

Abstract
Regular exercise and a physically active lifestyle have favorable effects on health. Several issues related to this theme are addressed in this report. A comment on the requirements of personalized exercise medicine and in-depth biological profiling along with the opportunities that they offer is presented. This is followed by a brief overview of the evidence for the contributions of genetic differences to the ability to benefit from regular exercise. Subsequently, studies showing that mutations in TP53 influence exercise capacity in mice and humans are succinctly described. The evidence for effects of exercise on endothelial function in health and disease also is covered. Finally, changes in cardiac and skeletal muscle in response to exercise and their implications for patients with cardiac disease are summarized. Innovative research strategies are needed to define the molecular mechanisms involved in adaptation to exercise and to translate them into useful clinical and public health applications.
Keywords: Personalized medicine, individual differences, genetics, mitochondria, endothelium
A. Introduction
As physical activity (PA) energy expenditure has diminished and sedentary pursuits have increased, a number of physical and psychosocial ailments related to inactivity have become manifest. The phenomenon was first documented by the seminal studies of Morris et al. (1,2) on occupational PA and risk of coronary heart disease (CHD) in London public transportation workers in the 1950s. Since then, a large number of epidemiological studies have confirmed that PA level and cardiorespiratory fitness (CRF) are associated with the risk of mortality, morbidities, and the risk factor profile for common chronic diseases (3–11). Moreover, a body of data from randomized, controlled exercise interventions and other experimental studies has established that regular exercise produces favorable changes in commonly recognized risk factors for cardiovascular disease (CVD), type 2 diabetes, and other conditions (12–16).
The health benefits of a physically active lifestyle are typically evaluated with reference to mean response of a given risk factor or outcome to an exercise program. This approach fails to recognize that there are considerable inter-individual differences in responses to any exercise program (17,18).
Issues related to this overarching theme organized around five topics are addressed in the present report. First, a brief expose of the requirements of personalized exercise medicine and of in-depth biological profiling is presented. This is followed by a summary of the evidence for the contributions of genetic differences to the ability to benefit from regular exercise. Third, studies showing that mutations in TP53 influence exercise capacity in mice and humans are succinctly described. This is followed by a section on the evidence for the effects of acute and regular exercise on endothelial function in healthy individuals and patients with CVD. Finally, the changes in cardiac and skeletal muscle in response to regular exercise and their implications for patients with cardiac disease are summarized.
B. Genomic implications for PA
The completion of the Human Genome Project and the subsequent expansion of genetic studies to genome-wide association studies (GWASs) and whole genome and whole exome sequencing have led to a new era in genetics. Though still in its infancy, personalized genomics and related personal omics profiling, encompassing epigenomics, pharmacogenomics, transcriptomics, proteomics, metabolomics, and antibody profiling, will allow for the deep biological profiling of humans in the face of PA.
1. Personalized genomics and beyond
Previous investigators have examined in a clinical context the whole genome sequence of an apparently healthy individual (19) and provided personalized results. Common disease variants were interpreted and estimated effect sizes aggregated to provide an estimate of the future risk of developing common diseases such as diabetes, CHD, hypertension, or osteoporosis. In addition, the genome was evaluated for evidence of rare variants associated with rare diseases associated with potential increased risk of sudden death, fracture, or metabolic disease. Evaluation of the whole genome sequence also allowed for elucidation of genetic differences, which may predispose to adverse or differential drug responses. Further work demonstrated the incremental value of phasing genomic information among a family to improve interpretability (20). Evaluation of common disease risk alleles showed that common alleles, as expected, can be differentially dispersed among family members so that high- and low-risk offspring may be seen from average-risk individuals, even in the absence of new genetic variation. Similarly, without targeted genotyping, individual family members who carry risk alleles of manifest rare diseases can be easily inferred.
Integrated personal omics profiling using a combination of genomic, transcriptomic, proteomic, metabolomics, and antibody profiling has been described in an apparently healthy individual (21). Repeated measures of metabolic, antibody, and transcript profiles allowed time-resolved discrimination of subject-environment interactions and identification of novel processes associated with changes in health status. Furthermore, cross-sample and cross-platform validation improved confidence of variant calls and in many cases proved the direct effects of genomic variants on transcript and proteomic profiles. While the extent of omic profiling demonstrated by Chen et al. has not been achieved at a cohort or population scale, the opportunities and limitations of multiscale phenotyping have yet to be fully evaluated. Efficient data reduction methods and validation to improve inference and causality will be required to allow omics profiling to reach common usage. Integrating genomic and omics profiling into research, clinical practice, and PA will require continued improvements in the efficiency of data generation as well as further optimization of nascent bioinformatics tools.
2. Genomic implications for PA and exercise training
Prior and current research on the genetics of exercise training is covered in other sections of this report. Findings from many disease-oriented GWASs, spanning for example obesity, diabetes, atherosclerosis, lung disease, psychiatric disorders, and neuromuscular disease [cataloged in (22)], may have implications for exercise training and health effects of PA. Exploring the interactions of these disease-associated variants with exercise (lifetime or recent burden) as a covariate may uncover heretofore unappreciated correlations. Comprehensive phenotypic and genetic information as available or under development in large population studies (China Kadoorie Biobank, Million Veteran Program, Women’s Health Initiative, etc.) may uncover further genetic variants with impacts on physical activity-related health.
In the realm of PA research and therapies, whole exome and genome sequencing and omics profiling will allow deep profiling of an individual’s genetic makeup that may impact the tolerance, effects, and performance expected from pursuing PA or training, in addition to tracking of the biological changes that occur with activity and intentional exercise. This personalization would be in line with the knowledge that considerations must be taken of the individual’s response to different types of training and doses when recommending or prescribing exercise (23).
Integrated personal omics profiling has the potential to inform effects of PA on health, training regimen selection, and athletic performance. This would start with focused interpretation of pertinent variants across the whole genome, including both rare and common variation. An athletic genome interpretation could delineate rare variants with negative or positive effects, such as a truncated erythropoietin receptor (24) suggesting inborn advantage in endurance sports or mutations associated with underlying cardiomyopathy, which if present could lead to restriction from athletic activity (25). In all individuals, common variants will impact disease risk and athletic potential; delineation of the impact and effect of these variants is and will be the subject of many ongoing and future studies. Integration of rare and common risk alleles will allow the generation of a complete genetic profile as it pertains to PA. This profile will give an integrated view of the person’s intrinsic base CRF level, response to PA, and physiological potential.
Athletic personal omics profiling would facilitate monitoring over time and integrate biological information with change in physiological states (e.g., activity intensity, duration, response to training or performance level). This could include proteomic, transcriptomic, and metabolomic analyses during a period of training or recovery. Certainly ascertainment of time- and tissue-resolved expression, protein, and metabolic programs would be critical for inferring biological causality. Novel insights could be gained by comparing the omic response to different exercise regimes. A reductionist approach would search for nonrandom patterns, either correlated patterns over time or single outlier events concordant across datatypes. Correlation network-based analytic methods would allow data congregation and facilitate identification of key nodal regulators (26). Analyses may for example show that training volume correlate with the translation or turnover of mitochondrial proteins, or that only training sessions of extremely high intensity will potentiate transcript and protein levels of skeletal muscle contractile and calcium-handling proteins. For the individual, these analyses could provide an explanatory model as to whether a specific regime was effective. For exercise scientists, these data could prove invaluable in exploring the coordinated molecular adaptations associated with exercise training.
