

To the Editor
Human leukocyte antigen-G (HLA-G) is a non-classical, class Ib, major histocompatibility complex antigen, encoded by a gene on chromosome 6p21 within the HLA complex [1]. HLA-G is constitutively expressed during pregnancy where it has a critical role in maintaining immune tolerance toward the allogenic fetus and placenta [2, 3], but has also been associated with inflammatory diseases such as psoriasis, multiple sclerosis, and ulcerative colitis, and with solid-organ transplantation [3, 4]. We recently reported associations between variation in HLA-G and risk for asthma in Chicago-area asthma families, in multigenerational Dutch asthma families, and in a birth cohort at high risk for developing asthma [1, 5]. A role for HLA-G in asthma pathogenesis was further suggested by the demonstration of expression of a soluble isoform of HLA-G, sHLA-G5, in airway epithelial cells [1] and of increased circulating plasma levels of sHLA-G in children with atopic asthma [6]. Because airway inflammation in asthma involves a Th2-skewing of lymphocytes similar to pregnancy, HLA-G is an attractive candidate molecule for promoting the immune profile characteristic of asthma. Localization of HLA-G to airway epithelium suggests that its dysregulation could contribute to airway inflammation in chronic asthma. To evaluate this further, we hypothesized that HLA-G abundance would be increased in asthmatic airways.
To test this hypothesis, we measured concentrations of sHLA-G in bronchoalveolar lavage (BAL) fluid obtained from 12 non-asthmatic control subjects and 15 subjects with mild persistent asthma. The use of human subjects was approved by the University of Chicago Institutional Review Board. Asthma was diagnosed using National Asthma Education and Prevention Program guidelines. Subjects with a smoking history of ≥10 pack/years, who had used oral cortico-steroids within 6 months of study, who had received emergent care or had been hospitalized for asthma within 6 months of study, were excluded. Bronchoscopy was done at a time of stability for each subject.
The demographic, clinical and pulmonary function data for the subjects in our study are presented in Table 1. As expected, subjects with asthma had a lower FEV1 percent predicted (P = 0.01), more atopy (P = 0.0001), and more peripheral blood eosinophils (P = 0.02) compared to control subjects. However, there were no significant differences in cells counts in BAL fluid between the two groups. Lavage fluid was concentrated approximately 30-fold using Centriprep ultra-filtration chambers (Millipore, Inc.) with a 3 kD molecular weight cut-off filter. The retentate was analyzed for the presence of sHLA-G using an ELISA assay (Exbio, Inc.). The capture antibody, MEM-G/9, recognizes shed G1 and secreted G5, and the secondary antibody, antiβ2m, ensures measurement of β2m-configured soluble G [7]. The limit of sensitivity was ~0.2 U/ml. Values were adjusted for the degree of concentration as noted above and expressed as U/ml BAL fluid.
Table 1.
Demographic and clinical characteristics of study subjects.
| Normal subjects | Asthmatic subjects | P = * | |
|---|---|---|---|
| Number of subjects | 12 | 15 | |
| Age (years) | 37 ± 3 | 32 ± 3 | 0.22 |
| Female (N, %) | 6 (50%) | 11 (73%) | 0.26 |
| White (N, %) | 11 (92%) | 9 (60%) | 0.09 |
| Smoking history (N, %) | 1 (8%) | 7 (47%) | 0.05 |
| History of nocturnal asthma (N, %) | N/A | 6 (40%) | N/A |
| History of inhaled corticosteroid use (N, %) | N/A | 10 (67%) | N/A |
| Past history of oral corticosteroid use (N, %) | N/A | 5 (33%) | N/A |
| Past hospitalization for asthma (N, %) | N/A | 8 (53%) | N/A |
| Past history of emergent care for asthma (N, %) | N/A | 10 (67%) | N/A |
| Presence of 1 or more positive skin prick tests to allergen (N, %) | 1 (8%) | 14 (93%) | 0.0001 |
| Peripheral blood eosinophil count (mean ± SEM, cells / μl) | 119 ± 24 | 218 ± 29 | 0.02 |
| FVC (mean ± SEM, L) | 4.54 ± 0.29 | 4.09 ± 0.30 | 0.26 |
| FEV1 (mean ± SEM, L) | 3.79 ± 0.23 | 3.15 ± 0.23 | 0.05 |
| FEV1 (mean ± SEM, % predicted) | 105 ± 3 | 91 ± 3 | 0.01 |
| FEV1 / FVC ratio (mean ± SEM, %) | 84 ± 1 | 78 ± 2 | 0.02 |
| Methacholine PD20 (mean ± SEM, mg/ml) | > 10 | 1.35 ± 0.47 | † |
P values calculated using Fisher’s exact 2 x 2 test. Values < 0.05 were considered significant.
