPC components for muscle resistance to contraction-induced injury.
By focussing this review on models with mutations/inactivations in the DAPC, we have selected models thought to be sensitive to contraction-induced damage to the muscle fibre membrane. There are a number of ways to assess this sarcolemmal damage in mice, such as evaluating the uptake of systemically administered Evans blue dye into muscle fibres (Hamer et al. 2002), with this phenomenon seen in the mdx (Matsuda et al. 1995), Sgca-null, gsg−/−A, gsg−/−B, adbn−/− and Scgb-null mouse models. Another method, which is also used in human patients, is assessment of MCK levels in the serum, with increased levels observed in human muscular dystrophy patients (Moat et al. 2013), the mdx mouse and the vast majority of the other muscular dystrophy models featured in this review, including Largemyd, Scgb-null, Largevls, postnatal fukutin null, Myf5-Cre-FKTN-KO, Sgcd-null, MCK-Cre-FKTN-KO, Scga-null, FKRP L276Ineo+/P448Lneo+, gsg−/−A, gsg −/−B FKRP P448L, dsg−/− FKRP P448Lneo+ mice, L276Ineo+/E310delneo+ and MCK-DG null. Given that all models, with the exception of the POMGnT1−/− mouse, displayed centrally nucleated fibres in histopathological analysis, indicating muscle regeneration following muscle fibre degeneration, it is not surprising that so many models display increased serum CK in the phenotype.
Measuring the levels of MCK in circulating blood is a crude, global measure of muscle fibre damage and does not provide any information about the specific pattern of muscle involvement or pathological progression of the disease. Instead, currently the most common method for evaluating the progression of muscle damage in human patients is by taking biopsies to evaluate the histology, which in itself is quite invasive and causes muscle damage, so focus has been on finding new, alternative biomarkers for evaluating the progression of muscular dystrophy. Promising biomarkers are changes in the composition of certain muscles, detectable by magnetic resonance imaging (MRI) (Fan et al. 2014), and alterations in the levels of skeletal muscle-specific microRNAs (myoMirs) in the serum of mutants such as the mdx in comparison with control mice, which may mirror pathological processes occurring within skeletal muscle (Roberts et al. 2012, 2013; Vignier et al. 2013). Further evaluation of disease progression in these other mouse models with inactivations/mutations in the other DAPC components will help to elucidate disease progression further, for example using miRNA biomarkers to identify whether and when crisis periods occur in other models and fMRI to explore the pattern of muscle involvement in these models.
Physical symptoms
A number of these mouse models are smaller than control littermates – Largemyd, Largeenr, FKRPMD, POMGnT1 null, POMGnT1−/−, fukutin chimera, dsg−/−, Myf5-Cre-Fktn-KO, gsg−/− FKRP P448L-neo+, Largevls and MORE-DG null. This could be due to reduced growth, a loss of body weight once the animal's phenotype worsens and it starts to lose muscle fibres and muscle mass, or a combination of the two. The IGF1-Akt signalling pathway is involved in regulation of skeletal muscle growth (reviewed in Schiaffino and Mammucari (2011)), and it is initiated by laminin binding to α-dystroglycan in the extracellular space, so a loss of this binding, seen in a number of these models, reduces Akt signalling and consequently muscle growth, contributing to smaller mutant mice (Langenbach & Rando 2002). Interestingly, these phenotypes are in marked contrast to the muscle-specific dystroglycan knockout and Sgca-null, which have increased weight compared to controls and the gsg−/−B mouse, which has no change in body weight compared to wild type. Similarly to mdx (Sharp et al. 2011), these models are reported to have a number of hypertrophic muscle fibres or an increased number of muscle fibres, so it is likely that the muscle fibre hypertrophy accounts for the increased body mass. Both the mdx and gsg−/−B models are reported to have increased levels of Akt activation (Peter & Crosbie 2006), demonstrating that loss of individual DAPC components differentially affects regulation of muscle size. Perhaps, a total loss of laminin binding to α-dystroglycan, as seen in the Largemyd or FKRPMD, results in smaller mutant mice because there is little Akt activation, whereas a loss of other DAPC components not directly involved in binding (dystrophin or γ-sarcoglycan) perturbs regulatory or compensatory signalling pathways, leading to increased Akt activation.
However, in Scga-null mouse, the increased mass was reported as due to increased water and connective tissue content, rather than contractile material, suggesting that in reality, things are not as simple as postulated above. Given that dystrophin binds directly to the contractile apparatus, it is not surprising to assume that altered expression of dystrophin would change the arrangement or amount of contractile proteins in the muscle fibre. Contrastingly, the sarcoglycan complex has been shown to affect aquaporin-4 expression, suggesting that loss of sarcoglycan affects water regulation in muscle fibres (Assereto et al. 2008). Furthermore, the sarcoglycan complex is associated with dystroglycan (Brenman et al. 1995; Chan et al. 1998), providing a mechanism by which a loss of sarcoglycan expression might affect the composition or arrangement of the extracellular matrix. This suggests different roles for α-sarcoglycan and dystrophin with respect to the organization of the intracellular muscle environment.
Some muscular dystrophy mouse models also have physical abnormalities, with kyphosis reported in mdx (Laws & Hoey 2004), Largemyd and Largeenr. Additionally and unsurprisingly, motor function and coordination is affected, with a number of mutants, reported to lose their hindlimb extension reflex – Largemyd, POMGnT1-null, Largeenr, fukutin chimera, FKRP L276Ineo+/P448Lneo+, FKRP L276Ineo+/E310delneo+, FKRP P448L, FKRP P448Lneo+ and MORE-DG null. In some models, these abnormalities have been further characterized, with a motion tremor and delayed righting reflex identified in the MORE-DG null; reduced endurance in gsg−/−; gait abnormalities in the Largemyd, gsg−/−A and fukutin chimera; reduced absolute force in Sgcb-null mice; a reduced specific force in mdx (Lynch et al. 2001), Sgca-null and Sgcb-null mice; reduced resistance to exercise in MCK-Cre-FKTN-KO, mdx (Dellorusso et al. 2001; Sharp et al. 2011) and Sgcd-null; and finally a reduction in forelimb grip strength identified in mdx (Connolly et al. 2001), Largeenr, Largevls, postnatal fukutin null and Myf5-Cre-FKTN-KO. However, histological indicators of muscular dystrophy do not necessarily translate into functional impairments, with gsg−/− mice not having reduced resistance to eccentric exercise and abdn−/− mice not having a reduction in isometric force, specific force or reduced resistance to eccentric muscle contraction. This suggests that the different parts of the complex play different roles in the regulation of muscle function, with α-dystrobrevin less important than dystrophin or dystroglycan with respect to resistance of the muscle fibre to contraction-induced damage.