Combining integrated personal omics profiles from a cohort of athletes could provide a larger catalogue to inform personalized training. Even though training load and performance level differs, the data and results from athletes may also be applicable to approaches for PA in the general population. A compendium of integrated omics profiles may be useful to predict the best choice of exercise on a personalized basis, without the need to perform extensive or repeated exercise testing of every individual.
C. Genetics and response to regular exercise
Human variation in responsiveness to regular exercise was first recognized more than 30 years ago (27). In a series of experiments conducted with groups of sedentary young (18 to 30 years) men and women, it was shown that there were large interindividual differences in trainability for CRF as assessed by maximal oxygen uptake (VO2Max) and submaximal exercise capacity, skeletal muscle oxidative potential, and adipose tissue lipid mobilization and storage markers (17,28–31). Subsequently, variation in trainability was appropriately quantified by the HERITAGE Family Study, in which 742 healthy, sedentary subjects followed a highly standardized, laboratory-based endurance-training program for 20 weeks. For example, the average increase in VO2Max was about 400 mL O2 with a standard deviation of about 200. The training responses varied from no change to increases of more than 1000 mL O2 per minute (18,32,33). The same pattern of variation was evident for several other training response traits (32,34).
In experimental studies with pairs of monozygotic (MZ) twins, it was established that individual differences in trainability were not randomly distributed, with consistently more variance in training responses between pairs of MZ twins than between brothers or sisters (within pairs) (30,35,36). Four twin studies performed with exercise programs that differed in duration, intensity, and control over dietary intake revealed that there was a strong genotype–training interaction effect contributing to variation in trainability (30,31,36–38). These observations were confirmed In the 99 families of European descent who were part of HERITAGE: the increase in VO2Max showed 2.5 times more variance between families than within families. A model-fitting analytical procedure yielded a maximal heritability estimate of 47% for VO2Max response level (18).
Genome-wide linkage analysis was used in HERITAGE to find genomic regions harboring genes implicated in the response to exercise training. Quantitative trait loci (QTLs) for training-induced changes in submaximal exercise (50 W) stroke volume and heart rate were found on chromosomes 10p11 and 2q33.3–q34, respectively (39,40). The QTL on 10p11 for the gains in stroke volume was narrowed down to a 7 Mb region. Among the linkage-positive families, the strongest associations were found with single nucleotide polymorphisms (SNPs) in the kinesin family member 5B (KIF5B) gene locus (41). Resequencing of KIF5B revealed several sequence variants. The SNP with the strongest association modified the KIF5B promoter activity in cell-based systems. Furthermore, inhibition and overexpression studies in C2C12 cells showed that changes in KIF5B expression level altered mitochondrial localization and biogenesis; KIF5B inhibition led to diminished biogenesis and perinuclear accumulation of mitochondria, while overexpression enhanced mitochondrial biogenesis (41).
A QTL for the changes in exercise heart rate at 50 W (HR50) on chromosome 2q33.3-q34 was localized within a 10 Mb region (42). The strongest evidence of association was detected with two SNPs located in the 5′-region of the cAMP responsive element binding protein 1 (CREB1) gene locus (P=1.6 × 10−5). The most significant SNP explained almost 5% of the variance in HR50 response, and the common allele homozygotes and heterozygotes had about 57% and 20%, respectively, greater decreases in HR50 than the minor allele homozygotes. Furthermore, one of these SNPs located about 2.6 kb upstream of the first exon of CREB1 was shown to modify promoter activity in vitro; the A-allele, which was associated with a blunted HR50 response, showed significantly greater promoter activity in a C2C12 cell model than the G-allele.
A GWAS of VO2Max training response was undertaken with more than 320,000 SNPs in HERITAGE (43). No SNP reached a genome-wide level of significance, likely because of the limited sample size of this exercise intervention study. A total of 39 individual SNPs were associated with the VO2Max training response at P<1.5 × 10−4. The strongest evidence of association (P=1.3 × 10−6) was observed with a SNP located in the first intron of the acyl-CoA synthetase long-chain family member 1 (ACSL1) gene. When all 39 SNPs were analyzed simultaneously in multivariate regression models, 16 SNPs accounted for 45% of the variance in VO2Max trainability, a value comparable to the heritability estimate of 47% reported previously in HERITAGE (18). A predictor score was constructed using all 21 SNPs that entered in the final regression model. Each SNP was coded based on the number of high-VO2Max training response alleles. While the theoretical range of the score was from zero (no beneficial alleles) to 42 (two copies of the beneficial alleles at all 21 loci), the observed scores ranged from 7 to 31. The difference in VO2Max training response between those with the lowest (9 or less, N = 36, mean = +221 mL/min) and the highest (19 or more, N = 52, mean = +604 mL/min) scores was 383 mL/min. It is essential that these observations be the focus of replication studies.
D. Single-gene TP53 mutation contributing to the determination of exercise capacity
The growing complexity with which tumor protein 53 (p53), encoded by the TP53 gene in humans, regulates the mitochondria has recently been highlighted by the delineation of a specific molecular mechanism by which it translocates into the mitochondria (44,45). p53 can affect mitochondrial function not only through its activities as a nuclear transcription factor but also by its direct presence in the organelle (Figure 1A) (46,47). The physiological significance of basal levels of p53 in promoting respiration in cell lines has been confirmed using mouse models. The absence of p53 in p53−/− (null state) mice results in significant depletion of mitochondrial DNA (mtDNA) in skeletal muscle and reduces endurance exercise capacity, both at baseline and with training (Figure 1B) (47–49).
Figure 1.
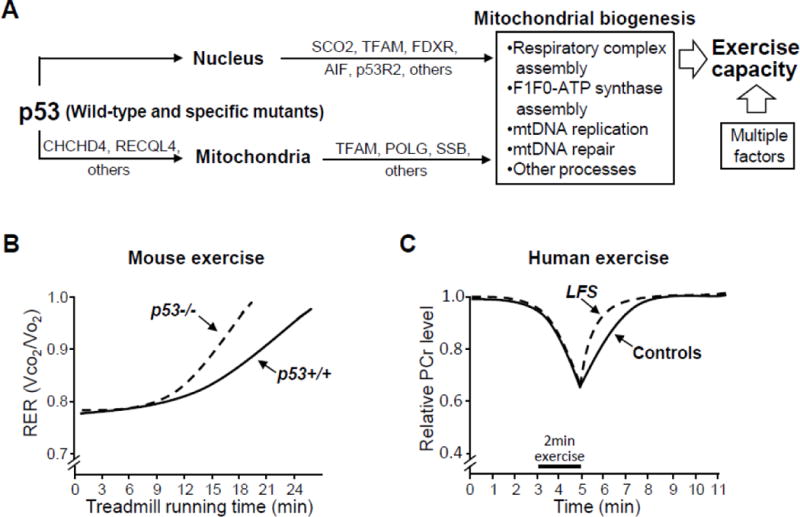
(A) Mechanisms and in vivo effects of tumor protein 53 (p53) regulation of mitochondrial function. p53 can regulate mitochondrial function through both its nuclear transcriptional target genes (such as SCO2, TFAM, FDXR, AIF, and p53R2 [RRM2B]) and its various post-translational interactions with proteins found inside the mitochondria (such as TFAM, POLG, and SSB). p53 can translocate into the mitochondria utilizing the disulfide relay system that depends on respiration and CHCHD4 and by binding to RECQL4 which has a mitochondrial targeting sequence. p53 promotes mitochondrial biogenesis activities such as respiratory complex I and IV assembly via AIF and SCO2, respectively, and ATP synthase formation. Mitochondrial genomic DNA (mtDNA) maintenance and repair can be mediated by the physical interaction of p53 with proteins found in the matrix such as TFAM, POLG, and SSB. These activities of p53 are likely to function in concert with a host of other factors to determine human exercise capacity. (B) The presence of p53 in p53+/+ versus p53−/− mice had a marked effect in improving the respiratory exchange ratio (RER, a marker of aerobic metabolic capacity) during treadmill running after exercise training. This phenomenon observed in p53−/− mice (null state with no p53 expression) should be distinguished from that in mice expressing mutated p53, such as in the Li-Fraumeni syndrome (LFS) mouse model. (C) Skeletal muscle mitochondrial function can be noninvasively assessed in vivo using P-31 magnetic resonance spectroscopy (31P-MRS). The recovery kinetics of phosphocreatine (PCr) levels in skeletal muscle after exercise-induced depletion can serve as an indicator of mitochondrial oxidative phosphorylation capacity. PCr recovery was faster in individuals carrying LFS mutations (LFS TP53) compared to noncarrier and healthy volunteer subjects (Controls).
