

Abstract
Ischemia/reperfusion (I/R) elicits renin release from cardiac mast cells (MC), thus activating a local renin-angiotensin system (RAS), culminating in ventricular fibrillation. We hypothesized that in I/R, neurogenic ATP could degranulate juxtaposed MC and that ecto-nucleoside triphosphate diphosphohydrolase 1/CD39 (CD39) on MC membrane could modulate ATP-induced renin release. We report that pharmacological inhibition of CD39 in a cultured human mastocytoma cell line (HMC-1) and murine bone marrow-derived MC with ARL67156 (100 µM) increased ATP-induced renin release (≥2-fold), whereas purinergic P2X7 receptors (P2X7R) blockade with A740003 (3 µM) prevented it. Likewise, CD39 RNA silencing in HMC-1 increased ATP-induced renin release (≥2-fold), whereas CD39 overexpression prevented it. Acetaldehyde, an I/R product (300 µM), elicited an 80% increase in ATP release from HMC-1, in turn, causing an autocrine 20% increase in renin release. This effect was inhibited or potentiated when CD39 was overexpressed or silenced, respectively. Moreover, P2X7R silencing prevented ATP- and acetaldehyde-induced renin release. I/R-induced RAS activation in ex vivo murine hearts, characterized by renin and norepinephrine overflow and ventricular fibrillation, was potentiated (∼2-fold) by CD39 inhibition, an effect prevented by P2X7R blockade. Our data indicate that by regulating ATP availability at the MC surface, CD39 modulates local renin release and thus, RAS activation, ultimately exerting a cardioprotective effect.—Aldi, S., Marino, A., Tomita, K., Corti, F., Anand, R., Olson, K. E., Marcus, A. J., Levi, R. E-NTPDase1/CD39 modulates renin release from heart mast cells during ischemia/reperfusion: a novel cardioprotective role.
Key Words: ATP, renin-angiotensin system, arrhythmias, P2X7 purinergic receptors, toxic aldehydes
We demonstrated that cardiac mast cells (MC) are a critical source of renin (1). Released by ischemia/reperfusion (I/R), MC-derived renin initiates the activation of a local renin-angiotensin system (RAS). This promotes excessive norepinephrine (NE) release, thus causing severe arrhythmic dysfunction (2). Sympathetic/purinergic nerve terminals are juxtaposed to cardiac MC (1, 3, 4) and release neurotransmitter ATP, which elicits MC degranulation (5, 6). ATP, which is also produced in MC (4) and released in greater amounts during I/R (7), could positively feedback in an autocrine mode on the MC, initiating a vicious cycle that would amplify renin release, activate a local RAS, and elicit cardiac dysfunction.
Cell-surface ecto-nucleoside triphosphate diphosphohydrolases (E-NTPDases) are the primary enzymes that control availability of extracellular nucleotides to purinergic receptors (8, 9). E-NTPDase1/CD39 (CD39), expressed on vascular endothelial cells (10, 11), plays a critical role in maintaining blood fluidity by metabolizing ADP and ATP released from activated platelets (12, 13). We had reported that CD39 is also present in sympathetic/purinergic nerve terminals (14), where it exerts a cardioprotective effect in I/R (7) by attenuating the autocrine NE-releasing effects of neurogenic ATP (15).
As MC express CD39 (16), the purpose of our study was to investigate whether, by regulating ATP availability at the MC surface, CD39 might modulate local renin release, thus limiting local RAS activation and ultimately exerting a cardioprotective effect. We present data indicating that the CD39 expression level in MC indeed determines the extent of ATP-induced renin release and that CD39 inhibition in ex vivo murine hearts subjected to I/R enhances MC renin release and arrhythmic dysfunction, an action mediated by activation of the purinergic P2X7 receptor subtype (P2X7R) on MC.
MATERIALS AND METHODS
HMC-1 cell line
The human mastocytoma cell line (HMC-1) was kindly provided by Dr. J. H. Butterfield (Mayo Clinic, Rochester, MN, USA). Cells were maintained in suspension culture as described previously (17).
MC differentiation from murine bone marrow
Murine bone marrow-derived MC (BMMC) were obtained as described previously (18). In brief, C57BL/6 wild-type (WT) mice were anesthetized with CO2 vapor and euthanized by cervical dislocation, as approved by the Weill Cornell Medical College Institutional Animal Care and Use Committee. Bone marrow was flushed out from femora and tibiae. BMMC were cultured in RPMI-1640 medium (Invitrogen Life Technologies, Carlsbad, CA, USA), containing antibiotics (10 U/ml penicillin/streptomycin), 10% heat-inactivated fetal calf serum, 2-ME (55 μmol/L), recombinant murine IL-3, and stem cell factor (both 20 ng/ml; PeproTech, Rocky Hill, NJ, USA). BMMC were cultured at a density of 0.5 × 106 cells/ml. Cell medium was changed every 3-4 d, and nonadherent cells were transferred to a new culture flask. Mature BMMC were obtained after 4-5 wk when >90% of cells were double positive for c-kit/CD117 and high-affinity IgE receptor.
β-Hexosaminidase and renin assay
β-Hexosaminidase (β-HEX) and renin release were measured as described previously (19). In brief, HMC-1 and BMMC were washed and resuspended in Ringer buffer (140 mM NaCl, 5 mM KCl, 10 mM HEPES, 1 mM MgCl2, 2 mM glucose, and 2 mM CaCl2, pH 7.4). Identical volumes of cells were aliquoted in Eppendorf tubes (HMC-1) or in 96-well plates (BMMC) and incubated with gentle oscillation at 37°C with ATP (1 μM–1 mM) or acetaldehyde (100–500 μM) for 20 min (both from Sigma-Aldrich, St. Louis, MO, USA). When reported, cells were preincubated with pharmacological agents before ATP was added. ARL67156 and A740003 (both from R&D Systems, Minneapolis, MN, USA) were used at concentrations of 100 μM for 10 min and 3 μM for 15 min, respectively (half-maximal inhibitory concentration values: 0.3 mM and 40 nM for ARL67156 and A740003, respectively) (20, 21). At the end of the incubation, samples were placed in ice and centrifuged at 500 g for 5 min. Supernatants were collected and used to measure the β-HEX content and renin release. For the renin assay, human and porcine angiotensinogen was used for HMC-1 and BMMC samples, respectively. Renin activity (angiotensin I formed) was determined by GammaCoat Plasma Renin Activity [125I] Radioimmunoassay (DiaSorin, Stillwater, MN, USA). Cell pellets were lysed with cell lysis buffer 1× (Cell Signaling Technology, Danvers, MA, USA), and total lysates were used to determine total β-HEX content and total protein concentration by the DC Protein Assay kit (Bio-Rad Laboratories, Hercules, CA, USA). Total β-HEX content and total protein concentration were used to normalize β-HEX and renin release, respectively. Results were expressed as percent increase above basal level.
