

Abstract
Tissue repair/wound healing, in which angiogenesis plays an important role, is a critical step in many diseases including chronic wound, myocardial infarction, stroke, cancer, and inflammation. Recently, we were the first to report that orphan nuclear receptor TR3/Nur77 is a critical mediator of angiogenesis and its associated microvessel permeability. Tumor growth and angiogenesis induced by VEGF-A, histamine, and serotonin are almost completely inhibited in Nur77 knockout mice. However, it is not known whether TR3/Nur77 plays any roles in wound healing. In these studies, skin wound-healing assay was performed in 3 types of genetically modified mice having various Nur77 activities. We found that ectopic induction of Nur77 in endothelial cells of mice is sufficient to improve skin wound healing. Although skin wound healing in Nur77 knockout mice is comparable to the wild-type control mice, the process is significantly delayed in the EC-Nur77-DN mice, in which a dominant negative Nur77 mutant is inducibly and specifically expressed in mouse endothelial cells. By a loss-of-function assay, we elucidate a novel feed-forward signaling pathway, integrin β4 → PI3K → Akt → FAK, by which TR3 mediates HUVEC migration. Furthermore, TR3/Nur77 regulates the expression of integrin β4 by targeting its promoter activity. In conclusion, expression of TR3/Nur77 improves wound healing by targeting integrin β4. TR3/Nur77 is a potential candidate for proangiogenic therapy. The results further suggest that TR3/Nur77 is required for pathologic angiogenesis but not for developmental/physiologic angiogenesis and that Nur77 and its family members play a redundant role in normal skin wound healing.—Niu, G., Ye, T., Qin, L., Bourbon, P. M., Chang, C., Zhao, S., Li, Y., Zhou, L., Cui, P., Rabinovitz, I., Mercurio, A. M., Zhao, D., Zeng, H. Orphan nuclear receptor TR3/Nur77 improves wound healing by upregulating the expression of integrin β4.
Keywords: angiogenesis, VEGF
Tissue repair/wound healing is a critical step in many diseases including chronic wound, myocardial infarction, stroke, cancer, and inflammation, in which angiogenesis plays an important role (1–4). Among many angiogenic factors, VEGF-A plays a central role in angiogenesis and associated microvessel permeability to plasma proteins (1–4). Recently, we reported that VEGF-A exerts much of its permeability and angiogenic activity by upregulating the expression of TR3/Nur77 (human TR3/mouse Nur77) (5, 6).
TR3/Nur77 (human: TR3, mouse: Nur77, rat: NGFI-B) belongs to nuclear receptor IV subfamily of transcription factors including TR3/Nur77, NOT1/Nurr1, and NOR1, all of which contain 3 functional domains, the transactivation domain, the DNA-binding domain, and the ligand-binding domain [see review by Hsu et al. (7)]. They are highly homologous in the DNA binding domain (>97%) but have much less similarity in the transactivation domains [<30%; reviewed by Hsu et al. (7)]. Due to their high homology in the DNA binding domain, they presumably regulate expression of similar genes, which may explain why they play redundant roles in T-cell receptor-mediated apoptosis and brown fat thermogenesis. However, they also play different roles in development [reviewed by Hsu et al. (7)].
Our recent studies demonstrated that TR3/Nur77 is important for tumor angiogenesis and its associated microvessel permeability (5, 6, 8). TR3/Nur77 is highly and transiently upregulated in cultured endothelial cells (ECs) and in angiogenesis in vivo induced by angiogenic factors having microvessel permeable activity, including VEGF-A, histamine, or serotonin, but not the angiogenic factors that do not have microvessel permeable activity, including bFGF, placental growth factor, and platelet-derived growth factor (5, 6, 8). Using loss-of-function assays, we found that inhibition of endogenous TR3/Nur77 by either antisense DNA or shRNA blocks endothelial cell proliferation, migration, and tube formation induced by VEGF-A, histamine, or serotonin in vitro (5, 6, 8). Growth of B16 melanoma, angiogenesis, and microvessel permeability induced by VEGF-A, histamine, and serotonin are largely inhibited in Nur77 knockout mice (5, 6, 8). In our gain-of-function assays, we found that overexpression of full-length TR3 is sufficient to induce endothelial cell proliferation, migration, and tube formation in vitro (5, 8). Furthermore, microvessel permeability in various organs and vessel area but not vessel density in heart tissues are increased in our transgenic mice (EC-Nur77-S mice), in which full-length Nur77 is selectively and inducibly expressed in mouse endothelium (6).
Considering the close relationship between angiogenesis and wound healing, we hypothesize that TR3/Nur77 plays important roles in the wound-healing process, which has no preceding studies. In the present study, we examine the expression and delineate the function of TR3/Nur77 in normal skin wound healing using our previously developed transgenic mouse models. The potential mechanism for this response is also investigated.
MATERIALS AND METHODS
Materials
Actinomycin D (Cat. No. A1410) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Antibodies against TR3/Nur77 (Cat. No. sc-5569), FAK (Cat. No. 1688), and integrin β4 (Cat. No. sc-9090) were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Anti-pAkt-S473 (Cat. No. 9271), anti-pAkt-T308 (Cat. No. 9275), anti-Akt (Cat. No. 9272), anti-PI3K (Cat. No. 4292), anti-phospho-p42/p44 MAPK (Cat. No. 9106S), and anti-p42/p44 MAPK (Cat. No. 9211) antibodies are products of Cell Signaling Technology (Danvers, MA, USA). Antibody against mouse CD31 (Cat. No. 553370) was purchased from BD Bioscience (San Jose, CA, USA).
Cell culture
Primary HUVECs purchased from Clonetics (Walkersville, MD, USA) were grown on plates coated with 30 µg/ml Pure Col (Cat. No. 5005-B; Advanced BioMatrix, San Diego, CA, USA) in endothelial basic medium (EBM) with EGM-MV BulletKit (5% fetal bovine serum, 12 µg/ml bovine brain extract, 1 µg/ml hydrocortisone, 1 µl/ml GA-1000 (gentamicin. amphotericin B), and 10 ng/ml epidermal growth factor) that was purchased from Clonetics. HUVECs were transduced with virus expressing TR3 cDNA, TR3 antisense DNA, or shRNAs for various experiments as indicated.
