

Abstract
Timely detection of cutaneous squamous cell carcinoma with non-invasive modalities like nonlinear spectral imaging (NLSI) can ensure efficient preventive or therapeutic measures for patients. In this study, in vivo NLSI was used to study spectral characteristics in murine skin treated with 7, 12-dimethylbenz(a)anthracene. The results show that NLSI could detect emission spectral changes during the early preclinical stages of skin carcinogenesis. Analyzing these emission spectra using simulated band-pass filters at 450-460 nm and 525-535 nm, gave parameters that were expressed as a ratio. This ratio was increased and thus suggestive of elevated metabolic activity in early stages of skin carcinogenesis.
OCIS codes: (180.4315) Nonlinear microscopy; (180.6900) Three-dimensional microscopy; (170.1870) Dermatology; (170.6510) Spectroscopy, tissue diagnostics; (170.3880) Medical and biological imaging; (300.6420) Spectroscopy, nonlinear
1. Introduction
The global incidence of cutaneous squamous cell carcinoma (cSCC) affecting the Caucasian population has increased considerably in the last few decades [1]. Increase in sun seeking behaviors, popularity of tanning salons and more efficient reporting of new cSCC cases in current health systems could be considered as the etiology of this observed upward trend [2]. If left undiagnosed, cSCC arising in face, neck or scalp can turn invasive and reduce the patient’s quality of life. The solution to this problem would be to enable earliest detection of cancer related skin changes such that preventive or therapeutic intervention is effective for the individual.
Suspect skin lesions are often detected by the general practitioner at a routine clinical examination. However the general practitioner is often left in a dilemma to decide whether a patient needs to be referred to a dermatologist, who later sends skin biopsies to histopathology for confirmation. Secondly, needless biopsies performed on benign skin lesions are not beneficial to the patient and leaves behind cosmetic scarring and waste pathologist’s time Furthermore, diagnostic reports are not immediate. However these problems may be solved by exploring the potential of non-invasive diagnostic modalities in these fields. The most commonly used non-invasive instrument by the dermatologist since the last two decades is the dermatoscope that aided in better clinical diagnosis of cSCC [3]. However dermatoscopes cannot clearly differentiate between benign and malignant lesions as it cannot provide subcellular details that are needed to do so. These subcellular details can be provided with high resolution non-invasively by nonlinear optical imaging.
Nonlinear optical imaging depends on the following optical processes a) Two-photon Excitation Fluorescence (TPEF) [4] and b) Second Harmonic Generation (SHG) [5]. As a result, this imaging modality possesses various advantages over other conventional imaging techniques such as confocal laser scanning microscopy. This includes (a) minimal out-of-focus images, (b) deeper tissue imaging (c) ability to perform 3-D optical sectioning in the imaged tissue and (d) reduced phototoxicity [6]. Nonlinear optical imaging also provides subcellular resolution without relying on exogenous fluorescent labels. Due to these merits as a label-free non-invasive modality, the applications for nonlinear optical imaging in cancer diagnostics has burgeoned significantly in the last two decades [7–9]
Nonlinear optical imaging has been successfully used to detect tissue structural changes and differentiate between normal and cancerous tissues in various organs with high sensitivity and specificity [10–13]. Besides altered tissue architecture, cancerous tissues have also been discovered to have modified biochemical composition, such as altered production of proteins, lipids or other subcellular components [14–16]. This could affect the concentrations of endogenous fluorophores which may in turn change the tissue spectral characteristics. These spectral changes may be detected by the combination of nonlinear optical imaging with spectroscopy, better known as nonlinear spectral imaging (NLSI). Due to this beneficial aspect, NLSI has already been used to evaluate the morphology and biochemistry of skin [17–19] and other organs [20, 21]. But those studies were mainly restricted to normal or non-diseased tissues. Studies that used nonlinear optical imaging for skin cancer have so far relied mainly on fluorescence lifetime measurements [22–24], instead of emission spectral characteristics. Among the few studies that did use NLSI for skin cancer diagnosis, the experiments had been performed on ex vivo human skin biopsies [25–27]. The only notable study that employed in vivo NLSI for cancer characterization was performed in the hamster oral cavity cancer model by Edward et al. [28]. The avenue of in vivo NLSI for skin cancer diagnostics, especially cSCC remains largely unexplored at present.
