

Abstract
Sepsis remains the leading cause of morbidity and mortality in critically ill patients. Excessive inflammation is a major cause of organ failure and mortality in sepsis. Ectonucleoside triphosphate diphosphohydrolase 1, ENTPDase1 (CD39) is a cell surface nucleotide-metabolizing enzyme, which degrades the extracellular purines ATP and ADP, thereby regulating purinergic receptor signaling. Although the role of purinergic receptor signaling in regulating inflammation and sepsis has been addressed previously, the role of CD39 in regulating the host’s response to sepsis is unknown. We found that the CD39 mimic apyrase (250 U/kg) decreased and knockout or pharmacologic blockade with sodium polyoxotungstate (5 mg/kg; IC50 ≈ 10 μM) of CD39 increased mortality of mice with polymicrobial sepsis induced by cecal ligation and puncture. CD39 decreased inflammation, organ damage, immune cell apoptosis, and bacterial load. Use of bone marrow chimeric mice revealed that CD39 expression on myeloid cells decreases inflammation in septic mice. CD39 expression is upregulated during sepsis in mice, as well as in both murine and human macrophages stimulated with Escherichia coli. Moreover, E. coli increases CD39 promoter activity in macrophages. Altogether, these data indicate CD39 as an evolutionarily conserved inducible protective pathway during sepsis. We propose CD39 as a novel therapeutic target in the management of sepsis.—Csóka, B., Németh, Z. H., Törő, G., Koscsó, B., Kókai, E., Robson, S. C., Enjyoji, K., Rolandelli, R. H., Erdélyi, K., Pacher, P., Haskó, G. CD39 improves survival in microbial sepsis by attenuating systemic inflammation.
Key Words: interleukin, TNF, MIP, lung, kidney
Sepsis is a leading cause of mortality in intensive care units and is the 10th leading cause of death overall in high-income countries (1). More than 2 decades ago, the term systemic inflammatory response syndrome (SIRS) was introduced to describe the underlying massive inflammatory reaction of the body that contributes to the development of sepsis (2). Although, since then, the term SIRS has been shown to lack specificity in predicting sepsis, the underlying concept that uncontrolled inflammation predisposes to sepsis remains valid (3). The SIRS, or hyperinflammatory, theory of sepsis states that, in response to an infectious or traumatic insult, the body unleashes an inflammatory reaction consisting of an overproduction of a host of inflammatory mediators by macrophages and other myeloid cells, which leads to endothelial injury, organ damage, mitochondrial dysfunction, and eventually death in critically ill patients (4). This idea is supported by a recent large-scale human study, which has provided evidence that, in critically ill patients after blunt trauma or burn injury, leukocytes display a prolonged and simultaneous increase in the expression of inflammatory genes (5). This increased inflammatory reaction is often complicated and exacerbated by an inability of the immune system to control bacterial growth, the main causes of which are immune cell apoptosis and dysfunction (6–9).
The purinergic system consists of cell surface purinergic receptors, which fall into the P1 or adenosine receptor family and the P2 or ATP/ADP receptor family, as well as metabolic enzymes, which convert extracellular purines to each other and nucleoside transporters, which terminate purinergic receptor signaling by purine uptake into the cell (10, 11). The purinergic system has been shown to fine tune immune cell functions, such as cytokine and chemokine secretion, intracellular pathogen removal, and apoptosis. Although the general view is that P1 receptors are anti-inflammatory, P2 receptors are viewed as mediating proinflammatory signaling. In this regard, ectonucleoside triphosphate diphosphohydrolase 1, ENTPDase1 (CD39) serves as a molecular switch in the purinergic system, as it converts proinflammatory ATP/ADP to inactive AMP, which is then further metabolized by another cell surface enzyme, ecto-5′-nucleotidase (CD73), to adenosine, which is anti-inflammatory (12). CD39 can therefore decrease inflammation both by decreasing ATP/ADP and increasing adenosine availability. In addition to hematopoietic immune cells such as macrophages (13) and regulatory T cells (14), CD39 on nonhematopoietic cells, such as endothelial (15) and epithelial (16) cells, has also been shown to regulate inflammation.
Multiple lines of evidence have demonstrated that CD39 can protect organs against noninfectious inflammatory, hypoxic, and ischemic insults (17–20), all of which can contribute to the development of sepsis (6, 7). For example, it was recently shown that LPS-induced neutrophil trafficking to the lung was exacerbated in the genetic absence or after pharmacological inhibition of CD39 and that this increased lung inflammation was attenuated by administering soluble CD39 (21). In addition, in CD39-deficient mice, the organ injury and inflammation that followed cardiac (18, 22, 23), renal (19, 23, 24), hepatic (20, 25), and intestinal (26, 27) ischemia-reperfusion injury were more severe than in the corresponding wild-type (WT) mice.
In contrast to its protective functions in noninfectious insults, the role of CD39 in infections is more complex. Indeed, many pathogens can use CD39 to generate an adenosine-rich milieu, which allows them to escape immune surveillance and which favors their invasion of and dissemination in the host (28, 29). Thus, the role of CD39 in sepsis, a disease that has both noninfectious and infectious components, is hard to predict. To address the role of CD39 in sepsis, we investigated the role of CD39 in mice undergoing cecal ligation and puncture (CLP), a widely used model of abdominal sepsis.
MATERIALS AND METHODS
Experimental animals and cell lines
CD73 knockout (KO) mice and their WT littermates were kindly provided by Dr. Linda F. Thompson (Oklahoma Medical Research Foundation, Oklahoma City, OK, USA). CD39 and CD73 KO mice were developed on the C57BL/6 background as previously described (30, 31). All mice were bred and all colonies were maintained in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals, and the experiments were approved by the New Jersey Medical School Animal Care Committee. Sodium polyoxotungstate (POM-1) and apyrase were purchased from Sigma-Aldrich (St. Louis, MO, USA). THP-1 and RAW 264.7 macrophage-like cell lines (ATCC, Manassas, VA, USA) were grown in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum, 50 U/ml penicillin, 50 µg/ml streptomycin, and 1.5 mg/ml sodium bicarbonate in a humidified atmosphere of 95% air and 5% CO2.
CLP
Polymicrobial sepsis was induced by subjecting mice to CLP (32, 33). CD39 KO or WT mice between the ages of 8 and 12 wk were anesthetized with intraperitoneally administered pentobarbital injection (50 mg/kg). Under aseptic conditions, a 2 cm midline laparotomy was performed to allow exposure of the cecum with adjoining intestine. Approximately two-thirds of the cecum was tightly ligated with a 3-0 silk suture, and the ligated part of the cecum was perforated twice (through and through) with a 20.5 gauge needle (BD Biosciences, San Jose, CA, USA). Then the ligated cecum was gently squeezed to extrude a small amount of feces through the perforation site and was returned to the peritoneal cavity. After this, the laparotomy was closed in 2 layers with 4-0 silk sutures. Following the operation, all mice were resuscitated with 1 ml of physiologic saline injected subcutaneously and returned to their cages, where they were provided free access to food and water. In studies where biochemical, immunologic, and bacteriologic analysis were performed, the mice (all male) were reanesthetized with pentobarbital (50 mg/kg, intraperitoneally) 6 or 16 h after the CLP procedure, and blood, peritoneal lavage fluid, and various organs were harvested. A separate set of female CD39 KO and WT mice were used in survival studies. The effect of POM-1 or apyrase was evaluated using male C57BL/6J mice in a similar fashion to that described for the CD39 KO or WT mice. In these experiments, the mice were injected intraperitoneally immediately before the CLP operation with POM-1 (5 mg/kg, intraperitoneally), apyrase (250 U/kg), or their vehicle (DMSO for POM-1 or physiologic saline for apyrase) as described previously (19).
