

Abstract
Heme oxygenase (HO)-2 deficiency impairs wound healing and exacerbates inflammation following injury. We examine the impact of HO-2 deficiency on macrophage function and the contribution of macrophage HO-2 to inflammatory and repair responses to injury. Corneal epithelial debridement was performed in control and macrophage-depleted HO-2−/− and wild-type (WT) mice and in bone marrow chimeras. Peritoneal macrophages were collected for determination of phagocytic activity and classically activated macrophage (M1)-alternatively activated macrophage (M2) polarization. Depletion of macrophages delayed corneal healing (13.2%) and increased neutrophil infiltration (54.1%) by day 4 in WT mice, whereas in HO-2−/− mice, it did not worsen the already impaired wound healing and exacerbated inflammation. HO-2−/− macrophages displayed an altered M1 phenotype with no significant expression of M2 or M2-like activated cells and a 31.3% reduction in phagocytic capacity that was restored by inducing HO-1 activity or supplementing biliverdin. Macrophage depletion had no effect, whereas adoptive transfer of WT bone marrow improved wound healing (34% on day 4) but did not resolve the exaggerated inflammatory response in HO-2−/− mice. These findings indicate that HO-2–deficient macrophages are dysfunctional and that macrophage HO-2 is required for proper macrophage function but is insufficient to correct the impaired healing of the HO-2−/− cornea, suggesting that corneal epithelial expression of HO-2 is a key to resolution and repair in wound healing.—Bellner, L., Marrazzo, G., van Rooijen, N., Dunn, M. W., Abraham, N. G., Schwartzman, M. L. Heme oxygenase-2 deletion impairs macrophage function: implication in wound healing.
Keywords: inflammation, bone marrow transfer, clodronate liposomes
Macrophages are key to the wound healing process because their phagocytic activity functions to clear debris and necrotic cells. Through their presence and activity, macrophages produce chemotactic factors that not only attract additional inflammatory cells to the injury site, but also activate tissue residing cells, e.g., fibroblasts and epithelial cells, thereby bringing about the resolution of inflammation and contributing to the healing process. Another equally important function of these inflammatory cells is to increase the innate immunity to prevent infection.
The cornea is a critical component of the refractive system of the eye and represents an initial barrier to the external environment and the continuous threat from UV irradiation, microorganisms, and toxins. Despite this challenge, the cornea remains avascular and transparent and shows an extraordinary capacity for epithelial regeneration while maintaining a unique immune-privileged environment. The corneal wound healing response is characterized by a complex interplay of corneal residential cells, in particular stromal keratocytes, and inflammatory cells to produce lipid and protein mediators that initiate, amplify, and ultimately resolve the inflammation. Excessive activation of these circuits can lead to tissue destruction, leukoma formation, perforation, and neovascularization, ultimately leading to a loss of vision (1, 2). The role of macrophages in corneal wound healing has been examined in a number of injury models, including epithelial debridement, alkali burn, and allogenic corneal transplantation (1–4); in these models, a deficiency of macrophages results in an increase in infiltration of neutrophils into the wound and a delayed healing response (5). The cornea, similar to most other tissues, contains a number of residential macrophages that work along with dendritic cells as sentinels tracking and responding to tissue injury and infection. On injury, circulating monocytes are recruited to the site of injury where they differentiate into macrophages (6, 7) and contribute to the phagocytosis of debris and the production of factors that regulate tissue repair and angiogenesis. They also contribute to the maintenance of lymphatic vessels in injured corneas.
Macrophages express multiple proinflammatory and anti-inflammatory proteins, including heme oxygenase (HO). HO, the rate-limiting enzyme in heme catabolism that cleaves heme into equimolar amounts of iron, carbon monoxide, and biliverdin (rapidly reduced to bilirubin by biliverdin reductase), has been implicated in the resolution of inflammation (8, 9). Bilverdin/bilirubin are powerful antioxidants, and both carbon monoxide and biliverdin/bilirubin exert anti-inflammatory properties (10–12). Two HO-isoforms, HO-1 and HO-2, are expressed in most tissues, including the cornea (11, 13). HO-1 is an inducible enzyme, whereas HO-2 is constitutively expressed (10).
We previously showed that HO-2 knockdown impairs epithelial cell wound closure in vitro (14) and corneal healing in vivo (15). We also reported that a deficiency in HO levels, such as in the HO-2 null (HO-2−/−) mouse, exacerbates inflammation in response to corneal epithelial injury, leading to massive infiltration of inflammatory cells, delayed healing, corneal scaring, edema, perforation, and neovascularization (12, 13, 15). On injury, neutrophils start to infiltrate the wound within hours and are followed by macrophages, which number peaks 4 d after injury (13). In the HO-2−/− mice, systemic neutrophil depletion worsened rather than improved wound healing, suggesting that the absence of HO-2 within other cells in the cornea contributes to the impaired corneal healing in HO-2−/− mice (14). Histologic staining and cytokine profiling indicated that the number of infiltrating macrophages after injury is increased in the corneas of HO-2−/− mice compared with wild-type (WT) mouse corneas, a direct result of the increased number of infiltrating neutrophils (11, 13). As indicated above, macrophages play a crucial role in the resolution phase (8), and one of their major functions is to phagocytose apoptotic neutrophils. Indeed, our previous studies suggest that the number of apoptotic cells in the injured HO-2−/− cornea is higher than in the injured WT cornea (12). The HO system plays a crucial role in the resolution of inflammation and wound repair, and inflammatory cells are major contributors of the observed increase in HO levels in injured tissue (9). Based on these findings, we hypothesize that the underlying cause of the impaired corneal wound healing and exaggerated and prolonged inflammatory response in the HO-2−/− mouse is, at least in part, a dysfunctional macrophage phenotype.
By use of experimental approaches of systemic macrophage depletion and bone marrow chimeras of HO-2−/− and WT mice, we examined the impact of HO-2 deficiency on macrophage polarization and phagocytic activity and the contribution of macrophage HO-2 to the inflammatory and repair responses in the cornea.
MATERIALS AND METHODS
Animals
All animal experiments were performed following a protocol approved by the Institutional Animal Care and Use Committee and in accordance with the U.S. National Institutes of Health Guide for Care and Use of Laboratory Animals. The HO-2 null (HO-2−/−) mice are direct descendants of the HO-2 mutants produced by Poss et al. (16) on a C57BL/6×129/Sv genetic background. HO-2+/− mice were bred and genotyped using HO-2– and neomycin (Neo)-specific primers as described previously (17). HO-2−/− and age-matched WT mice were used at 6–8 wk of age. There were no significant gender differences in healing; therefore, both genders were used in this study. Mice were anesthetized with ketamine (50 mg/kg) and xylazine (20 mg/kg) intramuscularly, and a drop of 0.5% tetracaine-HCl was applied to the eye to deliver local anesthesia before animals were subjected to injury. The corneal epithelium was removed up to the corneal/limbal border with a 0.5 mm corneal rust ring remover (Algerbrush II; Alger Equipment, Lago Vista, TX, USA). Wound closure (re-epithelialization) was monitored by fluorescein staining. Images of the anterior surface were taken with a dissecting microscope (Discovery V12; Carl Zeiss, Jena, Germany) coupled to a digital camera (AxioCam HRc; Carl Zeiss) and analyzed using Zeiss software (Axiovision 4.6). Mice were killed either 4 or 7 d after injury, corneas were removed, and corneas free of conjunctival tissue were dissected and processed for selected analyses.