Construction of integrin β4 shRNAs, shITGB4-498, and shITGB4-5719
This study utilized 2 integrin β4 shRNAs targeting different regions of the integrin β4 transcript. The shITGB4-498 targeted integrin β4 cDNA at nucleotides 498 with sense: 5′-AGCGACTACACTATTGGAT-3′. The shITGB4-5719 (Cat. No. TRCN0000057768) was purchased from Thermo Scientific Inc. (Pittsburgh, PA, USA) and targets nucleotide sequence 5719 of the human integrin β4 (NM_000213.3) with sense: 5′-CTCCTCAGCTACTCCATCCTT-3′. These shRNA oligos were cloned to lentiviral vector pLKO.1 following the instruction provided by AddGene (Cambridge, MA, USA).
Preparation and infection of retroviruses and lentiviruses
Preparation of retroviruses was carried out as described in detail in our previous publication (9, 10). Briefly, 293T cells were seeded at a density of 6 × 106 cells per 100 mm plate 24 h before transfection. DNA transfection was carried out with the Effectene Transfection Reagent (Qiagen, Valencia, CA, USA). Target gene (2 µg), pMD.MLV gag.pol (1.5 µg), and pMD.G DNA (0.5 µg), encoding the cDNAs of the proteins that are required for virus packaging (kindly provided by Dr. Richard C. Mulligan, Harvard Medical School, Boston, MA, USA), were mixed in 300 µl EC buffer. En-hancer (30 μl) was added to the DNA mixture. After incubation at room temperature for 5 min, Effectene (30 μl) was added to the DNA mixture and incubated at room temperature for at least 10 min. The DNA mixture was added to 293T cells. The medium was changed after 16 h. The retrovirus was collected 48 h after transfection and filtered with syringe filter (0.45 μm pore size) and used immediately for infection or frozen at −70°C. Lentiviruses were prepared in the similar way as retroviruses, except that the composition of DNA mixture is 2 µg shRNA, 1.78 μg pCMV-dR8.2dvpr, and 0.2 μg pCMV-VSVG.
Twenty-four hours before infection, HUVECs were seeded at a density of 0.4 × 106 cells per 100 mm plate. Retroviruses (1 ml) or lentivirus solution (3 ml; ∼ 2 × 107 plaque-forming unit/ml) and fresh medium (5 ml) were added to cells with 10 µg/ml Polybrene (Sigma-Aldrich Company, LLC, St. Louis, MO, USA) per 100 mm plate. The medium was changed after 16 h, and cells were ready for experiment 60 h after infection.
Proliferation assay
HUVECs (2 × 103 cells/well) were seeded in 96-well plates. Twenty-four hours later, cells were transduced with retroviruses or lentiviruses as indicated. Forty-eight hours later, cells were serum-starved with 0.1% fetal bovine serum (FBS) in EBM medium for 48 h, cells were washed with PBS twice then kept in a freezer at −80°C for several hours. The proliferation assay was carried out with CyQuant Cell Proliferation Assay Kit (Invitrogen, Carlsbad, CA, USA) as per the user guide. In brief, cells were taken out of the freezer, warmed up at room temperature for 30 min, incubated with premixed CyQuant reagent solution at 4°C overnight, and then the dye incorporation was measured with Spectra Max (Molecular Devices, Sunnyvale, CA, USA).
Migration assay
Briefly, HUVECs (3 × 104 cells/well) were seeded in 12-well plates. Twenty-four hours later, cells were transduced with retroviruses or lentiviruses as indicated. Forty-eight hours later, cells were serum-starved with 0.1% FBS in EBM medium for 24 h. Scratch wound was generated with a 20 μl pipette tip and photographed immediately as 0 h and at 16 h, respectively. Cells migrated to the wound area were counted. Results were expressed as mean ± sd.
Immunoblotting
Briefly, HUVECs (5 × 105 cells/plate) were seeded in 100 mm plates. Twenty-four hours later, cells were transduced with retroviruses or lentiviruses as indicated. Forty-eight hours later, cells were serum-starved with 0.1% FBS in EBM medium for 24 h. Cellular extracts were subjected to immunoblotting with antibodies as indicated.
Promoter assay
HUVECs (6 × 104 cells/well) were seeded in 12-well plate. Twenty-four hours later, cells were washed with Minimum Essential Media (MEM; Life Technologies, Grand Island, NY, USA) 3 times and transfected with promoter luciferase construct (4 wells per group) and infected with adenoviruses at the same time. Integrin β4 promoter construct (990 ng/well), pGL3ITGB4-1572 (11), was added to 80 μl MEM. pRT-SV40 luciferase vector, serving as internal control (10 ng/well), was added to 20 μl MEM, which was added to the mixture of promoter construct. TransIT2020 (1 μl; Mirus Bio Limited Liability Corporation, Madison, WI, USA) was then added to the DNA mixture. The transfection mixture was incubated at room temperature for 20 to 30 min and added to cells. Adenoviruses expressing Lac Z as a control or FLAG-fused TR3 (kindly provided by Dr. Jianxin Sun (Center for Translational Medicine, Thomas Jefferson University, Philadelphia, PA, USA) was added to cells. Six hours later, cells were changed to MEM. Twenty-four hours after transfection, cells were lysed and subjected to luciferase analysis with Dual-Luciferase Reporter Assay System (Promega Corporation, Madison, WI, USA), following the instruction provided by the company. The luciferase activity in each well was normalized to the internal luciferase activity. The data are expressed as average fold change of luciferase activity in TR3 expressing cells to lac Z-expressing cells.
Quantitative real-time RT-PCR
RNA was isolated from cells or wound tissues and reverse-transcribed and subjected to real-time RT-PCR. The sequences for real-time RT-PCR primers are Nur77 forward primer: 5′-GTGTTGATGTTCCCGCCTTT-3′; Nur77 reverse primer: 5′-GGAGCCCGTGTCGATCAGT-3′; integrin β4 forward primer: 5′-CTGGTCTTCTCCACCGAGTCA-3′; integrin β4 reverse primer: 5′-ATCGTTGCGGCTCATGATG-3′. GAPDH served as an internal control. Experiments were repeated 3 times in duplicate.
Mouse lines with various Nur77 transgenes
Nur77−/− mice in C57BL/6 background that were kindly provided by Dr. Jeffrey Milbrandt (Washington University School of Medicine, St. Louis, MO, USA) were backcrossed to wild-type Fvb mice for 10 generation to obtain inbred Nur77−/− (Fvb) mice. EC-Nur77-S and EC-Nur77-DN mice, in which the full-length Nur77cDNA or its dominant negative mutant is inducibly and specifically expressed in mouse endothelium, were described in detail in our previous report (6). Tetracycline was provided in drinking water (1.5 g tetracycline, 50 g sucrose per liter tap water) to prevent expression of the trans-gene. Tetracycline was removed from drinking water to induce the expression of transgenes as described previously (6).