Another key component that has been reported in cancerous tissue is its intrinsic metabolism profile. Detection and analysis of cellular metabolism is often based on the fluorescence measurements of endogenous metabolic biomarkers like nicotinamide adenine dinucleotide (NADH) in relation to flavin adenine dinucleotide (FAD) in a ratiometric form known as the metabolic redox ratio [29]. The use of redox ratio as a marker in cancer diagnostics is based on the hypothesis that increased cellular metabolism in cancer cells may cause the route of energy production to shift from the oxidative phosphorylation pathway to the glycolytic pathway [30, 31]. This can happen either in absence of oxygen (called diminished Pasteur effect) or in presence of oxygen (called aerobic glycolysis or Warburg effect) [32, 33]. This transition causes decrease in the rate of oxidative phosphorylation resulting in reduced FAD production and/or increased accumulation of NADH from glycolysis leading to altered intracellular dynamics between NADH and FAD. Various studies have tested this hypothesis of altered cellular metabolism or the metabolic redox ratio to differentiate cancer affected tissues from normal tissues in different organs such as oral cavity, breast, colon and ovaries [34–37]. However the metabolic activity in epidermal cells during skin cancer progression remains relatively unexplored.
This study primarily utilized in vivo NLSI that relied on TPEF, SHG and spectroscopy to uniquely detect any alteration in the spectral characteristics in carcinogen treated mice skin during various stages of skin cancer. This study was distinct as it mainly focussed on observing spectroscopic changes from the preclinical stages of skin cancer (i.e. in clinically normal mice skin prior to tumour appearance). In addition, the acquired emission spectra were further analyzed using simulated band-pass filters to indirectly estimate NADH and FAD levels in the epidermis. The quantified parameters were expressed as a ratio that was then compared during the various stages of skin carcinogenesis.
2. Materials and methods
2.1. DMBA skin carcinogenesis protocol
This experimental animal protocol was discussed and approved by the Animal Research Committee of the Erasmus University, Rotterdam. 36 female albino SKH1-hr strain hairless mice were ordered from Charles River Laboratories, Someren, the Netherlands. The mice were housed under standard housing conditions and fed ad libitum on a chlorophyll-free diet (Hope Farms BV, Woerden, The Netherlands). This diet was followed to prevent the autofluorescence emission from pheophorbide-a in mice skin at 674 nm [38]. The mice were randomly and equally distributed over three groups. In the first control group, the mice skin remained untreated, while mice in the second control group were treated with weekly topical application of acetone (Sigma Aldrich Chemie, Zwijndrecht, the Netherlands) on skin for 20 weeks. The test group mice were treated similarly with a weekly topical application of 0.15 mg 7,12-dimethylbenz(a)anthracene (DMBA) (Sigma Aldrich Chemie, Zwijndrecht, the Netherlands) dissolved per ml of acetone [39]. The mice were clinically examined every week post the first topical DMBA application, to assess skin changes and onset of tumor formation.
2.2. In vivo imaging protocol with the animal model
The imaging protocol was performed on a self assembled NLSI setup that has been described elaborately earlier [17]. A schematic diagram of the NLSI system is shown in Fig. 1 . The wavelength and laser power of the setup was assessed regularly to ensure consistent signal. The wavelength of the spectrograph was calibrated using red, green and blue optical RazorEdge filters (Semrock, Rochester, New York, U.S.A) and a multiple band-pass emission filter (FF01-390/482/563/640-25, Semrock, Rochester, New York, U.S.A). SHG peak measurement from dermal collagen in an ex vivo sample of pig skin was used to calibrate the wavelength up to 1 nm accuracy. The average power at sample stage (focal plane) was measured with a power meter (Coherent Europe B.V., Utrecht, the Netherlands). In addition, the flat-field calibration of the setup was done by analyzing the two-photon spectra from solutions of Lucifer Yellow (Life Technologies Europe, Bleiswijk, the Netherlands) and Coumarin-120 (Sigma Aldrich Chemie, Zwijndrecht, the Netherlands).
Fig. 1.