Measurement of ATP release
ATP levels were measured in HMC-1 supernatants, as described previously (22). In brief, HMC-1, processed as above, were incubated with acetaldehyde (100–500 μM) and gently oscillated at 37°C for 20 min. Supernatants were then collected by centrifugation at 500 g for 5 min, and ATP levels were assayed immediately by use of a firefly luciferin/luciferase-based commercial kit (ATP Bioluminescence Assay Kit HS II; Roche Diagnostics, Indianapolis, IN, USA). Luciferin/luciferase reagent (50 μl) was added to 50 μl of each diluted supernatant (1:100 in Ringer buffer), and the mixture was pipetted into appropriate test tubes and placed in a TD-20/20 luminometer (Turner Designs, Sunnyvale, CA, USA). ATP concentrations were calculated from a calibration curve constructed the same day by use of ATP standards included in the kit. The optimal detection range was between 10−10 and 10−16 mol of ATP. Cell lysates were assayed for protein content by the DC Protein Assay kit (Bio-Rad Laboratories) and used to normalize samples.
HMC-1 lentivirus transduction
Short hairpin (sh)RNA was delivered by a modified pCDH1 lentivirus expression vector to knock down the expression of endogenous CD39 and P2X7R in HMC-1 cells. Vector containing an shRNA sequence and lentiviral particles was prepared as described previously (22). We identified potential 19 bp small interfering (si)RNA target sequences within the human CD39 or P2X7R coding regions by use of the siRNA Target Finder application (available at http://www.genscript.com/siRNA_target_finder.html; GenScript, Piscataway, NJ, USA). The target sequence for human CD39 was AAGTACCTGAGTGAATACTGC, and the target sequence for human P2X7R was AAGGTGAAAGAGTGGATCGTG. HMC-1 cells were transduced at a multiplicity of infection (MOI) of 5. Seventy-two hours after transduction, cells were harvested, cell lysates prepared, and expression of human CD39 or P2X7R was assayed by Western blot. A lentiviral construct encoding a scrambled shRNA, which did not alter CD39 or P2X7R expression, served as the control virus. For overexpression of human CD39 in HMC-1, human CD39 was cloned from RNA, prepared from HUVECs, as reported previously (22). The primers used to generate the human CD39 open-reading frame from HUVEC-derived cDNA were 5′-ATGGAAGATACAAAGGAGTCTAACGTG and 3′-TACCATATCTTTCCAGAAATATGAAGGC. Lentiviral expression vector and particles were generated as described previously (22). HMC-1 were transduced at an MOI of 10. Seventy-two hours following transduction, total cell lysates and membrane proteins were prepared. Expression of CD39 protein was assessed by Western blot. Transduced cells were also sorted according to the intensity of green fluorescent protein by the FACSAriaII-SORP cell sorter (BD Biosciences, San Jose, CA, USA). As a control, HMC-1 were also transduced with the same expression vector lacking the human CD39 sequence and addressed as an empty vector.
Western blot
Total lysates from HMC-1 and BMMC were prepared with RIPA buffer (Sigma-Aldrich), and protein concentration was calculated by the DC Protein Assay kit (Bio-Rad Laboratories). Membrane proteins were isolated as described previously (22). Total lysates and membrane proteins were tested by electrophoresis and Western blot, as described previously (17). PVDF membranes were probed with anti-human CD39 (Ancell, Bayport, MN, USA) and anti-mouse CD39 (Epitomics, Burlingame, CA, USA), both at a dilution of 1:2000, and anti-P2X7R (Sigma-Aldrich), at a dilution of 1:1000. Anti-rabbit and anti-mouse IgG horseradish peroxidase (HRP)-linked secondary antibodies (Cell Signaling Technology) were used at a dilution of 1:5000. Proteins of interest were detected by Immobilon Western Chemiluminescent HRP Substrate (EMD Millipore, Billerica, MA, USA). β-Actin HRP-conjugated antibody (Alpha Diagnostic International, San Antonio, TX, USA) and Na+/K+-ATPase (Santa Cruz Biotechnology, Dallas, TX, USA) were used as controls (Supplemental Fig. S1).
Perfusion of mouse hearts ex vivo
WT C57BL/6J mice (male, 10–12 wk old; The Jackson Laboratory, Bar Harbor, ME, USA) were injected i.p. with heparin (100 IU), anesthetized with CO2, and euthanized by cervical dislocation, as approved by the Weill Cornell Medical College Institutional Animal Care and Use Committee. Hearts were isolated and transferred to a Langendorff apparatus (Radnoti, Monrovia, CA, USA). The aorta was cannulated with a flanged, 20-gauge stainless-steel needle. The heart was perfused through the aorta in a retrograde mode at a constant pressure of 100 cm H2O, with modified Krebs-Henseleit buffer containing (mM): NaCl, 120; KCl, 4.7; CaCl2, 2.5; MgSO4, 1.2; KH2PO4, 1.2; NaHCO3, 25; glucose, 11; pyruvic acid, 2; and EDTA, 0.5. The perfusion fluid was bubbled with 95% O2 and 5% CO2 at 37°C to give a pH of 7.4. After equilibration, all hearts were subjected to 40 min global ischemia, followed by 30 min reperfusion (I/R). To inhibit CD39, hearts were perfused with the CD39 inhibitor ARL67156 (30 µM) for 10 min before I/R. To block P2X7R, hearts were perfused with the selective P2X7R antagonist A740003 (3 µM) (21) for 5 min, followed by a combination with A740003 and ARL67156 for 10 min before ischemia and for the initial 15 min of reperfusion. Surface electrocardiogram was recorded and analyzed by use of PowerLab 8/SP (ADInstruments, Colorado Springs, CO, USA). Coronary flow was measured by timed collections of the effluent every 5 min; samples were assayed for renin and NE. Renin release was measured by incubating samples of coronary effluent for 1.5 h with porcine angiotensinogen. Renin activity (angiotensin I formed) was then determined by radioimmunoassay, as described above. NE overflow into the coronary effluent was measured by HPLC with electrochemical detection, as described previously (19).
