

Abstract
Insulin resistance may be linked to incomplete fatty acid β-oxidation and the subsequent increase in acylcarnitine species in different tissues including skeletal muscle. It is not known if acylcarnitines participate in muscle insulin resistance or simply reflect dysregulated metabolism. The aims of this study were to determine whether acylcarnitines can elicit muscle insulin resistance and to better understand the link between incomplete muscle fatty acid β-oxidation, oxidative stress, inflammation, and insulin-resistance development. Differentiated C2C12, primary mouse, and human myotubes were treated with acylcarnitines (C4:0, C14:0, C16:0) or with palmitate with or without carnitine acyltransferase inhibition by mildronate. Treatment with C4:0, C14:0, and C16:0 acylcarnitines resulted in 20–30% decrease in insulin response at the level of Akt phosphorylation and/or glucose uptake. Mildronate reversed palmitate-induced insulin resistance concomitant with an ∼25% decrease in short-chain acylcarnitine and acetylcarnitine secretion. Although proinflammatory cytokines were not affected under these conditions, oxidative stress was increased by 2–3 times by short- or long-chain acylcarnitines. Acylcarnitine-induced oxidative stress and insulin resistance were reversed by treatment with antioxidants. Results are consistent with the conclusion that incomplete muscle fatty acid β-oxidation causes acylcarnitine accumulation and associated oxidative stress, raising the possibility that these metabolites play a role in muscle insulin resistance.—Aguer, C., McCoin, C. S., Knotts, T. A., Thrush, A. B., Ono-Moore, K., McPherson, R., Dent, R., Hwang, D. H., Adams, S. H., Harper, M.-E. Acylcarnitines: potential implications for skeletal muscle insulin resistance.
Keywords: fatty acid β-oxidation, mitochondria, inflammation, oxidative stress, myotubes
Individuals with insulin resistance and type 2 diabetes mellitus often have impaired skeletal muscle oxidative capacity and inefficient or incomplete fatty acid β-oxidation (defined herein as a condition in which lipid fuel delivery to mitochondria is not well matched to oxidative capacity) (1–5). Typically, there is an inverse relationship between efficient muscle fatty acid β-oxidation and reactive oxygen species (ROS) production (6), and in type 2 diabetes mellitus, muscle mitochondrial dysfunction may be the consequence of oxidative stress that develops during fuel overload (7). The mechanisms responsible for the fatty acid β-oxidation-ROS relationship are unclear. However, it is likely that a greater proportion of reducing equivalents produced from fatty acid β-oxidation enter the electron transport chain at the electron transfer flavoprotein, thus bypassing complex I, the main ROS production site (8, 9).
It is also well recognized that chronic low-grade inflammation is a contributor to insulin resistance (10). In obesity-associated diseases, there is an infiltration of proinflammatory macrophages within white adipose tissue. Therein, macrophages secrete different cytokines, including TNF-α, which promotes whole-body insulin resistance (11) and inhibits insulin signaling in insulin target tissues, including skeletal muscle (12).
In metabolic situations of high fatty acid availability, the citric acid cycle is sometimes not able to increase its activity in response to increased fatty acid β-oxidation rates, resulting in cellular acyl-CoA accumulation (13). Acyl groups are exported from the mitochondria as acylcarnitines produced via carnitine palmitoyl transferase (CPT)1, CPT2, or the matrix enzyme carnitine acetyltransferase (CrAT). Interestingly, muscle insulin resistance is associated with increased acylcarnitine accumulation in the muscle or circulation (13–15). It has thus been hypothesized that acylcarnitines could interfere with insulin signaling by activating proinflammatory mechanisms (15), an idea recently reiterated (16). However, to our knowledge there is no direct evidence that acylcarnitines are responsible for insulin-resistance development, and the mechanisms linking acylcarnitine accumulation to insulin resistance have not yet been studied. The aims of the present study were to determine the role of acylcarnitines in insulin-resistance development and to better understand the link between inefficient muscle fatty acid β-oxidation, muscle oxidative stress, inflammation, and insulin resistance.
MATERIALS AND METHODS
Isolation of mouse primary muscle cells
Primary muscle cells were isolated from skeletal muscle of C57BL/6J mice as described elsewhere (17). Desmin immunofluorescence staining was used to verify purity of the cells (18), which were >90% desmin-positive (Supplemental Fig. 1A). Cells were grown and differentiated as described elsewhere (19). Myoblast capacity to differentiate was verified by troponin T Western blots and troponin T immunofluorescence staining (marker of differentiation; data not shown).
Human primary myotubes
Five lean and 5 obese nondiabetic women were recruited. The 5 lean participants were matched for sex, age, and physical activity (assessed by the International Physical Activity Questionnaire [IPAQ: 7-d]). All participants gave informed consent, and the experimental protocol was approved by the Human Research Ethics Committees of the Ottawa Hospital and the University of Ottawa Heart Institute. The characteristics of the participants are described in Table 1. The participants were not diabetic, so they were not taking any diabetic medications. One lean and 1 obese subject were taking an antidepressant that can cause weight gain. No other medications that could affect metabolism or weight gain were taken by the other participants. Body mass and body composition were assessed using bioelectrical impedance analysis (Tanita TBF-410GS Body Composition Analyzer, Arlington Heights, IL, USA). Fasting blood glucose, hemoglobin A1c, and fasting plasma insulin were measured on the Beckman Coulter LX 20 automated chemistry analyzer or the Beckman Coulter Dxi immunoassay analyzer using manufacturer’s reagents (Beckman Coulter, Brea, CA, USA). Resting metabolic rate was determined for 30 min by indirect calorimetry (MOXUS Modular VO2 System, AEI Technologies, Naperville, IL, USA) (20). For isolation of human primary muscle cells, biopsies of vastus lateralis were obtained in the fasted state using a 5 mm Bergstrom needle (Opitek, Glostrup, Denmark) (21). Muscle satellite cells were isolated, grown, and differentiated for 7 d as described elsewhere (21, 22). Cells used were >90% desmin-positive (Supplemental Fig. 1B).
TABLE 1.
Characteristics of human muscle biopsy donors
| Control (n = 5) | Obese, insulin-resistant (n = 5) | |
|---|---|---|
| Sex | Female | Female |
| Age (y) | 44.4 ± 7.2 | 45.0 ± 9.1 |
| BMI (kg/m2) | 19.9 ± 1.4 | 35.9 ± 2.3* |
| % fat | 23.1 ± 7.5 | 47.1 ± 1.8* |
| Resting metabolic rate (kJ) | 5255 ± 286 | 7207 ± 385* |
| Fasting blood glucose (mmol/L) | 4.8 ± 0.5 | 5.1 ± 0.3 |
| Fasting plasma insulin (pmol/L) | 19.0 ± 7.8 | 62.4 ± 13.4* |
| HOMA-IR | 0.6 ± 0.02 | 2.1 ± 0.52* |
| HbA1C (%) | 5.2 ± 0.4 | 5.3 ± 0.2 |
BMI, body mass index; HOMA-IR, homeostatic model assessment–insulin resistance; HbA1C, hemoglobin A1C.