Skin wound-healing assay
Mice were anesthetized with isoflurane inhalation and shaved with electric razor and hair remover 2 days before the wound preparation to minimize the skin irritation. On the day of wound creation (D0), animals were deeply anesthetized with i.p. administration of 9% ketamine HCl+ 1% xylazine in HBSS (10 μl/g body weight). After disinfection with iodine tincture, and deiodination with disposable alcohol pad, full-thickness, excisional skin wounds were created on the flank skin with a 4 mm diameter, sterile and disposable biopuncher (punch skin biopsy punch; Acuderm Incorporated, Ft. Lauderdale, FL, USA). Wound tissues were collected for isolation of proteins and RNA, or embedded in optimal cutting temperature compound, and stored at −80°C for hematoxylin and eosin (H&E) stain or immunohistochemical analysis at various days after wounding as indicated.
Wound closure rate
Wounds were photographed with Wild M400 Macroscope with Digital Camera Capability, Lightglass Optics (Cedar Crest, NM, USA) immediately after wounding as day 0 and daily until day 12 when most wounds healed or there was no obvious difference apparent. Wound area was measured with ImageJ software (Image Processing and Analysis in Java, U.S. NIH). Briefly, freehand selection tool of ImageJ was used to circle around the wound edge, and then the area in square inches was given automatically. The wound closure ratio was obtained by the wound area of each day divided by the wound area of the same mice in day 0. Average wound closure ratio obtained from 10 mice in each group were used to plot the wound closure graph.
Histologic studies
Optimal cutting temperature embedded wound tissues were cut into 5 μm thick sections, fixed with cold acetone for 10 min, air-dried, then stained with H&E or with an anti-CD31 antibody. Wound tissues on day 7 were selected to analyze re-epithelialization (i.e., the area of new epithelial tissues growing into the wounded area). Wound tissues on day 14 were used to analyze dermal gap as the shortest distance between the immigrating dermis on both sides of the wounded area. The measurements were performed with Image J software as mentioned previously. Four mice in each group were compared with respective controls.
Animal welfare
All animal experiments were performed in compliance with the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee.
Statistics
Unpaired Student’s t test was used to determine statistical significance.
RESULTS
Induction of angiogenesis in flank skin by ectopic expression of TR3/Nur77 in mouse endothelium
Recently, we generated the transgenic mice, EC-Nur77-S mice and EC-Nur77-DN mice, in which Nur77 full-length cDNA or its dominant-negative mutant is inducibly and specifically expressed in vascular EC under the control of a tetracycline-sensitive promoter (6). Microvessel permeability in flank skin induced by VEGF-A is almost completely inhibited in EC-Nur77-DN mice (6). Vessel area but not vessel density in heart tissues is increased in EC-Nur77-S mice after TR3/Nur77 is induced for 6 d with a tet-off system (6). We further studied whether angiogenesis is induced after long-term induction of TR3/Nur77. Tetracycline was withdrawn from the drinking water of EC-Nur77 mice and their wild-type control littermates for 6 d and 3 mo, respectively. Flank skin was dissected and photographed. As shown in Fig. 1, enlarged vessels but not vessel density are clearly visible in the flank skin of EC-Nur77 mice after tetracycline was withdrawn for 6 d (Fig. 1, arrowhead in A vs. arrow in B). Vessel density was increased in EC-Nur77 mice compared with their wild-type control mice at 3 mo after tetracycline was withdrawn from mouse drinking water (Fig. 1 C vs. D and E vs. F). The increase of vessel density was further confirmed by immunostaining with an antibody against CD31. CD31-positive vessels were increased in the flank skin of EC-Nur77 mice (Fig. 1 G vs. H, right panel, P < 0.001). Furthermore, the mice were relatively healthy after Nur77 had been induced for 3 mo. Our data clearly indicate that angiogenesis is induced in mouse flank skin after induction of TR3/Nur77 in mouse endothelium, suggesting that it might play a role in skin wound healing.
Figure 1.
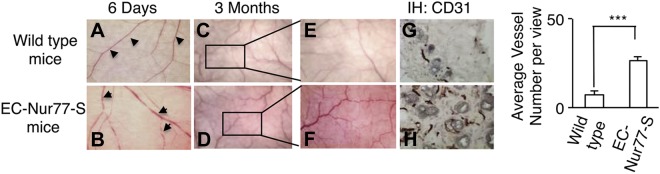
Induction of angiogenesis in the flank skin of the EC-Nur77-S mice. Macroscopic images of flank skin of the EC-Nur77-S mice (B, D, and F) and their respective wild-type control littermates (A, C, and E), in which tetracycline had been withdrawn from the drinking water for 6 days (A and B) or 3 months (C, D, E, and F). Four mice were used for each condition. Immunohistochemical staining of the sections obtained from the flank skin of E and F with an antibody against CD31 (G and H). Quantification of vessel number (right panel, n = 20 views, P < 0.001).
Upregulation of TR3/Nur77 in skin wound tissues
To study whether TR3/Nur77 plays a role in skin wound healing, we first studied whether expression of TR3/Nur77 is upregulated in skin wound tissues. Skin wounds were created on the flank skin of wild-type Fvb mice with a 4 mm biopuncher. The punched skin was collected as control for the wound tissues obtained from the same mouse. Starting from the next day (day 1), animals were sacrificed every day and wound tissues were punched and frozen in liquid nitrogen. On day 1 after wounding, wound tissues were punched by a 6 mm biopuncher because of the contraction of mouse wound. On the subsequent days, wound tissues were punched by a 4 mm biopuncher. As shown in Fig. 2, the level of TR3/Nur77 protein increased in wound tissues from day 1 to day 3 compared with that in their respective control tissues, then degraded on day 4, and returned to baseline level on day 5 (Fig. 2A, top panels, lanes w vs. c for each animal). The intensity of each band was measured with Quantity One (Bio-Rad Laboratories, Hercules, CA, USA) and plotted (Fig. 2B, P < 0.001). Similar to the protein expression profile, TR3/Nur77 mRNA reached a peak on day 1 in wound tissues, about 3 times of that in control tissues, then gradually dropped on subsequent days and returned to baseline level after day 3 (Fig. 2C, P < 0.001). To further determine whether TR3/Nur77 exerts its effect on wound healing by regulating angiogenesis, we examined whether TR3/Nur77 is expressed in vasculature of wound tissue. Immunofluorescence staining was performed with antibodies against endothelial marker CD31 and TR3/Nur77. As shown in Fig. 2D, TR3/Nur77 is partially colocalized with CD31 (Fig. 2D). Our data demonstrate that Nur77 is highly and transiently upregulated in wound vasculature.