Finally, histopathological changes in the heart and/or functional evidence of cardiac dysfunction has been observed in the mdx (Bridges 1986) and some of the other mouse models – Scgd−/−, Sgca-null, Sgcd-null, gsg−/−B, Sgcb-null, Scgb-null, abdn−/− and gsg−/−A mice, but not others – FKRP L276Ineo+/P448Lneo+, FKRP L276Ineo+/E310delneo+ mice and FKRP P448Lneo+. This further supports the idea that despite the same complex being functionally compromised, there are different degrees of severity, affecting different tissues, depending on which part of the complex is inactivated.
Reproduction
Mdx mice, which are bred as homozygous females with hemizygous males, are exceptional breeders (http://jaxmice.jax.org/strain/001801.html), so the mutation appears to have no effect on the mouse's reproductive capability. The other muscular dystrophy models discussed here are generally recessively inherited, generated by breeding of two heterozygote mice, and so very few studies have reported the effect of these mutations on reproduction. Where it has been commented on Largemyd, Largeenr, POMGnT1 mutant and FKRPMD, with the exception of the FKRPMD, reproduction has been affected by the mutation. Although in combination these data suggest that a loss of the signalling function at the DAPC through α-dystroglycan affects the reproduction phenotype, more studies on other mouse models are necessary to conclusively determine how, if at all, reproductive functioning is affected by loss or inactivation of the different DAPC components.
Lifespan
Although early work on mice up to 12 months of age suggested that mdx did not have a reduced lifespan (Carnwath & Shotton 1987), a more recent long-term study showed that mutants have a significantly shorter lifespan than C57Bl/10 mice (Chamberlain et al. 2007). Other models are also reported to have reduction in lifespan, including the MORE-DG null (very few animals survive beyond 4 weeks), Largemyd (mean lifespan 17 weeks of age, ranging from 5 to 37 weeks), gsg−/−A, dsg−/− (50% of animals dying by 5 months of age), Myf5-Cre-FKTN-KO (78% of mutants die before 35 weeks of age) and Largeenr (average lifespan 6-8 months, with maximum of 11 months). Associated with this reduced lifespan, these models are also reported to have preweaning losses. Interestingly, a number of other models, including POMGnT1−/−, POMGnT1-null, fukutin chimera and FKRP P448Lneo+, are reported to have a variable life expectancy, ranging from a few days to old age, suggesting a wide range of variation in phenotypic severity between individual animals. Contrastingly, there are other models that are not reported to have a reduced lifespan – FKRPMD, gsg−/−B, FKRP L276Ineo+/P448Lneo+, FKRP L276Ineo+/E310delneo+, abdn−/− and the MCK-DG null mouse. Some of these differences can be explained by the design of the models; for example, knocking out dystroglycan early in development (MORE-DG null) produces a markedly more severe phenotype than knocking it out just in the muscles later during development (MCK-DG null). Furthermore, as a number of the models with a reduced or variable lifespan have been shown to have cortical involvement, deaths in these animals, although connected to a loss of DAPC function, might be unrelated to dystrophic muscle pathology. However, it should be noted that in those where lifespan is not reported to be reduced, it might be that they simply have not looked at old enough mice or performed a survival study to demonstrate conclusively when the phenotype is lethal.
Disease modifiers
To study these different models further, a number of different transgenic crosses have been performed, leading to interesting and somewhat surprising results. It seems that the genetic background of the original mouse model plays an important role in modifying disease phenotype. Due to the nature of generating transgenic mouse models, a number of these mutants are on mixed genetic mouse backgrounds, but even when taking this into account, crossing mice onto the DBA2/J background worsens the disease phenotype, seen with both mdx (Fukada et al. 2010) and gsg−/− mice (Heydemann et al. 2005). This suggests that muscle development, structure and/or maintenance varies between the different laboratory mouse strains, which, whilst interesting, has implications for comparisons of different mouse models or studies, particularly those on mixed or different genetic backgrounds.
These mouse mutants are caused by reduced expression or inactivation of one of the components of the DAPC, so transgenic restoration of this component improves or ameliorates the phenotype, as shown with dystrophin in the mdx mouse (Cox et al. 1993a; Wang et al. 2000; Harper et al. 2002; Gregorevic et al. 2004) and large in the Largemyd mouse (Gumerson et al. 2013). However, as studies in the δ-sarcoglycan-deficient chimeric mice show (Vitale et al. 2012), a certain level of protein expression is required, with histological and functional improvement observed only when wild-type embryonic stem cell incorporation was over 60%. Interestingly, this is different to the mdx, where the phenotype was improved with <30% embryonic stem cell incorporation into mdx chimera (Stillwell et al. 2009), and furthermore, significant functional improvement in mdx is seen when only 20% of the fibres express dystrophin (Sharp et al. 2011). Furthermore, the level of protein expression needs to be controlled, because transgenic overexpression of γ-sarcoglycan induced a dystrophic phenotype in wild-type mice (Zhu et al. 2001). However, these mice were generated on a mixed background of BL6/DBA2 – as discussed earlier; the DBA2/J background is generally a poor background for muscle function, which would affect the results of this study. Again, these further highlight that the different components of the DAPC are not functionally equivalent and support the idea that the loss of individual proteins from the complex affects it function in different ways.
Unsurprisingly, crossing two models with inactivations in two DAPC components together to create a double mutant worsens the phenotype, seen when the mdx was bred independently with the abdn−/− (Grady et al. 1999), Largemyd (Martins et al. 2013) and δ-sarcoglycan (Li et al. 2009) mice, or when the Scga-null mouse was bred with the Scgae-null mouse (Lancioni et al. 2011). Similarly, mdx mice that are also utrophin deficient exhibit a worsened pathology (Deconinck et al. 1997).
In muscular dystrophies where the function of the DAPC is perturbed, it is thought that a loss of adhesion between the muscle fibre and the extracellular matrix contributes to the phenotype, because the resulting muscle fibre is less resistant to contraction-induced membrane damage. However, the DAPC is not the only adhesion complex in muscle. A mutant with a double inactivation of the DAPC and integrin α7 (affecting the integrin adhesion complexes) had an exacerbated phenotype compared to the single gsg−/−A (Allikian et al. 2004) and mdx mutant mice (Rooney et al. 2006), whilst upregulation of integrin α7 (theoretically increasing muscle fibre adhesion) in the mdxutr−/− mouse increased the longevity of the mouse and reduced the dystrophic histopathology (Burkin et al. 2001). Furthermore, inactivation of dysferlin (a protein linked to skeletal muscle repair) through crossing the mdx mouse model with a dysferlin-null mouse created a double knockout with a worsened phenotype than either of the single-gene knockout mice (Han et al. 2011). However, contrastingly, transgenic upregulation of dysferlin (which would in theory increase the rate of membrane repair) (Millay et al. 2009) or integrin α7 (Milner & Kaufman 2007) did not improve the phenotype of Sgcd-null mice.