Generation of CD39 bone marrow chimeric mice
Bone marrow chimeras were generated as previously described (34). In brief, male donor mice (8- to 10-wk-old CD39 WT or KO) were euthanized, and bone marrow from the femur was harvested by flushing the marrow cavity with sterile isotonic NaCl solution. The bone marrow cells were centrifuged at 400 g for 5 min, resuspended, and counted. Recipient mice (8- to 10-wk-old CD39 WT or KO mice) were irradiated with a total dose (in 2 doses) of 12 Gray from a [137Cs] source. A total of 107 bone marrow cells per recipient were retro-orbitally injected in 0.2 ml 0.9% NaCl. The resulting chimeric mice were housed for at least 8 wk before experimentation and were fed with water containing tetracycline (100 μg/ml) in the first 2 wk following bone marrow transplantation. Chimeric mice were subjected to CLP and killed 16 h later as described above.
Collection of blood, peritoneal lavage fluid, and organs
After opening the chest of mice, blood samples were obtained aseptically by cardiac puncture using heparinized syringes. The blood samples were placed into heparinized microcentrifuge tubes and kept on ice until further processing for bacteriologic analysis. Serial dilutions for bacteriologic analysis were made as described previously (32–35). The blood samples were centrifuged at 2000 g for 10 min, and then the recovered plasma was stored at –70°C until further use. To collect peritoneal lavage fluid, first the abdominal skin was cleansed with 70% ethanol and the abdominal wall was exposed by opening the skin. Two milliliters of sterile physiologic saline was slowly injected into the peritoneal cavity via an 18 gauge needle. The abdomen was gently massaged for 1 min while keeping the tip of the needle in the peritoneum. In the next step, the peritoneal fluid was recovered through the needle, and the recovered peritoneal lavage fluid was placed on ice until processed for bacteriologic examination. After making serial dilutions of the peritoneal lavage fluid to determine the number of the colony-forming units (CFUs), the peritoneal lavage fluid was centrifuged at 5000 g for 10 min, and the supernatant was stored at –70°C until further analysis. Heart, lung, kidney, liver, and thymus samples were excised and snap frozen using liquid nitrogen.
Quantification of bacterial CFUs from blood and peritoneal lavage fluid
Blood or peritoneal lavage fluid was diluted serially in sterile physiologic saline. Fifty microliters of each dilution was aseptically plated and cultured on Trypticase blood agar plates (BD Biosciences) at 37°C. After 24 h of incubation, the number of bacterial colonies was counted. The number of cultures is expressed as CFUs per milliliter of blood or peritoneal lavage fluid.
Determination of blood urea nitrogen
Blood urea nitrogen (BUN) was analyzed using a clinical chemistry analyzer system (VetTest8008; IDEXX Laboratories), as previously described (33).
Hematoxylin and eosin staining and myeloperoxidase staining of the lung
For histologic analysis, lung specimens were fixed in 10% formalin solution overnight. After automated dehydration through a graded ethyl alcohol series, transverse lung slices were embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin and eosin. Morphologic assessment was performed by a pathologist who was blinded to the origin (WT or KO mice) of lungs.
For myeloperoxidase staining, antigen unmasking was achieved by heat-induced epitope retrieval in citrate buffer. Endogenous peroxidases were inactivated by 3% hydrogen peroxide. Sections were then blocked in normal goat serum for 1 h, followed by an overnight incubation with an myeloperoxidase antibody (Biocare Medical, Concord, CA, USA) in a moist chamber. Immune complexes were detected by using the Vectastain ABC Kit (Vector Laboratories, Burlingame, CA, USA) according to the manufacturer’s instructions. Color development was induced by incubation with the DAB kit (Vector Laboratories) for 4 min. Nuclear Fast Red was used as a counterstain. Finally, the sections were dehydrated in ethanol, cleared in xylene, and mounted. The staining was visualized, and images were acquired using BX-41 microscope and U-TV1x2 camera (Olympus, Tokyo, Japan) with ×200 magnification.
RNA extraction, cDNA synthesis, and real-time PCR
Total RNA was extracted from the lung, liver, and RAW 264.7 and THP-1 macrophage-like cells using TRI reagent (Molecular Research Center, Cincinnati, OH, USA) and reverse transcribed, as described previously (36, 37). For detecting human or murine CD39 mRNA transcripts, a commercially available real-time PCR kit was used (Applied Biosystems, Foster City, CA, USA), and all data were normalized to constitutive rRNA (cyclophylin or 18S) values. Primer sequences were as follows: human CD39 forward, 5′-AGCAGCTGAAATATGCTGGC-3′, reverse, 5′-GAGACAGTATCTGCCGAAGTCC-3′; human cyclophilin forward, 5-GTCTCCTTTGAGCTGTTTGCAGAC-3′, reverse, 5-CTTGCCACCAGTGCCATTATG-3′; mouse CD39 forward, 5′-AGCTGCCCCTTATGGAAGAT-3′, reverse, 5′- TCAGTCCCACAGCAATCAAA-3′; and mouse 18S forward, 5′-GGGAGCCTGAGAAACGGC-3′, reverse, 5′- GGGTCGGGAGTGGGTAATTTT-3′. The Applied Biosystems 7700 sequence detector was used for amplification of target sequences, and quantitation of differences between treatment groups was done using the comparative threshold cycle (CT) method.
Flow cytometric detection of CD39 on macrophages
RAW 264.7 macrophages were seeded at a density of 5 × 105 cells per well and were stimulated with heat-inactivated Escherichia coli (1:15 macrophage/E. coli ratio) for 12 h. After the incubation, macrophages were labeled with anti-CD39 antibody (eBioscience, San Diego, CA, USA) and subsequently with secondary phycoerythrin-conjugated anti-rat IgG2a antibody. Fluorescence-activated cell sorter acquisitions were performed as described previously (34).
Immunofluorescent detection of CD39
RAW 264.7 cells were plated onto glass coverslips at a density of 2 × 105 cells per well in a 24-well plate. The cells were stimulated with heat-killed E. coli (in a 15:1 bacteria/macrophage ratio) for 12 h. After the treatment, the cells were washed with PBS and blocked with 1% bovine serum albumin/PBS for 1 h and then incubated with Alexa Fluor 647-conjugated anti-mouse CD39 (Ebioscience) or Alexa Fluor 647 rat IgG2a isotype control antibody (BD Biosciences) for 1 h. After the staining, the cells were fixed with 4% paraformaldehyde for 10 min. The coverslips were rinsed with PBS and mounted in Mowiol-DABCO antifade media. Images were aquired with a LEICA DMI 6000 CS microscope using LAS AF software. A 63× oil immersion objective was used.
Protein extraction
Organs were homogenized in a Dounce homogenizer in modified radioimmunoprecipitation assay buffer (50 mM Tris HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.25% sodium deoxycholate, 1% Nonidet P-40, 1 μg/ml pepstatin, 1 μg/ml leupeptin, 1 mM PMSF, 1 mM Na3VO4). Then the lysates were centrifuged at 15,000 g for 15 min, and the supernatant was recovered. The Bio-Rad (Hercules, CA, USA) protein assay kit was used to determine the protein concentrations.