Macrophage depletion
Liposomes were encapsulated with clodronate/dichloromethylene diphosphonate (a kind gift from Roche Diagnostics GmbH, Mannheim, Germany). Macrophage depletion was achieved by intraperitoneal injection of a 0.2 ml suspension of clodronate- (5 mg/ml) or PBS-containing liposomes 24 h before epithelial injury and then every 2 d thereafter. Intraperitoneal injection of clodronate successfully depletes macrophages in the peritoneal cavity, spleen, liver, bone marrow, and blood (18). Successful depletion of macrophages was confirmed by immunostaining of spleen sections as outlined below. Some cells positive for cluster of differentiation (CD)68 were observed in the spleens of all mice, but the overall success of depletion of macrophages was estimated to be at least 80% effective.
Myeloperoxidase activity
Measurement of myeloperoxidase (MPO) activity was used to quantify polymorphonuclear cells (PMNs) in dissected corneas as described previously (13). In brief, tissues were homogenized in potassium phosphate buffer (pH 6.0) containing 0.5% hexadecyltrimethylammonium bromide, followed by 3 cycles of sonication and freeze-thaw. The particulate matter was removed by centrifugation, and MPO activity in the supernatant was measured by spectrophotometry using o-dianisidine dihydrochloride reduction as a colorimetric indicator. Calibration curves for conversion of MPO activities to PMN number were established with PMNs that were collected from zymosan A-induced peritonitis in mice.
Isolation and culture of macrophages
WT and HO-2−/− mice were administered sodium thioglycolate (TG; 0.1% in 100 µl PBS; Sigma-Aldrich, St. Louis, MO, USA) via intraperitoneal injection 4 d prior to collection of peritoneal exudate. TG has been widely used as a simple and reproducible method to elicit macrophages (19). Primary peritoneal residential and TG-elicited macrophages were isolated from WT and HO-2−/− mice by flushing of the peritoneal cavity with 5 ml of sterile PBS. Cells were collected by centrifugation at 300 × g for 5 min at 4°C, counted, and resuspended in RPMI 1640 medium containing 10% fetal bovine serum (FBS). Cells were plated at a density of 5 × 105 cells per well in a 6-well plate and allowed to adhere for 2 h. In some experiments, cells were treated with SnCl2 (1 µM; Sigma-Aldrich), biliverdin (10 µM; Frontier Scientific, Logan, UT, USA), and Tiron (1 mM; Sigma-Aldrich) for 12 h prior to assessing phagocytic activity as below. Nonadherent cells were removed by aspiration of medium and by carefully rinsing the plates twice with sterile PBS. RAW 264.7 macrophages were plated in a 6-well plate (5 × 105 cells per well) and incubated overnight prior to assessing phagocytic activity. For HO-2 knockdown, nontarget and target specific shRNA plasmids (Origene, Rockville, MD, USA) were used in combination with transfection reagent (X-tremeGENE 9; Roche Applied Science, Indianapolis, IN, USA) according to the instructions provided by the manufacturer. Successful knockdown was confirmed by quantitative PCR and shown to be on average 50%.
Phagocytosis assay
Medium was aspirated, and fresh medium containing Texas Red-conjugated Zymosan A particles (Life Technologies, Grand Island, NY, USA) at a ratio of 5:1 (Zymosan particles:cell) were added to the wells. Cells were incubated for 1 h at 37°C, after which the Zymosan-containing medium was removed, and cells were rinsed with PBS. Cells were then incubated for 10 min at 37°C with medium containing Lyticase (100 U/ml) to enzymatically remove Zymosan particles that had adhered to the cell surface. Cells were rinsed with PBS, fixed in 4% paraformaldehyde in PBS on ice for 10 min and then rinsed 2 times with PBS. Nuclei were counterstained with DAPI for 30 min. Images were captured using an AxioImager M1 fluorescence microscope (Carl Zeiss). Images were analyzed using the ImageJ software and the MBF plugin. The number of Zymosan particles per field was obtained by dividing the total fluorescent area with the area of an average Zymosan particle. The resulting number was then divided by the number of cells per field. A minimum of 8 separate fields per well was captured using a ×40 water immersion objective. Data are expressed as the number of Zymosan particles per cell relative to control.
Real-time PCR
Total RNA was isolated using Spinsmart RNA extraction kit (Denville Scientific, South Plainfield, NJ, USA), and RNA was quantified by the Take3 plate and BioTek plate reader (Biotek, Winooski, VT, USA). Reverse transcription reaction of total RNA was performed using the qScript cDNA synthesis kit (Quanta Bioscience, Gaithersburg, MD, USA). Quantitative real-time PCR was performed using PerfeCTa SYBR Green QPCR FastMix (Quanta Bioscience) and the Stratagene Mx3000p real-time PCR system (Agilent Technologies, Santa Clara, CA, USA). Specific primers were designed based on published sequences (GenBank, http://www.ncbi.nlm.nih.gov/) and were as follows: HO-1, sense 5′-AAGCCGAGAATGCTGAGTTCA-3′ and anti-sense 5′-GCCGTGTAGATATGGTACAAGGA-3′; HO-2, sense 5′-ACCGAGCAGAAAATACCCAGT-3′ and antisense, 5′-GTTGCGGTCCATTTCCTCCTC-3′; IL-1, sense 5′-GCACCTTACACCTACCAGAGT-3′ and antisense 5′-TGCAGGTCATTTAACCAAGTGG-3′; IL-6, sense 5′- TAGTCCTTCCTACCCCAATTTCC-3′ and antisense 5′-TTGGTCCTTAGCCACTCCTTC-3′; IL-10, sense 5′-GCTGGACAACATACTGCTAACC-3′ and antisense 5′-ATTTCCGATAAGGCTTGGCAA-3′; arginase-1, sense 5′-TGTCCCTAATGACAGCTCCTT-3′ and antisense 5′ GCATCCACCCAAATGACACAT-3′; iNOS, sense 5′-GCTCCCTATCTTGAAGCCCC-3′ and antisense 5′-GACACTTCGCACAAAGCAGG-3′; TNF, sense 5′-GAGAAGTTCCCAAATGGCCT-3′ and antisense 5′-GAGAACCTGGGAGTAGACAA-3′; CD54, sense 5′-GTGATGCTCAGGTATCCATCCA-3′ and antisense 5′-CACAGTTCTCAAAGCACAGCG-3′; CD80, sense 5′-GCTGTGTCGTTCAAAAGAAGGA-3′ and antisense 5′-TGGGAAATTGTCGTATTGATGCC-3′; CD163, sense 5′-CTGGCGGGTGGTGAAAACA-3′ and antisense 5′-CAGCCGTTACTGCACACTG-3′; CD206, sense 5′-CTCTGTTCAGCTATTGGACGC-3′ and antisense 5′-CGGAATTTCTGGGATTCAGCTTC-3′; arachidonate lipoxygenase (Alox)5, sense 5′-ACTACATCTACCTCAGCCTCATT-3′ and antisense 5′-GGTGACATCGTAGGAGTCCAC-3′; Alox15, sense 5′-GGGATCGGAGTACACGTTCC-3′ and antisense 5′-GAGGTCAGAGATACTGGTCGC-3′; cyclooxygenase 2 (COX-2), sense 5′-TGAGCAACTATTCCAAACCAGC-3′ and antisense 5′-GCACGTAGTCTTCGATCACTATC-3′; Ym1 sense 5′-CAGGTCTGGCAATTCTTCTGAA-3′ and antisense 5′-GTCTTGCTCATGTGTGTAAGTGA-3′; and Fizz1 sense 5′-CCAATCCAGCTAACTATCCCTCC-3′ and antisense 5′-ACCCAGTAGCAGTCATCCCA-3′; and 18s, sense 5′-GATGGGCGGCGGGAAAATAG-3′ and antisense 5′-GCGTGGATTCTGCATAATGG-3′. PCR efficiency for each primer pair was determined by quantifying amplification with increasing concentrations of template cDNA, and specific amplification was verified by subsequent analysis of the melt curve profile for each amplification. A nontemplate control served as a negative control to exclude the formation of primer dimers or any other nonspecific PCR products. RNA expression of target genes was calculated based on the real-time PCR efficiency and the threshold crossing point and is expressed in comparison to the reference gene 18s as described previously (13). Data are expressed as mRNA expression relative to WT peritoneal residential macrophages (PRMs).