The precise glycosylation arrangement on α-dystroglycan is important for binding of DAPC to the basement membrane, anchoring the muscle fibre to the extracellular environment. Looking beyond the DAPC, the carbohydrate arrangement in muscle seems to be important for progression and development of the phenotype, with overexpression of Galgt2, a cytotoxic T cell GalNAc transferase, shown to improve the phenotype of both mdx (Martin et al. 2009) and Sgca-null mice (Xu et al. 2009). Of interest is the role of CMAH, a gene encoding CMP-Neu5Ac cytidine-5′-monophospho-N-acetylneuraminic acid hydroxylase, which generates Neu5GC, a common sialic acid modification in mammalian muscle (Varki 2010), with the exception of humans, where the function of this gene has been lost (Chou et al. 1998; Irie et al. 1998). The loss of this gene worsened the phenotype of both the mdx mouse model (Chandrasekharan et al. 2010) and the Scga-null mouse (Martin et al. 2013), perhaps contributing to the phenotype differences between mouse models of muscular dystrophy and the human phenotype.
Relevance to human conditions
Human conditions are caused by a variety of genetic changes that affect the synthesis or functioning of a protein – these can be whole-scale genetic changes, seen in DMD with deletions or duplications of dystrophin exons (Muntoni et al. 2003), or they can be point mutations in individual proteins, such as the L276I mutation in Fkrp, which is associated with LGMD2I (Brockington et al. 2001). Furthermore, recent studies have shown a complex relationship between genotype and patient phenotype demonstrating genetic heterogeneity along with a broad phenotypic spectrum associated with mutations in one single gene. For example, this can be seen in the dystroglycanopathies, where mutations in each gene have been associated with both severe congenital neuromuscular conditions and adult onset limb girdle muscular dystrophies (Godfrey et al. 2011).
Interestingly, in mice, introducing some of these genetic changes identified in human patients does not generate a disease phenotype, for example FKRPTyr307Asn+/+ (Ackroyd et al. 2009) and α-sarcoglycanHis77Cys+/+ (Kobuke et al. 2008), with the phenotype emerging only once gene expression had also been reduced. Differences between mice and humans, particularly in their size and mechanisms of growth and regeneration [reviewed (Partridge 2013)], should thus be considered when using mice as models of human muscular dystrophies.
Conclusions
The functioning of the DAPC is essential for muscle integrity, and perturbation of this complex in mouse generates a dystrophic muscle phenotype. A vast number of different mouse models carrying mutations or inactivations in the different component of the DAPC show that interestingly, perturbing either different parts of the complex or the same part of the complex at different developmental time points generates a phenotype with slightly different characteristics, suggesting that some parts of the complex are more crucial than others for its functioning. However, it must be noted that all these models carry different mutations on different genetic backgrounds, with potential disease modifiers contributing to the phenotypic differences. Unsurprisingly, creating double-knockout models by either inactivating the function of more than one part of the DAPC or further affecting muscle function generates a mutant with a worsened phenotype, when compared to the original mice used in the cross. However, correcting or interfering with the disease process in another way, for example increasing membrane repair, does not improve disease phenotype, suggesting that perhaps the disease process is not as simple as first thought. Alternatively, with recent in vitro data identifying both novel proteins associated with dystroglycan and alterations in dystroglycan protein interactions in the absence of dystrophin (Yoon et al. 2012; Johnson et al. 2013), perhaps the structure of the DAPC and/or the arrangement of its binding partners varies far more widely than previously considered. Therefore, to fully explain the differences between the models highlighted here, we may need to rethink our view of DAPC structure and consequently function.
References
- Ackroyd MR, Skordis L, Kaluarachchi M, et al. Reduced expression of fukutin related protein in mice results in a model for fukutin related protein associated muscular dystrophies. Brain. 2009;132:439–451. doi: 10.1093/brain/awn335. [DOI] [PubMed] [Google Scholar]
- Adams ME, Kramarcy N, Krall SP, et al. Absence of alpha-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J. Cell Biol. 2000;150:1385–1398. doi: 10.1083/jcb.150.6.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allikian MJ, Hack AA, Mewborn S, Mayer U. McNally EM. Genetic compensation for sarcoglycan loss by integrin alpha7beta1 in muscle. J. Cell Sci. 2004;117:3821–3830. doi: 10.1242/jcs.01234. [DOI] [PubMed] [Google Scholar]
- Andersson DC, Meli AC, Reiken S, et al. Leaky ryanodine receptors in beta-sarcoglycan deficient mice: a potential common defect in muscular dystrophy. Skelet. Muscle. 2012;2:9. doi: 10.1186/2044-5040-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki E, Nakamura K, Nakao K, et al. Targeted disruption of exon 52 in the mouse dystrophin gene induced muscle degeneration similar to that observed in Duchenne muscular dystrophy. Biochem. Biophys. Res. Commun. 1997;238:492–497. doi: 10.1006/bbrc.1997.7328. [DOI] [PubMed] [Google Scholar]
- Assereto S, Mastrototaro M, Stringara S, et al. Aquaporin-4 expression is severely reduced in human sarcoglycanopathies and dysferlinopathies. Cell Cycle. 2008;7:2199–2207. doi: 10.4161/cc.7.14.6272. [DOI] [PubMed] [Google Scholar]
- Barton ER. Impact of sarcoglycan complex on mechanical signal transduction in murine skeletal muscle. Am. J. Physiol. Cell Physiol. 2006;290:C411–C419. doi: 10.1152/ajpcell.00192.2005. [DOI] [PubMed] [Google Scholar]