normal subjects with a methacholine PD20 < 10 mg/ml were excluded from study.
sHLA-G levels were increased in the BAL fluid of 15 asthmatic subjects (median 6.8 [in-terquartile range, 2.8, 7.8] U/ml) compared to 12 control subjects (median 1.6 [1.0, 3.0] U/ml, P = 0.01 by Mann-Whitney test) (Figure 1). One control value, and no asthmatic values, were below the limit of detection for the assay. We also examined whether racial background accounted for the observed differences in sHLA-G. There was no significant difference in sHLA-G levels in 9 Caucasian asthmatic subjects (median 5.6 [1.5, 6.9] U/ml) versus 6 African-American asthmatic subjects (median 6.9 [6.2, 8.6] U/ml, P = 0.24 by Mann-Whitney test). There were too few African-American control subjects for analysis in that group.
Figure 1.
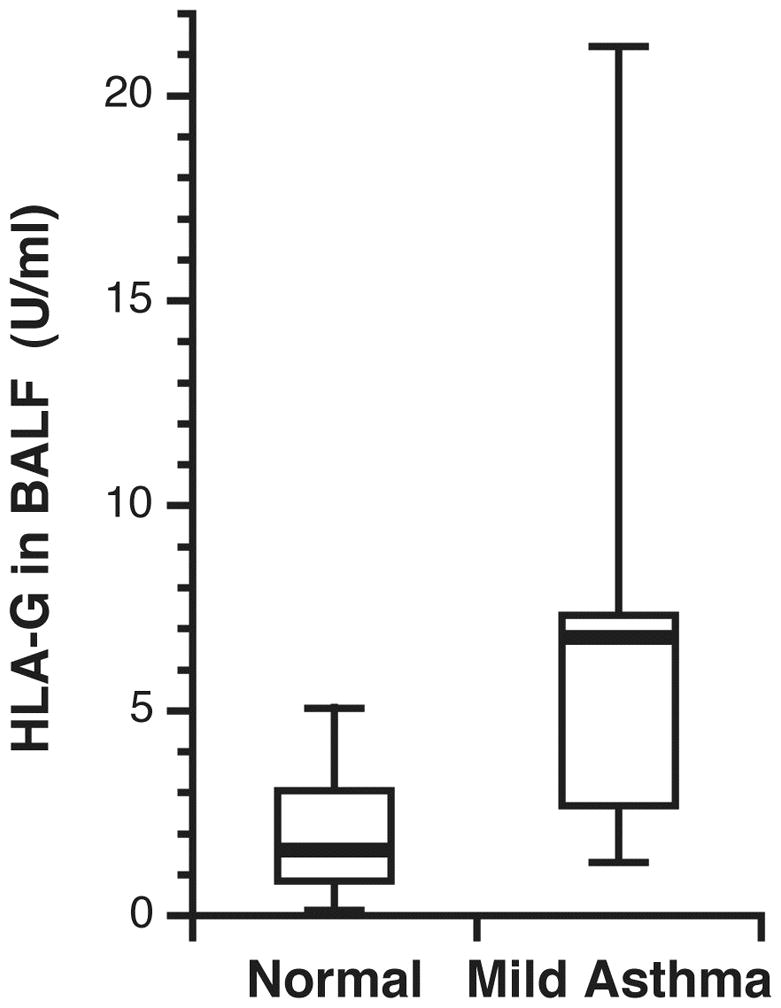
Level of sHLA-G in BAL fluid of 12 control subjects and 15 subjects with mild, persistent asthma. BAL fluid was concentrated and then analyzed by ELISA. The bar represents the median concentration. One control subject had a value below the limit of detection of the assay.