Determination of cytokine and chemokine levels
Concentrations of TNF-α, IL-1β, IL-10, IL-6, IL-12p40, MIP-1α, MCP-1, and MIP-2 in blood, peritoneal lavage fluid, and organ extracts (heart, kidney, and lung) were determined using commercially available ELISA kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions.
Generation of 5′ deletion mutants of the CD39 full promoter
For promoter analysis, we used a 1104 bp fragment containing 1039 bp of the 5′-flanking region and 65 bp of the untranslated first exon (full CD39-pGL3 construct; −1039/+65) of mouse CD39 that was cloned into the pGL3 basic vector (the construct was a kind gift from David J. Pinsky, University of Michigan Medical Center, Ann Arbor, MI, USA). For the generation of the 5′-deleted CD39 promoter-luciferase reporter constructs (1: −268/+65, 2: −136/+65), the desired sequences were amplified by PCR using the full CD39-pGL3 construct as a template. The PCR fragments were cloned into the SacI-HindIII cloning sites of the pGL3 basic vector (Promega, Madison, WI, USA). For the PCR amplification, we used the following forward primers: forward 1, 5′-ggcaggagctctataacggaggaaaagagg-3′; forward 2, 5′-ggcaggagctcctctcccaatctgtatttc-3′; and forward 3, 5′-ggcaggagctctgttatatctctgaaccgtg-3′. The SacI recognition site is highlighted by bold lettering in each forward oligonucleotide. The same pGL3 vector sequence-specific reverse oligonucleotide was applied in each PCR reaction, which was 5′-ggtggctttaccaacagtac-3′.
Transient transfection of RAW 264.7 cells with CD39 promoter-luciferase constructs and luciferase assay
RAW 264.7 cells were transiently transfected using FUGENE 6.0 transfection reagent (Roche, Indianapolis, IN, USA). For transfection, 1 × 106/ml cells were seeded in a 24-well plate. The next day, the cells were transfected with 0.3 µg of CD39 reporter constructs or pGL3 control reporter plasmid. All transfections were performed at 37°C overnight, and the following day the cells were washed with medium and stimulated with E. coli (1:15 macrophage/bacteria ratio) for 4, 8, or 16 h. Luciferase activities were determined using Luciferase Reporter Assay System (Promega). Luciferase activity was normalized to protein concentration.
Statistical analysis
Survival statistics were performed using a Kaplan-Meier curve and log-rank test. To compare cytokine concentrations, CFU numbers, and all other laboratory parameters, as well as densitometry readouts, the Student’s t test or Mann-Whitney U test was used. Statistical significance was assigned to P < 0.05.
RESULTS
CD39 improves survival in CLP-induced sepsis
To begin to study the role of CD39 in sepsis, we injected septic C57BL/6J mice with the CD39 mimic apyrase (250 U/kg) or vehicle. As Fig. 1 shows, apyrase improved the survival of mice with sepsis both when its administration was initiated before (Fig. 1A) and 6 h after (Fig. 1B) the septic insult. We then examined the effect of genetic CD39 deficiency on survival. We found that CD39 KO mice showed decreased survival compared with WT mice (Fig. 1C). We then further examined the role of CD39 in regulating sepsis by using the selective CD39 inhibitor, POM-1 (5 mg/kg). POM-1–treated C57BL/6J mice displayed decreased survival compared with vehicle-treated mice (Fig. 1D), confirming the deleterious effect of CD39 KO. The variability between the mortality of control animals from POM-1 and apyrase treatment was due to the fact that the vehicle of the drugs was different. As is well documented, DMSO can exhibit an anti-inflammatory effect by reducing NF-κB activation in association with decreased expression of proinflammatory mediators (38). The vehicle in POM-1–treated animals was DMSO, whereas apyrase-administered mice were injected with physiologic saline as vehicle. In summary, we established that CD39 protects mice against sepsis-induced mortality.
Figure 1.

CD39 improves survival in CLP-induced sepsis. Survival of vehicle- (Veh)- or apyrase-treated WT mice (A, B). Apyrase or its vehicle was administrated before CLP (A) or 6 h after CLP (B). Survival of CD39 KO and WT mice (C) and Veh- or POM-1–treated WT mice (D). Surviving mice were counted every day for 7 d after inducing polymicrobial sepsis by way of CLP. Numbers of bacterial CFUs in the peritoneum (E) or blood (F) of WT and CD39 KO animals 16 h after CLP. n = 8 mice/group. Data are presented as mean ± sem. *P < 0.05 and **P < 0.01 vs. WT littermates or sham-operated animals.
Because local bacterial load and systemic bacterial dissemination are central events contributing to death during CLP-induced sepsis, we evaluated the influence of CD39 on bacterial numbers at the primary site of infection and in the bloodstream. CD39 KO mice displayed higher bacterial burden in the peritoneum compared with WT animals after CLP (Fig. 1E). The numbers of bacteria in the blood were comparable in CD39 KO and WT mice (Fig. 1F).
CD39 decreases systemic and peritoneal cytokine and chemokine levels in CLP-induced sepsis
In the next series of experiments, we evaluated the effect of CD39 on sepsis-induced cytokine and chemokine production in the blood and peritoneal lavage fluid. We found that apyrase reduced the blood and peritoneal levels of TNF-α, IL-6, IL-10, IL-12p40, and MIP-2 (Fig. 2A–E). In contrast, we detected elevated TNF-α, IL-6, MIP-1α, and MCP-1 levels in plasma of CD39 KO mice compared with WT animals (Fig. 2F, G, K, L). Peritoneal fluid concentrations of TNF-α, IL-6, IL-1β, IL-10, IL-12p40, MIP-1α, and MIP-2 (Fig. 2F–L) were also higher in septic CD39 KO vs. WT mice. Similar to genetic CD39 KO, pharmacological CD39 inhibition also augmented blood levels of IL-6, IL-10, IL-12p40, MIP-1α, MCP-1, and MIP-2 and peritoneal lavage levels of TNF-α, IL-1β, and MIP-2 (Fig. 2N–U). CD39 KO animals and POM-1–treated animals in some cases show different cytokine/chemokine responses in the peritoneum and systemically in the bloodstream (Fig. 2G, O and Fig. 2I, Q, respectively) or even opposite responses (Fig. 2K, S). These contrasting responses may be due to the fact that these inflammatory mediators are produced by different cell populations locally (i.e., in the peritoneum) and systemically (i.e., blood stream). Taken together, these data show that CD39 prevents excessive inflammation during polymicrobial sepsis.
Figure 2.

CD39 decreases cytokine and chemokine production in CLP-induced sepsis. Blood and peritoneal lavage levels of TNF-α (A), IL-6 (B), IL-10 (C), IL-12 p40 (D), and MIP-2 (E) in apyrase- or vehicle (Veh)-treated mice 16 h after CLP. Results are representative of 2 experiments, n = 10 mice/group. Blood and peritoneal lavage levels of TNF-α (F), IL-6 (G), IL-1β (H), IL-10 (I), IL-12 p40 (J), MIP-1α (K), MIP-2 (L), and MCP-1 (M) in CD39 KO and WT mice 16 h after CLP. n = 8 mice/group. Blood and peritoneal lavage levels of TNF-α (N), IL-6 (O), IL-1β (P), IL-10 (Q), IL-12 p40 (R), MIP-1α (S), MIP-2 (T), and MCP-1 (U) in POM-1– or Veh-treated mice 16 h after CLP. Results are representative of 2 experiments, n = 10 mice/group. Cytokine and chemokine levels were determined using ELISA. Data are presented as mean ± sem. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. WT or Veh-treated mice.