Figure 2.
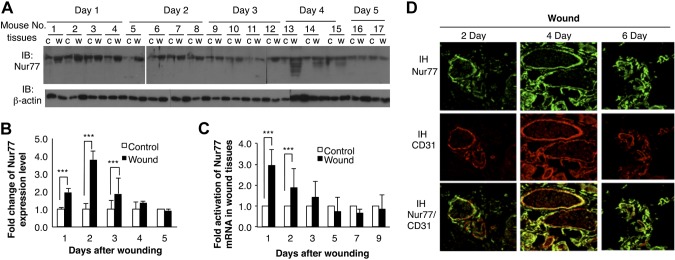
Expression of Nur77 in wound tissues. A) Protein extracts from wound tissues (w) and their respective normal control tissues (c) from the same mouse were immunoblotted with antibodies against Nur77 (top panel) and β-actin to confirm protein equal loading (bottom panel). B) Quantitative measurement of intensity in A (4, 3, or 2, P < 0.001). C) RNA were isolated from wound tissues and their control tissues, and subjected to real-time RT-PCR. Data are expressed as average fold changes of animals on each day (n = 4, P < 0.001). D) Wound tissues were immunostained with antibodies against Nur77 and CD31. Data are representative of the results from 4 mice.
Effects of various TR3/Nur77 activities on skin wound healing
Because angiogenesis is regulated in mice with various TR3/Nur77 activities (6), we studied whether TR3/Nur77 regulated skin wound healing. Skin wounds were created on the flank skin of the EC-Nur77-S mice, EC-Nur77-DN mice, and Nur77−/− mice, and their respective wild-type control littermates, 6 d after tetracycline was withheld from their drinking water. Wounds were photographed daily, and the wound area was measured and analyzed with ImageJ software. The rate of wound closure is expressed as the average ratio of wound area on each day after wounding to the initial wound area for each animal. A larger wound ratio indicates slower wound closure. The data show that wound closure was significantly accelerated in the EC-Nur77-S mice and delayed in the EC-DN-Nur77 mice, compared with their respective control mice (Fig. 3A, panels I and II, and B, graphs I and II, P < 0.05). To our surprise, the rate of wound closure was not defective in the first 5 d but was significantly delayed after day 5 in the Nur77−/− mice compared with wild-type mice (Fig. 3A, panels III and 3B, graph III). We then analyzed the epithelialization (i.e., the area of new epithelial tissues growing into the wounded area) of wound tissues collected on day 7 with H&E staining as described elsewhere (12). Re-epithelialization of wound tissues was improved in the EC-Nur77-S mice, was defective in EC-Nur77-DN mice, and showed no change in Nur77−/− mice compared with their respective control animals (Fig. 3C, area enclosed by dashed lines, P < 0.05). Analysis of dermal closure determined by the dermal gap as the shortest distance between the immigrating dermis on both sides of the wounded area, as described (12), on tissues collected on day 14 also indicated that dermal closure was significantly increased in EC-Nur77-S mice, delayed in EC-Nur77-DN mice but not affected in Nur77−/− mice (Fig. 3D, dashed lines, P < 0.05). We further analyzed the effect of Nur77 activities on CD31+ microvessel density. The data show that induction of Nur77 significantly increases the number of CD31+ microvessels but not average vessel area (Fig. 3E, a vs. b, and F, P < 0.001). The number of CD31+ microvessels but not average vessel area in the wound area in EC-DN-Nur77 mice was significantly lower than that in the control group (Fig. 3E, c, d and Fig. 3F, P < 0.001). There was no significant difference in both of the number and the area of CD31+ microvessels in the wound area between Nur77−/− mice and the control group (Fig. 3E, e, f and Fig. 3F). Skin wound healing was increased in EC-Nur77-S mice, delayed in EC-Nur77-DN, and not affected in Nur77−/− mice.
Figure 3.

Skin wound healing on mice with various TR3/Nur77 activities. A) Macroscopic images of skin wounds (representative of 10 mice/group). B) Average percentage of wound area related to day 0 (n = 10, P < 0.05). C) Re-epithelialization analysis (n = 4, P < 0.05). D) Dermal closure analysis (n = 4, P < 0.05). E) Immunostaining of wound tissues obtained from the transgenic mice with various TR3/Nur77 activity. F) Quantitative measurement of vessel number and vessel density from images in E (n = 20 views, P < 0.001).
Integrin β4 as a downstream target of TR3 in cell migration but not proliferation
It is well known that integrin β4 plays an important role in angiogenesis [review by Giancotti (13)]. Murgia et al. reported that mice lacking the entire β4 cytoplasmic domain die at birth as a result of extensive epidermal blistering (14). However, mice carrying a targeted deletion of the β4 signaling domain can survive, in which VEGF-A- and bFGF-induced angiogenesis is defective (15). We examined whether TR3 regulates the expression of integrin β4. RNA were isolated from HUVECs that were transduced with retrovirus expressing LacZ as control or TR3 sense cDNA and subjected to real-time RT-PCR. Overexpression of TR3 increased the RNA level of integrin β4 about 4-fold (Fig. 4A, P < 0.001). We further confirm the protein expression of integrin β4 with immunoblotting. HUVECs were transduced with LacZ as a control, TR3 sense cDNA, TR3 antisense DNA, or TR3 shRNA. Expression of integrin β4 proteins was upregulated by overexpression of TR3 cDNA but was decreased by expressing TR3 antisense DNA or shRNA (Fig. 4B).
Figure 4.

Regulation of integrin β4 by TR3/Nur77. A) HUVECs were transduced without (H) or with LacZ as control, TR3 full-length cDNA. RNA were isolated and subjected to real-time RT-PCR with integrin β4-specific primers (n = 2 for real-time PCR, P < 0.001). B) Cellular extracts from HUVECs that were transduced with LacZ as control, TR3 full-length cDNA, TR3 antisense DNA, or TR3 shRNA were immunoblotted with antibodies against integrin β4 (left top panel) and β-actin for protein equal loading control (left bottom panel). Quantitative measurement of integrin β4 expression (right panel). Experiments were repeated 3 times.