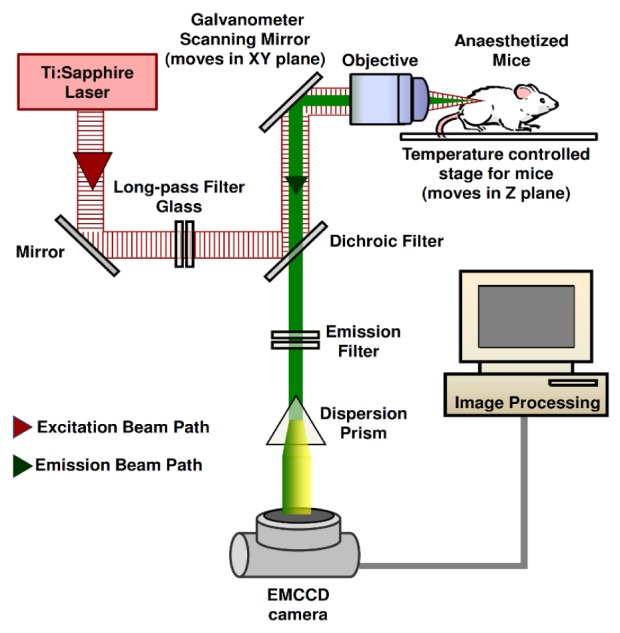
Schematic diagram of the NLSI setup. (EMCCD – Electron Multiplying Charge Coupled Device)
The reference emission spectral values were primary obtained from ex vivo rat tail tendon, 8.1 g/ml aqueous solution of human epidermal keratin, 0.2 mg/ml FAD in phosphate buffered saline (PBS) solution (pH 7.4) and 2.5 mM NADH (Sigma-Aldrich Chemie, Zwijndrecht, the Netherlands) in 1 ml of 3-(N-morpholino)propanesulfonic acid (MOPS) buffer solution (pH 7.4). From all three groups, mice were taken for imaging at weeks 11, 14, 17, 20 and 23 after the first topical application. Prior to imaging, a combination of ketamine (75 mg/kg of mouse body weight) and medetomidine (1 mg/kg of mouse body weight) in physiological saline solution (Janssen Pharmaceutica, Tilburg, the Netherlands) was administered intraperitoneally to the mice for anesthesia. Black ink was later used to pre-mark the skin area to be imaged on the mice.
The femtosecond laser functioned at a pulse repetition frequency of 80 MHz and pulse duration of 90 femtoseconds. Wavelength of 765 nm was selected as the excitation light source while spectral imaging was performed with an infinity corrected water-immersion objective (40 × , Numerical Aperture = 0.8, Nikon, Japan). In every imaged region, one transverse x-y scan (field of view: 100 μm × 100 μm) was performed per interval of 3 μm extending up to depth of 60 μm from the surface, along with an additional axial x-z scan (field of view: 100 μm × 60 μm). The average laser power ranged from 17 mW – 45 mW at the mice skin surface, and was gradually raised with increasing tissue depths. The pixel dwell time was kept at 128 μs, and scanned over 224 × 224 pixels. The acquisition time was 6.4 seconds/scan and ~2.25 minutes per skin spot. These imaging steps were repeated for at least two spots per mouse. All mice from untreated, acetone treated and DMBA treated groups underwent the same imaging protocol.
In vivo NLSI was performed on clinically normal skin or DMBA induced lesions preferentially located near the thigh region of the mice to (a) minimize motion artifacts from breathing and (b) eliminate anatomical variation in skin from different regions. Following imaging anaesthetized mice were euthanized by cervical dislocation. After euthanasia, skin biopsies from the pre-marked regions were taken and fixed in 10% buffered formalin solution and sent for histopathologic evaluation by a certified veterinarian pathologist. Based on the clinical macroscopic appearance and its corresponding histopathological evaluation, mice skin was later categorized as: (a) Normal (non-neoplastic): No microscopic or clinical skin changes, (b) Preclinical hyperplasia: Positive microscopic changes of hyperplasia with no clinical skin change and (c) Clinical visible tumors (pre-/neoplastic): Positive microscopic changes of preneoplasia or neoplasia with presence of clinically visible lesion. All experiments were performed in compliance with the relevant laws and institutional guidelines in accordance with the ethical standards of the Declaration of Helsinki.