CD39 protects against excessive organ inflammation and injury
To evaluate tissue inflammation, which contributes to organ dysfunction and mortality in sepsis, we measured cytokine and chemokine levels in the heart, lung, and kidney of septic CD39 WT and KO mice. The levels of TNF-α, IL-6, IL-10, MIP-1α, MCP-1, and MIP-2 were higher in the heart of CD39 KO than WT animals 16 h after CLP (Fig. 3A). Moreover, increased concentrations of MIP-1α and MCP-1 (Fig. 3B) were seen in the lung of CD39 KO mice in sepsis compared with WT animals. In agreement with the elevated chemokine levels, we found increased neutrophil infiltration in the lung of CD39 KO mice (Fig. 3C). The levels of MCP-1 in the kidney were higher in septic CD39 KO vs. WT mice after CLP (Fig. 3D). To evaluate kidney function, we measured BUN levels in septic animals. As Fig. 3E, F shows, genetic or pharmacologic inhibition of CD39 augmented BUN levels in animals with sepsis suggesting increased kidney injury compared with WT or vehicle-treated mice, respectively. Altogether, these results indicate a protective role of CD39 against sepsis-induced tissue inflammation and organ damage.
Figure 3.

CD39 protects against sepsis-induced organ injury and inflammation. A) Cytokine and chemokine levels in the heart of WT and CD39 KO animals 16 h after CLP, as determined by ELISA. B) Chemokine levels in the kidney of WT and CD39 KO animals 16 h after CLP. n = 8 mice/group. C) Tissue sections of lungs of septic CD39 KO and WT mice 6 h after CLP stained with hematoxylin and eosin (HE) (top) or with anti-myeloperoxidase antibody (bottom). A representative section is shown from slides from 8 WT and KO animals. The staining was visualized and images were acquired by BX-41 microscope and U-TV1x2 camera (Olympus) with UPlan FLN 20×/0.5 objective (f/1.1 1/240) at a final ×200 magnification, using cellSens Entry software (Olympus). For enhancing quality, TIF file images were further adjusted by Adobe Photoshop CS5 (Adobe Systems Incorporated, San Jose, CA, USA). Layers were duplicated with the mode changed to soft light. All images were processed with the same modifications. D) MCP-1 levels in the kidney of WT and CD39 KO mice 16 h after CLP. E and F) BUN levels in the plasma of CD39 KO vs. WT mice (E) and Veh- or POM-1–treated mice (F) 16 h after CLP. n = 8–10 mice/group. Data are presented as mean ± sem. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. WT or Veh-treated mice.
CD39 deficiency on myeloid cells is responsible for the increased inflammatory response of CD39 KO mice
We next examined whether the increase in cytokine and chemokine levels in CD39 KO mice was due to deficiency of CD39 on myeloid- or non–myeloid-derived cells. Therefore, we created bone marrow chimeric mice. CD39 KO→CD39 WT chimeras with CD39-deficient bone marrow and WT parenchymal cells displayed augmented IL-6 and IL-10 production (Fig. 4C, G) in the blood and increased TNF-α, IL-6, IL-1β, IL-10, MIP-1α, and MIP-2 levels in the peritoneum compared with CD39 WT→CD39 WT (WT bone marrow and WT parenchyma) mice (Fig. 4B, D, F, H, J, L). Furthermore, blood levels of TNF-α, IL-1β, and MIP-1α were comparable in CD39 WT→CD39 WT and CD39 KO→CD39 WT or CD39 KO→CD39 KO (KO bone marrow and KO parenchyma) chimeras (Fig. 4A, E, I, K). These observations suggest that CD39 on hematopoietic cells is pivotal in decreasing the production of cytokines and chemokines in polymicrobial sepsis.
Figure 4.
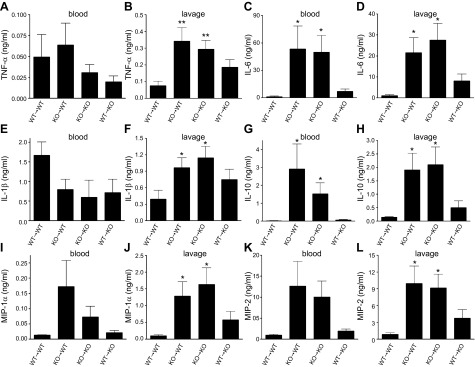
CD39 deficiency on myeloid cells is responsible for the increased inflammatory response of CD39 KO mice. Blood and peritoneal lavage levels of TNF-α (A, B), IL-6 (C, D), IL-1β (E, F), IL-10 (G, H), MIP-1α (I, J), and MIP-2 (K, L) in WT→WT (CD39 WT bone marrow and WT parenchyma), KO→WT (CD39 KO bone marrow and WT parenchyma), KO→KO (CD39 KO bone marrow and KO parenchyma), and WT→KO (CD39 WT bone marrow and KO parenchyma) mice 16 h after CLP. n = 4, 8, 6, and 8 mice/group, respectively. Data are presented as mean ± sem. *P < 0.05 and **P < 0.01 vs. WT→WT mice.
Sepsis upregulates CD39 expression
To further analyze the basis of the protective effects of CD39, we compared CD39 transcript levels in sham- vs. CLP-operated mice. The results showed that CD39 transcript levels increased in lung and liver of septic mice (Fig. 5A, B), emphasizing the potential role of CD39 in sepsis.
Figure 5.

Sepsis upregulates CD39 expression in vivo and in vitro. A, B) mRNA expression of CD39 in lung (A) and liver (B) of CLP- vs. sham-operated WT mice. n = 3–6 mice/group. C–F) Escherichia coli upregulates CD39 expression in murine (C–E) and human (F) THP-1 macrophages. RAW 264.7 macrophages were stimulated with heat-inactivated E. coli (15:1 bacteria/macrophage ratio) for 12 h. CD39 mRNA was detected using real-time PCR (C), and CD39 expression on the cell surface was determined using flow cytometry (D) or immune fluorescence (E). For the and immune fluorescent imaging (E), the fluorescence intensity was quantified using ImageJ software and was normalized to vehicle treatments. THP-1 macrophage-like cells were stimulated with heat-inactivated E. coli (15:1 bacteria/macrophage ratio) for 12 h. CD39 mRNA was detected using real-time PCR (F). G) TLR agonists increase CD39 gene expression in RAW264.7 macrophages. Cells were stimulated with 1 μg/ml PAM3CSK4, 1 μg/ml FSL-1, 10 μg/ml Poly(I:C), 10 μg/ml LPS, 10 μg/ml flagellin, 10 μg/ml ssRNA40, or 5 μM ODN1826 for 12 h. Murine CD39 mRNA was detected using real-time PCR. H) E. coli augments CD39 promoter activity in RAW 264.7 macrophages. RAW 264.7 cells were transiently transfected with full-length CD39 reporter (−1039/+65) or control pGL3 reporter plasmid. Transfected cells were treated with E. coli (1:15 macrophage/bacteria ratio) for 4, 8, or 16 h. Luciferase activities were determined using Dual-Luciferase Reporter Assay System. I) Role of promoter regions in the enhancing effect of E. coli on CD39 promoter activity in macrophages. RAW 264.7 cells were transfected with a series of CD39 promoter deletion mutants that were inserted in the pGL3 luciferase reporter vector. Transfected cells were stimulated with E. coli (1:15 macrophage/bacteria ratio) for 8 h. Luciferase activities are expressed as the mean activity and sem relative to the activity of the empty pGL3 vector after E. coli stimulation followed by normalization to protein concentration. Each result is representative of 3 experiments. Data are presented as mean ± sem. **P < 0.01 and ***P < 0.001 vs. vehicle or 0 h or pGL3.