Statistical analysis
Values are expressed as means ± sem. Unpaired t-test was used throughout the study. P < 0.05 was considered statistically significant.
RESULTS
ATP-induced degranulation and renin release in HMC-1 cells and BMMC
The administration of ATP to HMC-1 in culture elicited a concentration-dependent (1–100 µM) degranulation, as evidenced by the release of β-HEX, which was associated with renin release (Fig. 1A, C). In the presence of the selective P2X7R antagonist A740003 (3 µM) (23), the ATP-induced release of β-HEX and renin was markedly diminished (Fig. 1A, C). In contrast, in the presence of the CD39 inhibitor ARL67156 (100 µM) (15, 24), MC degranulation and renin release were both markedly potentiated (Fig. 1A, C). This suggested that P2X7R mediate ATP-induced MC degranulation and renin release and that CD39, by hydrolyzing ATP (25), negatively modulates ATP-induced MC degranulation and renin release.
Figure 1.

ATP elicits degranulation and renin release in HMC-1 and BMMC: potentiation by CD39 inhibition and antagonism by P2X7R blockade. Concentration-response curves for the degranulating (i.e., β-HEX release) and renin-releasing effects of ATP (20 min incubation at each concentration), alone or in the presence of the CD39 inhibitor ARL67156 (100 μM, 10 min preincubation) or of the selective P2X7R antagonist A740003 (3 μM, 15 min preincubation; P2X7R blocker). Points are means (±sem; n = 3–47). Basal β-HEX release was 3.63 ± 0.82 and 2.48 ± 0.15% in HMC-1 (n = 24) and BMMC (n = 34), respectively; basal renin release (angiotensin I formed) was 4.96 ± 1.09 (n = 16) and 4.42 ± 0.93 (n = 31) ng/h/mg protein in HMC-1 and BMMC, respectively. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. corresponding control (ATP alone) by unpaired t-test.
Similar to HMC-1, BMMC were also degranulated by ATP and released renin as a function of the ATP concentration to which they were exposed (10–1000 µM; Fig. 1B, D). Again, the degranulating and renin-releasing effect of ATP was blocked by A740003 (3 µM) and potentiated by ARL67156 (100 μM; Fig. 1B, D), indicating that CD39 negatively modulates the renin-releasing action of ATP also in native MC. As ATP is released from sympathetic nerve endings that lay juxtaposed to MC (1, 26), these findings suggest that MC CD39 is likely to modulate the renin-releasing effects of neurotransmitter ATP.
CD39 expression modulates ATP-induced degranulation and renin release in HMC-1 cells
As pharmacological inhibition of CD39 suggested that this ectonucleotidase modulates the renin-releasing action of ATP in HMC-1 and BMMC, we sought corroboration of this view by modifying the expression of CD39. For this, we overexpressed CD39 in HMC-1 by use of a lentivirus carrying a subcloned human CD39-coding sequence (CD39+) (22) (Fig. 2A). We found that an increase in membrane CD39 protein resulted in an ∼5- to 10-fold reduction in ATP-induced β-HEX and renin release (Fig. 2C). Conversely, we found that silencing of CD39 in HMC-1 cells by transducing them with lentivirus carrying shCD39 (22) (Fig.2B) caused a 2-fold increase in ATP-induced MC degranulation and renin release, which was prevented by pharmacological blockade of P2X7R with A740003 (3 µM) (Fig.2C). These findings highlighted the relevance of CD39 as a pivotal modulator of ATP-induced MC degranulation and renin release and its final mediation by P2X7R.
Figure 2.

CD39 expression modulates ATP-induced MC degranulation and renin release in HMC-1 cells. A) Representative Western blot of membrane fractions, prepared from WT and CD39-overexpressing (CD39+) HMC-1, probed with anti-CD39, anti-β-actin, and anti-Na+/K+ ATPase (membrane fraction marker, 40 and 5 μg/lane for WT and CD39+ cells, respectively). B) Western blot of HMC-1 total lysates transduced with lentivirus carrying sh scrambled (Scr) or shCD39 probed with anti-CD39 and anti-β-actin (50 μg/lane). C) ATP-induced degranulation and renin release are markedly reduced in CD39-overexpressing HMC-1. In contrast, the silencing of CD39 enhances degranulation and renin release. WT HMC-1 and HMC-1 infected with empty vector (Vector), CD39-overexpressing vector (CD39+), lentivirus carrying sh scrambled, and sh-silenced CD39 (shCD39) were incubated with ATP (100 μM) for 20 min in the absence or presence of A740003 (P2X7R blocker; 3 μM, 15 min preincubation); β-HEX and renin content was measured in the supernatants at the end of the incubation. Basal β-HEX release was 2.27 ± 0.30% (n = 22), and basal renin release (angiotensin I formed) was 3.20 ± 0.75 ng/h/mg protein (n = 17). Bars are means ± sem (n = 4–28). *P < 0.05, **P < 0.01 vs. WT cells by unpaired t-test.