AIF = apoptosis-inducing factor 1; CHCHD4 = mitochondrial intermembrane space import and assembly protein 40; FDXR = NADPH:adrenodoxin oxidoreductase, mitochondrial; p53R2 = p53-inducible ribonucleotide reductase small subunit 2 homolog; POLG = DNA polymerase subunit gamma-1; RECQL4 = ATP-dependent DNA helicase Q4; SCO2 = cytochrome oxidase deficient homolog 2; SSB = lupus La protein; TFAM = transcription factor A, mitochondrial;
Germline mutations of TP53 in humans causes Li-Fraumeni syndrome (LFS), a condition characterized by the early onset of a variety of cancers (50). As it is well established that skeletal muscle mitochondrial biogenesis contributes to improving exercise capacity, we speculated that LFS patients may have alterations in oxidative metabolism affecting exercise. Using P-31 magnetic resonance spectroscopy (31P-MRS), we noninvasively monitored the recovery kinetics of skeletal muscle phosphocreatine (PCr) levels after exercise as a measure of mitochondrial oxidative phosphorylation capacity (51,52). The sensitivity of this in vivo measure of mitochondrial function was especially useful because LFS is a rare disease. It would have been difficult to enroll the large numbers of patients required to demonstrate significant alterations in exercise capacity by standard treadmill testing, which can be affected by various factors including age, gender, CRF, and motivation.
Using a modified foot exercise apparatus, the recovery kinetics of PCr was monitored in the tibialis anterior muscle of noncarriers and carriers of LFS TP53 mutations after its partial depletion by submaximal exercise (52). We unexpectedly observed a shorter PCr recovery time constant (median value was 22% shorter compared to noncarriers), suggesting that individuals carrying an alteration in their TP53 gene had increased capacity for oxidative phosphorylation (Figure 1C). Within the group of individuals carrying TP53 mutations, the PCr recovery time constant was inversely correlated to VO2 at ventilatory threshold, validating the 31P-MRS technique as a measure of aerobic exercise capacity (52). We confirmed this in vivo finding of increased skeletal muscle oxidative metabolism in the LFS family participants by demonstrating higher mitochondrial respiration and biogenesis in their blood cells and skeletal muscle myoblasts. To further support the human data, we utilized a mouse model with the knock-in of a human LFS TP53 mutation to demonstrate a doubling of its running distance on a treadmill and increased mitochondrial biogenesis. Under conditions of genetic and environmental homogeneity, this constituted powerful evidence that an amino acid variation of a single gene can markedly affect exercise phenotype.
In summary, the study provides proof of principle that the genetic alteration of TP53 can contribute to determining individual exercise capacity in humans. Two important points need to be emphasized. Although LFS is considered to be a rare disease, its true prevalence remains unknown. We have examined only 10 LFS mutations out of more than 700 different reported germline TP53 sequence variations, not all of which are known to cause Li-Fraumeni syndrome (IARC TP53 database version R17). Whether the results of our pilot study are generally applicable to other LFS mutations remains to be determined. Secondly, the decrease in exercise capacity in a p53−/− mice model (null state) contrasts with the reported increase in exercise capacity in mice with a knock-in LFS mutation. Our investigations show that specific mutant p53 proteins can retain their mitochondrial biogenesis activity despite losing the cell cycle regulatory functions of wild-type p53 (52). This was further underscored at the post-transcriptional level whereby mutated p53 can retain its ability to translocate into the mitochondria and preserve mitochondrial genomic DNA integrity, as well as possibly contributing to ATP synthase assembly (45,53). With these observations, significant questions remain unanswered including whether non-LFS sequence variants of TP53 can also play a role in mitochondrial biogenesis and exercise capacity and how the upregulation of mitochondrial activities by specific LFS mutations affects cancer development.
E. The importance of PA for vascular wall health
Sedentary lifestyle is a risk factor for CVD and is preceded by deleterious structural and functional changes within the vascular endothelium (54). Endothelial cells regulate vascular tone by releasing a number of soluble mediators, including nitric oxide (NO) and other vasoactive compounds (55). Imbalances between pro- and antioxidants result in increased production of reactive oxygen species, impaired NO bioavailability, and enhanced production of contracting factors that reduce endothelium-dependent vasodilation (56). Since blood flow-induced shear stress is a major stimulus to vasodilation, perturbed flow or low shear stress is thought to be a primary mechanism by which endothelial function is altered with a sedentary lifestyle. Interestingly, a large majority of clinical and experimental studies suggest that habitual PA increases NO bioavailability and reduces oxidative stress (57), which underscores the overall importance of exercise and other forms of PA for enhancing vascular health through the endothelium.
1. Effects of acute exercise on endothelial function
Exercise training has been closely linked to lower morbidity and mortality rates from CHD (58). Habitual PA has also been shown to improve peripheral vascular endothelial function (59). Paradoxically, studies have demonstrated that acute aerobic exercise reduces endothelial function measured as brachial artery flow-mediated dilation (FMD) among overweight and obese inactive subjects while regularly active obese individuals experience increases in FMD (60). Furthermore, certain exercise modes (i.e., weight lifting) can induce sizeable, yet transient increases in arterial pressure that are known to reduce endothelial function in people who are unaccustomed to exercise (61,62). Jurva et al. observed impaired vascular endothelial function following a weight lifting episode in sedentary individuals but not in trained weightlifters (61). In another set of studies, Phillips et al. demonstrated impairments in brachial artery FMD among physically inactive subjects following acute hypertension induced by a relatively short weight lifting bout (63).
In light of these data, it is important to understand the exact mechanism(s) by which chronic exercise provides protection against acute exercise-related impairments in vascular function. In addition, these studies highlight the importance of investigating the ways in which other CVD risk factors (i.e., obesity) can impact endothelial responses to exercise. Indeed, recently Currie et al. (64) showed that high-intensity interval exercise acutely improved FMD in patients with CHD, making it important to consider how the dose, intensity, and disease status may impact endothelial responses to acute exercise.