Immunoperoxidase labeling of endobronchial biopsies fixed in 10% neutral buffered for-malin was done using a primary antibody directed against sHLA-G (clone 2A12, Exbio, Inc., Czech Republic), which targets intron 4 sequences present only in sHLA-G. sHLA-G was present in the epithelium of endobronchial biopsies collected from 6 of 9 control subjects and 9 of 11 subjects with mild asthma that had sufficient epithelium present in the mucosal layer for review. Staining was evident in each major epithelial cell type (not shown). No visible differences in labeling patterns or intensity were apparent between asthmatic and control subjects, and no other submucosal airway cells and structures were stained. No notable infiltration of inflammatory cells was present in any biopsy as judged by hematoxylin and eosin staining.
We report for the first time increased levels of sHLA-G in BAL fluid from subjects with asthma compared to control subjects, supporting a role for sHLA-G in asthma pathogenesis. Our results are consistent with our previous reports of associations between regulatory polymorphisms in HLA-G and asthma [1, 5], and a study showing higher plasma levels of sHLA-G in 27 Turkish school children with atopic asthma compared to either 26 non-atopic, asthmatic children or 16 normal controls [6]. We note that all but one of our subjects with asthma were atopic and that we previously reported an association between HLA-G genotype and atopy in Dutch children [1]. Thus, it is possible that HLA-G influences asthma susceptibility through atopic pathways.
The source of the differences in HLA-G concentrations that we observed is the airway epithelium, as there was no detectable HLA-G in other airway structures. We propose that epithelial-derived sHLA-G has a paracrine role in regulating the activity of key inflammatory cells found in asthmatic airways. We note that in other contexts HLA-G has been shown to suppress dendritic cells and T cells that participate in inflammation [8], and to activate FoxP3+CD4+CD25+ regulatory T cells that can suppress cells that participate in airway inflammation [9].
Our observation does not provide insights into cause and effect: is HLA-G driving the inflammation in asthmatic airways or is it a reactive attempt to suppress inflammation present in asthmatic airways? In pregnancy, HLA-G is thought to promote the skewing of T cells toward a Th2 phenotype and to activate T regulatory cells [2, 3], an immune phenotype that parallels that seen in asthma. It is tempting to speculate that some individuals are genetically predisposed to over-express HLA-G in response to specific signals. Once secreted, HLA-G could promote a cascade of events that result in worsening inflammation. Several polymorphisms in the promoter region of HLA-G coincide with transcription factor binding sites could account for inter-individual differences in expression of HLA-G [10]. We previously identified a polymorphism in the 3’UTR of HLA-G that disrupts a microRNA target site and demonstrated allele-specific expression of HLA-G in the presence of microRNAs that bind to that target [5]. Therefore either lack of suppression or over-expression of HLA-G could explain the association we report here with asthma. We note that the small numbers of subjects in this study precludes more detailed analysis of relationships between genetic variation and HLA-G expression. Future, larger studies are required to clarify the potential modulating role of HLA-G on the clinical manifestations of asthma and the role of genetic variation on expression levels.
In conclusion, sHLA-G is present in greater concentrations in BAL in mild asthma. We suggest that the over expression or lack of suppression of HLA-G contributes to the disease process and that sHLA-G represents a novel pathway of asthma pathogenesis.
Signed,
Steven R. White, M.D.
Dagan A Loisel, Ph.D.
John F. McConville, M.D.
Randi Stern, M.S.
YingLi Tu
Bertha A. Marroquin, M.S.
Imre Noth, M.D.
Carole Ober, Ph.D.