Because CD39 on hematopoietic cells was required for controlling inflammatory cytokine levels plus the fact that macrophages are the major cytokine-producing cell population in sepsis, we next studied how bacteria and hypoxia, both of which are sepsis-associated factors, regulate CD39 gene expression in macrophages. We challenged murine macrophages with heat-inactivated E. coli (1:15 macrophage/bacteria ratio) for 12 h and then measured mRNA and protein levels of CD39. We observed that E. coli augmented both the mRNA and protein level of CD39 in murine RAW 264.7 macrophages (Fig. 5C–E). We also observed that E. coli upregulated CD39 mRNA accumulation in human macrophage-like (THP-1) cells (Fig. 5F). Finally, hypoxia did not change the expression of CD39 on macrophages (data not shown).
To begin to identify the pattern recognition receptors through which E. coli can upregulate CD39 expression, we treated macrophages with selective TLR or nucleotide-binding oligomerization domain (NOD) agonists. The selective TLR2 agonists Pam3CSK4 and FSL-1, TLR3 agonist poly(I:C), TLR4 agonist LPS, TLR5 agonist flagellin, and TLR9 agonist ODN1826, but not the TLR7 agonist ssRNA40 (Fig. 5G) or NOD (data not shown) agonists, mimicked the effect of E. coli in upregulating CD39 expression. Thus, sepsis-associated factors upregulate CD39 expression on macrophages.
Escherichia coli increases CD39 promoter activity in macrophages
Because the molecular mechanisms by which the induction of CD39 occurs are likely to be vitally important to our understanding of clinical sepsis, we began exploring in detail the molecular mechanisms by which bacteria increase the expression of CD39 on macrophages. We first studied the effect of E. coli on CD39 promoter activity by transfecting RAW 264.7 cells with a construct in which luciferase expression was driven by the full-length CD39 promoter (−1039/+65). We found that E. coli increased CD39 promoter activity approximately 2-fold after 8 h of exposure, and its effect on CD39 promoter activity was more profound (∼6-fold) 16 h after E. coli challenge (Fig. 5H). To begin to identify the DNA region that is responsible for the E. coli increase of CD39 promoter activity in macrophages, a series of promoter mutants that contain successive deletions from the 5′ end was inserted upstream of the luciferase reporter gene. Following transfection of RAW 264.7 cells with these constructs, luciferase activity was measured after E. coli treatment. Analysis of luciferase activity from the 5′ deletion mutants revealed the effect of E. coli was markedly decreased by deletion of sequences between –1039 and –268 and completely abolished by deletion of sequences between −268 and –136 (Fig. 5I). These results show that multiple regions in the CD39 promoter are responsible for mediating full promoter activity after E. coli stimulation.
Adenosine generated by CD39 and CD73 on macrophages suppresses IL-12 and augments IL-10 production
To further investigate the anti-inflammatory role of CD39, we hypothesized that pericellular adenosine derived by CD39 and CD73 expressed on macrophages is important in limiting proinflammatory macrophage functions. We previously showed that extracellular ATP decreases macrophage IL-12 and increases IL-10 responses through its conversion to adenosine (39), which in turn binds to adenosine receptors on the surface of macrophages. Here, we tested the hypothesis that CD39 metabolizes ATP to AMP, which is then followed by conversion of AMP to adenosine by CD73, which in turn will decrease IL-12 and increase IL-10 production by macrophages. We exposed macrophages to ATP in the presence or absence of POM-1 and then activated the cells with LPS. Figure 6A,B shows that ATP inhibits IL-12 and augments IL-10 production by LPS-activated murine macrophages in the absence but not in the presence of POM-1. We then treated macrophages isolated from either WT or CD73 KO mice with AMP followed by activation with LPS and found that AMP suppressed IL-12 production by WT but not CD73 KO macrophages (Fig. 6C). In addition, although AMP increased IL-10 production by both WT and KO macrophages, AMP was less efficacious in CD73 KO than WT macrophages (Fig. 6D). However, CD73 KO macrophages produced reduced, but relatively high, levels of IL-10. One explanation for this observation could be that adenosine is produced by a CD73-independent way. Indeed, besides CD73 and CD39, alkaline phosphatases can also generate adenosine by sequential removal of phosphate groups from ATP (40). Another plausible explanation is that removal of extracellular ATP by ectonucleotidases might be more important in regulating IL-10 production than the generation of adenosine. It is also noteworthy that POM-1 at 50 µM can be an antagonist of P2Y12 receptors, and it has been shown that antagonism of this receptor can reduce IL-10 production (41). Taken together, these results establish that sequential metabolism of ATP to adenosine by CD39 and CD73 on macrophages suppresses IL-12 and augments IL-10 production.
Figure 6.

Effect of ATP and AMP on IL-12 and IL-10 production by macrophages with inactivated CD39 or CD73. A, B) The effect of ATP on IL-12 p40 (A) and IL-10 (B) production by LPS-stimulated peritoneal macrophages in the absence (vehicle) or presence of POM-1. Peritoneal macrophages were stimulated with 10 μg/ml LPS or 300 μM ATP + LPS for 24 h in the absence of presence of POM-1, and then cytokine levels were measured from the supernatants. C, D) The effect of AMP on IL-12 p40 (A) and IL-10 (B) production by LPS-stimulated WT or CD73 peritoneal macrophages. CD73 KO or WT macrophages were stimulated with LPS or 300 μM AMP + LPS for 24 h. Results are representative of 3 experiments. Data are presented as mean ± sem. ***P < 0.001 vs. control (con, no LPS) group and #P < 0.05 or ###P < 0.001 vs. LPS.
DISCUSSION
Our study demonstrates that CD39, which is upregulated during abdominal sepsis, enhances the survival of septic mice and that this enhanced survival is correlated with diminished inflammation and lower abdominal but not systemic bacterial burden. Bone marrow chimeric mouse studies showed that CD39 on myeloid cells decreases inflammation in sepsis. The role of myeloid CD39 was confirmed in vitro, as we demonstrated that bacteria increased CD39 expression on macrophages, thereby promoting the anti-inflammatory properties of these cells.
It is well known that uncontrolled inflammation leads to organ failure and consequently to death in sepsis (6, 42, 43). As our data demonstrate that CD39 suppresses sepsis-induced inflammation and intraorgan cytokine and chemokine production as well as organ injury, this anti-inflammatory effect is likely a major mechanism by which CD39 decreases mortality. The anti-inflammatory properties of CD39 can be due to a direct effect on cytokine production by immune cells. Conversely, CD39 may downregulate inflammation indirectly by decreasing the bacterial burden in the peritoneum. Further studies are needed to explore the exact mechanisms by which CD39 reduces inflammation in sepsis.