P < 0.001, obese, insulin resistant vs. control.
C2C12 cell culture
C2C12 murine myoblasts (CRL-1772, ATCC, Manassas, VA, USA) were grown in DMEM supplemented with 10% fetal bovine serum (FBS; Lot#K11050 Atlanta Biologicals, Lawrenceville, GA, USA), 1× penicillin-streptomycin, 1× Glutamax (both from Life Technologies, Grand Island, NY, USA) and 100 μmol/L l-carnitine (Advent Bio, Downer’s Grove, IL, USA). Cells were cultured until ∼90% confluence, then induced to differentiate into myotubes for 5 d by substituting 10% horse serum (HyClone, Logan, UT, USA) for the FBS.
Myotube treatment
For determinations of ROS, glucose uptake, oxidative stress, fatty acid β-oxidation, and acylcarnitine measurements, myotubes were treated for the last 24 h of differentiation with 1 mmol/L mildronate and/or 600 µmol/L palmitate bound to 2% bovine serum albumin (BSA) in the presence of 1 mmol/L carnitine. Human primary myotubes were also treated for the last 24 h of differentiation with 10 μmol/L C4:0, C14:0, C16:0 acylcarnitines or a mix of acylcarnitines (10 μmol/L each) ± 50 µmol/L ascorbate or 5 mmol/L N-acetyl cysteine (NAC) in serum-free DMEM. For glucose uptake experiments, myotubes were treated for 10 min with 100 nmol/L insulin. C2C12 myotubes were treated for the last 18 h of differentiation with DMEM containing 1% FBS, 2% BSA, different concentrations of C4:0, C14:0, or C16:0 acylcarnitine (5, 10, and 25 μmol/L), or palmitate (750 μmol/L). The purity of the acylcarnitines and the concentrations used in the acylcarnitine stock solutions and acylcarnitine-containing media were routinely checked through mass spectrometry (data not shown). Studies examining C2C12 myotube cytokine release employed 4 h 0.25% FBS serum-starvation followed by 18 h treatment with 0.25% FBS plus these acylcarnitines or LPS (100 ng/ml) used as a positive control. For Western blots, C2C12 myotubes were then treated for 2 h with DMEM containing 2% BSA followed by 100 nmol/L insulin stimulation for 10 min (23).
Myotube fatty acid β-oxidation
Myoblasts were grown to ∼90% confluency in a 12-well plate, and differentiated to myotubes for 7 d. Complete fatty acid β-oxidation and incomplete fatty acid β-oxidation [acid soluble products (ASPs) measured in cell lysate and in the medium] were measured as described elsewhere (19).
Western blots
Cell lysates were quantified for protein content and 20–30 µg of protein was separated by 10 or 12% SDS-PAGE and transferred to a nitrocellulose membrane. The following primary antibodies were used: polyclonal anti-p-Akt (Ser473), anti-p-Akt (Thr308), and Akt1/2 (Cell Signaling, Danvers, MA, USA) diluted 1:1000. The secondary antibody was anti-rabbit antibody coupled to horseradish peroxidase. Proteins were visualized using an enhanced luminescent reagent and exposed to autoradiograph film. Expression of proteins was quantified by density analysis using ImageJ Launcher (U.S. National Institutes of Health, Bethesda, MD, USA) and BioRad Image Lab 4.1 Software (Bio-Rad, Hercules, CA, USA).
Acylcarnitine measurement
Primary human muscle cells were grown and differentiated for 7 d in 25 cm2 flasks. Aliquots of cell culture media and cells were frozen on dry ice prior to delivery to Case Western Reserve University for acylcarnitine analyses. Cell culture media (10 µl) for acylcarnitine analysis and 10 µl of a 1:10 dilution of the media (equivalent of 1 µl) for total carnitine, free carnitine, and butyrobetaine were used. Acylcarnitines were also measured in the whole cell lysate. Acylcarnitines and carnitine were measured using HPLC-mass spectrometry methods described in detail elsewhere (15, 24).
Myotube glucose uptake
Cells (100,000 cells per well) were plated in 24-well plates. Myotubes were incubated in glucose-free DMEM ± 100 nM insulin or ± 5 µmol/L cytochalasin B (to determine nonspecific glucose uptake) for 10 min and then incubated in 0.25 μCi/well [3H]2-deoxyglucose (2-DG) and 100 μmol/L cold 2-DG (17).
IL-6 concentration measurements
Media levels of IL-6 were determined using the Mouse IL-6 Quantikine ELISA Kit (R&D Systems, Minneapolis, MN, USA). Conditioned media (50 μl) taken at the 18 h time point was assessed following the manufacturer’s protocol using a Biotek Synergy 2 Plate Reader (Winooski, VT, USA).
Gene transcript measurements
Total RNA was extracted using the RiboPure Kit (Life Technologies) according to the manufacturer’s instructions. Briefly, RNA was quantitated using the NanoDrop ND-1000 spectrophotometer and 1 μg/sample of RNA was utilized to synthesize cDNA using the SuperscriptIII First-Strand Synthesis RT-PCR System (Life Technologies). Gene expression, as measured by quantitative real-time PCR, was assessed using FAM-MGB-labeled probes on 20 ng of cDNA, in triplicate. cDNA was dried in each well prior to adding quantitative PCR reagents to facilitate an 8 μl/well assay, and expression values were assessed relative to control transcript (18s) levels using the ∆∆Ct method described previously (25). Primers/probe ABI identifiers for C2C12 gene expression experiments were IL-6 (Mm99999064_m1), MCP-1/CCL2 (monocyte chemoattractant protein-1) (Mm00441242_m1), PTGS-2/COX-2 (prostaglandin-endoperoxide synthase 2) (Mm01307329_m1), and TNF-α (Mm99999068_m1).
ROS content
Cells (30,000 per well) were plated in 96-well plates. Myotubes were incubated for 30 min with serum-free differentiation medium containing 20 μmol/L DCFH-DA (2′,7′-dichlorfluorescein-diacetate) at 37°C. Myotubes were then washed twice and 100 μl of PBS was added per well. The plate was read at 488/525 nm (excitation/emission). Myotubes were solubilized in 50 µl 0.05 M NaOH and quantified for protein.