Then we studied whether integrin β4 is a downstream target of TR3 in angiogenesis. We generated lentiviruses expressing integrin β4 shRNAs targeting 2 different regions of integrin β4 cDNA. HUVECs were transduced with lentiviruses expressing negative control shRNA (shNEG), 2 integrin shRNAs, shITGB4-498 and shITGB4-5719. Real-time RT-PCR analysis indicated that expression of integrin β4 was inhibited partially or greatly by shITGB4-498 or shITGB4-5719, respectively (Fig. 5A, P < 0.001). Previously, we reported that TR3 regulates endothelial cell proliferation and migration (8). Nikolopoulos et al. reported that integrin β4 is required for cell migration but not proliferation (15). We study whether integrin β4 is required for TR3-mediated HUVEC migration and proliferation. HUVECs that were transfected with LacZ as a control, LacZ + TR3, TR3+ shITGB4-498, TR3+ shITGB4-5719, were subjected to proliferation assay and scratch wound assay. In EC proliferation assay, cell proliferation was increased in HUVECs transduced with TR3 as compared with LacZ (Fig. 5B, lane 2 vs. 1, P < 0.001). Cotransduction of TR3 with either integrin β4 shRNA did not affect HUVEC proliferation (Fig. 5B, lanes 3 and 4 vs. lane 2). In the scratch wound migration assay, expression of TR3 increased cell migration (Fig. 5C, right panel, bar 2 vs. bar 1, P < 0.001) and knockdown of the expression of integrin β4 significantly inhibited cell migration induced by TR3 (Fig. 5C, right panel, bars 3 and 4 vs. bar 2, P < 0.001). We further studied whether TR3 regulates the signaling molecules. Cellular extracts isolated from HUVECs that were transduced with negative control shRNA, TR3, TR3 + shITGB4-498, or TR3+shITGB4-5719 were immunoblotted with antibodies as indicated. As shown in Fig. 5D, expression of TR3 downregulated the expression of Akt, hence its phosphorylation, PI3K, and FAK, and slightly increased the phosphorylation but not the expression of MAPK42/44 (Fig. 5D, lane 2 vs. 1). shITGB4-498 partially and shITGB4-5719 almost completely inhibited the effect of TR3 on the phosphorylation and expression of Akt, PI3K, and FAK (Fig. 5D, lanes 3 and 4 vs. lane 2). However, shITGB4s were unable to reverse the slight increase of phosphorylated MAPK42/44 induced by TR3 but further increased the phosphorylation of MAPK42/44 (Fig. 5D, panel VII, lane 4 vs. lane 2). Our data demonstrate that integrin β4 is a novel downstream target of TR3 in angiogenesis.
Figure 5.

Integrin β4 is a downstream target of TR3/Nur77 in migration but not proliferation. A) RNA isolated from HUVECs that were transduced with LacZ, as control, shITGB4-498, or shITGB4-5719 were subjected to real-time RT-PCR (n = 2, P < 0.001). B) HUVECs were transduced as indicated and subjected to proliferation assay (n = 6, P < 0.001). C) HUVECs were transduced as indicated and subjected to scratch wound migration assay (n = 10, P < 0.001). D) Cellular extracts from HUVECs that were transduced with negative control shRNA (lane 1), HA-tagged TR3 (lane 2), HA-TR3 + shITGB4-498 (lane 3), or HA-TR3 + ITGB4-5719 (lane 4) were immunoblotted with antibodies against HA to indicate the expression of TR3 (panel I), pAkt-S473 (panel II), pAkt-T308 (panel III), Akt (panel IV), PI3K (panel V), FAK (panel VI), pMAPK42/44 (panel VII), MAPK42/44 (panel VIII), and β-actin (panel IX) as protein equal loading control. Experiments were repeated 3 times.
Transcriptional regulation of integrin β4 expression by TR3
We reported previously that the transcriptional activity of TR3/Nur77 is required for its function in angiogenesis (5, 6, 8). However, at least some of its activities, such as those involved in neuronal differentiation and T-cell apoptosis, occur independently of its transcriptional activity (16). We studied the molecular mechanism by which TR3/Nur77 mediates the expression of integrin β4. HUVECs were transduced with viruses expressing LacZ as a control, TR3, and deletion mutants of transactivation domain (TR3ΔTAD), of DNA-binding domain (TR3ΔDBD), or of ligand-binding domain (TR3ΔLBD). RNA and proteins were isolated and subjected to real-time RT-PCR and immunoblot. As shown in Fig. 6, expression of TR3ΔTAD or TR3ΔDBD was unable to increase the expression of integrin β4, and expression of TR3ΔLBD increased the expression of integrin β4 at a level comparable to that induced by full-length TR3 (Fig. 6A, B, P < 0.001). These data demonstrate that transcriptional activity of TR3 is required for the upregulation of integrin β4.
Figure 6.

Transcriptional regulation of integrin β4 by TR3/Nur77. A) HUVECs were transduced as indicated. RNA were subjected to real-time RT-PCR with integrin β primers (n = 2 for real-time PCR, P < 0.001). B) Cellular extracts from HUVECs that were transduced as indicated were immunoblotted with antibodies against integrin β4 (left top panel) or β-actin as protein equal loading control (left bottom panel). Quantitative measurement of integrin β4 expression (right panel). C) HUVECs were infected with adenovirus expressing Lac Z as control and TR3, and transfected with integrin β4 promoter construct and internal luciferase construct. Data are presented as fold change of luciferase activity in cells expressing TR3 related to LacZ (n = 4, P < 0.001). Experiments were repeated 3 times.
We generated the integrin β4 promoter luciferase reporter construct (pGL3-ITGB4-1572) by cloning the −1572/+254 sequence of integrin β4 promoter to pGL3-basic luciferase vector. HUVECs were transduced with LacZ or TR3, and then transfected with pGL3-ITGB4-1572 with an internal luciferase control. As shown in Fig. 6, expression of TR3 increased the promoter activity of integrin β4 (Fig. 6C, P < 0.001).
DISCUSSION
Normal wound healing and pathologic wound healing are always accompanied by increased release of many angiogenic factors such as VEGF and bFGF that cause massive neovascularization and tissue remodeling. Blocking the angiogenic effect by VEGF-neutralizing antibody shows dramatic decrease in granulation tissue vessel density and granulation tissue growth in the model of abdominal wall wound, whereas administration of VEGF improves wound healing (17).