2. Effects of exercise training on mechanisms of endothelial function
Promotion of PA and exercise prescription are important components of CVD prevention and endothelial function. Exercise training promotes specific morphological and functional adaptations within the vascular wall that increase capacity to respond to physical stressors (65). While the mechanism(s) by which PA alters endothelial function have not been fully elucidated, several studies have shown a role for exercise intensity on the propensity for improved vascular health. Studies by Goto et al. found that moderate-intensity (50% VO2Max) aerobic exercise (but not high-intensity at 75% VO2Max) augments endothelium-dependent vasodilation in the forearm through increased production of NO (66). Interestingly, the effects of exercise training do not typically enhance vascular smooth muscle function, suggesting that the endothelium is the key modulator of exercise-related vascular effects (66,67).
The augmentation of NO bioavailability induced by increased vascular shear stress and blood flow during exercise may be related to increased activity and expression of endothelial NO synthase. Hambrecht et al. performed one of the first studies highlighting such beneficial effects of aerobic exercise training during cardiac rehabilitation and demonstrated improvements in endothelial function (68) and increased endothelial nitric oxide synthase expression in blood vessels of patients after 4 weeks of cardiac rehabilitation (69). Further studies have included similar interventions that reduced NADPH oxidase generation of reactive oxygen species in the vascular wall of patients participating in cardiac rehabilitation (70).
Recently, high-intensity interval training has shown promise in improving endothelial function compared to continuous aerobic exercise training (71), and there appears to be beneficial effects of alternative modes of exercise (i.e., waltz dancing) on improving endothelial function in heart failure (HF) patients (72). The mechanism of the improved endothelium-dependent dilation is not entirely clear. For example, in addition to NO, the reactive oxygen species H2O2 may be an important player in exercise-induced vascular relaxation (73). Increased H2O2 generation can compensate for decreased NO during CVD, suggesting a rapid switch between H2O2 and NO vasodilators (74). Previously, we found a similar switch from NO to H2O2 mechanisms involved in the acetylcholine vasodilation of isolated resistance arteries from habitually active subjects after acute exertion (75). In sedentary individuals, the dilation following acute exertion is severely reduced (75). Since exercise also upregulates antioxidant defense mechanisms to minimize oxidative stress (76), these data indicate substantial differences in the mechanisms for endothelium-dependent vasodilation after an acute stressor such as exercise.
Regardless of the intensity (i.e., low, moderate, or high), exercise training is associated with reduced risk for the development of CVD and subsequent events (58). However, it is well recognized that cardiovascular events are more likely to occur during or soon after vigorous exercise (77). In terms of athletic populations engaged in high-intensity exercise training, there appears to be a protection from endothelial dysfunction induced by acute exertion (63). The beneficial effects of exercise on endothelial function represent a sort of “paradox” where on the one hand chronic exercise maintains endothelial function but on the other hand acute, sudden exertion greatly impairs endothelial function. Indeed, the mechanism of this paradox (NO vs. other vasodilators) may represent an important risk marker in populations without overt CVD.
F. Cellular discoveries and implications for clinical practice
Exercise intolerance is the hallmark of patients with CVD. This physical limitation has been associated with a constellation of pathophysiological alterations. Cardiac dysfunction causes sympathetic nervous exacerbation and increases in the renin-angiotensin system. These neurohumoral changes provoke intense peripheral vasoconstriction. In the skeletal muscle, the reduction in blood flow supply causes proinflammation and oxidative stress, as well as imbalances in muscle protein synthesis and degradation. This skeletal myopathy explains, in great part, the exercise intolerance in patients with cardiac dysfunction.
Cell and molecular studies have immensely extended the knowledge regarding the effects of exercise training on CVD, especially facing both the cardiac muscle and skeletal muscle.
Exercise training improves the balance of myocardial Ca2+ handling during the cardiac cycle. Sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2 (SERCA2), phospho-Ser16-phospholamban, and phospho-Thr17-phospholamban expression are all increased by exercise training. Na+/Ca2+ exchanger and phosphatase 1 expression are restored in exercise-trained HF mice (78). Exercise training decreases heart weight and cardiomyocyte width, which has been attributed to the reduction in nuclear calcineurin/nuclear factor of activated T-cells (NFAT) and transcription factor GATA-4 expression and translocation (79). Calcineurin is a Ca2+/calmodulin-dependent phosphatase that dephosphorylates members of NFAT transcription factor family (NFATc3) in the cytoplasm. The result of such nuclear translocation is the activation of hypertrophic genes. Cardiac failure is often preceded by mitochondrial dysfunction and cytosolic protein quality control disruption. Exercise training restores cardiac mitochondrial function, proteasomal activity, and protein quality control, which contributes to the improvement in cardiac function (80) (Figure 2).
Figure 2.
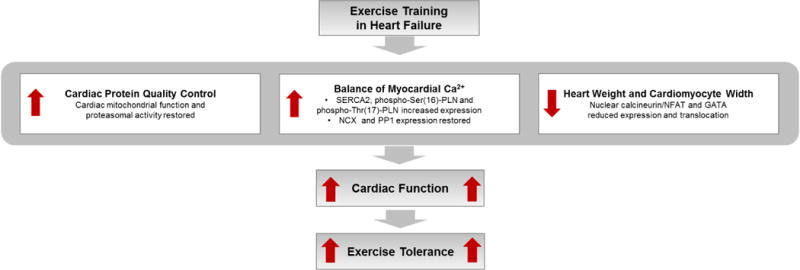
Exercise training effects on cardiac muscle in HF. SERCA2 = sarcoplasmic reticulum Ca2+ ATPase; phospho-Ser(16)-PLN = phospho-Ser16-phospholamban; phospho-Thr(17)-PLN = phospho-Thr17-phospholamban; NCX = Na+/Ca2+ exchanger; PP1 = phosphatase 1; NFAT = nuclear factor of activated T-cells.
During the past decade, several studies have provided evidence that exercise training provokes significant changes in the skeletal muscle in HF. Exercise training significantly reduces muscle sympathetic nerve activity and increases muscle blood flow at rest and during exercise in HF patients (81,82). The increase in muscle blood flow is essential for not only the reduction in proinflammation and oxidative stress, but also for the improvement in imbalance of muscle protein synthesis and degradation. Reductions in tumor necrosis factor-α (TNF-α) and in interleukin (IL) IL-6 and IL-1β gene expression in skeletal muscle have been demonstrated in exercise-trained HF patients (83). In addition, exercise training reduces skeletal muscle lipid hydroperoxidation and protein carbonylation in animal models of HF, which contributes to the restoration in the ubiquitin-proteasome system, the major proteolytic pathway (84,85). Exercise training significantly decreases ubiquitinated proteins and chymotrypsin-like proteasome activity in HF mice (85). On the other hand, exercise training increases insulin-like growth factor 1 (IGF-1) levels and reduces myostatin levels in the skeletal muscle in HF patients (86,87). Altogether, these changes demonstrate that exercise training improves muscle protein synthesis and degradation balance, which substantially contributes to the amelioration in skeletal myopathy in HF (Figure 3).
Figure 3.