Adoptive bone marrow transfer
Isolation of bone marrow
Bone marrow donor mice were killed with ketamine (50 mg/kg) and xylazine (20 mg/kg) intramuscularly, followed by cervical dislocation. Killed mice were immediately immersed in 70% ethanol for several minutes, the skin was removed from the lower extremities, and the hind legs were cut off. The skeletal muscles were removed from the tibias and femurs, which were placed in tubes containing ice-cold DMEM (Lonza, Walkersville, MD, USA) containing 10% FBS (Atlanta Biologicals, Lawrenceville, GA, USA) and 1% antibiotics (penicillin/streptomycin/amphotericin B; MP Biomedicals, Santa Ana, CA, USA). The bone marrow was exposed by cutting of the bone caps under sterile condition and collected into sterile centrifuge tubes by flushing the bones with DMEM containing 10% FBS and heparin (10 U/ml; APP Pharmaceuticals, Schaumburg, IL, USA). The cells were collected by centrifugation at 300 × g for 5 min at 4°C. The supernatant was aspirated, and the cell pellet was resuspended in 10 ml red cell lysis buffer (0.15 M NH4Cl; 0.01 M KHCO3; 0.1 M Na2EDTA, pH 7.2–7.4) and incubated on ice for 15 min. Cells were then collected by centrifugation, washed twice with PBS containing 1% albumin, and resuspended to a single cell suspension in PBS containing 1% albumin. Trypan blue was used to visualize dead cells. Overall, viability greater than 95% was observed. Cells were collected by centrifugation and resuspended in DMEM containing 1% albumin and heparin (5 U/ml) at a concentration of 5 × 106 cells/100 µl.
Irradiation and injection of bone marrow
Recipient mice received a lethal dose of γ-irradiation (900 rad) followed by injection of 10 × 106 bone marrow cells in 200 μl into the tail vein. One week before and 4 wk after irradiation, recipient mice were placed on acidified water (pH 3.0) supplemented with 0.17 mg/ml enrofloxacin (Baytril; Bayer Healthcare, Shawnee Mission, KS, USA). The resulting chimeric mice were subjected to corneal epithelium removal 6 wk after bone marrow transfer. Successful chimerism was confirmed by genotyping of blood collected at the time of death using the same primers as used for the original genotyping (17).
Histology and immunostaining
Dissected corneas and spleens were fixed in 4% paraformaldehyde at 4°C for a few hours, washed 3 times with PBS, incubated overnight in 30% sucrose, and then embedded in optimal cutting temperature compound (Sakura Finetek, Torrance, CA, USA). Cryostat sections were cut transversely into 5 µm thick sections. Immunofluorescence staining was performed using the following antibodies: rat anti-mouse CD68 (1:100; AbD Serotec, Raleigh, NC, USA), rabbit anti–HO-2 (1:100; Enzo Life Sciences, Farmingdale, NY, USA), Alexa Fluor-conjugated goat anti-rabbit and goat anti-rat antibodies (1:1000–1:2000; Life Technologies), and Cy3-conjugated goat anti-rat antibody (1:1000–1:2000; Jackson Immunoresearch Laboratories, West Grove, PA, USA). To further verify cellular entity, sections were counterstained for nuclei with DAPI for 15 min. Immunofluorescence was visualized using a Zeiss AxioImager M1 fluorescent microscope. Images were captured and analyzed using AxioVision multichannel image processing software (Carl Zeiss) (11).
Statistical analysis
For comparison between treatment groups, the null hypothesis was tested by either a single-factor ANOVA for multiple groups or unpaired t test for 2 groups, and the data are presented as mean ± sem; P < 0.05 was considered significant.
RESULTS
Macrophage depletion impairs wound healing in WT but not in HO-2−/− mice
We examined the effect of systemic macrophage depletion on corneal wound healing following epithelial injury in WT and HO-2−/− mice. As seen in Fig. 1A–B, epithelial removal produced a consistent wound in control WT mice (7.2 ± 0.1 mm2, n = 17) that exhibited 79.2 ± 2.4% and 98.0 ± 0.6% re-epithelialization 2 and 4 d after injury, respectively. In contrast, wound closure in macrophage-depleted WT mice (7.5 ± 0.1 mm2, n = 15) was delayed, displaying 71.1 ± 2.4% and 84.8 ± 4.8% re-epithelialization 2 and 4 d after injury, respectively. The impaired rate of re-epithelialization observed in HO-2−/− mice (7.5 ± 0.1 mm2, n = 17), 67.0 ± 2.4% and 77.0 ± 3.4% re-epithelialization by days 2 and 4 after injury, respectively, was not different from the re-epithelialization rate observed in macrophage-depleted HO-2−/− mice (7.6 ± 0.1 mm2, n = 14; 66.2 ± 2.9% and 80.9 ± 4.9% re-epithelialization 2 and 4 d after injury, respectively).
Figure 1.

Effect of macrophage (MØ) depletion on wound healing and neutrophil infiltration. A) Representative images of fluorescein-stained corneas at days 2 and 4 after injury in control and MØ-depleted HO-2+/+ (WT) and HO-2−/− mice. B) Wound closure as percent change from day 0. Results are mean ± sem; n = 10–20. *P < 0.05 and **P < 0.01 from control WT mice. C) Neutrophil infiltration into the cornea at day 4 after injury. Left, WT mice; right, HO-2−/− mice; white bars, control mice receiving PBS liposomes; black bars, MØ-depleted mice receiving clodronate liposomes. Results are mean ± sem; n = 6–10. *P < 0.05 and **P < 0.01 from control (PBS liposome-treated) WT mice.
Macrophage depletion increases neutrophil infiltration into injured corneas of WT but not HO-2−/− mice
The delayed healing that was observed in the macrophage-depleted WT mice was associated with a 50% increase of infiltrating neutrophils in the injured cornea at day 4 after injury compared with WT control mice (Fig. 1C). The number of neutrophils in the injured corneas of control and macrophage-depleted HO-2−/− mice was more than 2-fold greater compared with WT control mice. However, the number of neutrophils in macrophage-depleted HO-2−/− mice was not different from that in injured corneas of control HO-2−/− mice (Fig. 1C).
HO-2 deficiency impairs macrophage phagocytic activity in vitro
As shown in Fig. 2A, PRMs isolated from HO-2−/− mice exhibited a 30% reduction in phagocytic activity in vitro compared with PRMs isolated from WT mice. To examine whether deficiency in HO activity accounts for the reduction in phagocytic activity, we used interventions to increase HO-1 expression and amplify HO activity. Thus, pretreatment of HO-2−/− PRMs with SnCl2, an inducer of HO-1 expression and HO activity (20), increased (43%) their phagocytic capacity compared with control, whereas preincubation of WT PRMs with SnCl2 had little effect on their phagocytic activity. Biliverdin, a product of HO activity, increased phagocytic activity by 40% and 60% in PRMs from WT and HO-2−/− mice, respectively. Because biliverdin has antioxidative activity in addition to its anti-inflammatory capacity (10), we assessed whether addition of an antioxidant such as Tiron (21) has a similar effect. As seen in Fig. 2B, Tiron had no effect on the phagocytic activity of either HO-2−/− or WT PRMs. That HO-2 deficiency impairs phagocytic function was further confirmed using the mouse macrophage cell line RAW 264.7. RAW 264.7 cells treated with HO-2 shRNA lost 40% of their phagocytic activity, whereas treatment with nontarget shRNA had no effect (Fig. 2C). Furthermore RAW 264.7 cells treated with SnCl2 exhibited a 35% increase in phagocytic activity (Fig. 2D).