Based on our recent findings that Nur77 is an important transcriptional mediator in angiogenesis, cultured endothelial cells, Matrigel angiogenesis assay, and xenograft model of melanoma, we anticipated that Nur77 also plays an important role on wound healing. In this study, we found that angiogenesis in skin was induced significantly after Nur77 was induced in mouse endothelium. Nur77 was upregulated in wound tissues and regulated skin wound healing. Overexpression of Nur77 in endothelial cells accelerated wound healing. Although skin wound healing was almost not defective in Nur77 knockout mice, it was greatly delayed in transgenic mice (EC-Nur77-DN mice), in which the dominant negative mutant of Nur77 was inducibly expressed in mouse endothelium. Mechanistically, TR3/Nur77 upregulated the expression of integrin β4. Knocking down the expression of integrin β4 by its shRNA significantly inhibited endothelial cell migration and the downregulation of signaling molecules but not proliferation mediated by TR3/Nur77. Further, TR3/Nur77 regulated the transcription of integrin β4.
Our data showed that angiogenesis was induced in mouse skin wound 3 months after Nur77 was induced in mouse endothelium (Fig. 1). However, there was no obvious health problem in Nur77-S transgenic mice after Nur77 was been induced for 3 months. Further, skin wound healing was significantly improved by overexpression of Nur77 in mouse endothelium (Fig. 3). Therefore, TR3/Nur77 has a potential to be developed for proangiogenic therapy.
The wound healing process involves multiple cell types. Our data showed that TR3/Nur77 was induced in cells other than vessels (Fig. 2C), suggesting that, in addition to angiogenesis, TR3/Nur77 also regulates other steps in skin wound healing, such as macrophage polarization, because it was reported that TR3/Nur77 is expressed in macrophages and modulate macrophage polarization (18, 19).
In contrast to tumor growth, skin wound healing and wound angiogenesis is not defective in Nur77 knockout mice, although there is somehow a delay in the wound closure rate (Fig. 3). However, wound closure rate, re-epithelialization of wound tissues, and wound angiogenesis are greatly delayed in the EC-Nur77-DN transgenic mice, in which the dominant negative mutant of Nur77 is inducibly and specifically expressed in mouse endothelium (Fig. 3). It is known that all of the 3 TR3 family members interact with the same NGFI-B (nerve growth factor-induced gene-B) response element in the promoters of their targeted genes, because the DNA binding domains of all 3 TR3 family members are homologous (>97%) (20). In the Nur77-DN mutant, the transactivation domain of Nur77 was deleted (6). The Nur77-DN may bind to the NGFI-B (nerve growth factor-induced gene-B) response element to prevent the binding of all 3 TR3/Nur77 members, hence block the transcription regulated by all 3 TR3/Nur77 members. Our results demonstrate that TR3/Nur77 family members have a redundant role in the early stage of normal skin wound healing, in addition to their redundant roles in T-cell receptor-mediated apoptosis and brown fat thermogenesis (21, 22). Nur77 null mice are viable and fertile and appear to develop a normal adult vasculature (22). However, our previous reports clearly demonstrated that angiogenesis induced by VEGF-A and histamine and growth of tumors are greatly inhibited in Nur77 knockout mice. All the observations demonstrate that expression of TR3/Nur77 is required for pathologic angiogenesis but not for developmental/physiologic angiogenesis, suggesting that TR3/Nur77 is an excellent specific target for pathologic angiogenesis.
In normal adult vessels, vascular integrity is maintained by endothelial cell-endothelial cell junctions and endothelial cell-basement interactions. To induce angiogenesis, both of these interactions must be modified to facilitate endothelial cell proliferation and migration. We already reported that TR3/Nur77 destabilizes endothelial cell-endothelial cell junctions to induce microvessel permeability and angiogenesis (6). However, it is not known whether TR3/Nur77 regulates endothelial cell-basement interaction. In quiescent vessels, the main components in basement membrane are laminin and collagen. In activated/angiogenic vessels found in tumors and inflammation, basement membrane changes to form a “provisional” extracellular matrix that mainly consists of fibronectin and vitronectin. The expression profile of integrins in angiogenic vessels differs from that in quiescent vessels [reviewed by Stupack and Cheresh (23)]. Furthermore, integrins have long been known to play important roles in angiogenesis. We found that TR3 increases the expression of integrin β4 that is known to play an important role in endothelial cell-basement interaction and cell migration. Our results, that knocking down the expression of integrin β4 by its shRNAs almost completely inhibits migration but not proliferation induced by TR3/Nur77, correlate well with the previous report that integrin β4 regulates endothelial cell migration but not proliferation (15).
It was reported that Akt phosphorylates TR3 to trigger the cytosolic translocation of TR3, resulting in loss of the TR3 transcriptional activity (24). Our data that TR3 downregulates the expression of PI3K-Akt axis indicate that TR3 induces a feed-forward signal pathway. Our results correlate very well with our previous report that PI3K mediates an inhibitory pathway in VEGF-induced HUVEC proliferation via phosphorylation of VEGF receptor 1 (Flt1) (9), although it is well accepted that PI3K-Akt promotes endothelial cell proliferation and migration. The conflicting conclusions may be due to the difference in culture condition (i.e., culture mediums and the Matrix that the cells were cultured on) (25–30). Furthermore, conflict results of Akt in heart diseases were reported, which may be due to the short- or long-term Akt activation [reviewed elsewhere (31, 32)]. Our results indicate that TR3 slightly increases the phosphorylation of MAPK42/44, which is not reversed by knocking down the expression of integrin β4. Further shITGB4s increase the phosphorylation of MAPK42/44. It is well known that phosphorylation of MAPK plays an important role in cell proliferation. These results further confirm that integrin β4 is required for HUVEC migration but not proliferation induced by TR3. Previously, we reported that TR3 mediates HUVEC proliferation via regulation of the expression of cell cycle genes, and permeability via downregulation of the expression of endothelial cell-endothelial cell junction proteins (5, 6). Here, our studies elucidate a novel signaling pathway, TR3 → integrin β4 → PI3K → Akt → FAK, by which TR3 regulates HUVEC migration. It was reported that integrin β4 activates AKT and PI3K in several cell types, including several basal breast cell lines, keratinocytes, pancreatic cell lines, among others (33–37). Our data indicate that TR3 downregulates the expression of Akt and PI3K in HUVECs, which is inhibited by integrin β4 shRNA, suggesting that integrin β4 plays different roles in endothelial cells, epithelial cells, and fibroblasts.