Total protein carbonyls
Protein carbonyls in human primary myotubes were measured using the OxyBlot Protein Oxidation Detection Kit (Millipore, Billerica, MA, USA), following the manufacturer’s instructions.
CrAT activity
CrAT activity was measured in duplicate in cell lysates with a method adapted from Aguer et al. (19).
Statistics
Results are presented as mean ± sem. Statistical analyses were performed using Statview 5.0 (SAS Institute, Cary, NC, USA) or Prism 5.0 (Graphpad, San Diego, CA, USA). Unpaired 2-tailed t tests, paired t tests, 1-way and 2-way ANOVAs with post hoc tests were used to assess statistical differences. P < 0.05 was considered significant.
RESULTS
Decreased complete fatty acid β-oxidation and increased ASP release from myotubes of obese subjects
Muscle insulin resistance is known to be associated with incomplete fatty acid β-oxidation (13) and metabolic inflexibility (i.e., the capacity to switch from fatty acid β-oxidation to glucose oxidation) (26). We measured fatty acid β-oxidation in human primary myotubes derived from lean or obese insulin-resistant subjects, in the basal state or in the presence of glucose/pyruvate to test metabolic flexibility, as well as with or without 24 h palmitate pretreatment to induce insulin resistance (23, 27) to phenotype the human primary myotubes used in this study. Complete fatty acid β-oxidation (carboxyl-carbon radiolabel in evolved CO2) was significantly lower in myotubes from obese insulin-resistant subjects in the basal state (Fig. 1A). Palmitate pretreatment decreased complete fatty acid β-oxidation only in myotubes from lean subjects. The addition of glucose/pyruvate decreased complete fatty acid β-oxidation in both groups of myotubes. Interestingly, ASP exported to the medium were significantly higher in every condition with myotubes from obese patients (Fig. 1B), whereas ASP measured in cell lysates were not different between groups (Fig. 1C). Palmitate pretreatment decreased medium-radiolabeled ASP, and palmitate and/or glucose treatments increased radiolabeled ASP measured in cell lysates in both groups (Fig. 1B, C). Total fatty acid β-oxidation was not significantly different between groups and as expected was decreased in the presence of glucose/pyruvate as well as with palmitate pretreatment (Fig. 1D). Metabolic flexibility (percent inhibition of basal fatty acid β-oxidation in the presence of glucose/pyruvate) was significantly higher in myotubes derived from lean compared with obese insulin-resistant subjects (Fig. 1E). Interestingly, after palmitate pretreatment, metabolic flexibility was unchanged in myotubes from obese patients but was decreased in myotubes from lean subjects to the level observed in myotubes from obese individuals, consistent with the possibility that palmitate pretreatment was inducing metabolic inflexibility in myotubes from lean subjects (Fig. 1E). ASP would primarily represent radiolabeled acetylcarnitine or acetate in myotubes treated with 1-14C-palmitate, and 14CO2 marks combustion of fatty acid-derived acetyl-CoA in the citric acid cycle. Thus, the findings may be indicative of increased flux of fatty acid-derived acetyl-CoA toward nonoxidative fates (i.e., acetylcarnitine or acetate production) relative to complete oxidation in myotubes derived from obese insulin-resistant subjects, concurrent with a mismatch between fatty acid β-oxidation and citric acid cycle activity. Such a mismatch promotes accumulation of medium- to long-chain acylcarnitines, which under our experimental paradigm could not be detected because these metabolites would not have been radiolabeled.
Figure 1.

Myotubes derived from obese insulin-resistant subjects release more metabolites associated with incomplete fatty acid β-oxidation and are metabolically inflexible. A) Complete fatty acid β-oxidation (14CO2 production from radiolabeled 1-14C-palmitate) measured in human primary myotubes derived from lean (white bars) and obese (black bars) subjects. §P < 0.05: lean vs. obese. ***P < 0.001: basal vs. glucose. ††P < 0.01: basal vs. palmitate. Group × condition effect: P < 0.05. B) Incomplete fatty acid β-oxidation (14C ASP released in the medium from radiolabeled palmitate) measured in human primary myotubes derived from lean (white bars) and obese (black bars) subjects. §P < 0.05: lean vs. obese. ††P < 0.01: basal vs. palmitate. C) Incomplete fatty acid β-oxidation (14C ASP measured in cell lysate) measured in human primary myotubes derived from lean (white bars) and obese (black bars) subjects. *P < 0.05: treatment effect. D) Total fatty acid β-oxidation (14CO2 + 14C-ASP) measured in human primary myotubes derived from lean (white bars) and obese (black bars) subjects. *P < 0.05: basal vs. glucose. ††P < 0.01, basal vs. palmitate. E) Metabolic flexibility measured as the percentage of fatty acid β-oxidation inhibition in presence of glucose/pyruvate. §P < 0.05: lean vs. obese. A–E) See Materials and Methods for details concerning myotube treatments. n = 5, each measurement done in triplicate. Data are shown as mean ± sem.
Acylcarnitine treatments induce insulin resistance in C2C12 myotubes
To test the hypothesis that increased acylcarnitines could interfere with insulin sensitivity, we exposed myotubes to acylcarnitines and measured different markers of insulin sensitivity. Murine C2C12 myotubes were treated for 18 h with C4:0, C14:0, or C16:0 acylcarnitine at 3 different concentrations (5, 10, and 25 μmol/L). Treatment with C4:0 acylcarnitine at all concentrations resulted in a significant decreased Akt (Ser473) phosphorylation in response to insulin (Fig. 2A). Treatment with 5–10 μmol/L C14:0 or C16:0 acylcarnitines significantly decreased Akt (Ser473) phosphorylation in response to insulin stimulation (Fig. 2B, C). As expected, Akt (Thr308) phosphorylation was also decreased by acylcarnitine treatments (data not shown). These effects were equivalent to that induced by palmitate treatment (positive control; Fig. 2C). The 5–10 μmol/L acylcarnitine effective range corresponds to levels of muscle acylcarnitines estimated from tissue concentrations for insulin-resistant rodents (13, 14).
Figure 2.