Figure 2.

Effect of HO-2 deficiency on macrophage phagocytosis. A) Number of Zymosan particles per cell relative to WT control with and without SnCl2 (1 µM) preincubation. Results are the mean ± sem; n = 2–4. ***P < 0.001. White bars, WT; black bars, HO-2−/− cells. B) Number of Zymosan particles per cell relative to vehicle control with and without biliverdin (BVD; 10 µM) or Tiron (1 mM) preincubation. Results are the mean ± sem; n = 1–3. ***P < 0.001. White bars, vehicle control cells; black bars, biliverdin-treated cells; gray bars, Tiron-treated cells. C) Number of Zymosan particles per cell relative to control. Results are the mean ± sem; n = 3. *P < 0.05 for HO-2 shRNA-treated (black bar) from control (white bar); ‡P < 0.05 for nontarget shRNA (gray bar). D) Number of Zymosan particles per cell relative to vehicle control with and without SnCl2 (1 or 5 µM) preincubation. Results are the mean ± sem; n = 2–3. ***P < 0.05 for SnCl2 1 µM treated (black bar) from vehicle control (white bar).
HO-2 deficiency alters cytokine production by macrophages
The finding of reduced phagocytic activity in macrophages lacking the HO-2 gene suggested that these macrophages are functionally impaired and phenotypically distinct. Analysis of expression of cytokines that are key for the control of the initiation and resolution of inflammation indicated that levels of IL-10 and TNF-α mRNA were markedly suppressed in HO-2−/− compared with WT PRMs (Fig. 3A–B), whereas mRNA levels of IL-1 and IL-6 were not significantly different (Fig. 3C–D). The expression levels of these cytokines were increased by manyfold (10–1000) in both WT and HO-2−/− TG-elicited macrophages (Fig. 3). Notably, the expression of IL-6 in activated HO-2−/− was 5 times higher than in activated WT macrophages (Fig. 3D). The finding of significant differences in the expression of inflammatory and anti-inflammatory cytokines between nonactivated and activated WT and HO-2−/− macrophages suggest that HO-2−/− macrophages have a compromised M1/M2 phenotype and/or transition.
Figure 3.

Real-time PCR analysis of mRNA expression of IL-10 (A), TNF (B), IL-1 (C), and IL-6 (D) in PRMs and TG-elicited peritoneal macrophages isolated from WT (white bars) and HO-2−/− (black bars) mice. Results are presented as mRNA expression relative to WT PRMs and are the mean ± sem; n = 5–10. *P < 0.05, **P < 0.01, and ***P < 0.001 from WT PRMs. Comparison between WT and HO-2−/− PRMs using unpaired t test: †P < 0.05 and ‡P < 0.001.
HO-2 deficiency alters the expression of classically activated macrophage and alternatively activated macrophage surface and cellular markers
Figure 4 depicts the relative expression of markers associated with classically activated macrophage (M1) and alternatively activated macrophage (M2) polarization (22) in PRMs and TG-elicited macrophages from WT and HO-2−/− mice. Expression levels of the M1-associated markers CD80 and iNOS were lower in HO-2−/− compared with WT PRMs (Fig. 4B–C), whereas the mRNA level of CD54, an M1-associated marker, was not significantly different (Fig. 4A). HO-2−/− macrophages also displayed a distinct expression pattern for M2-associated markers that generally suggested an impaired transition to M2 macrophages. Although CD163 expression was elevated in HO-2−/− PRMs, it failed to increase on activation (Fig. 4D). Conversely, mRNA levels of CD206 and arginase-1 were lower in HO-2−/− PRMs and their induction following activation was significantly impaired compared with WT macrophages (Fig. 4E, F). Interestingly, neither Ym1 nor Fizz1 mRNA levels were altered in HO-2−/− macrophages compared with WT macrophages (Fig. 4G–H). However, Fizz1 mRNA levels were significantly elevated in TG-elicited macrophages compared with nonactivated macrophages (Fig. 4H).
Figure 4.

Real-time PCR analysis of mRNA expression of macrophage M1-M2 polarization associated genes. A) CD54, B) CD80, C) iNOS, D) arginase 1, E) CD206, F) CD163, G) Ym1, and H) Fizz1 in PRMs and TG-elicited peritoneal macrophages isolated from WT (white bars) and HO-2−/− (black bars) mice. Results are presented as mRNA expression relative to WT PRMs and are the mean ± sem; n = 4–10. *P < 0.05, **P < 0.01, and ***P < 0.001 from WT PRMs (i.e., nonelicited macrophages).
HO-2 deficiency impairs HO-1 expression and inducibility in macrophages
Expression and induction of HO-1 in macrophages have been shown to modulate cytokine production, surface receptor expression, maturation state, and polarization toward pro- (M1) and anti- (M2) inflammatory phenotypes (23). Interestingly, HO-1 expression in HO-2−/− PRMs was markedly suppressed compared with WT PRMs. Moreover, HO-1 inducibility in activated TG-elicited HO-2−/− macrophages was significantly impaired compared with corresponding WT macrophages (Fig. 5A). Other genes that are linked to HO-1 induction and to the resolution of inflammation, including Alox15, Alox5, and COX-2 (24–26), showed similar pattern of low expression and activation in HO-2−/− macrophages (Fig. 5B–D). We previously reported that HO-2 deficiency greatly influences the expression and inducibility of HO-1 following injury (11, 13, 27). It is unclear, however, exactly how HO-2 affects HO-1 expression and activity.
Figure 5.

Real-time PCR analysis of mRNA expression of HO-1 (A), Alox15 (B), Alox5 (C), and COX-2 (D) in PRMs and TG-elicited peritoneal macrophages isolated from WT (white bars) and HO-2−/− (black bars) mice. Results are presented as mRNA expression relative to WT PRMs and are the mean ± sem; n = 5–10; *P < 0.05, **P < 0.01, and ***P < 0.001 from WT PRMs.