- Beastrom N, Lu H, Macke A, et al. mdx((5)cv) mice manifest more severe muscle dysfunction and diaphragm force deficits than do mdx Mice. Am. J. Pathol. 2011;179:2464–2474. doi: 10.1016/j.ajpath.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beedle AM, Turner AJ, Saito Y, et al. Mouse fukutin deletion impairs dystroglycan processing and recapitulates muscular dystrophy. J. Clin. Investig. 2012;122:3330–3342. doi: 10.1172/JCI63004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaeser A, Keramaris E, Chan YM, et al. Mouse models of fukutin-related protein mutations show a wide range of disease phenotypes. Hum. Genet. 2013;132:923–934. doi: 10.1007/s00439-013-1302-7. [DOI] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Xia H, Aldape K. Bredt DS. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell. 1995;82:743–752. doi: 10.1016/0092-8674(95)90471-9. [DOI] [PubMed] [Google Scholar]
- Bridges LR. The association of cardiac-muscle necrosis and inflammation with the degenerative and persistent myopathy of mdx mice. J. Neurol. Sci. 1986;72:147–157. doi: 10.1016/0022-510x(86)90003-1. [DOI] [PubMed] [Google Scholar]
- Brockington M, Yuva Y, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum. Mol. Genet. 2001;10:2851–2859. doi: 10.1093/hmg/10.25.2851. [DOI] [PubMed] [Google Scholar]
- Brunig I, Suter A, Knuesel I, Luscher B. Fritschy JM. GABAergic terminals are required for postsynaptic clustering of dystrophin but not of GABA(A) receptors and gephyrin. J. Neurosci. 2002;22:4805–4813. doi: 10.1523/JNEUROSCI.22-12-04805.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulfield G, Siller WG, Wight PA. Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl Acad. Sci. USA. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnell TM, Jaeger MA, Fitzsimons DP, Prins KW. Ervasti JM. Destabilization of the dystrophin-glycoprotein complex without functional deficits in alpha-dystrobrevin null muscle. PLoS One. 2008;3:e2604. doi: 10.1371/journal.pone.0002604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkin DJ, Wallace GQ, Nicol KJ, Kaufman DJ. Kaufman SJ. Enhanced expression of the alpha 7 beta 1 integrin reduces muscular dystrophy and restores viability in dystrophic mice. J. Cell Biol. 2001;152:1207–1218. doi: 10.1083/jcb.152.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai B, Spencer MJ, Nakamura G, Tseng-Ong L. Tidball JG. Eosinophilia of dystrophin-deficient muscle is promoted by perforin-mediated cytotoxicity by T cell effectors. Am. J. Pathol. 2000;156:1789–1796. doi: 10.1016/S0002-9440(10)65050-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnwath JW. Shotton DM. Muscular dystrophy in the mdx mouse: histopathology of the soleus and extensor digitorum longus muscles. J. Neurol. Sci. 1987;80:39–54. doi: 10.1016/0022-510x(87)90219-x. [DOI] [PubMed] [Google Scholar]
- Chamberlain JS, Phelps SF, Cox GA, Maichele AJ. Greenwood AD. PCR analysis of muscular dystrophy in mdx mice. Mol. Cell Biol. Hum. Dis. Ser. 1993;3:167–189. doi: 10.1007/978-94-011-1528-5_7. [DOI] [PubMed] [Google Scholar]
- Chamberlain JS, Metzger J, Reyes M, Townsend DW. Faulkner JA. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J. 2007;21:2195–2204. doi: 10.1096/fj.06-7353com. [DOI] [PubMed] [Google Scholar]
- Chan YM, Bonnemann CG, Lidov HG. Kunkel LM. Molecular organization of sarcoglycan complex in mouse myotubes in culture. J. Cell Biol. 1998;143:2033–2044. doi: 10.1083/jcb.143.7.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YM, Keramaris-Vrantsis E, Lidov HG, et al. Fukutin-related protein is essential for mouse muscle, brain and eye development and mutation recapitulates the wide clinical spectrums of dystroglycanopathies. Hum. Mol. Genet. 2010;19:3995–4006. doi: 10.1093/hmg/ddq314. [DOI] [PubMed] [Google Scholar]
- Chandrasekharan K, Yoon JH, Xu Y, et al. A human-specific deletion in mouse Cmah increases disease severity in the mdx model of Duchenne muscular dystrophy. Sci. Transl. Med. 2010;2:42ra54. doi: 10.1126/scitranslmed.3000692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman VM, Miller DR, Armstrong D. Caskey CT. Recovery of induced mutations for X chromosome-linked muscular dystrophy in mice. Proc. Natl Acad. Sci. USA. 1989;86:1292–1296. doi: 10.1073/pnas.86.4.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou HH, Takematsu H, Diaz S, et al. A mutation in human CMP-sialic acid hydroxylase occurred after the Homo-Pan divergence. Proc. Natl Acad. Sci. USA. 1998;95:11751–11756. doi: 10.1073/pnas.95.20.11751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn RD, Henry MD, Michele DE, et al. Disruption of DAG1 in differentiated skeletal muscle reveals a role for dystroglycan in muscle regeneration. Cell. 2002;110:639–648. doi: 10.1016/s0092-8674(02)00907-8. [DOI] [PubMed] [Google Scholar]
- Connolly AM, Keeling RM, Mehta S, Pestronk A. Sanes JR. Three mouse models of muscular dystrophy: the natural history of strength and fatigue in dystrophin-, dystrophin/utrophin-, and laminin alpha2-deficient mice. Neuromuscul. Disord. 2001;11:703–712. doi: 10.1016/s0960-8966(01)00232-2. [DOI] [PubMed] [Google Scholar]
- Consolino CM, Duclos F, Lee J, Williamson RA, Campbell KP. Brooks SV. Muscles of mice deficient in alpha-sarcoglycan maintain large masses and near control force values throughout the life span. Physiol. Genomics. 2005;22:244–256. doi: 10.1152/physiolgenomics.00311.2004. [DOI] [PubMed] [Google Scholar]
- Constantin B. Dystrophin complex functions as a scaffold for signalling proteins. Biochim. Biophys. Acta. 2014;1838:635–642. doi: 10.1016/j.bbamem.2013.08.023. [DOI] [PubMed] [Google Scholar]
- Coral-Vazquez R, Cohn RD, Moore SA, et al. Disruption of the sarcoglycan-sarcospan complex in vascular smooth muscle: a novel mechanism for cardiomyopathy and muscular dystrophy. Cell. 1999;98:465–474. doi: 10.1016/s0092-8674(00)81975-3. [DOI] [PubMed] [Google Scholar]
- Cote PD, Moukhles H, Lindenbaum M. Carbonetto S. Chimaeric mice deficient in dystroglycans develop muscular dystrophy and have disrupted myoneural synapses. Nat. Genet. 1999;23:338–342. doi: 10.1038/15519. [DOI] [PubMed] [Google Scholar]
- Coulton GR, Morgan JE, Partridge TA. Sloper JC. The mdx mouse skeletal muscle myopathy: I. A histological, morphometric and biochemical investigation. Neuropathol. Appl. Neurobiol. 1988;14:53–70. doi: 10.1111/j.1365-2990.1988.tb00866.x. [DOI] [PubMed] [Google Scholar]