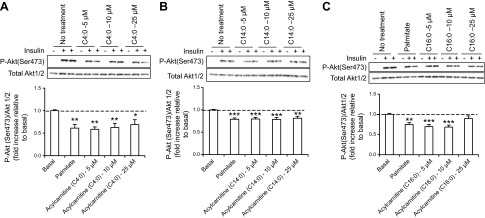
Acylcarnitine treatments result in insulin resistance at the level of Akt phosphorylation in C2C12 myotubes. A–C) Top panel: representative Western blot of p-Akt (Ser473) expression in C2C12 myotubes. Bottom panel: quantification by density analysis of fold increase in p-Akt (Ser473) relative to basal. C2C12 myotubes were pretreated for 18 h with 750 μmol/L palmitate or 5, 10, or 25 μmol/L (A) C4:0, (B) C14:0, or (C) C16:0 acylcarnitine. p-Akt was calculated relative to total Akt 1/2, and results are expressed as fold of insulin-stimulation of p-Akt in vehicle controls. Akt 1/2 is shown as a loading control. *P < 0.05, **P < 0.01, ***P < 0.001, treated cells vs. basal. Data are shown as mean ± sem.
Reduction of short-chain acylcarnitine and acetylcarnitine formation protects against palmitate-induced insulin resistance
We then tested whether inhibition of acylcarnitine formation in vitro was able to reverse palmitate-induced insulin resistance. Primary myotubes (mouse and human) were treated with 1 mmol/L mildronate (an inhibitor of acylcarnitine transferases) (28, 29), which decreased CrAT activity by 30% (Supplemental Fig. 2). Mildronate treatment tended to decrease palmitate-induced acetylcarnitine secreted by human primary myotubes (Fig. 3A, P = 0.07), whereas it significantly decreased palmitate-induced short-chain acylcarnitine secretion (Fig. 3B). Palmitate treatment increased levels of medium/long-chain acylcarnitines 10- to 12-fold (C8:0–C14:0 acylcarnitines, Fig. 3C), and mildronate slightly attenuated this effect but it did not reach significance (see individual C8:0–C14:0 acylcarnitine values in Supplemental Tables 1 and 2). Palmitate treatment clearly increased palmitoyl-carnitine secretion (by ∼80 times), but mildronate treatment had no effect on C16:0 acylcarnitine (Fig. 3D), showing that mildronate, under our experimental conditions, acted as an inhibitor of CrAT more than an inhibitor of CPT1. The majority of acylcarnitines synthetized in response to palmitate treatment was exported from the cells, as assessed by higher levels of acylcarnitines measured in the medium compared with those in the cell lysate (Supplemental Tables 1 and 2). Interestingly, palmitate-induced insulin resistance was restored by mildronate treatment in myotubes from lean subjects (Fig. 3E, top panel). The outcome showing little to no impact of mildronate on medium- and long-chain acylcarnitines does not support the idea that palmitate-associated insulin resistance occurs via accumulation of medium- or long-chain fatty acylcarnitines: mildronate rescued palmitate-associated insulin resistance without altering longer-chain acylcarnitines. In this model, therefore, it is possible that mildronate rescues palmitate-induced insulin resistance in part via a moderate decrease in short-chain acylcarnitines. However, because reductions in the latter were modest, the results do not rule out other mildronate-associated mechanisms.
Figure 3.

Moderate carnitine acyltransferase inhibition by mildronate restores palmitate-induced insulin resistance in human primary myotubes. A–D) Acylcarnitine measured in the medium of human primary myotubes pretreated with 600 μmol/L palmitate ± 1 mmol/L mildronate for 24 h. n = 3, each individual culture done in duplicate. A) Acetylcarnitine. P = 0.07 palmitate vs. palmitate + mildronate. B) Sum of short-chain acylcarnitines (C4:0 and C6:0 acylcarnitines). **P < 0.01: palmitate vs. palmitate + mildronate. C) Sum of C8:0 to C14:0 acylcarnitines. D) Palmitoyl-carnitine (C16:0). E) Glucose uptake measured in human primary myotubes derived from lean (top panel, white bars) and obese subjects (bottom panel, black bars) pretreated with 600 μmol/L palmitate ± 1 mmol/L mildronate for 24 h. Glucose uptake is presented as the fold increase in response to insulin. *P < 0.05: basal vs. palmitate, #P < 0.05: palmitate vs. palmitate + mildronate. n = 5, each measurement done in triplicate. A–E) See Materials and Methods for details concerning myotube treatments. Data are shown as mean ± sem.
Myotubes from obese patients were insulin resistant as attested by the lower response of insulin-stimulated glucose uptake (Fig. 3E, bottom panel). Perhaps because myotubes from obese subjects were already insulin resistant in the basal state, mildronate and/or palmitate treatment did not affect insulin-stimulated glucose uptake in these myotubes (Fig. 3E, bottom panel).
Acylcarnitine-induced murine myotube insulin resistance is not correlated with cytokine release or gene markers of inflammation
To test the hypothesis that acylcarnitine treatment induces insulin resistance through inflammatory processes, we measured different markers of inflammation in C2C12 myotubes treated with C14:0 and C16:0 acylcarnitines (5, 10, and 25 μmol/L). LPS was used as a positive control. Secretion of IL-6 in the medium by C2C12 myotubes was increased 20-fold in response to LPS treatment (Fig. 4A). C14:0 acylcarnitine treatment, at these concentrations, did not significantly affect IL-6 secretion (Fig. 4A). Lower levels of C16:0 acylcarnitine treatment (5–10 μmol/L) did not significantly affect IL-6 secretion, whereas 25 μmol/L C16:0 acylcarnitine treatment significantly increased IL-6 secretion (Fig. 4A). A similar pattern was observed for IL-6 gene expression (Fig. 4B). IL-6 secretion was also assessed in human primary myotubes treated with C4:0, C14:0, or C16:0 acylcarnitines (10 μmol/L each). These acylcarnitine treatments did not affect IL-6 secretion (n = 2, data not shown). MCP-1, PTGS-2, and TNF-α mRNA abundances were measured in C2C12 cells in response to acylcarnitine treatment (Fig. 4C–E). None of these proinflammatory markers were significantly affected by the acylcarnitine treatments. Taken together, results indicate that acylcarnitine-associated insulin resistance in cultured myotubes can be dissociated from inflammatory cytokine release.
Figure 4.

Acylcarnitine treatment does not affect C2C12 myotube cytokine secretion at concentrations that elicit insulin resistance. A) IL-6 secretion of C2C12 myotubes. B) IL-6, (C) iMCP1, (D) PTGS-2, (E) TNF-α gene expression in C2C12 myotubes. A–E) See Materials and Methods for details concerning myotube treatments. ***P < 0.001, treated cells vs. basal. Data are shown as mean ± sem.