Reactive aldehydes promote ATP release from HMC-1 cells, in turn, eliciting autocrine renin release, an action controlled by MC CD39
MC are themselves an important source of ATP, particularly in I/R, a condition in which toxic aldehydes [e.g., acetaldehyde, 4-hydroxynonenal (4-HNE)] and reactive oxygen species (ROS) are formed in large quantities (27–32). Hence, we next investigated whether acetaldehyde might be capable of releasing ATP from MC and that this ATP might then act in an autocrine mode to cause MC degranulation and renin release. We found that, as a function of its concentration (100–500 µM), acetaldehyde elicited an ∼25–100% increase in ATP release from HMC-1 cells (Fig. 3A). At a 300 µM concentration, at which acetaldehyde elicited an ∼80% increase in ATP release, β-HEX and renin release was also increased by ∼20% (Fig. 3B). Notably, acetaldehyde-induced release of β-HEX and renin was decreased by ∼80% in HMC-1 overexpressing CD39. In contrast, acetaldehyde-induced release of β-HEX and renin was increased by ∼22% and ∼80%, respectively, in cells in which CD39 had been silenced (Fig. 3B); this increase was prevented by blockade of P2X7R with A740003 (3 µM). These findings suggested that MC-derived ATP acted in an autocrine mode to release renin from the same cells, an action controlled by CD39 and mediated by P2X7R.
Figure 3.
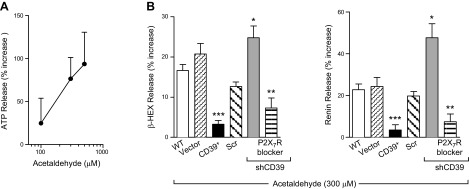
Acetaldehyde elicits release of ATP, β-HEX, and renin in HMC-1 cells. A) Incubation of HMC-1 with acetaldehyde for 20 min elicits a concentration-dependent release of endogenous ATP (n = 6–12). Basal ATP release was 374.3 ± 51.08 nmol/L (n = 11). B) Expression of CD39 in MC modulates acetaldehyde-induced release of β-HEX and renin. WT HMC-1 and HMC-1 infected with empty vector, CD39-overexpressing vector, lentivirus carrying sh scrambled, and sh-silenced CD39 sequence were incubated with acetaldehyde (300 μM) for 20 min in the absence or presence of A740003 (P2X7R blocker; 3 μM, 15 min preincubation); β-HEX and renin content was measured in the supernatants at the end of incubation. Basal β-HEX release was 2.28 ± 0.32% (n = 21), and basal renin release (angiotensin I formed) was 5.16 ± 1.07 ng/h/mg protein (n = 16). Bars are mean ± sem (n = 5–16). *P < 0.05, **P < 0.01, and ***P < 0.001 vs. WT cells, respectively, by unpaired t test.
P2X7R mediate paracrine and autocrine ATP-induced release of renin from HMC-1 cells
The finding that A740003 inhibited the ATP-induced MC renin release in HMC-1 and BMMC (see Fig. 1) revealed a likely involvement of P2X7R, a subtype that is thought to play a major role in HMC-1 purinergic activation (4). To strengthen these pharmacologic data, we silenced P2X7R in HMC-1 cells by transducing them with lentivirus carrying shRNA against P2X7R (22) (Fig. 4A). We found that incubation of P2X7R-silenced HMC-1 with ATP at a supramaximal concentration (100 µM) elicited a release of β-HEX and renin that was only 20% of that elicited in WT and scrambled control cells (Fig. 4B). Notably, P2X7R silencing also abolished the degranulating and renin-releasing effects of acetaldehyde (300 µM; Fig.4C). Collectively, these findings suggested that the MC renin-releasing effects of ATP, whether released from neighboring cells or from MC themselves, involve P2X7R activation.
Figure 4.

P2X7R silencing in HMC-1 cells abrogates ATP- and acetaldehyde-induced degranulation and renin release. A) Representative Western blot detecting P2X7R in total lysates of HMC-1, transduced with sh scrambled or sh P2X7R-silencing lentivirus (shP2X7R), probed with anti-P2X7R and anti-β-actin antibodies (40 μg/lane). B) ATP-induced MC degranulation and renin release are attenuated in P2X7R-silenced HMC-1. WT, scrambled, and shP2X7R HMC-1 were incubated with ATP (100 μM, for 20 min); β-HEX and renin content was measured in the supernatants at the end of incubation. Basal β-HEX release was 2.27 ± 0.30% (n = 22), and basal renin release (angiotensin I formed) was 3.20 ± 0.75 ng/h/mg protein (n = 17). Bars are means ± sem (n = 5–19). *P < 0.05, and **P < 0.01 vs. WT control by unpaired t-test. C) Acetaldehyde-induced MC degranulation and renin release are attenuated in P2X7R-silenced HMC-1. WT, scrambled, and shP2X7R HMC-1 were incubated with acetaldehyde (300 μM, for 20 min), and β-HEX and renin content was measured in the supernatants at the end of incubation. Basal β-HEX release was 2.28 ± 0.32% (n = 21), and basal renin (angiotensin I formed) was 5.16 ± 1.07 ng/h/mg protein (n = 16). Bars are mean ± sem (n = 6–15). **P < 0.01, and ***P < 0.001 vs. WT cells by unpaired t-test, respectively.
CD39 inhibition in ex vivo mouse hearts subjected to I/R potentiates renin release and RAS activation
As our data suggested that CD39 expression and activity modulate ATP-induced MC renin release and given that I/R enhances ATP and renin release in the heart (2, 7, 17), we next investigated whether CD39 inhibition might have an impact on the I/R-induced release of MC renin. We found that when Langendorff-perfused murine hearts were subjected to normothermic global ischemia for 40 min, followed by 30 min reperfusion, this resulted in a large increase in renin and NE overflow (i.e., 30- and 6-fold, respectively) during the first 5 min of reperfusion and the development of ventricular tachycardia/fibrillation (VT/VF; Fig. 5). We had determined previously that the release of MC renin contributes to the activation of a local RAS, which leads to NE release and severe arrhythmias (2). When hearts were perfused with the CD39 inhibitor ARL67156 (30 µM) and subjected to I/R, renin and NE overflow further increased 2-fold and ∼60%, respectively, and VT/VF lasted twice as long (Fig. 5). Notably, all of the effects caused by CD39 inhibition were abolished in hearts perfused with the selective P2X7R antagonist A740003 (Fig. 5). Therefore, during I/R, ATP derived from various cardiac cells is likely to promote MC renin release and activate a local RAS, culminating in arrhythmic dysfunction, a process mediated by P2X7R and likely controlled by CD39. Furthermore, P2X7R blockade reduced NE release to a level significantly lower than in I/R controls (Fig. 5), suggesting that P2X7R may also facilitate NE release directly at sympathetic nerve endings.