2.4. Data analysis of NLSI experimental data
Spectral width of SHG obtained in vivo from mice skin was broader than that obtained from the reference sample. This was also observed by Palero et al. [19] and was attributed to spectral resolution degradation of the setup due to increased scattering of detected light. This generally occurs more during deeper in vivo imaging of thicker specimens like skin, as compared to ex vivo imaging of relatively thinner monolayers of reference samples. This spectral broadening of SHG is a setup artifact and was found to be inconsequential for the data analyzed in this study
Transverse x-y scans deeper than 48 μm were not evaluated due to marked signal attenuation and decreased signal-to-noise ratio beyond this depth. In addition, spectral scans that were affected heavily by breathing artifacts from the anaesthetized mice were excluded. Moreover NLSI had also been performed on skin lesions that had a diameter > 4 mm and were extensively keratinized. These types of lesions gave no detectable signal from any fluorophores besides keratin throughout the imaged depth and were therefore omitted from further data analysis. Eventually a sample size of 32 skin regions was considered, comprising of 17 normal skin (from 6 untreated and 6 acetone treated mice), 8 skin with preclinical hyperplasia (from 6 DMBA treated mice) and 7 skin with clinically visible tumors (from 6 DMBA treated mice). Analysis was performed for one axial x-z scan and a stack of transverse x-y scans for each skin region. For each individual scan, spectral analysis was performed by averaging over the entire image. Microscopic and spectral information of scans deeper than 36 μm were similar to that obtained from scans at 36 μm and therefore did not contribute any additional valuable information.
Spectral data acquisition and processing into RGB images was performed by software written in V + + (Digital Optics, Auckland, New Zealand). The raw data file was processed in V + + by a script written in the built-in programming language called Vpascal. This script executed the following functions: (a) averaging and subtraction of background spectra from the fluorescence emission spectrum, (b) applying a threshold, (c) creating 8-bit auto-scaled intensity images by summation over the whole emission spectrum and (d) applying 8-bit RGB coloring. The spectral images were then converted to real color RGB images by multiplying the emission spectrum by the spectra for the red, green and blue sensitivities of the human eye (flat-field correction is included). The sums of these multiplied spectra gave the R, G, and B values, respectively. The R/G/B values were then scaled from 0 to 1 (the latter being the largest of R, G, and B) and multiplied by the intensity (8-bit) of the pixel [17, 40].
The microscopic features seen in NLSI RGB images were then compared with the findings seen in hematoxylin and eosin (H&E) stained slides that had been evaluated by the pathologist. The acquisition of spectral details from the image data file was done on the ImageJ software (available at http://imagej.nih.gov/ij/) by using the approach mentioned in previous studies [17, 18]. The acquired emission spectra was obtained from axial x-z scans and then normalized to the maximal intensity. The normalized emission spectra were categorized under the assigned clinico-pathologic grade, averaged and then compared. This process was also repeated for emission spectra obtained from transverse x-y scans at varying depths from skin surface, to observe spectral variation with depth in all three clinico-pathologic grades of mice skin.
The original (un-normalized) emission spectral plot values of axial x-z scans were then divided by the square of the average laser power used during imaging. This was done to compensate for nonlinear fluctuation in fluorescence intensities that arises due to differences in laser powers used during the imaging experiments. Following this, the area under curve (AUC) for the spectral plot was integrated from 410 nm – 650 nm to generate total autofluorescence intensity and from 370 nm – 410 nm for SHG intensity for the imaged skin region. These two parameters were averaged to obtain the mean total autofluorescence intensity and the mean total SHG intensity for each clinico-pathologic grade, which were then compared later.