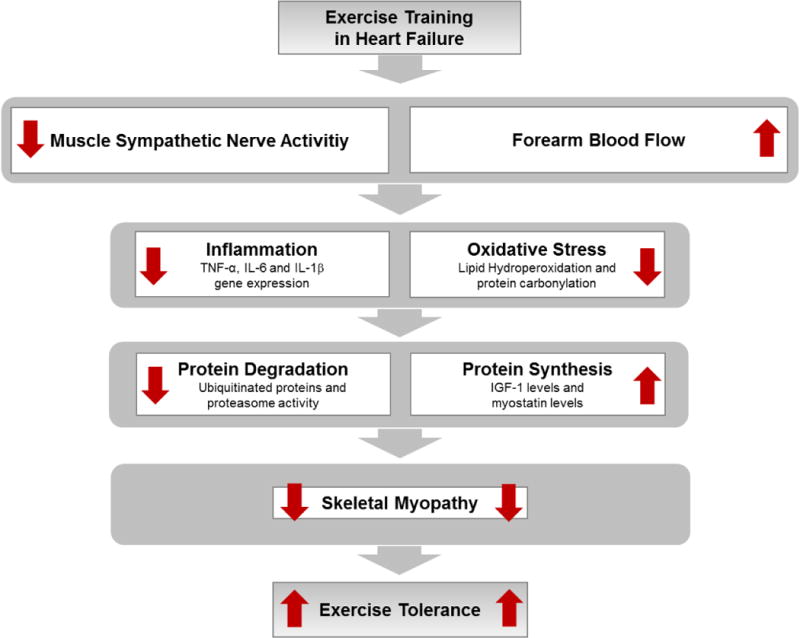
Exercise training effects on skeletal muscle in HF. TNF-α = tumor necrosis factor− α; IL−6 = interleukin−6; IL−1β = interleukin−1β; and IGF−1 = insulin-like growth factor−1.
The changes in cardiac and skeletal muscles provoked by exercise training have important clinical implications. They improve functional capacity and quality of life in patients suffering of HF. In addition, muscle sympathetic nerve activity and muscle blood flow are independent predictors of mortality in patients with HF (88). Thus, nonpharmacological therapy based on exercise training should be strongly recommended in the treatment of patients with CVD.
G. Summary and conclusions
There is overwhelming evidence that regular exercise and PA have beneficial effects on disease prevention when considered at a population level and in a public health context. Some of the exercise-associated cardioprotective and other health benefits are well known, and the mechanisms underlying them have been the topics of recent reviews (89,90). Skeletal muscle adaptation to exercise plays an important role in disease prevention, and molecular events taking place as a result of exposure to exercise are beginning to be better understood (91). As this short review has emphasized, individual differences in responsiveness play an important role in the magnitude of the benefits accrued from regular exercise. Even though we have made significant progress in acquiring technologies that are necessary to probe genomic features and metabolic pathways and systems responsible for the individuality of adaptation to exercise, and despite the progress made to date in defining these genomic markers and pathways (43,92), creative research strategies are needed to define the molecular mechanisms involved and to translate them into useful clinical and public health applications.
Acknowledgments
C Bouchard is partially supported by the John W. Barton, Sr. Chair in Genetics and Nutrition. Research on the HERITAGE Family Study was supported for over 20 years by the National Institutes of Health (HL-45670). PY Wang and PM Hwang are supported by the Division of Intramural Research, National Heart, Lung, and Blood Institutes (NHLBI) and by a Bench-to-Bedside of the National Institutes of Health. LM Antunes-Correa was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo FAPESP (2013/15651-7 and 2013/07607-8). CE Negrao was supported by CNPq (301867/2010-0), FAPESP (2010/50048-1 and 2013/07607-8), and, in part, by Fundação Zerbini.
Abbreviations
- ACSL1
Acyl-CoA synthetase long-chain family member 1
- CREB1
cAMP responsive element binding protein 1
- CHD
Coronary heart disease
- CRF
Cardiorespiratory fitness
- CVD
Cardiovascular disease
- GWAS
Genome-Wide Association Studies
- FMD
Flow-mediated dilation
- HF
Heart failire
- HR50
Heart rate at 50 W
- IGF-1
Insulin-like growth factor 1
- IL
Interleukin
- KIF5B
Kinesin family member 5B
- LFS
Li-Fraumeni syndrome
- mtDNA
Mitochondrial DNA
- MZ
Monozygotic
- NO
Nitric oxide
- NFAT
Nuclear calcineurin/nuclear factor of activated T-cells
- 31P-MRS
P-31 magnetic resonance spectroscopy
- PA
Physical activity
- PCr
Phosphocreatine
- QTL
Quantitative trait loci
- SERCA2
Sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2
- SNPs
Single nucleotide polymorphisms
- TNF-α
Tumor necrosis factor-α
- p53
Tumor protein 53
- VO2Max
Maximal oxygen consumption
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Morris JN, Heady JA, Raffle PA, et al. Coronary heart-disease and physical activity of work. Lancet. 1953;265:1111–1120. doi: 10.1016/s0140-6736(53)91495-0. concl. [DOI] [PubMed] [Google Scholar]
- 2.Morris JN, Heady JA, Raffle PA, et al. Coronary heart-disease and physical activity of work. Lancet. 1953;265:1053–1057. doi: 10.1016/s0140-6736(53)90665-5. contd. [DOI] [PubMed] [Google Scholar]
- 3.Blair SN, Kohl HW, III, Barlow CE, et al. Changes in physical fitness and all-cause mortality. A prospective study of healthy and unhealthy men. JAMA. 1995;273:1093–1098. [PubMed] [Google Scholar]
- 4.Blair SN, Kohl HW, III, Paffenbarger RS, Jr, et al. Physical fitness and all-cause mortality. A prospective study of healthy men and women. JAMA. 1989;262:2395–2401. doi: 10.1001/jama.262.17.2395. [DOI] [PubMed] [Google Scholar]
- 5.Hooker SP, Sui X, Colabianchi N, et al. Cardiorespiratory fitness as a predictor of fatal and nonfatal stroke in asymptomatic women and men. Stroke. 2008;39:2950–2957. doi: 10.1161/STROKEAHA.107.495275. [DOI] [PubMed] [Google Scholar]
- 6.Jones LW, Watson D, Herndon JE, 2nd, et al. Peak oxygen consumption and long-term all-cause mortality in nonsmall cell lung cancer. Cancer. 2010;116:4825–4832. doi: 10.1002/cncr.25396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katzmarzyk PT, Church TS, Craig CL, et al. Sitting time and mortality from all causes, cardiovascular disease, and cancer. Med Sci Sports Exerc. 2009;41:998–1005. doi: 10.1249/MSS.0b013e3181930355. [DOI] [PubMed] [Google Scholar]
- 8.Kodama S, Saito K, Tanaka S, et al. Cardiorespiratory fitness as a quantitative predictor of all-cause mortality and cardiovascular events in healthy men and women: a meta-analysis. JAMA. 2009;301:2024–2035. doi: 10.1001/jama.2009.681. [DOI] [PubMed] [Google Scholar]
- 9.McAuley P, Pittsley J, Myers J, et al. Fitness and fatness as mortality predictors in healthy older men: the veterans exercise testing study. J Gerontol A Biol Sci Med Sci. 2009;64:695–699. doi: 10.1093/gerona/gln039. [DOI] [PubMed] [Google Scholar]
- 10.Paffenbarger RS, Jr, Hyde RT, Wing AL, et al. Physical activity, all-cause mortality, and longevity of college alumni. NEnglJ Med. 1986;314:605–613. doi: 10.1056/NEJM198603063141003. [DOI] [PubMed] [Google Scholar]
- 11.Sui X, Lee DC, Matthews CE, et al. Influence of cardiorespiratory fitness on lung cancer mortality. Med Sci Sports Exerc. 2010;42:872–878. doi: 10.1249/MSS.0b013e3181c47b65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Church TS, Blair SN, Cocreham S, et al. Effects of aerobic and resistance training on hemoglobin A1c levels in patients with type 2 diabetes: a randomized controlled trial. JAMA. 2010;304:2253–2262. doi: 10.1001/jama.2010.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Church TS, Earnest CP, Skinner JS, et al. Effects of different doses of physical activity on cardiorespiratory fitness among sedentary, overweight or obese postmenopausal women with elevated blood pressure: a randomized controlled trial. JAMA. 2007;297:2081–2091. doi: 10.1001/jama.297.19.2081. [DOI] [PubMed] [Google Scholar]