Adoptive transfer of bone marrow from HO-2−/− to WT mice does not affect the rate of wound healing or inflammation
On the basis of the findings that HO-2 deficiency negatively impacts macrophage phagocytosis and evidence, suggesting their important role in the wound healing process, we further assessed whether transfer of WT bone marrow to HO-2−/− mice would ameliorate the inflammatory response and accelerate the repair response and vice versa: whether transfer of HO-2−/− bone marrow would confer some degree of impaired healing and exaggerated inflammatory response on corneal epithelial injury in WT mice. As seen in Fig. 6A, the rate of wound healing in WT mice receiving HO-2−/− bone marrow (61.5 ± 2.1%, 97.3 ± 1.2%, and 99.1 ± 0.4% at days 2, 4, and 7, respectively) was not different from the rate observed in WT mice receiving WT bone marrow (61.7 ± 2.58%, 96.5 ± 2.0%, and 100 ± 0.0% at days 2, 4, and 7, respectively). Likewise, neutrophil infiltration into the healing cornea of WT mice was not impacted by transfer of either WT or HO-2−/− bone marrow (39,158 ± 3,523 vs. 42,640 ± 6174 neutrophils/cornea for syngeneic vs. allogeneic bone marrow, respectively; Fig. 6B). However, the rate of re-epithelialization in HO-2−/− mice receiving WT bone marrow (46.2 ± 1.2%, 79.1 ± 3.0%, and 90.1 ± 3.1% at days 2, 4, and 7, respectively) was accelerated at days 4 and 7 after injury compared with HO-2−/− mice receiving HO-2−/− bone marrow (50.8 ± 3.2%, 58.9 ± 1.3%, and 81.2 ± 4.2% at days 2, 4, and 7, respectively; Fig. 6A). Notably, corneal re-epithelialization in HO-2−/− bone marrow chimeras remained impaired compared with WT bone marrow chimeras (Fig. 6A). Similarly, injury-stimulated neutrophil infiltration remained exaggerated in corneas of HO-2−/− bone marrow chimeras compared with WT bone marrow chimeras (Fig. 6B). Moreover, the number of infiltrating neutrophils at day 7 after injury was not altered by allogeneic bone marrow in either HO-2−/− mice or WT mice (Fig. 6B). However, the number of infiltrating neutrophils into the injured corneas of HO-2−/− mice receiving HO-2−/− bone marrow was increased compared with injured corneas of WT mice receiving WT bone marrow. These results clearly indicate that adoptive transfer of WT bone marrow to HO-2−/− mice did not fully correct the impaired repair response of the HO-2−/− cornea. Likewise, adoptive transfer of HO-2−/− bone marrow to WT did not confer exaggerated inflammatory response and impaired healing in WT cornea.
Figure 6.
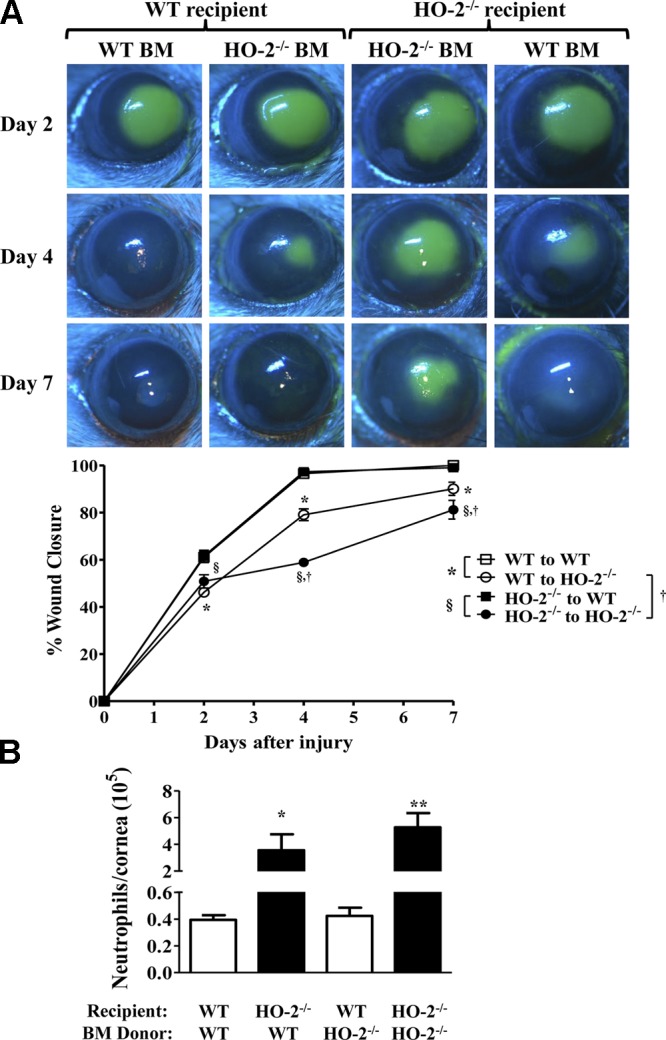
Effect of allogeneic bone marrow transfer on wound healing and neutrophil infiltration. A) Representative images of fluorescein-stained corneas at days 2, 4, and 7 after injury in syngeneic and allogeneic WT and HO-2−/− mice. B) Wound closure as percent change from day 0. Open squares, WT mice receiving WT bone marrow; closed squares, WT mice receiving HO-2−/− bone marrow; open circles, HO-2−/− mice receiving WT bone marrow; closed circles, HO-2−/− mice receiving HO-2−/− bone marrow. Results are mean ± sem; n = 5–10. *P < 0.05 comparison of the effect of WT bone marrow on wound healing in WT vs. HO-2−/− recipients; §P < 0.05 comparison of the effect of HO-2−/− bone marrow on wound healing in WT vs. HO-2−/− recipients; and †P < 0.05 comparison between HO-2−/− recipients of WT vs. HO-2−/− bone marrow and its effect on wound healing. B) Neutrophil infiltration into the cornea at day 7 after injury. White bars, WT recipients; black bars, HO-2−/− recipients. Results are mean ± sem; n = 5–10. *P < 0.05 comparison of the effect of WT bone marrow on corneal neutrophil infiltration in WT vs. HO-2−/− recipients; **P < 0.01 comparison of the effect of HO-2−/− bone marrow on corneal neutrophil infiltration in WT vs. HO-2−/− recipient mice.
HO-2 deficiency increases the number of macrophages in injured cornea
We further analyzed for the presence of macrophages in injured corneas of WT and HO-2−/− mice receiving syngeneic bone marrow at day 7 after injury by immunofluorescence staining of corneal sections for CD68. As seen in Fig. 7A, CD68 immunofluorescence was more pronounced in HO-2−/− bone marrow chimeras than in WT bone marrow chimeras. Quantitative analysis of the number of CD68 positive macrophages expressed as total CD68-positive fluorescent area showed an increase in the corneas of both groups of HO-2−/− mice regardless of bone marrow donor strain compared with WT mice receiving WT bone marrow (Fig. 7B).
Figure 7.
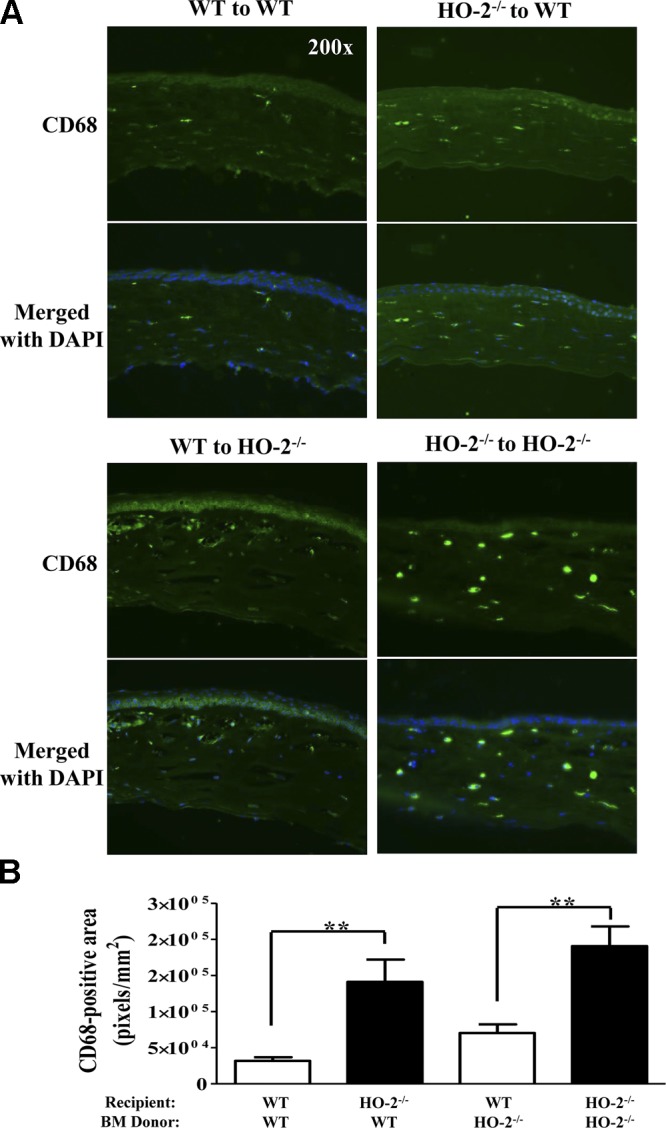
Corneas of HO-2–deficient mice maintain an increased macrophage population following injury. A) Representative images of frozen corneal sections stained for CD68 (green). Nuclei are counterstained with DAPI. Corneal sections day 7 after injury from WT mouse receiving WT bone marrow (top left); WT mouse receiving HO-2−/− bone marrow (top right); WT mouse receiving HO-2−/− bone marrow (bottom left); and HO-2−/− receiving HO-2−/− bone marrow (bottom right). B) Indirect measurement of CD68-positive cells expressed as total number of CD68-positive (green) pixels per square millimeter. Results are the mean ± sem; n = 5. **P < 0.01. Pairwise comparison of the effect of WT bone marrow on corneal macrophage infiltration in WT and HO-2−/− recipient mice (left) and HO-2−/− bone marrow in WT vs. HO-2−/− recipient mice (right).