CD39 can suppress inflammation by decreasing ATP/ADP concentrations and therefore P2 receptor signaling, increasing adenosine availability and adenosine receptor signaling, or both. It is unclear whether decreased P2 receptor signaling is a factor in the anti-inflammatory effects of CD39 in CLP-induced sepsis, as there are no studies in the literature addressing this question. In contrast, there is evidence that adenosine signaling can decrease inflammation in sepsis. We recently demonstrated that CD73 decreases inflammatory cytokine levels in both plasma and vital organs (lung, heart, and kidney) and increases survival (33). As CD73 converts the inactive metabolite AMP to adenosine, the similar effects of CD39 and CD73 indicate that the anti-inflammatory effects of CD39 are mediated by the downstream CD73-adenosine pathway. Using alternative approaches to increase endogenous adenosine levels, such as inhibition of adenosine deaminase (44) or adenosine kinase (45), others have shown improved survival in sepsis, supporting the beneficial effect of increasing adenosine. In addition, A1 (46), A2A (47), A2B (34), and A3 (48) receptors can all decrease inflammation and improve survival in sepsis.
Our data with bone marrow chimeric mice suggest the involvement of CD39 on hematopoietic cells in downregulating cytokine production in microbial sepsis. Although the particular CD39 expressing cell populations that downregulate inflammation are unknown, macrophages are good candidates because they express CD39 (49–51) and are major regulators of inflammation in sepsis (4, 52, 53). In addition, a recent study showed that CD39 on macrophages is anti-inflammatory in LPS-elicited endotoxemia (49).
It has been shown that the protective effects of CD39 in various inflammatory conditions, such as cardiac (18) and renal ischemia-reperfusion injury (19), during ischemic preconditioning of the liver (20), and in E. coli-induced peritonitis (54), coincide with increased expression of CD39 during these conditions. In accordance with these findings, we also observed upregulated CD39 expression in the lung and liver of septic animals compared with sham-operated mice. These results indicate that CD39 is a universal inducible protective pathway in inflammation.
Our in vitro results together with a recent study (49) with macrophages confirm this inducibility of CD39 in an inflammatory environment, as we observed increased CD39 expression on macrophages after stimulation with bacteria or bacterial products. The cellular mechanisms of CD39 induction in response to bacterial stimulation in macrophages have not been studied in detail. Our results with 5′-deletion mutants of the CD39 promoter indicate the role of a well-described CREB binding region between –210/203 in the CD39 promoter (50) in mediating the bacterial upregulation of CD39 gene expression. That is because the CD39 promoter activity increasing effect of E. coli was completely abolished in deletion mutants missing this region. It is also noteworthy that other regulatory regions may also be important in mediating the effect of E. coli in upregulating CD39 promoter activity. Our results show that in a reporter construct where the promoter region between –1039 and –268 nucleotides is deleted, E. coli-induced promoter activity was decreased to 25% compared with the promoter activity of the full-length CD39 promoter construct. Altogether, further studies are needed to evaluate the precise role of the various promoter regions and particular transcription factors in regulating CD39 expression in response to bacteria and bacterial products.
Because hypoxia also occurs during septic conditions and hypoxia has been shown to cause CD39 mRNA accumulation in human microvascular endothelial cells (HMECs) (55), we also studied the effect of hypoxia in macrophages. However, we found that in macrophages, hypoxia failed to affect CD39 expression, indicating that in septic conditions, bacteria-derived compounds may have a preeminent role in inducing CD39 expression.
We previously proposed targeting particular adenosine receptors for the treatment of sepsis (34, 35). However, targeting individual receptors may not recapture the evolutionary protective role of endogenous adenosine against tissue injury and sepsis. Our data showing that CD39 decreases inflammation and organ injury and improves survival in sepsis indicate that increasing adenosine production by administration of adenosine-producing ectonucleotidases may be superior to targeting individual adenosine receptors in treating clinical sepsis. Before this can be realized, it is important to emphasize that further studies are warranted that will more precisely define the possible clinical utility of ectonucleotidases in sepsis. Perhaps one of the most important outstanding issues is that of antibiotics. Indeed, the treatment with broad-spectrum antibiotics is standard care for sepsis. Thus, in the future, the issue of how CD39 regulates sepsis in conjunction with antibiotics will need to be examined. We propose CD39 supplementation as a novel approach, as it also has the advantage of locally releasing adenosine and targeting adenosine receptors at sites of injury where nucleotides (i.e., ATP and ADP) are released, providing an opportunity for interventions with limited side effects. In addition, CD39 may also work by terminating ATP/ADP signaling, which could be another advantage over adenosine receptor ligands. Further studies will determine the efficacy and the precise mechanisms of the protective effect of CD39 in sepsis.
Acknowledgments
This work was supported by U.S. National Institutes of Health (NIH) Grant R01GM66189 (to G.H.), U.S. Army Medical Research and Materiel Command Grants 09065004/W81XWH-10-1-1015 and W81XWH1210091 (to G.H.), Hungarian Scientific Research Fund (OTKA) Grants CK 78275 and K109178 (to G.H.) and human MB08-1-2011-0015 OTKA 84685 (to E.K.), and the Intramural Research Program of the U.S. NIH/National Institute on Alcohol Abuse and Alcoholism (to P.P.).