Antioxidant treatments partially ameliorate insulin resistance and oxidative stress induced by acylcarnitine treatments in human myotubes
To evaluate if ROS production is associated with acylcarnitine-associated responses, ROS levels were measured in mouse primary myotubes in the presence of mildronate to inhibit carnitine acyltransferases. We also treated myotubes with a high level of palmitate (600 µmol/L) as a positive control to increase ROS production (30). When carnitine acyltransferases were moderately inhibited, palmitate-induced ROS was inhibited (Fig. 5A). We checked whether acylcarnitine treatment increased oxidative stress in human primary myotubes (Fig. 5B), finding that 10 μmol/L C4:0 or C16:0 acylcarnitines significantly increased protein carbonyls; this was prevented when myotubes were treated with antioxidants NAC or ascorbate. C14:0 acylcarnitine treatment also increased protein carbonyls but it was more variable and thus did not achieve statistical significance. Finally, we measured glucose uptake in human primary myotubes treated with C4:0 or C16:0 acylcarnitines. Acylcarnitine treatments resulted in decreased insulin sensitivity as shown by the diminished insulin-stimulated glucose uptake (Fig. 5C). Interestingly, insulin sensitivity was partially restored, albeit modestly, in C16:0 acylcarnitine-treated myotubes in the presence of the antioxidant ascorbate (Fig. 5C).
Figure 5.
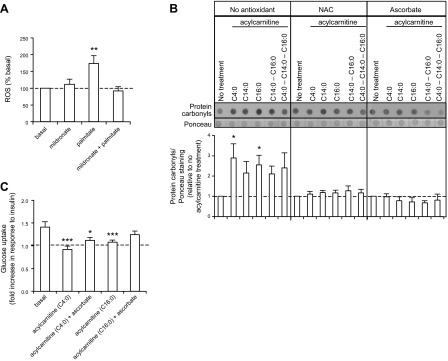
Acylcarnitine treatment of primary myotubes results in increased oxidative stress and insulin resistance, which is partially restored by antioxidant treatment. A) ROS emission measured in mouse primary myotubes with DCFH-DA. **P < 0.01, treated cells vs. basal. n = 4, each measurement done in 5 replicates. B) Top panel: representative dot blot of total protein carbonyl expression in primary myotubes derived from lean subjects. Ponceau staining was used as a loading control. Bottom panel: quantification of protein carbonyls by density analysis. *P < 0.05, treated cells vs. basal. n = 5. C) Glucose uptake measured in primary myotubes derived from lean subjects. Glucose uptake is presented as the fold increase in response to insulin. Basal absolute values (mean; range): 319; 210–447 pmol/min/mg. n = 5, each measurement done in triplicate. A–C) See Materials and Methods for details concerning myotube treatments. Data are shown as mean ± sem.
DISCUSSION
Acylcarnitines are increased in the circulation and in insulin-resistant skeletal muscle (13–15), but the role of acylcarnitines in the etiology of skeletal muscle insulin resistance has not been investigated. Based on proof-of-principle studies suggesting a proinflammatory activity of mixed stereoisomer dl-medium-chain acylcarnitines (activation of an NF-κB promoter construct), it was hypothesized that these metabolites contribute to insulin resistance (15). In the present study, we extended these observations in cultured murine and human myotubes, showing that treatment with physiologic l-acylcarnitine isomers induces insulin resistance and oxidative stress. Insulin-resistant primary myotubes from obese individuals had lower complete fatty acid β-oxidation and a greater release of ASP into the medium compared with myotubes from lean healthy subjects, suggesting higher incomplete fatty acid β-oxidation and acylcarnitine secretion. Inhibition of the acylcarnitine transferases with reduction in shorter-chain acylcarnitine formation in mouse or human primary myotubes was protective against palmitate-induced insulin resistance and ROS production. Moreover, acylcarnitine treatment of human primary myotubes resulted in increased oxidative stress, which was prevented by antioxidant treatment. Taken together, our results support the concept that inefficient muscle fatty acid β-oxidation leads to acylcarnitine accumulation, which in turn could contribute to muscle oxidative stress and insulin resistance.
Myotubes from insulin-resistant obese subjects showed increased media-radiolabeled ASP as well as decreased complete fatty acid β-oxidation compared with myotubes from lean healthy subjects, a scenario typically associated with increased acylcarnitine production and export (acetylcarnitine or acetate production in myotubes treated with 1-14C-palmitate) (31). A recent study showed that human myotubes secrete acylcarnitines during fatty acid β-oxidation (32). The results of the present study and others (26, 32) suggest that an increased incomplete fatty acid β-oxidation is linked to increased secretion of acylcarnitines, as well as decreased insulin sensitivity, and these defects are maintained in vitro in a cell culture model. We acknowledge that the current experiments involved primary myotubes from a relatively small number of biopsy donors; however, all data from each group responded consistently. Nonetheless, some of our results could be considered as hypothesis-generating, and future studies examining myotube phenotypes in a larger number of subjects would strengthen interpretations.
Greater long-chain acylcarnitine accumulation in tissues and the circulation is associated with insulin-resistant states (5, 13–15, 33). In the present study, we have directly demonstrated that treatment of myotubes with C16:0, C14:0, and/or C4:0 acylcarnitines elicits insulin resistance in C2C12 and in human primary myotubes. We recognize, however, that the effect of acylcarnitine treatment on p-Akt is not necessarily indicative of defective glucose uptake. Indeed, it has been previously demonstrated that impaired insulin-stimulated glucose uptake can be disconnected from the insulin response at the insulin signaling pathway level (34). However, it is notable that acylcarnitine treatments did result in decreased insulin-stimulated glucose uptake in human myotubes.
Previous research has shown that acylcarnitines can induce a proinflammatory response in a murine monocyte/macrophage cell line, leading to the hypothesis that these metabolites affect inflammatory systems that impair insulin signaling (15). Increased long-chain acylcarnitine accumulation in the serum, muscle, and adipose tissue have been reported during the development of insulin resistance in rats fed a “cafeteria diet,” and a proinflammatory response was seen in macrophages treated with C12:0 acylcarnitines (33). Very recently, proinflammatory effects of acylcarnitines in immune cells were further elaborated (35). In the present research, we did not detect increased cytokine gene expression or release in myotubes treated with acylcarnitines at concentrations that elicited insulin-resistance phenotypes. This suggests that acylcarnitine-associated mechanisms that impact insulin signaling are independent of muscle cytokine release. It remains to be established if other inflammation- or cell stress-associated pathways are engaged by acylcarnitines in muscle cells. Inefficient fatty acid β-oxidation is also linked to increased muscle accumulation of bioactive lipids such as ceramides, diacylglycerol, and acyl-CoA (36). These metabolites activate conventional protein kinase C (PKC), which in turn activates proinflammatory molecules including NF-κB, TNF-α, and IL-6 and interfere with the insulin signaling pathway by serine-phosphorylating insulin receptor substrate 1, thereby inhibiting insulin-stimulated Akt phosphorylation (37, 38). However, it has recently been demonstrated that oral high-fat loading or intravenous lipid infusion increased diacylglycerol and PKCθ levels in skeletal muscle in relation with decreased whole-body insulin sensitivity, without affecting inflammation (39). Taken together with the results of the present study, this suggests that inflammation is not always necessary to induce insulin resistance and that multiple lipid-derived metabolites might participate in this process.