Confirmation of chimerism by genotyping of whole blood and by immunohistological analysis of corneal HO-2 expression after injury
To confirm that successful repopulation of bone marrow occurred, we performed genotyping on whole blood obtained at death. Fig. 8A shows an agarose gel with PCR amplicons from 3 WT mice receiving HO-2−/− bone marrow, followed by 3 HO-2−/− mice receiving WT bone marrow. Two PCR reactions per mouse were performed: 1 using primers specific for the WT HO-2 (lower band, smaller amplicon) and 1 using primers specific for the knockout (Neo; upper band, larger amplicon). Because the DNA yields were less than optimal and some PCR bands barely visible, we confirmed the chimerism through histologic analysis of frozen corneal sections. Fig. 8B depicts representative images of frozen corneal sections stained for HO-2 (red) and CD68 (green). In the cornea of a WT mouse receiving HO-2−/− bone marrow (left), the corneal epithelium is positively stained for HO-2, whereas several CD68-positive macrophages are negative for HO-2. Conversely, the corneal epithelium in the HO-2−/− mouse receiving WT bone marrow (right) is devoid of HO-2 staining, whereas several CD68-positive cells in the underlying stroma are stained positive for HO-2 protein.
Figure 8.

Confirmation of chimerism through genotyping of whole blood and immunohistological analysis of corneal HO-2 expression after injury in chimeric mice. A) PCR fragments on 1% agarose gel from 3 WT mice receiving HO-2−/− bone marrow, followed by 3 HO-2−/− mice receiving WT bone marrow. Two PCR reactions for each mouse: 1 for the WT (HO-2) fragment followed by the HO-2−/− (Neo) fragment. B) Representative images of frozen corneal sections stained for HO-2 (red) and CD68 (green). Nuclei are counterstained with DAPI. Left, WT mouse receiving HO-2−/− bone marrow; right, HO-2−/− mouse receiving WT bone marrow. HO-2–positive staining in red (top), CD68-positive staining in green (middle), and merged images with DAPI (bottom).
DISCUSSION
This study demonstrates, for the first time, that HO-2 is a critical factor in the determination of macrophages polarization and phagocytic activity in inflammation. However, its expression within macrophages is not sufficient to correct for the exaggerated inflammation and aberrant repair of the HO-2–null cornea in response to injury. Consequently, we propose that HO-2 within the injured tissue is necessary for controlling the magnitude of the ensuing inflammation and, consequently, for promoting the healing of the injured tissue. Four key findings substantiate this conclusion.
The first key finding is that depletion of macrophages impairs corneal epithelial wound healing and exacerbates the inflammatory response, i.e., increased neutrophil infiltration, in the WT mouse, whereas in the HO-2−/− mouse, macrophage depletion has no impact on the already impaired corneal epithelial wound healing and the increased infiltration of neutrophils into the cornea (13). Our results are in agreement with previous reports that show the adverse effect of macrophage depletion on wound healing in rodents (5, 28, 29). Impaired wound healing, as often observed in diabetes, has also been attributed to a reduced number and activation of infiltrating macrophages (30).
The second key finding is that phagocytosis of fluorophore-conjugated Zymosan particles is significantly impaired in HO-2–deficient macrophages. Dysfunctional phagocytosis increases the risk of secondary necrosis, which in turn facilitates prolonged inflammation. We previously demonstrated that the number of CD68-positive cells, i.e., macrophages in the stroma during the healing process of the injured cornea of the HO-2−/− mice, is higher compared with corresponding corneas of WT mice (11, 13). We also showed that treatment of HO-2−/− corneas with biliverdin reduced the number of TUNEL-positive cells, suggesting that the HO activity is critical to the ability of macrophages to remove apoptotic cells (12). To this end, we previously showed that injury/stress-stimulated HO-1 induction is impaired in the HO-2−/− mice, resulting in deficient HO activity (11, 13). HO-1 has been implicated as a master regulator of mononuclear phagocyte function in health and disease (23). A recent study by Chiang et al. (31), substantiated the proresolving role of HO-1 in acute inflammation and further showed that adding HO metabolic products such as CO significantly increased macrophages carrying ingested apoptotic PMN in exudates and enhanced PMN apoptosis in a murine model of peritonitis. In the current study, expression of HO-1 in the HO-2−/− macrophages was shown to be more than 5-fold lower than that expressed in the WT macrophages. Together, these reports, along with our observations that the phagocytic capacity of HO-2–deficent macrophages is impaired and that this impairment can be ameliorated or even completely prevented by either inducing HO-1 expression and activity by SnCl2 or by adding the HO product biliverdin, clearly indicates that HO-1/HO-2 expression and HO activity play an important role in the phagocytic capacity of macrophages.
The third key finding is that HO-2 deficiency alters the functional phenotype of macrophages. The macrophages play a major role in the orchestration of the repair process during inflammation. They go through dynamic changes during different phases of wound healing with classically activated M1 cells implicated in initiating and maintaining inflammation and alternatively activated M2 or M2-like cells associated with resolution of inflammation and repair through their efficient phagocytic activity (25). The macrophage functional phenotype depends on the wound microenvironment, changing from an early phase proinflammatory environment to a later stage proresolving and tissue-regenerating favoring milieu. Hence, during the early stages of wound healing, macrophages exert a proinflammatory typical M1-associated function, whereas at the later stages, macrophages exert an anti-inflammatory, proapoptotic, and tissue-regenerating M2-associated function (32). Here we show that the HO-2−/− macrophages could not be simply characterized as either M1 or M2/M2-like phenotypes. In fact, the data appear to imply that the HO-2−/− macrophages are more likely representing an impaired M1 phenotype and a failure to undergo M1-M2 activation. Hence, expression of IL-1 and IL-6, which are associated with the M1 phenotype, was elevated in the HO-2−/− macrophages. Conversely, the mRNA levels of TNF and iNOS, 2 genes that are normally associated with proinflammatory M1 polarized macrophages, were markedly suppressed in the PRMs from HO-2−/− mice. TNF plays a crucial role in the activation of macrophages during the initial stages of inflammation, whereas iNOS plays an important role in the death of microbes through the formation of peroxynitrite in the lysosome-phagolysosome during phagocytosis. Deficiency in their expression may contribute to impaired immune response and activation (33).