- 14.Kelley GA, Kelley KS. Efficacy of aerobic exercise on coronary heart disease risk factors. Prev Cardiol. 2008;11:71–75. doi: 10.1111/j.1751-7141.2008.08037.x. [DOI] [PubMed] [Google Scholar]
- 15.Nikander R, Sievanen H, Heinonen A, et al. Targeted exercise against osteoporosis: A systematic review and meta-analysis for optimising bone strength throughout life. BMC Med. 2010;8:47. doi: 10.1186/1741-7015-8-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valkeinen H, Aaltonen S, Kujala UM. Effects of exercise training on oxygen uptake in coronary heart disease: a systematic review and meta-analysis. Scand J Med Sci Sports. 2010;20:545–555. doi: 10.1111/j.1600-0838.2010.01133.x. [DOI] [PubMed] [Google Scholar]
- 17.Lortie G, Simoneau JA, Hamel P, et al. Responses of maximal aerobic power and capacity to aerobic training. Int J Sports Med. 1984;5:232–236. doi: 10.1055/s-2008-1025911. [DOI] [PubMed] [Google Scholar]
- 18.Bouchard C, An P, Rice T, et al. Familial aggregation of VO(2max) response to exercise training: results from the HERITAGE Family Study1. JApplPhysiol. 1999;87:1003–1008. doi: 10.1152/jappl.1999.87.3.1003. [DOI] [PubMed] [Google Scholar]
- 19.Ashley EA, Butte AJ, Wheeler MT, et al. Clinical assessment incorporating a personal genome. Lancet. 2010;375:1525–1535. doi: 10.1016/S0140-6736(10)60452-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dewey FE, Chen R, Cordero SP, et al. Phased whole-genome genetic risk in a family quartet using a major allele reference sequence. PLoS Genet. 2011;7:e1002280. doi: 10.1371/journal.pgen.1002280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen R, Mias GI, Li-Pook-Than J, et al. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell. 2012;148:1293–1307. doi: 10.1016/j.cell.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Welter D, MacArthur J, Morales J, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buford TW, Roberts MD, Church TS. Toward Exercise as Personalized Medicine. Sports Med. 2013;43:157–165. doi: 10.1007/s40279-013-0018-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de la Chapelle A, Traskelin AL, Juvonen E. Truncated erythropoietin receptor causes dominantly inherited benign human erythrocytosis. Proc Natl Acad Sci U S A. 1993;90:4495–4499. doi: 10.1073/pnas.90.10.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation. 2004;109:2807–2816. doi: 10.1161/01.CIR.0000128363.85581.E1. [DOI] [PubMed] [Google Scholar]
- 26.Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12:56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bouchard C. Human Adaptability May Have a Genetic Basis. In: Landry F, editor. Health risk estimation, risk reduction and health promotion Proceedings of the 18th annual meeting of the Society of Prospective Medicine. Vol. 1983. Ottawa: Canadian Public Health Association; pp. 463–476. [Google Scholar]
- 28.Despres JP, Bouchard C, Savard R, et al. The effect of a 20-week endurance training program on adipose-tissue morphology and lipolysis in men and women. Metabolism. 1984;33:235–239. doi: 10.1016/0026-0495(84)90043-x. [DOI] [PubMed] [Google Scholar]
- 29.Savard R, Despres JP, Marcotte M, et al. Endurance training and glucose conversion into triglycerides in human fat cells. J Appl Physiol (1985) 1985;58:230–235. doi: 10.1152/jappl.1985.58.1.230. [DOI] [PubMed] [Google Scholar]
- 30.Hamel P, Simoneau JA, Lortie G, et al. Heredity and muscle adaptation to endurance training. Med Sci Sports Exerc. 1986;18:690–696. [PubMed] [Google Scholar]
- 31.Simoneau JA, Lortie G, Boulay MR, et al. Inheritance of human skeletal muscle and anaerobic capacity adaptation to high-intensity intermittent training. Int J Sports Med. 1986;7:167–171. doi: 10.1055/s-2008-1025756. [DOI] [PubMed] [Google Scholar]
- 32.Bouchard C, Rankinen T. Individual differences in response to regular physical activity. MedSciSports Exerc. 2001;33:S446–S451. doi: 10.1097/00005768-200106001-00013. [DOI] [PubMed] [Google Scholar]
- 33.Skinner JS, Wilmore KM, Krasnoff JB, et al. Adaptation to a standardized training program and changes in fitness in a large, heterogeneous population: the HERITAGE Family Study. Med SciSports Exerc. 2000;32:157–161. doi: 10.1097/00005768-200001000-00023. [DOI] [PubMed] [Google Scholar]
- 34.Wilmore JH, Stanforth PR, Gagnon J, et al. Cardiac output and stroke volume changes with endurance training: the HERITAGE Family Study. Med Sci Sports Exerc. 2001;33:99–106. doi: 10.1097/00005768-200101000-00016. [DOI] [PubMed] [Google Scholar]
- 35.Despres JP, Bouchard C, Savard R, et al. Adaptive changes to training in adipose tissue lipolysis are genotype dependent. Int J Obes. 1984;8:87–95. [PubMed] [Google Scholar]
- 36.Prud’homme D, Bouchard C, Leblanc C, et al. Sensitivity of maximal aerobic power to training is genotype-dependent. Med Sci Sports Exerc. 1984;16:489–493. doi: 10.1249/00005768-198410000-00012. [DOI] [PubMed] [Google Scholar]
- 37.Bouchard C, Tremblay A, Despr‚s JP, et al. The Response to Exercise with Constant Energy Intake in Identical Twins. Obesity Research. 1994;2:400–410. doi: 10.1002/j.1550-8528.1994.tb00087.x. [DOI] [PubMed] [Google Scholar]
- 38.Boulay MR, Lortie G, Simoneau JA, et al. Sensitivity of maximal aerobic power and capacity to anaerobic training is partly genotype dependent. In: Malina RM, Bouchard C, editors. Sport and Human Genetics. Vol. 1986. Champaign, IL: Human Kinetics; pp. 173–181. [Google Scholar]
- 39.Rankinen T, An P, Perusse L, et al. Genome-wide linkage scan for exercise stroke volume and cardiac output in the HERITAGE Family Study1. Physiol Genomics. 2002;10:57–62. doi: 10.1152/physiolgenomics.00043.2002. [DOI] [PubMed] [Google Scholar]
- 40.Spielmann N, Leon AS, Rao DC, et al. Genome-wide linkage scan for submaximal exercise heart rate in the HERITAGE family study. AmJPhysiol Heart CircPhysiol. 2007;293:H3366–H3371. doi: 10.1152/ajpheart.00042.2007. [DOI] [PubMed] [Google Scholar]