Many recent studies demonstrated that Nur77 plays different roles depending on its cellular localization. When present in the nucleus, Nur77 functions as a transcription factor that regulates gene expression and promotes cell growth. In the cytosol, Nur77 is hyperphosphorylated and does not have transcriptional activity but associates with other proteins, such as PKC to inhibit PKC activity (38) or Bcl2 to promote cancer cell apoptosis (39). Our previous studies demonstrated that the transcriptional activity of TR3/Nur77 is required for its function in angiogenesis and downregulation of the proteins in VE-cadherin-associated adherence. Here, we extend our finding that deletion of the transactivation domain or the DNA-binding domain but not the ligand-binding domain of TR3/Nur77 is required for the upregulation of integrin β4 by TR3/Nur77. We further demonstrated that TR3/Nur77 increases the promoter activity of integrin β4.
Collectively, we demonstrated that TR3/Nur77 is required for tumor angiogenesis and tumor growth but not for normal skin wound healing. Overexpression of TR3/Nur77 favors skin wound healing. TR3 mediates cell migration by a novel feed-forward signaling pathway. In the future, we will study whether TR3/Nur77 plays a role in chronic skin wound healing and develop TR3/Nur77 as a proangiogenic therapy. We will further study the mechanism by which TR3/Nur77 regulates endothelial cell-basement interaction.
Acknowledgments
The authors acknowledge Dr. Jianxin Sun (Center for Translational Medicine, Thomas Jefferson University, Philadelphia, PA, USA) for kindly providing the adenovirus expressing TR3. This work was supported by the U.S. National Institutes of Health Grants R01CA133235 (H.Z.), R01DK095873 and R21DK080970 (D.Z.), by the American Cancer Society Grant RSG CSM 113297 (D.Z.), and by the Scholarships from China Scholarship Council (G.N., S.Z., L.Z., Y.L., P.C.) and Renji Hospital, People’s Republic of China (T.Y.).
Glossary
- bFGF
basic fibroblast growth factor;
- EBM
endothelial basic medium;
- HA
hemagglutinin;
- H&E
hematoxylin and eosin;
- shRNA
short-hairpin RNA;
- TR3/Nur77
orphan nuclear receptor TR3 (human homolog, Nur77, mouse homolog)
REFERENCES
- 1.Folkman J. (1995) Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1, 27–31 [DOI] [PubMed] [Google Scholar]
- 2.Folkman J. (1996) Fighting cancer by attacking its blood supply. Sci. Am. 275, 150–154 [DOI] [PubMed] [Google Scholar]
- 3.Folkman J. (1997) Angiogenesis and angiogenesis inhibition: an overview. EXS 79, 1–8 [DOI] [PubMed] [Google Scholar]
- 4.Folkman J., D’Amore P. A. (1996) Blood vessel formation: what is its molecular basis? Cell 87, 1153–1155 [DOI] [PubMed] [Google Scholar]
- 5.Zeng H., Qin L., Zhao D., Tan X., Manseau E. J., Van Hoang M., Senger D. R., Brown L. F., Nagy J. A., Dvorak H. F. (2006) Orphan nuclear receptor TR3/Nur77 regulates VEGF-A-induced angiogenesis through its transcriptional activity. J. Exp. Med. 203, 719–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao D., Qin L., Bourbon P. M., James L., Dvorak H. F., Zeng H. (2011) Orphan nuclear transcription factor TR3/Nur77 regulates microvessel permeability by targeting endothelial nitric oxide synthase and destabilizing endothelial junctions. Proc. Natl. Acad. Sci. USA 108, 12066–12071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsu H. C., Zhou T., Mountz J. D. (2004) Nur77 family of nuclear hormone receptors. Curr. Drug Targets Inflamm. Allergy 3, 413–423 [DOI] [PubMed] [Google Scholar]
- 8.Qin L., Zhao D., Xu J., Ren X., Terwilliger E. F., Parangi S., Lawler J., Dvorak H. F., Zeng H. (2013) The vascular permeabilizing factors histamine and serotonin induce angiogenesis through TR3/Nur77 and subsequently truncate it through thrombospondin-1. Blood 121, 2154–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeng H., Dvorak H. F., Mukhopadhyay D. (2001) Vascular permeability factor (VPF)/vascular endothelial growth factor (VEGF) peceptor-1 down-modulates VPF/VEGF receptor-2-mediated endothelial cell proliferation, but not migration, through phosphatidylinositol 3-kinase-dependent pathways. J. Biol. Chem. 276, 26969–26979 [DOI] [PubMed] [Google Scholar]
- 10.Zeng H., Sanyal S., Mukhopadhyay D. (2001) Tyrosine residues 951 and 1059 of vascular endothelial growth factor receptor-2 (KDR) are essential for vascular permeability factor/vascular endothelial growth factor-induced endothelium migration and proliferation, respectively. J. Biol. Chem. 276, 32714–32719 [DOI] [PubMed] [Google Scholar]
- 11.Chang C., Yang X., Pursell B., Mercurio A. M. (2013) Id2 complexes with the SNAG domain of Snai1 inhibiting Snai1-mediated repression of integrin β4. Mol. Cell. Biol. 33, 3795–3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braiman-Wiksman L., Solomonik I., Spira R., Tennenbaum T. (2007) Novel insights into wound healing sequence of events. Toxicol. Pathol. 35, 767–779 [DOI] [PubMed] [Google Scholar]
- 13.Giancotti F. G. (2007) Targeting integrin beta4 for cancer and anti-angiogenic therapy. Trends Pharmacol. Sci. 28, 506–511 [DOI] [PubMed] [Google Scholar]
- 14.Murgia C., Blaikie P., Kim N., Dans M., Petrie H. T., Giancotti F. G. (1998) Cell cycle and adhesion defects in mice carrying a targeted deletion of the integrin beta4 cytoplasmic domain. EMBO J. 17, 3940–3951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nikolopoulos S. N., Blaikie P., Yoshioka T., Guo W., Giancotti F. G. (2004) Integrin beta4 signaling promotes tumor angiogenesis. Cancer Cell 6, 471–483 [DOI] [PubMed] [Google Scholar]