- Cox GA, Cole NM, Matsumura K, et al. Overexpression of dystrophin in transgenic mdx mice eliminates dystrophic symptoms without toxicity. Nature. 1993a;364:725–729. doi: 10.1038/364725a0. [DOI] [PubMed] [Google Scholar]
- Cox GA, Phelps SF, Chapman VM. Chamberlain JS. New mdx mutation disrupts expression of muscle and nonmuscle isoforms of dystrophin. Nat. Genet. 1993b;4:87–93. doi: 10.1038/ng0593-87. [DOI] [PubMed] [Google Scholar]
- Danko I, Chapman V. Wolff JA. The frequency of revertants in mdx mouse genetic models for Duchenne muscular dystrophy. Pediatr. Res. 1992;32:128–131. doi: 10.1203/00006450-199207000-00025. [DOI] [PubMed] [Google Scholar]
- Deconinck AE, Rafael JA, Skinner JA, et al. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell. 1997;90:717–727. doi: 10.1016/s0092-8674(00)80532-2. [DOI] [PubMed] [Google Scholar]
- Dellorusso C, Crawford RW, Chamberlain JS. Brooks SV. Tibialis anterior muscles in mdx mice are highly susceptible to contraction-induced injury. J. Muscle Res. Cell Motil. 2001;22:467–475. doi: 10.1023/a:1014587918367. [DOI] [PubMed] [Google Scholar]
- Dowling P, Culligan K. Ohlendieck K. Distal mdx muscle groups exhibiting up-regulation of utrophin and rescue of dystrophin-associated glycoproteins exemplify a protected phenotype in muscular dystrophy. Naturwissenschaften. 2002;89:75–78. doi: 10.1007/s00114-001-0289-4. [DOI] [PubMed] [Google Scholar]
- Duclos F, Straub V, Moore SA, et al. Progressive muscular dystrophy in alpha-sarcoglycan-deficient mice. J. Cell Biol. 1998;142:1461–1471. doi: 10.1083/jcb.142.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbeej M, Cohn RD, Hrstka RF, et al. Disruption of the beta-sarcoglycan gene reveals pathogenetic complexity of limb-girdle muscular dystrophy type 2E. Mol. Cell. 2000;5:141–151. doi: 10.1016/s1097-2765(00)80410-4. [DOI] [PubMed] [Google Scholar]
- Echigoya Y, Lee J, Rodrigues M, et al. Mutation types and aging differently affect revertant fiber expansion in dystrophic mdx and mdx52 mice. PLoS One. 2013;8:e69194. doi: 10.1371/journal.pone.0069194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans NP, Misyak SA, Robertson JL, Bassaganya-Riera J. Grange RW. Immune-mediated mechanisms potentially regulate the disease time-course of Duchenne muscular dystrophy and provide targets for therapeutic intervention. PM R. 2009;1:755–768. doi: 10.1016/j.pmrj.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z, Wang J, Ahn M, et al. Characteristics of magnetic resonance imaging biomarkers in a natural history study of golden retriever muscular dystrophy. Neuromuscul. Disord. 2014;24:178–191. doi: 10.1016/j.nmd.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farini A, Meregalli M, Belicchi M, et al. T and B lymphocyte depletion has a marked effect on the fibrosis of dystrophic skeletal muscles in the scid/mdx mouse. J. Pathol. 2007;213:229–238. doi: 10.1002/path.2213. [DOI] [PubMed] [Google Scholar]
- Fukada S, Morikawa D, Yamamoto Y, et al. Genetic background affects properties of satellite cells and mdx phenotypes. Am. J. Pathol. 2010;176:2414–2424. doi: 10.2353/ajpath.2010.090887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey C, Foley AR, Clement E. Muntoni F. Dystroglycanopathies: coming into focus. Curr. Opin. Genet. Dev. 2011;21:278–285. doi: 10.1016/j.gde.2011.02.001. [DOI] [PubMed] [Google Scholar]
- Grady RM, Grange RW, Lau KS, et al. Role for alpha-dystrobrevin in the pathogenesis of dystrophin-dependent muscular dystrophies. Nat. Cell Biol. 1999;1:215–220. doi: 10.1038/12034. [DOI] [PubMed] [Google Scholar]
- Gregorevic P, Blankinship MJ, Allen JM, et al. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat. Med. 2004;10:828–834. doi: 10.1038/nm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal PK, McLaughlan JM, Moore CJ, Browning CA. Hewitt JE. Characterization of the LARGE family of putative glycosyltransferases associated with dystroglycanopathies. Glycobiology. 2005;15:912–923. doi: 10.1093/glycob/cwi094. [DOI] [PubMed] [Google Scholar]
- Gumerson JD, Davis CS, Kabaeva ZT, Hayes JM, Brooks SV. Michele DE. Muscle-specific expression of LARGE restores neuromuscular transmission deficits in dystrophic LARGE(myd) mice. Hum. Mol. Genet. 2013;22:757–768. doi: 10.1093/hmg/dds483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack AA, Ly CT, Jiang F, et al. Gamma-sarcoglycan deficiency leads to muscle membrane defects and apoptosis independent of dystrophin. J. Cell Biol. 1998;142:1279–1287. doi: 10.1083/jcb.142.5.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack AA, Cordier L, Shoturma DI, Lam MY, Sweeney HL. McNally EM. Muscle degeneration without mechanical injury in sarcoglycan deficiency. Proc. Natl Acad. Sci. USA. 1999;96:10723–10728. doi: 10.1073/pnas.96.19.10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack AA, Lam MY, Cordier L, et al. Differential requirement for individual sarcoglycans and dystrophin in the assembly and function of the dystrophin-glycoprotein complex. J. Cell Sci. 2000;113(Pt 14):2535–2544. doi: 10.1242/jcs.113.14.2535. [DOI] [PubMed] [Google Scholar]
- Hamer PW, McGeachie JM, Davies MJ. Grounds MD. Evans Blue Dye as an in vivo marker of myofibre damage: optimising parameters for detecting initial myofibre membrane permeability. J. Anat. 2002;200:69–79. doi: 10.1046/j.0021-8782.2001.00008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han R, Rader EP, Levy JR, Bansal D. Campbell KP. Dystrophin deficiency exacerbates skeletal muscle pathology in dysferlin-null mice. Skelet. Muscle. 2011;1:35. doi: 10.1186/2044-5040-1-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper SQ, Hauser MA, Dellorusso C, et al. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat. Med. 2002;8:253–261. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- Heydemann A, Huber JM, Demonbreun A, Hadhazy M. McNally EM. Genetic background influences muscular dystrophy. Neuromuscul. Disord. 2005;15:601–609. doi: 10.1016/j.nmd.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- Hoffman EP, Morgan JE, Watkins SC. Partridge TA. Somatic reversion/suppression of the mouse mdx phenotype in vivo. J. Neurol. Sci. 1990;99:9–25. doi: 10.1016/0022-510x(90)90195-s. [DOI] [PubMed] [Google Scholar]