- 41.Argyropoulos G, Stutz AM, Ilnytska O, et al. KIF5B gene sequence variation and response of cardiac stroke volume to regular exercise. Physiol Genomics. 2009;36:79–88. doi: 10.1152/physiolgenomics.00003.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rankinen T, Argyropoulos G, Rice T, et al. CREB1 is a strong genetic predictor of the variation in exercise heart rate response to regular exercise: the HERITAGE Family Study. CircCardiovascGenet. 2010;3:294–299. doi: 10.1161/CIRCGENETICS.109.925644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bouchard C, Sarzynski MA, Rice TK, et al. Genomic predictors of the maximal O uptake response to standardized exercise training programs. J Appl Physiol. 2011;110:1160–1170. doi: 10.1152/japplphysiol.00973.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De S, Kumari J, Mudgal R, et al. RECQL4 is essential for the transport of p53 to mitochondria in normal human cells in the absence of exogenous stress. J Cell Sci. 2012;125:2509–2522. doi: 10.1242/jcs.101501. [DOI] [PubMed] [Google Scholar]
- 45.Zhuang J, Wang PY, Huang X, et al. Mitochondrial disulfide relay mediates translocation of p53 and partitions its subcellular activity. Proc Natl Acad Sci U S A. 2013;110:17356–17361. doi: 10.1073/pnas.1310908110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lago CU, Sung HJ, Ma W, et al. p53, aerobic metabolism, and cancer. Antioxid Redox Signal. 2011;15:1739–1748. doi: 10.1089/ars.2010.3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matoba S, Kang JG, Patino WD, et al. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 48.Park JY, Wang PY, Matsumoto T, et al. p53 improves aerobic exercise capacity and augments skeletal muscle mitochondrial DNA content. Circ Res. 105:705–712. 2009. doi: 10.1161/CIRCRESAHA.109.205310. 711 p following 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saleem A, Adhihetty PJ, Hood DA. Role of p53 in mitochondrial biogenesis and apoptosis in skeletal muscle. Physiol Genomics. 2009;37:58–66. doi: 10.1152/physiolgenomics.90346.2008. [DOI] [PubMed] [Google Scholar]
- 50.Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- 51.Chance B, Im J, Nioka S, et al. Skeletal muscle energetics with PNMR: personal views and historic perspectives. NMR Biomed. 2006;19:904–926. doi: 10.1002/nbm.1109. [DOI] [PubMed] [Google Scholar]
- 52.Wang PY, Ma W, Park JY, et al. Increased oxidative metabolism in the Li-Fraumeni syndrome. N Engl J Med. 2013;368:1027–1032. doi: 10.1056/NEJMoa1214091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bergeaud M, Mathieu L, Guillaume A, et al. Mitochondrial p53 mediates a transcription-independent regulation of cell respiration and interacts with the mitochondrial F(1)F0-ATP synthase. Cell Cycle. 2013;12:2781–2793. doi: 10.4161/cc.25870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Laufs U, Wassmann S, Czech T, et al. Physical inactivity increases oxidative stress, endothelial dysfunction, and atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:809–814. doi: 10.1161/01.ATV.0000158311.24443.af. [DOI] [PubMed] [Google Scholar]
- 55.Gokce N, Keaney JF, Jr, Hunter LM, et al. Risk stratification for postoperative cardiovascular events via noninvasive assessment of endothelial function: a prospective study. Circulation. 2002;105:1567–1572. doi: 10.1161/01.cir.0000012543.55874.47. [DOI] [PubMed] [Google Scholar]
- 56.Vanhoutte PM, Shimokawa H, Tang EH, et al. Endothelial dysfunction and vascular disease. Acta Physiol (Oxf) 2009;196:193–222. doi: 10.1111/j.1748-1716.2009.01964.x. [DOI] [PubMed] [Google Scholar]
- 57.Nyberg M, Blackwell JR, Damsgaard R, et al. Lifelong physical activity prevents an age-related reduction in arterial and skeletal muscle nitric oxide bioavailability in humans. J Physiol. 2012;590:5361–5370. doi: 10.1113/jphysiol.2012.239053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Myers J, Prakash M, Froelicher V, et al. Exercise capacity and mortality among men referred for exercise testing. N Engl J Med. 2002;346:793–801. doi: 10.1056/NEJMoa011858. [DOI] [PubMed] [Google Scholar]
- 59.Clarkson P, Montgomery HE, Mullen MJ, et al. Exercise training enhances endothelial function in young men. J Am Coll Cardiol. 1999;33:1379–1385. doi: 10.1016/s0735-1097(99)00036-4. [DOI] [PubMed] [Google Scholar]
- 60.Harris RA, Padilla J, Hanlon KP, et al. The flow-mediated dilation response to acute exercise in overweight active and inactive men. Obesity (Silver Spring) 2008;16:578–584. doi: 10.1038/oby.2007.87. [DOI] [PubMed] [Google Scholar]
- 61.Jurva JW, Phillips SA, Syed AQ, et al. The effect of exertional hypertension evoked by weight lifting on vascular endothelial function. J Am Coll Cardiol. 2006;48:588–589. doi: 10.1016/j.jacc.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 62.MacDougall JD, Tuxen D, Sale DG, et al. Arterial blood pressure response to heavy resistance exercise. J Appl Physiol (1985) 1985;58:785–790. doi: 10.1152/jappl.1985.58.3.785. [DOI] [PubMed] [Google Scholar]
- 63.Phillips SA, Das E, Wang J, et al. Resistance and aerobic exercise protects against acute endothelial impairment induced by a single exposure to hypertension during exertion. J Appl Physiol (1985) 2011;110:1013–1020. doi: 10.1152/japplphysiol.00438.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Currie KD, McKelvie RS, Macdonald MJ. Flow-mediated dilation is acutely improved after high-intensity interval exercise. Med Sci Sports Exerc. 2012;44:2057–2064. doi: 10.1249/MSS.0b013e318260ff92. [DOI] [PubMed] [Google Scholar]
- 65.Tinken TM, Thijssen DH, Hopkins N, et al. Shear stress mediates endothelial adaptations to exercise training in humans. Hypertension. 2010;55:312–318. doi: 10.1161/HYPERTENSIONAHA.109.146282. [DOI] [PubMed] [Google Scholar]
- 66.Goto C, Higashi Y, Kimura M, et al. Effect of different intensities of exercise on endothelium-dependent vasodilation in humans: role of endothelium-dependent nitric oxide and oxidative stress. Circulation. 2003;108:530–535. doi: 10.1161/01.CIR.0000080893.55729.28. [DOI] [PubMed] [Google Scholar]
- 67.Vona M, Rossi A, Capodaglio P, et al. Impact of physical training and detraining on endothelium-dependent vasodilation in patients with recent acute myocardial infarction. Am Heart J. 2004;147:1039–1046. doi: 10.1016/j.ahj.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 68.Hambrecht R, Fiehn E, Weigl C, et al. Regular physical exercise corrects endothelial dysfunction and improves exercise capacity in patients with chronic heart failure. Circulation. 1998;98:2709–2715. doi: 10.1161/01.cir.98.24.2709. [DOI] [PubMed] [Google Scholar]