The expression levels of markers for the M2/M2-like phenotype were markedly lower in the HO-2−/− macrophages. Hence, HO-1 and IL-10 mRNA levels were severalfold lower in PRMs isolated from HO-2−/− mice compared with PRMs isolated from WT mice. This is in agreement with previous reports showing that HO-1 is involved in the M2 polarization of macrophages (23, 34, 35). Furthermore, HO-1 potentiates the effects of IL-10 and vice versa (23, 36). IL-10–producing macrophages preferentially phagocytose early apoptotic cells, thereby preventing inflammation caused by late apoptotic and necrotic cells (37). IL-10 is a potent inducer of HO-1 in primary and cultured macrophages and protects mice against death on injection of a lethal dose of LPS (23, 38). Another well-characterized regulatory circuit that promotes macrophage activation and resolution of inflammation is the lipoxygenase-derived proresolving lipid mediators including lipoxins, resolvins, and maresins (39–41). Their mechanism of action includes in part induction of HO-1 and activation of the HO system. Interestingly, HO-2−/− macrophages displayed markedly lower levels of 12/15-lipoxygenase and 5-lipoxygenase; both enzymes participate in the production of these proresolving mediators and their low expression may contribute not only to the impaired induction of HO-1 but also to the dysfunction of the HO-2−/− macrophages. We previously documented that in the absence of HO-2, the expression level of HO-1 and the HO activity in the cornea, as well as the levels of HO-1 protein in macrophages infiltrating the cornea after injury, are reduced (11, 13). It is unclear, however, exactly how HO-2 affects HO-1 expression and activity. Colocalization studies suggested interplay between the HO-1 and HO-2 enzymes that may regulate their expression, as well as HO activity (42). It is important to note that the source of the mRNA is nonelicited peritoneum residing cells and thus potentially phenotypically different from the macrophages entering the corneal tissue following injury. Under these conditions, the HO-2−/− PRM population cannot be simply classified. What is clear, however, is that compared with WT, the HO-2−/− PRMs are potentially impaired with regard to microbial killing, endotoxin resistance, and phagocytosis of microbes and apoptotic cells.
The fourth key finding revealed that, although HO-2–null macrophages exhibit decreased phagocytic function, corneal wound healing in the WT mouse receiving HO-2–deficient bone marrow is not impaired compared with the WT mouse receiving WT bone marrow. It is noteworthy that transfer of HO-2–deficient bone marrow to the WT mouse did not cause an exaggerated inflammatory response. In contrast, in the HO-2−/− mouse receiving WT bone marrow, wound healing was accelerated compared with the HO-2–null mice receiving HO-2–null bone marrow; whereas the rate of wound healing increased, wound closure remained incomplete, suggesting that grafting functionally normal macrophages to repopulate the HO-2–deficient cornea is, in itself, insufficient for the completion of the repair response to injury. Finally, the number of macrophages in injured corneas of HO-2−/− mice was elevated regardless of bone marrow donor strain compared with WT mice receiving WT bone marrow, indicating that inflammation was not lessened by transfer of WT HO-2–expressing bone marrow cells. These results are in line with our previous results documenting increased levels of monocyte chemotactic protein 1 and macrophage inflammatory protein 2 in the debrided and sutured corneas of HO-2−/− mice (11, 13).
In summary, the present study details a compromised macrophage phenotype in the HO-2–deficient mouse that is characterized by a defective phagocytic function and M1-M2 polarization that may contribute, in part, to the impaired resolution of inflammation after injury. However, the failure to improve wound healing in the HO-2−/− bone marrow chimeras points out that it is HO-2 expression in the injured tissue rather than infiltrating macrophages or neutrophils (15) that is crucial for an ordered inflammatory and reparative response. Excessive inflammation delays wound healing, and our results clearly indicate that the absence of HO-2 halts the wound healing response in a proinflammatory milieu, thereby preventing the subsequent resolution and tissue regeneration.
Acknowledgments
The authors thank Dr. Jason Lee for assistance with the analysis of macrophage phagocytosis. Research reported in this publication was supported by the National Eye Institute of the U.S. National Institutes of Health (NIH) under award R01EY06513 (to M.L.S.) and Fight for Sight Grant-in-Aid under award GA13000 (to L.B.). The authors declare no conflicts of interest. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Glossary
- Alox
arachidonate lipoxygenase
- CD
cluster of differentiation
- COX-2
cyclooxygenase 2
- FBS
fetal bovine serum
- HO
heme oxygenase
- HO-2−/−
knockout for HO-2
- MPO
myeloperoxidase
- M1
classically activated macrophage
- M2
alternatively activated macrophage
- Neo
neomycin
- PMN
polymorphonuclear leukocyte
- PRM
peritoneal residential macrophage
- TG
thioglycolate
- WT
wild-type
REFERENCES
- 1.Lu P., Li L., Liu G., van Rooijen N., Mukaida N., Zhang X. (2009) Opposite roles of CCR2 and CX3CR1 macrophages in alkali-induced corneal neovascularization. Cornea 28, 562–569 [DOI] [PubMed] [Google Scholar]
- 2.Sivak J. M., Ostriker A. C., Woolfenden A., Demirs J., Cepeda R., Long D., Anderson K., Jaffee B. (2011) Pharmacologic uncoupling of angiogenesis and inflammation during initiation of pathological corneal neovascularization. J. Biol. Chem. 286, 44965–44975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slegers T. P., Torres P. F., Broersma L., van Rooijen N., van Rij G., van der Gaag R. (2000) Effect of macrophage depletion on immune effector mechanisms during corneal allograft rejection in rats. Invest. Ophthalmol. Vis. Sci. 41, 2239–2247 [PubMed] [Google Scholar]
- 4.Slegers T. P., van der Gaag R., van Rooijen N., van Rij G., Streilein J. W. (2003) Effect of local macrophage depletion on cellular immunity and tolerance evoked by corneal allografts. Curr. Eye Res. 26, 73–79 [DOI] [PubMed] [Google Scholar]
- 5.Leibovich S. J., Ross R. (1975) The role of the macrophage in wound repair. A study with hydrocortisone and antimacrophage serum. Am. J. Pathol. 78, 71–100 [PMC free article] [PubMed] [Google Scholar]
- 6.Ross R., Odland G. (1968) Human wound repair. II. Inflammatory cells, epithelial-mesenchymal interrelations, and fibrogenesis. J. Cell Biol. 39, 152–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Odland G., Ross R. (1968) Human wound repair. I. Epidermal regeneration. J. Cell Biol. 39, 135–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Willoughby D. A., Moore A. R., Colville-Nash P. R., Gilroy D. (2000) Resolution of inflammation. Int. J. Immunopharmacol. 22, 1131–1135 [DOI] [PubMed] [Google Scholar]
- 9.Wagener F. A., van Beurden H. E., von den Hoff J. W., Adema G. J., Figdor C. G. (2003) The heme-heme oxygenase system: a molecular switch in wound healing. Blood 102, 521–528 [DOI] [PubMed] [Google Scholar]
- 10.Abraham N. G., Kappas A. (2008) Pharmacological and clinical aspects of heme oxygenase. Pharmacol. Rev. 60, 79–127 [DOI] [PubMed] [Google Scholar]