Figure 5.

I/R in Langendorff-perfused mouse hearts ex vivo: CD39 inhibition enhances the overflow of renin and NE and prolongs arrhythmia (VT/VF) duration; these effects are prevented by selective blockade of P2X7R. Coronary overflow of renin [angiotensin (ANG) I formed] and NE and duration of VT/VF in control ischemia (i.e., 40 min normothermic global ischemia), followed by 30 min reperfusion (n = 9). Other hearts were perfused with the CD39 inhibitor ARL67156 (30 µM; 10 min before ischemia and during the initial 15 min of reperfusion; n = 9), alone or together with the selective P2X7R antagonist A740003 (3 µM; 5 min before ARL67156 and then together with it during the initial 15 min of reperfusion; n = 9). Bars represent means ± sem of renin; NE overflows during the first 5 min of reperfusion and duration of VT/VF measured during the entire 30 min of reperfusion. *P < 0.05 vs. I/R; #P < 0.05, ##P < 0.01, and ###P < 0.001 vs. I/R + ARL67156; †P < 0.05 vs. I/R by unpaired t-test, respectively.
DISCUSSION
The purpose of our study was to investigate whether, by regulating ATP availability at the MC surface, CD39 would modulate ATP-induced MC degranulation and renin release, thus limiting local RAS activation, ultimately exerting a so-far unidentified cardioprotective effect. Toward this goal, we first ascertained that exogenously administered ATP elicited degranulation and renin release in HMC-1 cells and BMMC, demonstrating the feasibility of both cell models. Second, pharmacological inhibition of CD39 with ARL67156 potentiated ATP-induced degranulation and renin release in both of these cell models, whereas blockade of the purinergic receptor subtype, known to be expressed in MC (i.e., P2X7R) (33), prevented these effects. This suggested that MC CD39 functions to regulate ATP-induced degranulation and renin release and that P2X7R activation mediates both of these ATP actions. Indeed, overexpression of CD39 in HMC-1 cells prevented ATP-induced degranulation and renin release, whereas silencing of CD39 potentiated both effects. This silencing-induced potentiation was prevented by blockade of P2X7R. Moreover, the silencing of P2X7R in HMC-1 prevented ATP-induced degranulation and renin release. All of these findings corroborated the pivotal role of P2X7R and CD39 in the mediation and modulation, respectively, of ATP-induced MC degranulation and renin release.
We had shown previously that renin-containing MC lay in close proximity to ATP-releasing neurons (1, 26); our present data indicate that activation of sympathetic/purinergic nerves, which enhance the release of neurogenic ATP in I/R (7), very likely provoke MC degranulation and renin release, a paracrine process controlled by CD39 present at the MC membrane. Indeed, the results of our experiments in ex vivo murine hearts subjected to I/R demonstrate that pharmacological blockade of CD39 magnifies the consequences of I/R-induced RAS activation, as indicated by an enhanced release of renin and NE into the coronary effluent, which was associated with an increased duration of reperfusion arrhythmias, such as VT/VF. Notably, arrhythmias, resulting from RAS activation, are caused, in part, by NE (released via the angiotensin II subtype 1 receptor-mediated effect of angiotensin II) (34–37) and, in part, by the direct arrhythmogenic effects of angiotensin on cardiac cells (38–44). That an enhanced ATP availability at the MC surface was responsible for the increased renin release and its downstream consequences was verified by the finding that blockade of P2X7R prevented the effects of CD39 inhibition.
Aside from the paracrine effects of neurotransmitter ATP on juxtaposed MC, we considered that MC are themselves an important source of ATP, particularly in I/R, a condition in which toxic aldehydes (e.g., acetaldehyde and 4-HNE) and ROS are formed in large quantities (27–31). We postulated that toxic aldehydes formed in I/R would elicit ATP release from MC and that this ATP might then activate P2X7R on MC in an autocrine mode, causing degranulation and renin release. If this were the case, then MC CD39 should play a modulatory role in the release of MC renin elicited by MC-derived ATP. Indeed, the administration of acetaldehyde to HMC-1 cells caused a release of ATP, which increased progressively as a function of acetaldehyde’s concentration. Notably, acetaldehyde elicited MC degranulation and renin release, both of which were prevented by CD39 overexpression or potentiated by CD39 silencing. The silencing-induced potentiation was prevented by P2X7R blockade. This demonstrated that ATP, produced by MC under the influence of acetaldehyde, acted in an autocrine mode, degranulating the MC and releasing their renin content, and that this action of MC-produced ATP was under CD39 control and ultimately mediated by P2X7R. The renin-releasing effect of exogenously applied acetaldehyde exemplifies the effect of endogenously produced acetaldehyde in I/R (17, 19). Indeed, renin overflow from hearts subjected to I/R and consequent local RAS activation were both greatly enhanced when CD39 was pharmacologically inhibited.
ATP, whether released from sympathetic/purinergic neurons during I/R (7) or produced by MC under the influence of toxic aldehydes, initiated degranulation and renin release via P2X7R activation. Indeed, the silencing of P2X7R prevented the degranulating and renin-releasing effects of ATP and acetaldehyde. Furthermore, the pivotal P2X7R dependence of local RAS activation in the heart was demonstrated by the finding that pharmacological antagonism of P2X7R prevented the increase in renin and NE release and the increased severity of reperfusion arrhythmias, all caused by inhibition of CD39. Notably, in the ex vivo hearts, P2X7R blockade reduced NE release to a level significantly lower than in I/R controls, whereas renin release and VT/VF duration were not significantly different from I/R controls. This suggests that P2X7R, seemingly expressed on sympathetic nerve endings (45), may participate in the mediation of NE release during I/R, whereas renin release and consequent RAS activation are predominantly modulated at the MC level.
We had demonstrated previously that facilitation of ATP-induced NE release from cardiac sympathetic nerves is a composite of two autocrine components: positive (mediated by high levels of ATP activating P2XR) and negative (mediated by low levels of ATP activating P2YR) (15). As P2YR are also present in MC (4), it is conceivable that a similar mechanism may operate in physiologic conditions. It is unlikely, however, that this may occur in a pathophysiological setting, such as I/R, in which purinergic signaling is prevalently ascribable to P2XR, given the greater ATP availability.