- Hu H, Li J, Gagen CS, et al. Conditional knockout of protein O-mannosyltransferase 2 reveals tissue-specific roles of O-mannosyl glycosylation in brain development. J. Comp. Neurol. 2011;519:1320–1337. doi: 10.1002/cne.22572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang PL, Dawson TM, Bredt DS, Snyder SH. Fishman MC. Targeted disruption of the neuronal nitric oxide synthase gene. Cell. 1993;75:1273–1286. doi: 10.1016/0092-8674(93)90615-w. [DOI] [PubMed] [Google Scholar]
- Im WB, Phelps SF, Copen EH, Adams EG, Slightom JL. Chamberlain JS. Differential expression of dystrophin isoforms in strains of mdx mice with different mutations. Hum. Mol. Genet. 1996;5:1149–1153. doi: 10.1093/hmg/5.8.1149. [DOI] [PubMed] [Google Scholar]
- Irie A, Koyama S, Kozutsumi Y, Kawasaki T. Suzuki A. The molecular basis for the absence of N-glycolylneuraminic acid in humans. J. Biol. Chem. 1998;273:15866–15871. doi: 10.1074/jbc.273.25.15866. [DOI] [PubMed] [Google Scholar]
- Jakubiec-Puka A, Biral D, Krawczyk K. Betto R. Ultrastructure of diaphragm from dystrophic alpha-sarcoglycan-null mice. Acta Biochim. Pol. 2005;52:453–460. [PubMed] [Google Scholar]
- Johnson EK, Li B, Yoon JH, et al. Identification of new dystroglycan complexes in skeletal muscle. PLoS One. 2013;8:e73224. doi: 10.1371/journal.pone.0073224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanagawa M, Nishimoto A, Chiyonobu T, et al. Residual laminin-binding activity and enhanced dystroglycan glycosylation by LARGE in novel model mice to dystroglycanopathy. Hum. Mol. Genet. 2009;18:621–631. doi: 10.1093/hmg/ddn387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly D, Chancellor K, Milatovich A, Francke U, Suzuki K. Popko B. Autosomal recessive neuromuscular disorder in a transgenic line of mice. J. Neurosci. 1994;14:198–207. doi: 10.1523/JNEUROSCI.14-01-00198.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobuke K, Piccolo F, Garringer KW, et al. A common disease-associated missense mutation in alpha-sarcoglycan fails to cause muscular dystrophy in mice. Hum. Mol. Genet. 2008;17:1201–1213. doi: 10.1093/hmg/ddn009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konieczny P, Swiderski K. Chamberlain JS. Gene and cell-mediated therapies for muscular dystrophy. Muscle Nerve. 2013;47:649–663. doi: 10.1002/mus.23738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudoh H, Ikeda H, Kakitani M, et al. A new model mouse for Duchenne muscular dystrophy produced by 2.4 Mb deletion of dystrophin gene using Cre-loxP recombination system. Biochem. Biophys. Res. Commun. 2005;328:507–516. doi: 10.1016/j.bbrc.2004.12.191. [DOI] [PubMed] [Google Scholar]
- Kurahashi H, Taniguchi M, Meno C, et al. Basement membrane fragility underlies embryonic lethality in fukutin-null mice. Neurobiol. Dis. 2005;19:208–217. doi: 10.1016/j.nbd.2004.12.018. [DOI] [PubMed] [Google Scholar]
- Lancioni A, Rotundo IL, Kobayashi YM, et al. Combined deficiency of alpha and epsilon sarcoglycan disrupts the cardiac dystrophin complex. Hum. Mol. Genet. 2011;20:4644–4654. doi: 10.1093/hmg/ddr398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane PW, Beamer TC. Myers DD. Myodystrophy, a new myopathy on chromosome 8 of the mouse. J. Hered. 1976;67:135–138. doi: 10.1093/oxfordjournals.jhered.a108687. [DOI] [PubMed] [Google Scholar]
- Langenbach KJ. Rando TA. Inhibition of dystroglycan binding to laminin disrupts the PI3K/AKT pathway and survival signaling in muscle cells. Muscle Nerve. 2002;26:644–653. doi: 10.1002/mus.10258. [DOI] [PubMed] [Google Scholar]
- Laws N, Hoey A. Progression of kyphosis in mdx mice. J. Appl. Physiol. (1985) 2004;97:1970–1977. doi: 10.1152/japplphysiol.01357.2003. [DOI] [PubMed] [Google Scholar]
- Lebakken CS, Venzke DP, Hrstka RF, et al. Sarcospan-deficient mice maintain normal muscle function. Mol. Cell. Biol. 2000;20:1669–1677. doi: 10.1128/mcb.20.5.1669-1677.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Kameya S, Cox GA, et al. Ocular abnormalities in Large(myd) and Large(vls) mice, spontaneous models for muscle, eye, and brain diseases. Mol. Cell. Neurosci. 2005;30:160–172. doi: 10.1016/j.mcn.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Levedakou EN, Chen XJ, Soliven B. Popko B. Disruption of the mouse Large gene in the enr and myd mutants results in nerve, muscle, and neuromuscular junction defects. Mol. Cell. Neurosci. 2005;28:757–769. doi: 10.1016/j.mcn.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Li DJ, Long C, Yue YP. Duan DS. Sub-physiological sarcoglycan expression contributes to compensatory muscle protection in mdx mice. Hum. Mol. Genet. 2009;18:1209–1220. doi: 10.1093/hmg/ddp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Ball SL, Yang Y, et al. A genetic model for muscle-eye-brain disease in mice lacking protein O-mannose 1,2-N-acetylglucosaminyltransferase (POMGnT1) Mech. Dev. 2006;123:228–240. doi: 10.1016/j.mod.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Lu QL, Morris GE, Wilton SD, et al. Massive idiosyncratic exon skipping corrects the nonsense mutation in dystrophic mouse muscle and produces functional revertant fibers by clonal expansion. J. Cell Biol. 2000;148:985–996. doi: 10.1083/jcb.148.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch GS, Hinkle RT, Chamberlain JS, Brooks SV. Faulkner JA. Force and power output of fast and slow skeletal muscles from mdx mice 6-28 months old. J. Physiol. 2001;535:591–600. doi: 10.1111/j.1469-7793.2001.00591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson PC, Schork MA. Faulkner JA. Contraction-induced injury to single fiber segments from fast and slow muscles of rats by single stretches. Am. J. Physiol. 1996;271:C1438–C1446. doi: 10.1152/ajpcell.1996.271.5.C1438. [DOI] [PubMed] [Google Scholar]
- Mann CJ, Perdiguero E, Kharraz Y, et al. Aberrant repair and fibrosis development in skeletal muscle. Skelet. Muscle. 2011;1:21. doi: 10.1186/2044-5040-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques MJ, Ferretti R, Vomero VU, Minatel E. Neto HS. Intrinsic laryngeal muscles are spared from myonecrosis in the mdx mouse model of Duchenne muscular dystrophy. Muscle Nerve. 2007;35:349–353. doi: 10.1002/mus.20697. [DOI] [PubMed] [Google Scholar]