Glossary
Abbreviations:
- BUN
blood urea nitrogen
- CD39
ectonucleoside triphosphate diphosphohydrolase 1, ENTPDase1
- CD73
ecto-5′-nucleotidase
- CFU
colony-forming unit
- CLP
cecal ligation and puncture
- KO
knockout
- MIP
macrophage inflammatory protein
- MCP
monocyte chemoattractant protein
- NOD
nucleotide-binding oligomerization domain
- POM-1
sodium polyoxotungstate
- SIRS
systemic inflammatory response syndrome
- WT
wild type
REFERENCES
- 1.Van der Poll T., and Opal S. M. (2008) Host-pathogen interactions in sepsis. Lancet Infect. Dis. 8, 32–43 [DOI] [PubMed] [Google Scholar]
- 2.Bone R. C., Balk R. A., Cerra F. B., Dellinger R. P., Fein A. M., Knaus W. A., Schein R. M., and Sibbald W. J.; ACCP/SCCM Consensus Conference Committee (2009) Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. 1992. Chest 136(5, Suppl)e28. [DOI] [PubMed] [Google Scholar]
- 3.Robertson C. M., and Coopersmith C. M. (2006) The systemic inflammatory response syndrome. Microbes Infect. 8, 1382–1389 [DOI] [PubMed] [Google Scholar]
- 4.Stearns-Kurosawa D. J., Osuchowski M. F., Valentine C., Kurosawa S., and Remick D. G. (2011) The pathogenesis of sepsis. Annu. Rev. Pathol. 6, 19–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiao W., Mindrinos M. N., Seok J., Cuschieri J., Cuenca A. G., Gao H., Hayden D. L., Hennessy L., Moore E. E., Minei J. P., Bankey P. E., Johnson J. L., Sperry J., Nathens A. B., Billiar T. R., West M. A., Brownstein B. H., Mason P. H., Baker H. V., Finnerty C. C., Jeschke M. G., López M. C., Klein M. B., Gamelli R. L., Gibran N. S., Arnoldo B., Xu W., Zhang Y., Calvano S. E., McDonald-Smith G. P., Schoenfeld D. A., Storey J. D., Cobb J. P., Warren H. S., Moldawer L. L., Herndon D. N., Lowry S. F., Maier R. V., Davis R. W., and Tompkins R. G.; Inflammation and Host Response to Injury Large-Scale Collaborative Research Program (2011) A genomic storm in critically injured humans. J. Exp. Med. 208, 2581–2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hotchkiss R. S., and Karl I. E. (2003) The pathophysiology and treatment of sepsis. N. Engl. J. Med. 348, 138–150 [DOI] [PubMed] [Google Scholar]
- 7.Riedemann N. C., Guo R. F., and Ward P. A. (2003) The enigma of sepsis. J. Clin. Invest. 112, 460–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ayala A., and Chaudry I. H. (1996) Immune dysfunction in murine polymicrobial sepsis: mediators, macrophages, lymphocytes and apoptosis. Shock 6(Suppl 1), S27–S38 [PubMed] [Google Scholar]
- 9.Oberholzer A., Oberholzer C., and Moldawer L. L. (2001) Sepsis syndromes: understanding the role of innate and acquired immunity. Shock 16, 83–96 [DOI] [PubMed] [Google Scholar]
- 10.Junger W. G. (2011) Immune cell regulation by autocrine purinergic signalling. Nat. Rev. Immunol. 11, 201–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Antonioli L., Pacher P., Vizi E. S., and Haskó G. (2013) CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 19, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colgan S. P., Eltzschig H. K., Eckle T., and Thompson L. F. (2006) Physiological roles for ecto-5′-nucleotidase (CD73). Purinergic Signal. 2, 351–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lévesque S. A., Kukulski F., Enjyoji K., Robson S. C., and Sévigny J. (2010) NTPDase1 governs P2X7-dependent functions in murine macrophages. Eur. J. Immunol. 40, 1473–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deaglio S., Dwyer K. M., Gao W., Friedman D., Usheva A., Erat A., Chen J. F., Enjyoji K., Linden J., Oukka M., Kuchroo V. K., Strom T. B., and Robson S. C. (2007) Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 204, 1257–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaczmarek E., Koziak K., Sévigny J., Siegel J. B., Anrather J., Beaudoin A. R., Bach F. H., and Robson S. C. (1996) Identification and characterization of CD39/vascular ATP diphosphohydrolase. J. Biol. Chem. 271, 33116–33122 [DOI] [PubMed] [Google Scholar]
- 16.Lu W., Reigada D., Sévigny J., and Mitchell C. H. (2007) Stimulation of the P2Y1 receptor up-regulates nucleoside-triphosphate diphosphohydrolase-1 in human retinal pigment epithelial cells. J. Pharmacol. Exp. Ther. 323, 157–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eltzschig H. K., Thompson L. F., Karhausen J., Cotta R. J., Ibla J. C., Robson S. C., and Colgan S. P. (2004) Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood 104, 3986–3992 [DOI] [PubMed] [Google Scholar]
- 18.Köhler D., Eckle T., Faigle M., Grenz A., Mittelbronn M., Laucher S., Hart M. L., Robson S. C., Müller C. E., and Eltzschig H. K. (2007) CD39/ectonucleoside triphosphate diphosphohydrolase 1 provides myocardial protection during cardiac ischemia/reperfusion injury. Circulation 116, 1784–1794 [DOI] [PubMed] [Google Scholar]
- 19.Grenz A., Zhang H., Hermes M., Eckle T., Klingel K., Huang D. Y., Müller C. E., Robson S. C., Osswald H., and Eltzschig H. K. (2007) Contribution of E-NTPDase1 (CD39) to renal protection from ischemia-reperfusion injury. FASEB J. 21, 2863–2873 [DOI] [PubMed] [Google Scholar]
- 20.Hart M. L., Gorzolla I. C., Schittenhelm J., Robson S. C., and Eltzschig H. K. (2010) SP1-dependent induction of CD39 facilitates hepatic ischemic preconditioning. J. Immunol. 184, 4017–4024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reutershan J., Vollmer I., Stark S., Wagner R., Ngamsri K. C., and Eltzschig H. K. (2009) Adenosine and inflammation: CD39 and CD73 are critical mediators in LPS-induced PMN trafficking into the lungs. FASEB J. 23, 473–482 [DOI] [PubMed] [Google Scholar]
- 22.Eckle T., Krahn T., Grenz A., Köhler D., Mittelbronn M., Ledent C., Jacobson M. A., Osswald H., Thompson L. F., Unertl K., and Eltzschig H. K. (2007) Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation 115, 1581–1590 [DOI] [PubMed] [Google Scholar]
- 23.Grenz A., Osswald H., Eckle T., Yang D., Zhang H., Tran Z. V., Klingel K., Ravid K., and Eltzschig H. K. (2008) The reno-vascular A2B adenosine receptor protects the kidney from ischemia. PLoS Med. 5, e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grenz A., Zhang H., Eckle T., Mittelbronn M., Wehrmann M., Köhle C., Kloor D., Thompson L. F., Osswald H., and Eltzschig H. K. (2007) Protective role of ecto-5′-nucleotidase (CD73) in renal ischemia. J. Am. Soc. Nephrol. 18, 833–845 [DOI] [PubMed] [Google Scholar]
- 25.Hart M. L., Much C., Gorzolla I. C., Schittenhelm J., Kloor D., Stahl G. L., and Eltzschig H. K. (2008) Extracellular adenosine production by ecto-5′-nucleotidase protects during murine hepatic ischemic preconditioning. Gastroenterology 135, 1739–1750 [DOI] [PubMed] [Google Scholar]
- 26.Hart M. L., Grenz A., Gorzolla I. C., Schittenhelm J., Dalton J. H., and Eltzschig H. K. (2011) Hypoxia-inducible factor-1α-dependent protection from intestinal ischemia/reperfusion injury involves ecto-5′-nucleotidase (CD73) and the A2B adenosine receptor. J. Immunol. 186, 4367–4374 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Hart M. L., Henn M., Köhler D., Kloor D., Mittelbronn M., Gorzolla I. C., Stahl G. L., and Eltzschig H. K. (2008) Role of extracellular nucleotide phosphohydrolysis in intestinal ischemia-reperfusion injury. FASEB J. 22, 2784–2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santos R. F., Pôssa M. A., Bastos M. S., Guedes P. M., Almeida M. R., Demarco R., Verjovski-Almeida S., Bahia M. T., and Fietto J. L. (2009) Influence of Ecto-nucleoside triphosphate diphosphohydrolase activity on Trypanosoma cruzi infectivity and virulence. PLoS Negl. Trop. Dis. 3, e387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sansom F. M., Newton H. J., Crikis S., Cianciotto N. P., Cowan P. J., d’Apice A. J., and Hartland E. L. (2007) A bacterial ecto-triphosphate diphosphohydrolase similar to human CD39 is essential for intracellular multiplication of Legionella pneumophila. Cell. Microbiol. 9, 1922–1935 [DOI] [PubMed] [Google Scholar]