The synthesis of acylcarnitines is catalyzed within mitochondria by different enzymes. CPT1 converts long-chain acyl-CoAs into long-chain acylcarnitines. CPT2 also has a strong capacity to synthesize medium- to long-chain acylcarnitines (40). If provision of acyl-CoA exceeds the capacity for fatty acid β-oxidation, CrAT, a mitochondrial matrix enzyme, can convert shorter-chain acyl-CoA moieties to acylcarnitines, thus regenerating CoA and allowing acylcarnitines to be effluxed from mitochondria. To test the hypothesis that decreased fatty acid β-oxidation or increased acylcarnitine synthesis is responsible for restoration of insulin sensitivity in palmitate-treated muscle, we partially inhibited carnitine acyltransferase activities in primary mouse and human myotubes using the carnitine mimetic mildronate. This had a protective effect on palmitate-induced insulin resistance at the level of glucose uptake. Our results are consistent with previous reports showing that acute blockade of CPT1 by etomoxir reversed fatty acid-induced insulin resistance in myotubes (13) and that CPT1 inhibition by oxfenicine improved whole-body glucose tolerance and insulin sensitivity in mice subjected to a high-fat diet (41). Taken with our finding that C16:0 acylcarnitine elicits insulin-resistance phenotypes, one conclusion is that mildronate blunts palmitate-induced insulin resistance via reduction in C16:0 acylcarnitine accumulation. However, we found that in primary human myotubes treated with palmitate, mildronate led to reduced shorter-chain acylcarnitine secretion only, suggestive of inhibition of CrAT and not CPT1 in this model. Along with the observation that C4:0 acylcarnitine inhibited insulin-stimulated glucose uptake, p-Akt, and increased oxidative stress, the results in aggregate support the idea that both longer- and shorter-chain acylcarnitines can impact insulin action. This concept requires further evaluation, because a recent study demonstrated that muscle-specific CrAT deletion in mice resulted in increased insulin-resistance development but also in higher accumulation of long-chain acylcarnitines in the muscle (5). In enzyme assays, CrAT activity was decreased by only 30% with mildronate treatment. It is thus possible that a moderate decrease in CrAT activity protects against insulin-resistance development, whereas a complete CrAT deletion is deleterious. The specific contributions of various chain lengths of acylcarnitines to their bioactivities warrant further research.
Studies have concluded that increased oxidative stress causes muscle insulin resistance (6, 42) and that oxidative stress develops in response to lipid overload (7). A recent study by our team (35) demonstrated that acylcarnitine increases ROS production in RAW 264.7 cells (mouse monocyte macrophages). Similarly, in the present study, acylcarnitine treatment was linked to increased oxidative stress in myotubes, and interestingly, antioxidant treatment partially restored insulin resistance induced by C16:0 acylcarnitine treatment. Furthermore, moderate CrAT inhibition restored acylcarnitine-induced ROS production and insulin resistance in myotubes. Taken together, these results suggest that insulin resistance in response to acylcarnitines is, at least in part, due to increased oxidative stress. How ROS induce insulin resistance in skeletal muscle is still unclear, but it seems that excess ROS can induce serine phosphorylation of IRS-1 directly or indirectly through the activation of JNK or inhibitor of NF-κB kinase, which leads to an inhibition of the insulin signaling pathway (43).
The exact mechanism(s) linking high acylcarnitine to insulin-resistance development, and the (patho)physiologic relevance of these pathways have yet to be determined. In subjects with a long-chain fatty acid oxidation defect (long-chain acyl-CoA dehydrogenase), for instance, glucose tolerance was found to be normal despite elevated plasma long-chain acylcarnitine concentrations (44). However, interpretations in persons with fatty acid oxidation disorders are confounded by an innate increased reliance on glucose as fuel. Under conditions of more typical insulin-resistance paradigms, we speculate that reducing chronic acylcarnitine accumulation in skeletal muscle and improving efficiency of fatty acid β-oxidation might protect against free fatty acid-induced insulin resistance, with the caveat that severe inhibition of carnitine acyltransferases is detrimental (45). Future in vivo studies of the effects of acylcarnitines are needed to directly prove that our model is valid at the whole organism level.
Supplementary Material
Acknowledgments
The authors thank Mahmoud Salkhordeh, Department of Biochemistry, Microbiology and Immunology, University of Ottawa (ON, Canada) for human primary cell isolation and technical assistance, and Dr. Charles Hoppel’s laboratory, Department of Pharmacology, Case Western Reserve University (Cleveland, OH, USA) for acylcarnitine measurements. This work was funded by Canadian Institutes of Health Research (Institute of Nutrition, Metabolism and Diabetes; MOP57810; M.E.H.), U.S. Department of Agriculture-Agriculture Research Service (USDA-ARS) Intramural Projects 5306-51530-016-00D and 5306-51530-019-00 (S.H.A.), U.S. National Institutes of Health-NIDDK Grants R01DK078328-01 (S.H.A., M.E.H.), and R01DK078328-02S1 (S.H.A., D.H.H.), ILSI Future Leader Award (S.H.A.), American Diabetes Association Grant 1-12-BS-02 (S.H.A., D.H.H.), and a Heart and Stroke Foundation of Canada Postdoctoral Fellowship (C.A.). The USDA is an equal opportunity provider and employer. Contributions to conception and design, acquisition of data, and analysis and interpretation of data: C.A., C.S.M., T.A.K., R.D., K.O.-M., R.M., D.H.H., S.H.A., M.E.H.; drafting the article or revising it critically for important intellectual content: C.A., C.S.M., T.A.K., R.D., R.M., D.H.H., S.H.A., M.E.H.; final approval of the version to be published: C.A., C.S.M., T.A.K., R.D., R.M., D.H.H., S.H.A., M.E.H. The authors declare no conflicts of interest.