- Martin PT, Xu R, Rodino-Klapac LR, et al. Overexpression of Galgt2 in skeletal muscle prevents injury resulting from eccentric contractions in both mdx and wild-type mice. Am. J. Physiol. Cell Physiol. 2009;296:C476–C488. doi: 10.1152/ajpcell.00456.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PT, Camboni M, Xu R, et al. N-Glycolylneuraminic acid deficiency worsens cardiac and skeletal muscle pathophysiology in alpha-sarcoglycan-deficient mice. Glycobiology. 2013;23:833–843. doi: 10.1093/glycob/cwt020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins PC, Ayub-Guerrieri D, Martins-Bach AB, et al. Dmdmdx/Largemyd: a new mouse model of neuromuscular diseases useful for studying physiopathological mechanisms and testing therapies. Dis. Models Mech. 2013;6:1167–1174. doi: 10.1242/dmm.011700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews KD, Mills KA, Bailey HL, Schelper RL, Murray JC. Mouse myodystrophy (myd) mutation: refined mapping in an interval flanked by homology with distal human 4q. Muscle Nerve Suppl. 1995;9:8–102. [PubMed] [Google Scholar]
- Matsuda R, Nishikawa A. Tanaka H. Visualization of dystrophic muscle fibers in mdx mouse by vital staining with Evans blue: evidence of apoptosis in dystrophin-deficient muscle. J. Biochem. 1995;118:959–964. doi: 10.1093/jb/118.5.959. [DOI] [PubMed] [Google Scholar]
- Michele DE. Campbell KP. Dystrophin-glycoprotein complex: post-translational processing and dystroglycan function. J. Biol. Chem. 2003;278:15457–15460. doi: 10.1074/jbc.R200031200. [DOI] [PubMed] [Google Scholar]
- Millay DP, Maillet M, Roche JA, et al. Genetic manipulation of dysferlin expression in skeletal muscle: novel insights into muscular dystrophy. Am. J. Pathol. 2009;175:1817–1823. doi: 10.2353/ajpath.2009.090107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner DJ. Kaufman SJ. Alpha7beta1 integrin does not alleviate disease in a mouse model of limb girdle muscular dystrophy type 2F. Am. J. Pathol. 2007;170:609–619. doi: 10.2353/ajpath.2007.060686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagoe-Suzuki Y, Masubuchi N, Miyamoto K, et al. Reduced proliferative activity of primary POMGnT1-null myoblasts in vitro. Mech. Dev. 2009;126:107–116. doi: 10.1016/j.mod.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Moat SJ, Bradley DM, Salmon R, Clarke A. Hartley L. Newborn bloodspot screening for Duchenne muscular dystrophy: 21 years experience in Wales (UK) Eur. J. Hum. Genet. 2013;21:1049–1053. doi: 10.1038/ejhg.2012.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison J, Lu QL, Pastoret C, Partridge T. Bou-Gharios G. T-cell-dependent fibrosis in the mdx dystrophic mouse. Lab. Invest. 2000;80:881–891. doi: 10.1038/labinvest.3780092. [DOI] [PubMed] [Google Scholar]
- Muntoni F, Torelli S. Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2:731–740. doi: 10.1016/s1474-4422(03)00585-4. [DOI] [PubMed] [Google Scholar]
- Nahirney PC, Dow PR. Ovalle WK. Quantitative morphology of mast cells in skeletal muscle of normal and genetically dystrophic mice. Anat. Rec. 1997;247:341–349. doi: 10.1002/(SICI)1097-0185(199703)247:3<341::AID-AR5>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Pagel CN. Partridge TA. Covert persistence of mdx mouse myopathy is revealed by acute and chronic effects of irradiation. J. Neurol. Sci. 1999;164:103–116. doi: 10.1016/s0022-510x(99)00061-1. [DOI] [PubMed] [Google Scholar]
- Parames SF, Coletta-Yudice ED, Nogueira FM, et al. Altered acetylcholine release in the hippocampus of dystrophin-deficient mice. Neuroscience. 2014;269:173–183. doi: 10.1016/j.neuroscience.2014.03.050. [DOI] [PubMed] [Google Scholar]
- Partridge TA. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. FEBS J. 2013;280:4177–4186. doi: 10.1111/febs.12267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge TA, Morgan JE, Coulton GR, Hoffman EP. Kunkel LM. Conversion of mdx myofibres from dystrophin-negative to -positive by injection of normal myoblasts. Nature. 1989;337:176–179. doi: 10.1038/337176a0. [DOI] [PubMed] [Google Scholar]
- Patel ND, Jannapureddy SR, Hwang W, Chaudhry I, Boriek AM. Altered muscle force and stiffness of skeletal muscles in alpha-sarcoglycan-deficient mice. Am. J. Physiol.Cell Physiol. 2003;284:C962–C968. doi: 10.1152/ajpcell.00326.2002. [DOI] [PubMed] [Google Scholar]
- Peter AK. Crosbie RH. Hypertrophic response of Duchenne and limb-girdle muscular dystrophies is associated with activation of Akt pathway. Exp. Cell Res. 2006;312:2580–2591. doi: 10.1016/j.yexcr.2006.04.024. [DOI] [PubMed] [Google Scholar]
- Porter JD, Rafael JA, Ragusa RJ, Brueckner JK, Trickett JI. Davies KE. The sparing of extraocular muscle in dystrophinopathy is lost in mice lacking utrophin and dystrophin. J. Cell Sci. 1998;111(Pt 13):1801–1811. doi: 10.1242/jcs.111.13.1801. [DOI] [PubMed] [Google Scholar]
- Reimers CD, Schlotter B, Eicke BM. Witt TN. Calf enlargement in neuromuscular diseases: a quantitative ultrasound study in 350 patients and review of the literature. J. Neurol. Sci. 1996;143:46–56. doi: 10.1016/s0022-510x(96)00037-8. [DOI] [PubMed] [Google Scholar]
- Roberts TC, Blomberg KE, McClorey G, et al. Expression analysis in multiple muscle groups and serum reveals complexity in the microRNA transcriptome of the mdx mouse with implications for therapy. Mol. Ther. Nucleic Acids. 2012;1:e39. doi: 10.1038/mtna.2012.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts TC, Godfrey C, McClorey G, et al. Extracellular microRNAs are dynamic non-vesicular biomarkers of muscle turnover. Nucleic Acids Res. 2013;41:9500–9513. doi: 10.1093/nar/gkt724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney JE, Welser JV, Dechert MA, Flintoff-Dye NL, Kaufman SJ. Burkin DJ. Severe muscular dystrophy in mice that lack dystrophin and alpha7 integrin. J. Cell Sci. 2006;119:2185–2195. doi: 10.1242/jcs.02952. [DOI] [PubMed] [Google Scholar]