In summary (Fig. 6), sympathetic/purinergic nerve terminals are juxtaposed to cardiac MC and release ATP (5), which activates P2X7R on MC, eliciting degranulation and renin release (26). ATP, which is also produced in MC and released in greater amounts in I/R (4), is likely to feed back positively in an autocrine mode on the MC, initiating a vicious cycle that amplifies renin release, activates a local RAS, and elicits cardiac dysfunction. We postulate that, by regulating ATP availability at the MC surface, CD39 modulates local renin release, ultimately exerting a cardioprotective effect. Until now, CD39 had been regarded as playing roles at the platelet/endothelial interface and sympathetic nerve terminals (10–15), where it exerts a cardioprotective effect in I/R (7) by attenuating the autocrine NE-releasing effects of neurogenic ATP. We have now discovered that, by limiting MC renin release, CD39 plays an additional cardioprotective role in I/R, ultimately preventing the dysfunctional consequences of RAS activation, including severe arrhythmias, such as VT/VF. Moreover, the CD39-induced modulation of MC renin release is likely to impact other organs (e.g., brain, kidney, and liver) that have renin-containing MC (46–49) and can suffer ischemic episodes (50–54). Thus, our findings may contribute to the identification of new therapeutic targets, not only in myocardial ischemia but also in ischemic syndromes of other organs.
Figure 6.

Proposed role of CD39 in ATP-induced MC degranulation and renin release.
Supplementary Material
Acknowledgments
Support for this work was provided by U.S. National Institutes of Health Grants HL034215, HL047073, HL047073-23S1, and HL089521 and by a Grant-in-Aid from the U.S. American Heart Association. This work was also supported, in part, by a MERIT Review grant from the U.S. Department of Veterans Affairs (to A.J.M.). The authors declare no conflicts of interest.
Glossary
Abbreviations:
- 4-HNE
4-hydroxynonenal
- β-HEX
β-hexosaminidase
- BMMC
murine bone marrow-derived mast cell(s)
- CD39
ecto-nucleoside triphosphate diphosphohydrolase 1/CD39
- E-NTPDase
ecto-nucleoside triphosphate wdiphosphohydrolase
- HMC-1
human mastocytoma cell line
- HRP
horseradish peroxidase
- I/R
ischemia/reperfusion
- MC
mast cell(s)
- MOI
multiplicity of infection
- NE
norepinephrine
- P2X7R
purinergic P2X7 receptors
- RAS
renin-angiotensin system
- ROS
reactive oxygen species
- sh
short hairpin
- si
small interfering
- VT/VF
ventricular tachycardia/fibrillation
- WT
wild-type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Silver R. B., Reid A. C., Mackins C. J., Askwith T., Schaefer U., Herzlinger D., and Levi R. (2004) Mast cells: a unique source of renin. Proc. Natl. Acad. Sci. USA 101, 13607–13612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mackins C. J., Kano S., Seyedi N., Schäfer U., Reid A. C., Machida T., Silver R. B., and Levi R. (2006) Cardiac mast cell-derived renin promotes local angiotensin formation, norepinephrine release, and arrhythmias in ischemia/reperfusion. J. Clin. Invest. 116, 1063–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bauer O., and Razin E. (2000) Mast cell-nerve interactions. News Physiol. Sci. 15, 213–218 [DOI] [PubMed] [Google Scholar]
- 4.Bulanova E., and Bulfone-Paus S. (2010) P2 receptor-mediated signaling in mast cell biology. Purinergic Signal. 6, 3–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiernan J. A. (1972) The involvement of mast cells in vasodilatation due to axon reflexes in injured skin. Q. J. Exp. Physiol. Cogn. Med. Sci. 57, 311–317 [DOI] [PubMed] [Google Scholar]
- 6.Kiernan J. A. (1974) Action of adenosine triphosphate on mast cells in normal and denervated skin. Arch. Dermatol. Forsch. 251, 83–86 [DOI] [PubMed] [Google Scholar]
- 7.Sesti C., Koyama M., Broekman M. J., Marcus A. J., and Levi R. (2003) Ectonucleotidase in sympathetic nerve endings modulates ATP and norepinephrine exocytosis in myocardial ischemia. J. Pharmacol. Exp. Ther. 306, 238–244 [DOI] [PubMed] [Google Scholar]
- 8.Knowles A. F. (2011) The GDA1_CD39 superfamily: NTPDases with diverse functions. Purinergic Signal. 7, 21–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deaglio S., and Robson S. C. (2011) Ectonucleotidases as regulators of purinergic signaling in thrombosis, inflammation, and immunity. Adv. Pharmacol. 61, 301–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaczmarek E., Koziak K., Sévigny J., Siegel J. B., Anrather J., Beaudoin A. R., Bach F. H., and Robson S. C. (1996) Identification and characterization of CD39/vascular ATP diphosphohydrolase. J. Biol. Chem. 271, 33116–33122 [DOI] [PubMed] [Google Scholar]
- 11.Marcus A. J., Broekman M. J., Drosopoulos J. H. F., Islam N., Alyonycheva T. N., Safier L. B., Hajjar K. A., Posnett D. N., Schoenborn M. A., Schooley K. A., Gayle R. B., and Maliszewski C. R. (1997) The endothelial cell ecto-ADPase responsible for inhibition of platelet function is CD39. J. Clin. Invest. 99, 1351–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marcus A. J., Broekman M. J., Drosopoulos J. H., Islam N., Pinsky D. J., Sesti C., and Levi R. (2003) Heterologous cell-cell interactions: thromboregulation, cerebroprotection and cardioprotection by CD39 (NTPDase-1). J. Thromb. Haemost. 1, 2497–2509 [DOI] [PubMed] [Google Scholar]