- 69.Hambrecht R, Adams V, Erbs S, et al. Regular physical activity improves endothelial function in patients with coronary artery disease by increasing phosphorylation of endothelial nitric oxide synthase. Circulation. 2003;107:3152–3158. doi: 10.1161/01.CIR.0000074229.93804.5C. [DOI] [PubMed] [Google Scholar]
- 70.Adams V, Linke A, Krankel N, et al. Impact of regular physical activity on the NAD(P)H oxidase and angiotensin receptor system in patients with coronary artery disease. Circulation. 2005;111:555–562. doi: 10.1161/01.CIR.0000154560.88933.7E. [DOI] [PubMed] [Google Scholar]
- 71.Wisloff U, Stoylen A, Loennechen JP, et al. Superior cardiovascular effect of aerobic interval training versus moderate continuous training in heart failure patients: a randomized study. Circulation. 2007;115:3086–3094. doi: 10.1161/CIRCULATIONAHA.106.675041. [DOI] [PubMed] [Google Scholar]
- 72.Belardinelli R, Lacalaprice F, Ventrella C, et al. Waltz dancing in patients with chronic heart failure: new form of exercise training. Circ Heart Fail. 2008;1:107–114. doi: 10.1161/CIRCHEARTFAILURE.108.765727. [DOI] [PubMed] [Google Scholar]
- 73.Miura H, Bosnjak JJ, Ning G, et al. Role for hydrogen peroxide in flow-induced dilation of human coronary arterioles. Circ Res. 2003;92:e31–40. doi: 10.1161/01.res.0000054200.44505.ab. [DOI] [PubMed] [Google Scholar]
- 74.Matoba T, Shimokawa H, Nakashima M, et al. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest. 2000;106:1521–1530. doi: 10.1172/JCI10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Phillips SA, Bian JT, Church EC, et al. Hydrogen Peroxide Prevents Impaired Endothelium-dependent Dilation Following Acute Exertion in Chronic Exercising but Not in Sedentary Subjects. Circulation. 2009;120:S1013–S1013. [Google Scholar]
- 76.Rush JW, Laughlin MH, Woodman CR, et al. SOD-1 expression in pig coronary arterioles is increased by exercise training. Am J Physiol Heart Circ Physiol. 2000;279:H2068–2076. doi: 10.1152/ajpheart.2000.279.5.H2068. [DOI] [PubMed] [Google Scholar]
- 77.Albert CM, Mittleman MA, Chae CU, et al. Triggering of sudden death from cardiac causes by vigorous exertion. N Engl J Med. 2000;343:1355–1361. doi: 10.1056/NEJM200011093431902. [DOI] [PubMed] [Google Scholar]
- 78.Rolim NP, Medeiros A, Rosa KT, et al. Exercise training improves the net balance of cardiac Ca2+ handling protein expression in heart failure. Physiol Genomics. 2007;29:246–252. doi: 10.1152/physiolgenomics.00188.2006. [DOI] [PubMed] [Google Scholar]
- 79.Oliveira RS, Ferreira JC, Gomes ER, et al. Cardiac anti-remodelling effect of aerobic training is associated with a reduction in the calcineurin/NFAT signalling pathway in heart failure mice. J Physiol. 2009;587:3899–3910. doi: 10.1113/jphysiol.2009.173948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Campos JC, Queliconi BB, Dourado PM, et al. Exercise training restores cardiac protein quality control in heart failure. PLoS One. 2012;7:e52764. doi: 10.1371/journal.pone.0052764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Antunes-Correa LM, Kanamura BY, Melo RC, et al. Exercise training improves neurovascular control and functional capacity in heart failure patients regardless of age. Eur J Prev Cardiol. 2012;19:822–829. doi: 10.1177/1741826711414626. [DOI] [PubMed] [Google Scholar]
- 82.Soares-Miranda L, Franco FG, Roveda F, et al. Effects of exercise training on neurovascular responses during handgrip exercise in heart failure patients. Int J Cardiol. 2011;146:122–125. doi: 10.1016/j.ijcard.2010.09.091. [DOI] [PubMed] [Google Scholar]
- 83.Gielen S, Adams V, Mobius-Winkler S, et al. Anti-inflammatory effects of exercise training in the skeletal muscle of patients with chronic heart failure. J Am Coll Cardiol. 2003;42:861–868. doi: 10.1016/s0735-1097(03)00848-9. [DOI] [PubMed] [Google Scholar]
- 84.Bacurau AV, Jardim MA, Ferreira JC, et al. Sympathetic hyperactivity differentially affects skeletal muscle mass in developing heart failure: role of exercise training. J Appl Physiol (1985) 2009;106:1631–1640. doi: 10.1152/japplphysiol.91067.2008. [DOI] [PubMed] [Google Scholar]
- 85.Cunha TF, Bacurau AV, Moreira JB, et al. Exercise training prevents oxidative stress and ubiquitin-proteasome system overactivity and reverse skeletal muscle atrophy in heart failure. PLoS One. 2012;7:e41701. doi: 10.1371/journal.pone.0041701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hambrecht R, Schulze PC, Gielen S, et al. Effects of exercise training on insulin-like growth factor-I expression in the skeletal muscle of non-cachectic patients with chronic heart failure. Eur J Cardiovasc Prev Rehabil. 2005;12:401–406. doi: 10.1097/01.hjr.0000173106.68485.b7. [DOI] [PubMed] [Google Scholar]
- 87.Lenk K, Erbs S, Hollriegel R, et al. Exercise training leads to a reduction of elevated myostatin levels in patients with chronic heart failure. Eur J Prev Cardiol. 2012;19:404–411. doi: 10.1177/1741826711402735. [DOI] [PubMed] [Google Scholar]
- 88.Barretto AC, Santos AC, Munhoz R, et al. Increased muscle sympathetic nerve activity predicts mortality in heart failure patients. Int J Cardiol. 2009;135:302–307. doi: 10.1016/j.ijcard.2008.03.056. [DOI] [PubMed] [Google Scholar]
- 89.Fiuza-Luces C, Garatachea N, Berger NA, et al. Exercise is the real polypill. Physiology (Bethesda) 2013;28:330–358. doi: 10.1152/physiol.00019.2013. [DOI] [PubMed] [Google Scholar]
- 90.Powers SK, Smuder AJ, Kavazis AN, et al. Mechanisms of exercise-induced cardioprotection. Physiology (Bethesda) 2014;29:27–38. doi: 10.1152/physiol.00030.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hoppeler H, Baum O, Lurman G, et al. Molecular mechanisms of muscle plasticity with exercise. Compr Physiol. 2011;1:1383–1412. doi: 10.1002/cphy.c100042. [DOI] [PubMed] [Google Scholar]
- 92.Ghosh S, Vivar JC, Sarzynski MA, et al. Integrative pathway analysis of a genome-wide association study of (V)O(2max) response to exercise training. J Appl Physiol (1985) 2013;115:1343–1359. doi: 10.1152/japplphysiol.01487.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]