Acknowledgments
This work was supported by AI056352, HL072414, HL080417, HL007605, HL095268 and RR024999. An abstract of a preliminary version of this manuscript was presented at the 2009 International meeting of the American Thoracic Society, San Diego, CA, on May 19, 2009. We thank Spring Maleckar, B.A., and Erika Low, B.A., for patient recruitment and technical assistance. We thank Jerry Krishnan, M.D., Ph.D., for advice on statistics. We thank Jacqueline Imperiale, R.N., the staff in the University of Chicago General Clinical Research Center, and the pulmonary and critical care fellows at the University of Chicago for assistance with bronchoscopy.
References
- 1.Nicolae D, Cox NJ, Lester LA, Schneider D, Tan Z, Billstrand C, Kuldanek S, Donfack J, Kogut P, Patel NM, Goodenbour J, Howard T, Wolf R, Koppelman GH, White SR, Parry R, Postma DS, Meyers D, Bleecker ER, Hunt JS, Solway J, Ober C. Fine mapping and positional candidate studies identify HLA-G as an asthma susceptibility gene on chromosome 6p21. Am J Hum Genet. 2005;76(2):349–357. doi: 10.1086/427763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hunt JS, Petroff MG, McIntire RH, Ober C. HLA-G and immune tolerance in pregnancy. FASEB J. 2005;19(7):681–693. doi: 10.1096/fj.04-2078rev. [DOI] [PubMed] [Google Scholar]
- 3.Rouas-Freiss N, Naji A, Durrbach A, Carosella ED. Tolerogenic functions of human leukocyte antigen G: from pregnancy to organ and cell transplantation. Transplantation. 2007;84(1 Suppl):S21–25. doi: 10.1097/01.tp.0000269117.32179.1c. [DOI] [PubMed] [Google Scholar]
- 4.Carosella ED, Moreau P, Lemaoult J, Rouas-Freiss N. HLA-G: from biology to clinical benefits. Trends Immunol. 2008;29(3):125–132. doi: 10.1016/j.it.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Tan Z, Randall G, Fan J, Camoretti-Mercado B, Brockman-Schneider R, Pan L, Solway J, Gern JE, Lemanske RF, Nicolae D, Ober C. Allele-specific targeting of microRNAs to HLA-G and risk of asthma. Am J Hum Genet. 2007;81(4):829–834. doi: 10.1086/521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tahan F, Patiroglu T. Plasma soluble human leukocyte antigen G levels in asthmatic children. Int Arch Allergy Immunol. 2006;141(3):213–216. doi: 10.1159/000095290. [DOI] [PubMed] [Google Scholar]
- 7.Fournel S, Huc X, Aguerre-Girr M, Solier C, Legros M, Praud-Brethenou C, Moussa M, Chaouat G, Berrebi A, Bensussan A, Lenfant F, Le Bouteiller P. Comparative reactivity of different HLA-G monoclonal antibodies to soluble HLA-G molecules. Tissue Antigens. 2000;55(6):510–518. doi: 10.1034/j.1399-0039.2000.550602.x. [DOI] [PubMed] [Google Scholar]
- 8.Naji A, Le Rond S, Durrbach A, Krawice-Radanne I, Creput C, Daouya M, Caumartin J, LeMaoult J, Carosella ED, Rouas-Freiss N. CD3+CD4low and CD3+CD8low are induced by HLA-G: novel human peripheral blood suppressor T-cell subsets involved in transplant acceptance. Blood. 2007;110(12):3936–3948. doi: 10.1182/blood-2007-04-083139. [DOI] [PubMed] [Google Scholar]
- 9.Lee JH, Yu HH, Wang LC, Yang YH, Lin YT, Chiang BL. The levels of CD4+CD25+ regulatory T cells in paediatric patients with allergic rhinitis and bronchial asthma. Clin Exp Immunol. 2007;148(1):53–63. doi: 10.1111/j.1365-2249.2007.03329.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan Z, Shon AM, Ober C. Evidence of balancing selection at the HLA-G promoter region. Hum Mol Genet. 2005;14(23):3619–3628. doi: 10.1093/hmg/ddi389. [DOI] [PubMed] [Google Scholar]