- 13.Jennings L. K. (2009) Mechanisms of platelet activation: need for new strategies to protect against platelet-mediated atherothrombosis. Thromb. Haemost. 102, 248–257 [DOI] [PubMed] [Google Scholar]
- 14.Machida T., Heerdt P. M., Reid A. C., Schaefer U., Silver R. B., Broekman M. J., Marcus A. J., and Levi R. (2005) Ectonucleoside triphosphate diphosphohydrolase 1/CD39, localized in neurons of human and porcine heart, modulates ATP-Induced norepinephrine exocytosis. J. Pharmacol. Exp. Ther. 313, 570–577 [DOI] [PubMed] [Google Scholar]
- 15.Sesti C., Broekman M. J., Drosopoulos J. H. F., Islam N., Marcus A. J., and Levi R. (2002) EctoNucleotidase in cardiac sympathetic nerve endings modulates ATP-mediated feedback of norepinephrine release. J. Pharmacol. Exp. Ther. 300, 605–611 [DOI] [PubMed] [Google Scholar]
- 16.Kurashima Y., Amiya T., Nochi T., Fujisawa K., Haraguchi T., Iba H., Tsutsui H., Sato S., Nakajima S., Iijima H., Kubo M., Kunisawa J., and Kiyono H. (2012) Extracellular ATP mediates mast cell-dependent intestinal inflammation through P2X7 purinoceptors. Nat. Commun. 3, 1034–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koda K., Salazar-Rodriguez M., Corti F., Chan N. Y.-K., Estephan R., Silver R. B., Mochly-Rosen D., and Levi R. (2010) Aldehyde dehydrogenase activation prevents reperfusion arrhythmias by inhibiting local renin release from cardiac mast cells. Circulation 122, 771–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aldi S., Robador P. A., Tomita K., Di Lorenzo A., and Levi R. (2014) IgE receptor-mediated mast-cell renin release. Am. J. Pathol. 184, 376–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aldi S., Takano K., Tomita K., Koda K., Chan N. Y., Marino A., Salazar-Rodriguez M., Thurmond R. L., and Levi R. (2014) Histamine H4-receptors inhibit mast cell renin release in ischemia/reperfusion via protein kinase C ε-dependent aldehyde dehydrogenase type-2 activation. J. Pharmacol. Exp. Ther. 349, 508–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crack B. E., Pollard C. E., Beukers M. W., Roberts S. M., Hunt S. F., Ingall A. H., McKechnie K. C., IJzerman A. P., and Leff P. (1995) Pharmacological and biochemical analysis of FPL 67156, a novel, selective inhibitor of ecto-ATPase. Br. J. Pharmacol. 114, 475–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Honore P., Donnelly-Roberts D., Namovic M. T., Hsieh G., Zhu C. Z., Mikusa J. P., Hernandez G., Zhong C., Gauvin D. M., Chandran P., Harris R., Medrano A. P., Carroll W., Marsh K., Sullivan J. P., Faltynek C. R., and Jarvis M. F. (2006) A-740003 [N-(1-[(cyanoimino)(5-quinolinylamino) methyl]amino-2,2-dimethylpropyl)-2-(3,4-dimethoxyphenyl)acetamide], a novel and selective P2X7 receptor antagonist, dose-dependently reduces neuropathic pain in the rat. J. Pharmacol. Exp. Ther. 319, 1376–1385 [DOI] [PubMed] [Google Scholar]
- 22.Corti F., Olson K. E., Marcus A. J., and Levi R. (2011) The expression level of ecto-NTP diphosphohydrolase1/CD39 modulates exocytotic and ischemic release of neurotransmitters in a cellular model of sympathetic neurons. J. Pharmacol. Exp. Ther. 337, 524–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Donnelly-Roberts D. L., and Jarvis M. F. (2007) Discovery of P2X7 receptor-selective antagonists offers new insights into P2X7 receptor function and indicates a role in chronic pain states. Br. J. Pharmacol. 151, 571–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schaefer U., Machida T., Broekman M. J., Marcus A. J., and Levi R. (2007) Targeted deletion of ectonucleoside triphosphate diphosphohydrolase 1/CD39 leads to desensitization of pre- and post-synaptic purinergic P2-receptors. J. Pharmacol. Exp. Ther. 322, 1269–1277 [DOI] [PubMed] [Google Scholar]
- 25.Maliszewski C. R., Delespesse G. J., Schoenborn M. A., Armitage R. J., Fanslow W. C., Nakajima T., Baker E., Sutherland G. R., Poindexter K., Birks C., Alpert A., Friend D., Gimpel S. D., and Gayle R. B., III. (1994) The CD39 lymphoid cell activation antigen. Molecular cloning and structural characterization. J. Immunol. 153, 3574–3583 [PubMed] [Google Scholar]
- 26.Morrey C., Brazin J., Seyedi N., Corti F., Silver R. B., and Levi R. (2010) Interaction between sensory C-fibers and cardiac mast cells in ischemia/reperfusion: activation of a local renin-angiotensin system culminating in severe arrhythmic dysfunction. J. Pharmacol. Exp. Ther. 335, 76–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Esterbauer H., Schaur R. J., and Zollner H. (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 11, 81–128 [DOI] [PubMed] [Google Scholar]
- 28.Cordis G. A., Maulik N., Bagchi D., Engelman R. M., and Das D. K. (1993) Estimation of the extent of lipid peroxidation in the ischemic and reperfused heart by monitoring lipid metabolic products with the aid of high-performance liquid chromatography. J. Chromatogr. A 632, 97–103 [DOI] [PubMed] [Google Scholar]
- 29.Eaton P., Li J. M., Hearse D. J., and Shattock M. J. (1999) Formation of 4-hydroxy-2-nonenal-modified proteins in ischemic rat heart. Am. J. Physiol. 276, H935–H943 [DOI] [PubMed] [Google Scholar]
- 30.Steiner D. R. S., Gonzalez N. C., and Wood J. G. (2003) Mast cells mediate the microvascular inflammatory response to systemic hypoxia. J. Appl. Physiol. 94, 325–334 [DOI] [PubMed] [Google Scholar]
- 31.Swindle E. J., and Metcalfe D. D. (2007) The role of reactive oxygen species and nitric oxide in mast cell-dependent inflammatory processes. Immunol. Rev. 217, 186–205 [DOI] [PubMed] [Google Scholar]