- 11.Bellner L., Vitto M., Patil K. A., Dunn M. W., Regan R., Laniado-Schwartzman M. (2008) Exacerbated corneal inflammation and neovascularization in the HO-2 null mice is ameliorated by biliverdin. Exp. Eye Res. 87, 268–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bellner L., Wolstein J., Patil K. A., Dunn M. W., Laniado-Schwartzman M. (2011) Biliverdin Rescues the HO-2 Null Mouse Phenotype of Unresolved Chronic Inflammation Following Corneal Epithelial Injury. Invest. Ophthalmol. Vis. Sci. 52, 3246–3253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seta F., Bellner L., Rezzani R., Regan R. F., Dunn M. W., Abraham N. G., Gronert K., Laniado-Schwartzman M. (2006) Heme oxygenase-2 is a critical determinant for execution of an acute inflammatory and reparative response. Am. J. Pathol. 169, 1612–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marrazzo G., Bellner L., Halilovic A., Li Volti G., Drago F., Dunn M. W., Schwartzman M. L. (2011) The role of neutrophils in corneal wound healing in HO-2 null mice. PLoS ONE 6, e21180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bellner L., Patil K. A., Castellano K., Halilovic A., Dunn M. W., Schwartzman M. L. (2011) Targeted suppression of HO-2 gene expression impairs the innate anti-inflammatory and repair responses of the cornea to injury. Mol. Vis. 17, 1144–1152 [PMC free article] [PubMed] [Google Scholar]
- 16.Poss K. D., Thomas M. J., Ebralidze A. K., O’Dell T. J., Tonegawa S. (1995) Hippocampal long-term potentiation is normal in heme oxygenase-2 mutant mice. Neuron 15, 867–873 [DOI] [PubMed] [Google Scholar]
- 17.Rogers B., Yakopson V., Teng Z. P., Guo Y., Regan R. F. (2003) Heme oxygenase-2 knockout neurons are less vulnerable to hemoglobin toxicity. Free Radic. Biol. Med. 35, 872–881 [DOI] [PubMed] [Google Scholar]
- 18.Van Rooijen N., Sanders A. (1997) Elimination, blocking, and activation of macrophages: three of a kind? J. Leukoc. Biol. 62, 702–709 [DOI] [PubMed] [Google Scholar]
- 19.Gallily R., Warwick A., Bang F. B. (1964) Effect of Cortisone of Genetic Resistance to Mouse Hepatitis Virus in Vivo and in Vitro. Proc. Natl. Acad. Sci. USA 51, 1158–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patil K., Bellner L., Cullaro G., Gotlinger K. H., Dunn M. W., Schwartzman M. L. (2008) Heme oxygenase-1 induction attenuates corneal inflammation and accelerates wound healing after epithelial injury. Invest. Ophthalmol. Vis. Sci. 49, 3379–3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ito Y., Pagano P. J., Tornheim K., Brecher P., Cohen R. A. (1996) Oxidative stress increases glyceraldehyde-3-phosphate dehydrogenase mRNA levels in isolated rabbit aorta. Am. J. Physiol. 270, H81–H87 [DOI] [PubMed] [Google Scholar]
- 22.Mosser D. M. (2003) The many faces of macrophage activation. J. Leukoc. Biol. 73, 209–212 [DOI] [PubMed] [Google Scholar]
- 23.Hull T. D., Agarwal A., George J. F. (2014) The mononuclear phagocyte system in homeostasis and disease: a role for heme oxygenase-1. Antioxid. Redox. Signal. 20, 1770.– [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brogliato A. R., Antunes C. A., Carvalho R. S., Monteiro A. P., Tinoco R. F., Bozza M. T., Canetti C., Peters-Golden M., Kunkel S. L., Vianna-Jorge R., Benjamim C. F. (2012) Ketoprofen impairs immunosuppression induced by severe sepsis and reveals an important role for prostaglandin E2. Shock 38, 620–629 [DOI] [PubMed] [Google Scholar]
- 25.Sica A., Mantovani A. (2012) Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122, 787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uderhardt S., Kronke G. (2012) 12/15-lipoxygenase during the regulation of inflammation, immunity, and self-tolerance. J. Mol. Med. (Berl) 90, 1247–1256 [DOI] [PubMed] [Google Scholar]
- 27.Bellner L., Martinelli L., Halilovic A., Patil K., Puri N., Dunn M. W., Regan R. F., Schwartzman M. L. (2009) Heme oxygenase-2 deletion causes endothelial cell activation marked by oxidative stress, inflammation, and angiogenesis. J. Pharmacol. Exp. Ther. 331, 925–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li S., Li B., Jiang H., Wang Y., Qu M., Duan H., Zhou Q., Shi W. (2013) Macrophage depletion impairs corneal wound healing after autologous transplantation in mice. PLoS ONE 8, e61799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mirza R., DiPietro L. A., Koh T. J. (2009) Selective and specific macrophage ablation is detrimental to wound healing in mice. Am. J. Pathol. 175, 2454–2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maruyama K., Asai J., Ii M., Thorne T., Losordo D. W., D’Amore P. A. (2007) Decreased macrophage number and activation lead to reduced lymphatic vessel formation and contribute to impaired diabetic wound healing. Am. J. Pathol. 170, 1178–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiang N., Shinohara M., Dalli J., Mirakaj V., Kibi M., Choi A. M., Serhan C. N. (2013) Inhaled carbon monoxide accelerates resolution of inflammation via unique proresolving mediator-heme oxygenase-1 circuits. J. Immunol. 190, 6378–6388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahdavian Delavary B., van der Veer W. M., van Egmond M., Niessen F. B., Beelen R. H. (2011) Macrophages in skin injury and repair. Immunobiology 216, 753–762 [DOI] [PubMed] [Google Scholar]
- 33.Marino M. W., Dunn A., Grail D., Inglese M., Noguchi Y., Richards E., Jungbluth A., Wada H., Moore M., Williamson B., et al. (1997) Characterization of tumor necrosis factor-deficient mice. Proc. Natl. Acad. Sci. USA 94, 8093–8098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abraham N. G., Drummond G. (2006) CD163-Mediated hemoglobin-heme uptake activates macrophage HO-1, providing an antiinflammatory function. Circ. Res. 99, 911–914 [DOI] [PubMed] [Google Scholar]
- 35.Weis N., Weigert A., von Knethen A., Brüne B. (2009) Heme oxygenase-1 contributes to an alternative macrophage activation profile induced by apoptotic cell supernatants. Mol. Biol. Cell 20, 1280–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee T. S., Chau L. Y. (2002) Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat. Med. 8, 240–246 [DOI] [PubMed] [Google Scholar]
- 37.Xu W., Roos A., Schlagwein N., Woltman A. M., Daha M. R., van Kooten C. (2006) IL-10-producing macrophages preferentially clear early apoptotic cells. Blood 107, 4930–4937 [DOI] [PubMed] [Google Scholar]
- 38.Howard M., Muchamuel T., Andrade S., Menon S. (1993) Interleukin 10 protects mice from lethal endotoxemia. J. Exp. Med. 177, 1205–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Serhan C. N., Yang R., Martinod K., Kasuga K., Pillai P. S., Porter T. F., Oh S. F., Spite M. (2009) Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J. Exp. Med. 206, 15–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwab J. M., Chiang N., Arita M., Serhan C. N. (2007) Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 447, 869–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Serhan C. N. (2010) Novel lipid mediators and resolution mechanisms in acute inflammation: to resolve or not? Am. J. Pathol. 177, 1576–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weng Y. H., Yang G., Weiss S., Dennery P. A. (2003) Interaction between heme oxygenase-1 and -2 proteins. J. Biol. Chem. 278, 50999–51005 [DOI] [PubMed] [Google Scholar]