- 30.Enjyoji K., Sévigny J., Lin Y., Frenette P. S., Christie P. D., Esch J. S. II, Imai M., Edelberg J. M., Rayburn H., Lech M., Beeler D. L., Csizmadia E., Wagner D. D., Robson S. C., and Rosenberg R. D. (1999) Targeted disruption of cd39/ATP diphosphohydrolase results in disordered hemostasis and thromboregulation. Nat. Med. 5, 1010–1017 [DOI] [PubMed] [Google Scholar]
- 31.Thompson L. F., Eltzschig H. K., Ibla J. C., Van De Wiele C. J., Resta R., Morote-Garcia J. C., and Colgan S. P. (2004) Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J. Exp. Med. 200, 1395–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Csóka B., Németh Z. H., Mukhopadhyay P., Spolarics Z., Rajesh M., Federici S., Deitch E. A., Bátkai S., Pacher P., and Haskó G. (2009) CB2 cannabinoid receptors contribute to bacterial invasion and mortality in polymicrobial sepsis. PLoS ONE 4, e6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haskó G., Csóka B., Koscsó B., Chandra R., Pacher P., Thompson L. F., Deitch E. A., Spolarics Z., Virág L., Gergely P., Rolandelli R. H., and Nemeth Z. H. (2011) Ecto-5′-nucleotidase (CD73) decreases mortality and organ injury in sepsis. J. Immunol. 187, 4256–4267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Csóka B., Németh Z. H., Rosenberger P., Eltzschig H. K., Spolarics Z., Pacher P., Selmeczy Z., Koscsó B., Himer L., Vizi E. S., Blackburn M. R., Deitch E. A., and Haskó G. (2010) A2B adenosine receptors protect against sepsis-induced mortality by dampening excessive inflammation. J. Immunol. 185, 542–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Németh Z. H., Csóka B., Wilmanski J., Xu D., Lu Q., Ledent C., Deitch E. A., Pacher P., Spolarics Z., and Haskó G. (2006) Adenosine A2A receptor inactivation increases survival in polymicrobial sepsis. J. Immunol. 176, 5616–5626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Csóka B., Selmeczy Z., Koscsó B., Németh Z. H., Pacher P., Murray P. J., Kepka-Lenhart D., Morris S. M. Jr., Gause W. C., Leibovich S. J., and Haskó G. (2012) Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. 26, 376–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Csoka B., Koscso B., Toro G., Kokai E., Virag L., Nemeth Z. H., Pacher P., Bai P., and Hasko G. (2013) A2B adenosine receptors prevent insulin resistance by inhibiting adipose tissue inflammation via maintaining alternative macrophage activation. Diabetes [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hollebeeck S., Raas T., Piront N., Schneider Y. J., Toussaint O., Larondelle Y., and During A. (2011) Dimethyl sulfoxide (DMSO) attenuates the inflammatory response in the in vitro intestinal Caco-2 cell model. Toxicol. Lett. 206, 268–275 [DOI] [PubMed] [Google Scholar]
- 39.Haskó G., Kuhel D. G., Salzman A. L., and Szabó C. (2000) ATP suppression of interleukin-12 and tumour necrosis factor-alpha release from macrophages. Br. J. Pharmacol. 129, 909–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peters E., Heemskerk S., Masereeuw R., and Pickkers P. (2014) Alkaline phosphatase: a possible treatment for sepsis-associated acute kidney injury in critically ill patients. Am. J. Kidney Dis. 63, 1038–1048 [DOI] [PubMed] [Google Scholar]
- 41.Garcia A. E., Mada S. R., Rico M. C., Dela Cadena R. A., and Kunapuli S. P. (2011) Clopidogrel, a P2Y12 receptor antagonist, potentiates the inflammatory response in a rat model of peptidoglycan polysaccharide-induced arthritis. PLoS ONE 6, e26035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leibovici L., Drucker M., Konigsberger H., Samra Z., Harrari S., Ashkenazi S., and Pitlik S. D. (1997) Septic shock in bacteremic patients: risk factors, features and prognosis. Scand. J. Infect. Dis. 29, 71–75 [DOI] [PubMed] [Google Scholar]
- 43.Martin G. S., Mannino D. M., Eaton S., and Moss M. (2003) The epidemiology of sepsis in the United States from 1979 through 2000. N. Engl. J. Med. 348, 1546–1554 [DOI] [PubMed] [Google Scholar]
- 44.Cohen E. S., Law W. R., Easington C. R., Cruz K. Q., Nardulli B. A., Balk R. A., Parrillo J. E., and Hollenberg S. M. (2002) Adenosine deaminase inhibition attenuates microvascular dysfunction and improves survival in sepsis. Am. J. Respir. Crit. Care Med. 166, 16–20 [DOI] [PubMed] [Google Scholar]
- 45.Firestein G. S., Boyle D., Bullough D. A., Gruber H. E., Sajjadi F. G., Montag A., Sambol B., and Mullane K. M. (1994) Protective effect of an adenosine kinase inhibitor in septic shock. J. Immunol. 152, 5853–5859 [PubMed] [Google Scholar]
- 46.Gallos G., Ruyle T. D., Emala C. W., and Lee H. T. (2005) A1 adenosine receptor knockout mice exhibit increased mortality, renal dysfunction, and hepatic injury in murine septic peritonitis. Am. J. Physiol. Renal Physiol. 289, F369–F376 [DOI] [PubMed] [Google Scholar]
- 47.Sullivan G. W., Fang G., Linden J., and Scheld W. M. (2004) A2A adenosine receptor activation improves survival in mouse models of endotoxemia and sepsis. J. Infect. Dis. 189, 1897–1904 [DOI] [PubMed] [Google Scholar]
- 48.Lee H. T., Kim M., Joo J. D., Gallos G., Chen J. F., and Emala C. W. (2006) A3 adenosine receptor activation decreases mortality and renal and hepatic injury in murine septic peritonitis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 291, R959–R969 [DOI] [PubMed] [Google Scholar]
- 49.Cohen H. B., Briggs K. T., Marino J. P., Ravid K., Robson S. C., and Mosser D. M. (2013) TLR stimulation initiates a CD39-based autoregulatory mechanism that limits macrophage inflammatory responses. Blood 122, 1935–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liao H., Hyman M. C., Baek A. E., Fukase K., and Pinsky D. J. (2010) cAMP/CREB-mediated transcriptional regulation of ectonucleoside triphosphate diphosphohydrolase 1 (CD39) expression. J. Biol. Chem. 285, 14791–14805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baek A. E., Kanthi Y., Sutton N. R., Liao H., and Pinsky D. J. (2013) Regulation of ecto-apyrase CD39 (ENTPD1) expression by phosphodiesterase III (PDE3). FASEB J. 27, 4419–4428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cavaillon J. M., and Adib-Conquy M. (2005) Monocytes/macrophages and sepsis. Crit. Care Med. 33(12, Suppl)S506–S509 [DOI] [PubMed] [Google Scholar]
- 53.Munoz C., Carlet J., Fitting C., Misset B., Blériot J. P., and Cavaillon J. M. (1991) Dysregulation of in vitro cytokine production by monocytes during sepsis. J. Clin. Invest. 88, 1747–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nakav S., Naamani O., Chaimovitz C., Shaked G., Czeiger D., Zlotnik M., and Douvdevani A. (2010) Regulation of adenosine system at the onset of peritonitis. Nephrol. Dial. Transplant. 25, 931–939 [DOI] [PubMed] [Google Scholar]
- 55.Eltzschig H. K., Köhler D., Eckle T., Kong T., Robson S. C., and Colgan S. P. (2009) Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood 113, 224–232 [DOI] [PMC free article] [PubMed] [Google Scholar]