- 32.Chen C.-H., Ferreira J. C., Gross E. R., and Mochly-Rosen D. (2014) Targeting aldehyde dehydrogenase 2: new therapeutic opportunities. Physiol. Rev. 94, 1–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuhny M., Hochdorfer T., Ayata C., Idzko M., and Huber M. (2014) CD39 is a negative regulator of P2X7-mediated inflammatory cell death in mast cells. Cell Commun. Signal. 12, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schömig A. (1990) Catecholamines in myocardial ischemia. Systemic and cardiac release. Circulation 82 (3Suppl) II13–II22 [PubMed] [Google Scholar]
- 35.Meredith I. T., Broughton A., Jennings G. L., and Esler M. D. (1991) Evidence of a selective increase in cardiac sympathetic activity in patients with sustained ventricular arrhythmias. N. Engl. J. Med. 325, 618–624 [DOI] [PubMed] [Google Scholar]
- 36.Grassi G., Arenare F., Pieruzzi F., Brambilla G., and Mancia G. (2009) Sympathetic activation in cardiovascular and renal disease. J. Nephrol. 22, 190–195 [PubMed] [Google Scholar]
- 37.Tan A. Y., and Verrier R. L. (2013) The role of the autonomic nervous system in cardiac arrhythmias. Handb. Clin. Neurol. 117, 135–145 [DOI] [PubMed] [Google Scholar]
- 38.Fleetwood G., Boutinet S., Meier M., and Wood J. M. (1991) Involvement of the renin-angiotensin system in ischemic damage and reperfusion arrhythmias in the isolated perfused rat heart. J. Cardiovasc. Pharmacol. 17, 351–356 [DOI] [PubMed] [Google Scholar]
- 39.Harada K., Komuro I., Hayashi D., Sugaya T., Murakami K., and Yazaki Y. (1998) Angiotensin II type 1a receptor is involved in the occurrence of reperfusion arrhythmias. Circulation 97, 315–317 [DOI] [PubMed] [Google Scholar]
- 40.Jalowy A., Schulz R., and Heusch G. (1999) AT1 receptor blockade in experimental myocardial ischemia/reperfusion. J. Am. Soc. Nephrol. 11 (10 Suppl), S129–S136 [PubMed] [Google Scholar]
- 41.Yahiro E., Ideishi M., Wang L. X., Urata H., Kumagai K., Arakawa K., and Saku K. (2003) Reperfusion-induced arrhythmias are suppressed by inhibition of the angiotensin II type 1 receptor. Cardiology 99, 61–67 [DOI] [PubMed] [Google Scholar]
- 42.Garg S., Narula J., Marelli C., and Cesario D. (2006) Role of angiotensin receptor blockers in the prevention and treatment of arrhythmias. Am. J. Cardiol. 97, 921–925 [DOI] [PubMed] [Google Scholar]
- 43.Iravanian S., and Dudley S. C. Jr (2008) The renin-angiotensin-aldosterone system (RAAS) and cardiac arrhythmias. Heart Rhythm 5 (6Suppl) S12–S17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.von Lewinski D., Kockskämper J., Rübertus S. U., Zhu D., Schmitto J. D., Schöndube F. A., Hasenfuss G., and Pieske B. (2008) Direct pro-arrhythmogenic effects of angiotensin II can be suppressed by AT1 receptor blockade in human atrial myocardium. Eur. J. Heart Fail. 10, 1172–1176 [DOI] [PubMed] [Google Scholar]
- 45.Calvert J. A., and Evans R. J. (2004) Heterogeneity of P2X receptors in sympathetic neurons: contribution of neuronal P2X1 receptors revealed using knockout mice. Mol. Pharmacol. 65, 139–148 [DOI] [PubMed] [Google Scholar]
- 46.Reid A. C., Silver R. B., and Levi R. (2007) Renin: at the heart of the mast cell. Immunol. Rev. 217, 123–140 [DOI] [PubMed] [Google Scholar]
- 47.Biran V., Cochois V., Karroubi A., Arrang J. M., Charriaut-Marlangue C., and Héron A. (2008) Stroke induces histamine accumulation and mast cell degranulation in the neonatal rat brain. Brain Pathol. 18, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Veerappan A., Reid A. C., Estephan R., O’Connor N., Thadani-Mulero M., Salazar-Rodriguez M., Levi R., and Silver R. B. (2008) Mast cell renin and a local renin-angiotensin system in the airway: role in bronchoconstriction. Proc. Natl. Acad. Sci. USA 105, 1315–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Veerappan A., Reid A. C., O’Connor N., Mora R., Brazin J. A., Estephan R., Kameue T., Chen J., Felsen D., Seshan S. V., Poppas D. P., Maack T., and Silver R. B. (2012) Mast cells are required for the development of renal fibrosis in the rodent unilateral ureteral obstruction model. Am. J. Physiol. Renal Physiol. 302, F192–F204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaneko H., Koshi S., Hiraoka T., Miyauchi Y., Kitamura N., and Inoue M. (1998) Inhibition of post-ischemic reperfusion injury of the kidney by diamine oxidase. Biochim. Biophys. Acta 1407, 193–199 [DOI] [PubMed] [Google Scholar]
- 51.Vural K. M., Liao H., Oz M. C., and Pinsky D. J. (2000) Effects of mast cell membrane stabilizing agents in a rat lung ischemia-reperfusion model. Ann. Thorac. Surg. 69, 228–232 [DOI] [PubMed] [Google Scholar]
- 52.Walther T., Olah L., Harms C., Maul B., Bader M., Hörtnagl H., Schultheiss H. P., and Mies G. (2002) Ischemic injury in experimental stroke depends on angiotensin II. FASEB J. 16, 169–176 [DOI] [PubMed] [Google Scholar]
- 53.Guo L., Richardson K. S., Tucker L. M., Doll M. A., Hein D. W., and Arteel G. E. (2004) Role of the renin-angiotensin system in hepatic ischemia reperfusion injury in rats. Hepatology 40, 583–589 [DOI] [PubMed] [Google Scholar]
- 54.Chen S., Li G., Zhang W., Wang J., Sigmund C. D., Olson J. E., and Chen Y. (2009) Ischemia-induced brain damage is enhanced in human renin and angiotensinogen double-transgenic mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R1526–R1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.