- 16.Lin B., Kolluri S. K., Lin F., Liu W., Han Y. H., Cao X., Dawson M. I., Reed J. C., Zhang X. K. (2004) Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell 116, 527–540 [DOI] [PubMed] [Google Scholar]
- 17.Howdieshell T. R., Callaway D., Webb W. L., Gaines M. D., Procter C. D. Jr, Sathyanarayana, Pollock J. S., Brock T. L., McNeil P. L. (2001) Antibody neutralization of vascular endothelial growth factor inhibits wound granulation tissue formation. J. Surg. Res. 96, 173–182 [DOI] [PubMed] [Google Scholar]
- 18.McMorrow J. P., Murphy E. P. (2011) Inflammation: a role for NR4A orphan nuclear receptors? Biochem. Soc. Trans. 39, 688–693 [DOI] [PubMed] [Google Scholar]
- 19.Pols T. W., Bonta P. I., de Vries C. J. (2007) NR4A nuclear orphan receptors: protective in vascular disease? Curr. Opin. Lipidol. 18, 515–520 [DOI] [PubMed] [Google Scholar]
- 20.Paulsen R. F., Granas K., Johnsen H., Rolseth V., Sterri S. (1995) Three related brain nuclear receptors, NGFI-B, Nurr1, and NOR-1, as transcriptional activators. J. Mol. Neurosci. 6, 249–255 [DOI] [PubMed] [Google Scholar]
- 21.Kanzleiter T., Schneider T., Walter I., Bolze F., Eickhorst C., Heldmaier G., Klaus S., Klingenspor M. (2005) Evidence for Nr4a1 as a cold-induced effector of brown fat thermogenesis. Physiol. Genomics 24, 37–44 [DOI] [PubMed] [Google Scholar]
- 22.Lee S. L., Wesselschmidt R. L., Linette G. P., Kanagawa O., Russell J. H., Milbrandt J. (1995) Unimpaired thymic and peripheral T cell death in mice lacking the nuclear receptor NGFI-B (Nur77). Science 269, 532–535 [DOI] [PubMed] [Google Scholar]
- 23.Stupack D. G., Cheresh D. A. (2004) Integrins and angiogenesis. Curr. Top. Dev. Biol. 64, 207–238 [DOI] [PubMed] [Google Scholar]
- 24.Chen H. Z., Zhao B. X., Zhao W. X., Li L., Zhang B., Wu Q. (2008) Akt phosphorylates the TR3 orphan receptor and blocks its targeting to the mitochondria. Carcinogenesis 29, 2078–2088 [DOI] [PubMed] [Google Scholar]
- 25.Bagli E., Stefaniotou M., Morbidelli L., Ziche M., Psillas K., Murphy C., Fotsis T. (2004) Luteolin inhibits vascular endothelial growth factor-induced angiogenesis; inhibition of endothelial cell survival and proliferation by targeting phosphatidylinositol 3′-kinase activity. Cancer Res. 64, 7936–7946 [DOI] [PubMed] [Google Scholar]
- 26.Cattaneo M. G., Chini B., Vicentini L. M. (2008) Oxytocin stimulates migration and invasion in human endothelial cells. Br. J. Pharmacol. 153, 728–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cattaneo M. G., Lucci G., Vicentini L. M. (2009) Oxytocin stimulates in vitro angiogenesis via a Pyk-2/Src-dependent mechanism. Exp. Cell Res. 315, 3210–3219 [DOI] [PubMed] [Google Scholar]
- 28.Ilan N., Mahooti S., Madri J. A. (1998) Distinct signal transduction pathways are utilized during the tube formation and survival phases of in vitro angiogenesis. J. Cell Sci. 111, 3621–3631 [DOI] [PubMed] [Google Scholar]
- 29.Lal B. K., Varma S., Pappas P. J., Hobson R. W. II, Durán W. N. (2001) VEGF increases permeability of the endothelial cell monolayer by activation of PKB/akt, endothelial nitric-oxide synthase, and MAP kinase pathways. Microvasc. Res. 62, 252–262 [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y., Hu G., Lin H. C., Hong S. J., Deng Y. H., Tang J. Y., Seto S. W., Kwan Y. W., Waye M. M., Wang Y. T., Lee S. M. (2009) Radix Astragali extract promotes angiogenesis involving vascular endothelial growth factor receptor-related phosphatidylinositol 3-kinase/Akt-dependent pathway in human endothelial cells. Phytother. Res. 23, 1205–1213 [DOI] [PubMed] [Google Scholar]
- 31.Abel E. D., Doenst T. (2011) Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc. Res. 90, 234–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Neill B. T., Abel E. D. (2005) Akt1 in the cardiovascular system: friend or foe? J. Clin. Invest. 115, 2059–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baril P., Gangeswaran R., Mahon P. C., Caulee K., Kocher H. M., Harada T., Zhu M., Kalthoff H., Crnogorac-Jurcevic T., Lemoine N. R. (2007) Periostin promotes invasiveness and resistance of pancreatic cancer cells to hypoxia-induced cell death: role of the beta4 integrin and the PI3k pathway. Oncogene 26, 2082–2094 [DOI] [PubMed] [Google Scholar]
- 34.Gilcrease M. Z., Zhou X., Welch K. (2004) Adhesion-independent alpha6beta4 integrin clustering is mediated by phosphatidylinositol 3-kinase. Cancer Res. 64, 7395–7398 [DOI] [PubMed] [Google Scholar]
- 35.Shaw L. M. (2001) Identification of insulin receptor substrate 1 (IRS-1) and IRS-2 as signaling intermediates in the alpha6beta4 integrin-dependent activation of phosphoinositide 3-OH kinase and promotion of invasion. Mol. Cell. Biol. 21, 5082–5093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shaw L. M., Rabinovitz I., Wang H. H., Toker A., Mercurio A. M. (1997) Activation of phosphoinositide 3-OH kinase by the alpha6beta4 integrin promotes carcinoma invasion. Cell 91, 949–960 [DOI] [PubMed] [Google Scholar]
- 37.Tang K., Nie D., Cai Y., Honn K. V. (1999) The beta4 integrin subunit rescues A431 cells from apoptosis through a PI3K/Akt kinase signaling pathway. Biochem. Biophys. Res. Commun. 264, 127–132 [DOI] [PubMed] [Google Scholar]
- 38.Kim H., Kim B. Y., Soh J. W., Cho E. J., Liu J. O., Youn H. D. (2006) A novel function of Nur77: physical and functional association with protein kinase C. Biochem. Biophys. Res. Commun. 348, 950–956 [DOI] [PubMed] [Google Scholar]
- 39.Wingate A. D., Arthur J. S. (2006) Post-translational control of Nur77. Biochem. Soc. Trans. 34, 1107–1109 [DOI] [PubMed] [Google Scholar]