- Sasaoka T, Imamura M, Araishi K, et al. Pathological analysis of muscle hypertrophy and degeneration in muscular dystrophy in gamma-sarcoglycan-deficient mice. Neuromuscul. Disord. 2003;13:193–206. doi: 10.1016/s0960-8966(02)00220-1. [DOI] [PubMed] [Google Scholar]
- Satz JS, Barresi R, Durbeej M, et al. Brain and eye malformations resembling Walker-Warburg syndrome are recapitulated in mice by dystroglycan deletion in the epiblast. J. Neurosci. 2008;28:10567–10575. doi: 10.1523/JNEUROSCI.2457-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiaffino S. Mammucari C. Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: insights from genetic models. Skelet. Muscle. 2011;1:4. doi: 10.1186/2044-5040-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PS, Bye-A-jee H. Wells DJ. Physiological characterization of muscle strength with variable levels of dystrophin restoration in mdx mice following local antisense therapy. Mol. Ther. 2011;19:165–171. doi: 10.1038/mt.2010.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicinski P, Geng Y, Ryder-Cook AS, Barnard EA, Darlison MG. Barnard PJ. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- Spurney CF, Gordish-Dressman H, Guerron AD, et al. Preclinical drug trials in the mdx mouse: assessment of reliable and sensitive outcome measures. Muscle Nerve. 2009;39:591–602. doi: 10.1002/mus.21211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stillwell E, Vitale J, Zhao Q, et al. Blastocyst injection of wild type embryonic stem cells induces global corrections in mdx mice. PLoS One. 2009;4:e4759. doi: 10.1371/journal.pone.0004759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Kondo M, Sasaki J, et al. Fukutin is required for maintenance of muscle integrity, cortical histiogenesis and normal eye development. Hum. Mol. Genet. 2003;12:1449–1459. doi: 10.1093/hmg/ddg153. [DOI] [PubMed] [Google Scholar]
- Torres LF, Duchen LW. The mutant mdx: inherited myopathy in the mouse. Morphological studies of nerves, muscles and end-plates. Brain. 1987;110(Pt 2):269–299. doi: 10.1093/brain/110.2.269. [DOI] [PubMed] [Google Scholar]
- Tozawa T, Itoh K, Yaoi T, et al. The shortest isoform of dystrophin (Dp40) interacts with a group of presynaptic proteins to form a presumptive novel complex in the mouse brain. Mol. Neurobiol. 2012;45:287–297. doi: 10.1007/s12035-012-8233-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A. Colloquium paper: uniquely human evolution of sialic acid genetics and biology. Proc. Natl Acad. Sci. USA. 2010;107(Suppl 2):8939–8946. doi: 10.1073/pnas.0914634107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignier N, Amor F, Fogel P, et al. Distinctive serum miRNA profile in mouse models of striated muscular pathologies. PLoS One. 2013;8:e55281. doi: 10.1371/journal.pone.0055281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalta SA, Nguyen HX, Deng B, Gotoh T. Tidball JG. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet. 2009;18:482–496. doi: 10.1093/hmg/ddn376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale JM, Schneider JS, Beck AJ, et al. Dystrophin-compromised sarcoglycan-delta-knockout diaphragm requires full wild-type embryonic stem cell reconstitution for correction. J. Cell Sci. 2012;125:1807–1813. doi: 10.1242/jcs.100537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite A, Tinsley CL, Locke M. Blake DJ. The neurobiology of the dystrophin-associated glycoprotein complex. Ann. Med. 2009;41:344–359. doi: 10.1080/07853890802668522. [DOI] [PubMed] [Google Scholar]
- Wang B, Li J. Xiao X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc. Natl Acad. Sci. USA. 2000;97:13714–13719. doi: 10.1073/pnas.240335297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wansapura JP, Millay DP, Dunn RS, Molkentin JD. Benson DW. Magnetic resonance imaging assessment of cardiac dysfunction in delta-sarcoglycan null mice. Neuromusc. Disord. 2011;21:68–73. doi: 10.1016/j.nmd.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz K. Fuchtbauer EM. Dmd(mdx-beta geo): a new allele for the mouse dystrophin gene. Dev. Dyn. 1998;212:229–241. doi: 10.1002/(SICI)1097-0177(199806)212:2<229::AID-AJA7>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Whitehead NP, Yeung EW. Allen DG. Muscle damage in mdx (dystrophic) mice: role of calcium and reactive oxygen species. Clin. Exp. Pharmacol. Physiol. 2006;33:657–662. doi: 10.1111/j.1440-1681.2006.04394.x. [DOI] [PubMed] [Google Scholar]
- Whitmore C, Fernandez-Fuente M, Booler H, et al. The transgenic expression of LARGE exacerbates the muscle phenotype of dystroglycanopathy mice. Hum. Mol. Genet. 2014;23:1842–1855. doi: 10.1093/hmg/ddt577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer T, Prados B, Falcon-Perez JM, et al. Targeted disruption of the Walker-Warburg syndrome gene Pomt1 in mouse results in embryonic lethality. Proc. Natl Acad. Sci. USA. 2004;101:14126–14131. doi: 10.1073/pnas.0405899101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson RA, Henry MD, Daniels KJ, et al. Dystroglycan is essential for early embryonic development: disruption of Reichert's membrane in Dag1-null mice. Hum. Mol. Genet. 1997;6:831–841. doi: 10.1093/hmg/6.6.831. [DOI] [PubMed] [Google Scholar]
- Willmann R, Possekel S, Dubach-Powell J, Meier T. Ruegg MA. Mammalian animal models for Duchenne muscular dystrophy. Neuromuscul. Disord. 2009;19:241–249. doi: 10.1016/j.nmd.2008.11.015. [DOI] [PubMed] [Google Scholar]
- Xu R, Devries S, Camboni M. Martin PT. Overexpression of Galgt2 reduces dystrophic pathology in the skeletal muscles of alpha sarcoglycan-deficient mice. Am. J. Pathol. 2009;175:235–247. doi: 10.2353/ajpath.2009.080967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Johnson E, Xu R, Martin LT, Martin PT. Montanaro F. Comparative proteomic profiling of dystroglycan-associated proteins in wild type, mdx, and Galgt2 transgenic mouse skeletal muscle. J. Proteome Res. 2012;11:4413–4424. doi: 10.1021/pr300328r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Hadhazy M, Groh ME, Wheeler MT, Wollmann R. McNally EM. Overexpression of gamma-sarcoglycan induces severe muscular dystrophy. Implications for the regulation of Sarcoglycan assembly. J. Biol. Chem. 2001;276:21785–21790. doi: 10.1074/jbc.M101877200. [DOI] [PubMed] [Google Scholar]