Glossary
- 2-DG
2-deoxyglucose
- ASP
acid soluble products
- CrAT
carnitine acetyltransferase
- CPT
carnitine palmitoyl transferase
- DCFH-DA
2′,7′-dichlorfluorescein-diacetate)
- DTNB
5,5′-dithiobis-(2-nitrobenzoic acid)
- FAM-MGB
5′6-carboxyfluorescein and 3′-minor groove binder
- MCP-1
monocyte chemoattractant protein-1
- NAC
N-acetyl cysteine
- PKC
protein kinase C
- ROS
reactive oxygen species
- PTGS-2
prostaglandin endoperoxide synthase 2
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Befroy D. E., Petersen K. F., Dufour S., Mason G. F., de Graaf R. A., Rothman D. L., and Shulman G. I. (2007) Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes 56, 1376–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kelley D. E., He J., Menshikova E. V., and Ritov V. B. (2002) Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 51, 2944–2950 [DOI] [PubMed] [Google Scholar]
- 3.Mogensen M., Sahlin K., Fernström M., Glintborg D., Vind B. F., Beck-Nielsen H., and Højlund K. (2007) Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 56, 1592–1599 [DOI] [PubMed] [Google Scholar]
- 4.Petersen K. F., Dufour S., Befroy D., Garcia R., and Shulman G. I. (2004) Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 350, 664–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muoio D. M., Noland R. C., Kovalik J. P., Seiler S. E., Davies M. N., DeBalsi K. L., Ilkayeva O. R., Stevens R. D., Kheterpal I., Zhang J., Covington, J. D., Bajpeyi, S., Ravussin, E., Kraus, W., Koves, T. R., and Mynatt, R. L. (2012) Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab. 15, 764–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson E. J., Lustig M. E., Boyle K. E., Woodlief T. L., Kane D. A., Lin C. T., Price J. W. III, Kang L., Rabinovitch P. S., Szeto H. H., Houmard, J. A., Cortright, R. N., Wasserman, D. H., and Neufer, P. D. (2009) Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Invest. 119, 573–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonnard C., Durand A., Peyrol S., Chanseaume E., Chauvin M. A., Morio B., Vidal H., and Rieusset J. (2008) Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J. Clin. Invest. 118, 789–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fisher-Wellman K. H., and Neufer P. D. (2012) Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol. Metab. 23, 142–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brand M. D. (2010) The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 45, 466–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glass C. K., and Olefsky J. M. (2012) Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 15, 635–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krogh-Madsen R., Plomgaard P., Møller K., Mittendorfer B., and Pedersen B. K. (2006) Influence of TNF-alpha and IL-6 infusions on insulin sensitivity and expression of IL-18 in humans. Am. J. Physiol. Endocrinol. Metab. 291, E108–E114 [DOI] [PubMed] [Google Scholar]
- 12.Plomgaard P., Bouzakri K., Krogh-Madsen R., Mittendorfer B., Zierath J. R., and Pedersen B. K. (2005) Tumor necrosis factor-alpha induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes 54, 2939–2945 [DOI] [PubMed] [Google Scholar]
- 13.Koves T. R., Ussher J. R., Noland R. C., Slentz D., Mosedale M., Ilkayeva O., Bain J., Stevens R., Dyck J. R., Newgard C. B., Lopaschuk, G. D., and Muoio, D. M. (2008) Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 7, 45–56 [DOI] [PubMed] [Google Scholar]
- 14.Noland R. C., Koves T. R., Seiler S. E., Lum H., Lust R. M., Ilkayeva O., Stevens R. D., Hegardt F. G., and Muoio D. M. (2009) Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control. J. Biol. Chem. 284, 22840–22852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adams S. H., Hoppel C. L., Lok K. H., Zhao L., Wong S. W., Minkler P. E., Hwang D. H., Newman J. W., and Garvey W. T. (2009) Plasma acylcarnitine profiles suggest incomplete long-chain fatty acid beta-oxidation and altered tricarboxylic acid cycle activity in type 2 diabetic African-American women. J. Nutr. 139, 1073–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schooneman M. G., Vaz F. M., Houten S. M., and Soeters M. R. (2013) Acylcarnitines: reflecting or inflicting insulin resistance? Diabetes 62, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mailloux R. J., Dumouchel T., Aguer C., deKemp R., Beanlands R., and Harper M. E. (2011) Hexokinase II acts through UCP3 to suppress mitochondrial reactive oxygen species production and maintain aerobic respiration. Biochem. J. 437, 301–311 [DOI] [PubMed] [Google Scholar]
- 18.Aguer C., Mercier J., Man C. Y., Metz L., Bordenave S., Lambert K., Jean E., Lantier L., Bounoua L., Brun J. F., Raynaud de Mauverger, E., Andreelli, F., Foretz, M., and Kitzmann, M. (2010) Intramyocellular lipid accumulation is associated with permanent relocation ex vivo and in vitro of fatty acid translocase (FAT)/CD36 in obese patients. Diabetologia 53, 1151–1163 [DOI] [PubMed] [Google Scholar]
- 19.Aguer C., Fiehn O., Seifert E. L., Bézaire V., Meissen J. K., Daniels A., Scott K., Renaud J. M., Padilla M., Bickel D. R., Dysart, M., Adams, S. H., and Harper, M. E. (2013) Muscle uncoupling protein 3 overexpression mimics endurance training and reduces circulating biomarkers of incomplete β-oxidation. FASEB J. 27, 4213–4225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aguer C., Pasqua M., Thrush A. B., Moffat C., McBurney M., Jardine K., Zhang R., Beauchamp B., Dent R., McPherson R., and Harper M. E. (2013) Increased proton leak and SOD2 expression in myotubes from obese non-diabetic subjects with a family history of type 2 diabetes. Biochim. Biophys. Acta 1832, 1624–1633 [DOI] [PubMed] [Google Scholar]
- 21.Costford S. R., Crawford S. A., Dent R., McPherson R., and Harper M. E. (2009) Increased susceptibility to oxidative damage in post-diabetic human myotubes. Diabetologia 52, 2405–2415 [DOI] [PubMed] [Google Scholar]
- 22.Aguer C., Gambarotta D., Mailloux R. J., Moffat C., Dent R., McPherson R., and Harper M. E. (2011) Galactose enhances oxidative metabolism and reveals mitochondrial dysfunction in human primary muscle cells. PLoS ONE 6, e28536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chavez J. A., Knotts T. A., Wang L. P., Li G., Dobrowsky R. T., Florant G. L., and Summers S. A. (2003) A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J. Biol. Chem. 278, 10297–10303 [DOI] [PubMed] [Google Scholar]
- 24.Minkler P. E. S. M., Ingalls S. T, Hoppel C. L. (2012) Selective, accurate, and precise quantification of acylcarnitines by UHPLC-MS/MS for detailed analysis in metabolic disease research. In 60th ASMS Conference on Mass Spectrometry and Allied Topics, Vancouver, BC, Canada [Google Scholar]
- 25.Knotts T. A., Lee H. W., Kim J. B., Oort P. J., McPherson R., Dent R., Tachibana K., Doi T., Yu S., Reddy J. K., Uno, K., Katagiri, H., Pasarica, M., Smith, S. R., Sears, D. D., Grino, M., and Adams, S. H. (2009) Molecular Characterization of the Tumor Suppressor Candidate 5 Gene: Regulation by PPARgamma and Identification of TUSC5 Coding Variants in Lean and Obese Humans. PPAR Res. 2009, Article ID 867678, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ukropcova B., McNeil M., Sereda O., de Jonge L., Xie H., Bray G. A., and Smith S. R. (2005) Dynamic changes in fat oxidation in human primary myocytes mirror metabolic characteristics of the donor. J. Clin. Invest. 115, 1934–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmitz-Peiffer C., Craig D. L., and Biden T. J. (1999) Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J. Biol. Chem. 274, 24202–24210 [DOI] [PubMed] [Google Scholar]
- 28.Jaudzems K., Kuka J., Gutsaits A., Zinovjevs K., Kalvinsh I., Liepinsh E., Liepinsh E., and Dambrova M. (2009) Inhibition of carnitine acetyltransferase by mildronate, a regulator of energy metabolism. J. Enzyme Inhib. Med. Chem. 24, 1269–1275 [DOI] [PubMed] [Google Scholar]
- 29.Georges B., Le Borgne F., Galland S., Isoir M., Ecosse D., Grand-Jean F., and Demarquoy J. (2000) Carnitine transport into muscular cells. Inhibition of transport and cell growth by mildronate. Biochem. Pharmacol. 59, 1357–1363 [DOI] [PubMed] [Google Scholar]
- 30.Lambertucci R. H., Hirabara S. M., Silveira Ldos. R., Levada-Pires A. C., Curi R., and Pithon-Curi T. C. (2008) Palmitate increases superoxide production through mitochondrial electron transport chain and NADPH oxidase activity in skeletal muscle cells. J. Cell. Physiol. 216, 796–804 [DOI] [PubMed] [Google Scholar]
- 31.Pourfarzam M., Schaefer J., Turnbull D. M., and Bartlett K. (1994) Analysis of fatty acid oxidation intermediates in cultured fibroblasts to detect mitochondrial oxidation disorders. Clin. Chem. 40, 2267–2275 [PubMed] [Google Scholar]
- 32.Wolf M., Chen S., Zhao X., Scheler M., Irmler M., Staiger H., Beckers J., de Angelis M. H., Fritsche A., Häring H. U., Schleicher, E. D., Xu, G., Lehmann, R., and Weigert, C. (2013) Production and release of acylcarnitines by primary myotubes reflect the differences in fasting fat oxidation of the donors. J. Clin. Endocrinol. Metab. 98, E1137–E1142 [DOI] [PubMed] [Google Scholar]
- 33.Sampey B. P., Freemerman A. J., Zhang J., Kuan P. F., Galanko J. A., O’Connell T. M., Ilkayeva O. R., Muehlbauer M. J., Stevens R. D., Newgard C. B., Brauer, H. A., Troester, M. A., and Makowski, L. (2012) Metabolomic profiling reveals mitochondrial-derived lipid biomarkers that drive obesity-associated inflammation. PLoS ONE 7, e38812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoehn K. L., Hohnen-Behrens C., Cederberg A., Wu L. E., Turner N., Yuasa T., Ebina Y., and James D. E. (2008) IRS1-independent defects define major nodes of insulin resistance. Cell Metab. 7, 421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rutkowsky J. M., Knotts T. A., Ono-Moore K. D., McCoin C. S., Huang S., Schneider D., Singh S., Adams S. H., and Hwang D. H. (2014) Acylcarnitines activate proinflammatory signaling pathways. Am. J. Physiol. Endocrinol. Metab. 306, E1378–E1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang L., Keung W., Samokhvalov V., Wang W., and Lopaschuk G. D. (2010) Role of fatty acid uptake and fatty acid beta-oxidation in mediating insulin resistance in heart and skeletal muscle. Biochim. Biophys. Acta 1801, 1–22 [DOI] [PubMed] [Google Scholar]
- 37.Coll T., Eyre E., Rodríguez-Calvo R., Palomer X., Sánchez R. M., Merlos M., Laguna J. C., and Vázquez-Carrera M. (2008) Oleate reverses palmitate-induced insulin resistance and inflammation in skeletal muscle cells. J. Biol. Chem. 283, 11107–11116 [DOI] [PubMed] [Google Scholar]
- 38.Weigert C., Brodbeck K., Staiger H., Kausch C., Machicao F., Häring H. U., and Schleicher E. D. (2004) Palmitate, but not unsaturated fatty acids, induces the expression of interleukin-6 in human myotubes through proteasome-dependent activation of nuclear factor-kappaB. J. Biol. Chem. 279, 23942–23952 [DOI] [PubMed] [Google Scholar]
- 39.Nowotny B., Zahiragic L., Krog D., Nowotny P. J., Herder C., Carstensen M., Yoshimura T., Szendroedi J., Phielix E., Schadewaldt P., Schloot, N. C., Shulman, G. I., and Roden, M. (2013) Mechanisms underlying the onset of oral lipid-induced skeletal muscle insulin resistance in humans. Diabetes 62, 2240–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baillet L., Mullur R. S., Esser V., and McGarry J. D. (2000) Elucidation of the mechanism by which (+)-acylcarnitines inhibit mitochondrial fatty acid transport. J. Biol. Chem. 275, 36766–36768 [DOI] [PubMed] [Google Scholar]
- 41.Keung W., Ussher J. R., Jaswal J. S., Raubenheimer M., Lam V. H., Wagg C. S., and Lopaschuk G. D. (2013) Inhibition of carnitine palmitoyltransferase-1 activity alleviates insulin resistance in diet-induced obese mice. Diabetes 62, 711–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Houstis N., Rosen E. D., and Lander E. S. (2006) Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 440, 944–948 [DOI] [PubMed] [Google Scholar]
- 43.Henriksen E. J., Diamond-Stanic M. K., and Marchionne E. M. (2011) Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic. Biol. Med. 51, 993–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gillingham M. B., Harding C. O., Schoeller D. A., Matern D., and Purnell J. Q. (2013) Altered body composition and energy expenditure but normal glucose tolerance among humans with a long-chain fatty acid oxidation disorder. Am. J. Physiol. Endocrinol. Metab. 305, E1299–E1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Melegh B., Seress L., Bedekovics T., Kispál G., Sümegi B., Trombitás K., and Méhes K. (1999) Muscle carnitine acetyltransferase and carnitine deficiency in a case of mitochondrial encephalomyopathy. J. Inherit. Metab. Dis. 22, 827–838 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.