In order to estimate NADH and FAD from the epidermis, we used an indirect method by applying simulated band-pass filters on the emission spectra of transverse x-y scans. As described earlier, the emission spectra of transverse x-y scans were also divided by the square of the average laser power to compensate for laser power variation. This method later involved integrating AUC of the emission spectra from 450 nm – 460 nm to provide a relative estimate for NADH in the epidermis and denoted as AUC 450nm-460nm. Similarly the emission spectra were integrated from 525 nm – 535 nm to obtain a relative estimate of FAD in the epidermis and indicated as AUC525nm-535nm. The simulated band-pass filters had a bandwidth of 10 nm and were based on emission peak values of NADH and FAD obtained from literature [41]. The ratios were calculated as AUC450nm-460nm/AUC525nm-535nm and then averaged for the corresponding clinico-pathologic grade and compared. To keep AUC450nm-460nm and AUC525nm-535nm as a reliable indicator for NADH and FAD respectively, we tried to minimize autofluorescence from other fluorophores such as keratin (stratum corneum and hair follicle), elastin and collagen (dermis) at 450 nm – 460 nm, and lipids and lipofuscin (stratum corneum) at 525 nm – 535 nm. This was done by performing the analysis in a spatially discriminated and depth specific manner in the transverse x-y scans. Therefore ratio calculations were limited to epidermal cells at depth of 6 – 12 μm for normal skin, 6 – 24 μm for skin with preclinical hyperplasia and 9 – 36 μm for clinically visible tumors. Statistical analysis was performed for all data while comparison, using two-sample t-test of unequal variance with a p-value < 0.05 being considered statistically significant.
It should additionally noted that analysis of the reference NADH (2.5mM) and FAD (0.2 mg/ml) solutions showed spectral overlap at the imaging wavelength 765 nm used in this study. The spectral overlap was about 11% and 29% at the simulated band-pass filter of 450 nm – 460 nm and 525 nm – 535 nm respectively. It is also known that the degree of spectral overlap between two fluorophores also depends on the individual fluorophore concentration. Therefore it is not known if the degree of spectral overlap observed ex vivo in reference solutions of known fluorophore concentrations can be extrapolated reliably for an ‘in vivo’ environment, where the fluorophore concentrations are unknown. Therefore for the simplicity of calculations in an in vivo scenario, the spectral overlap between NADH and FAD at 450 nm – 460 nm and 525 nm – 535 nm shall be considered negligible, while calculating the ratio AUC450nm-460nm/AUC525nm-535nm.
3. Results
3.1 Clinical and histopathological findings of DMBA induced skin carcinogenesis
Untreated and acetone treated mice exhibited no skin related symptoms or signs throughout the experiment. In DMBA treated mice the first skin lesion was observed after 11 weeks of topical DMBA application. Following its onset, DMBA induced lesions presented initially as non keratinized lesions of diameter < 1 mm and > 2 mm. These lesions increased in diameter and got heavily keratinized in the successive weeks.
Upon histopathological evaluation, normal skin was identified in biopsies from all three groups of mice. However skin regions with preclinical changes or clinically visible tumors were graded only from skin biopsies of DMBA treated mice. It was further observed that DMBA treated mice skin exhibited normal, preclinical hyperplasia and clinical pre-/neoplastic stages concurrently in the same mouse at all evaluated time points starting from week 11. Skins with preclinical changes were basically characterized by epidermal or squamous hyperplasia. Clinically visible early tumors (1 – 4 mm diameter) were identified as: (a) squamous cell papilloma (with or without aypia) and (b) exophytic and endophytic acanthoma (with or without atypia). The highly advanced and heavily keratinized skin tumors (diameter > 4 mm; not considered for further spectral analysis as mentioned earlier) were found to be (a) keratoacanthoma (with or without invasion), (b) squamous cell carcinoma in situ (Bowenoid lesion) or (c) squamous cell carcinoma (well or moderately differentiated).
3.2. In vivo nonlinear spectral images compared with standard H&E stained images
Since in vivo NLSI were taken from a microscopic field of view (100 × 100 μm), it was not possible to match the exact imaged spot with the corresponding H&E stained section of the skin biopsy. Hence, our analysis relies on the assumption that there are no major histological changes between the imaged spot and adjacent regions. As shown in Fig. 2 , NLSI scan of normal mice skin provided RGB images (Fig. 2(a)) that were quite comparable to the corresponding H&E stained section as seen in Fig. 2(b). In normal skin, stratum corneum was visualized as the greenish-yellow colored layer in the NLSI scan. The underlying stratum spinosum and basale appears as a blue layer, while SHG from the dermis was observed as the bright violet layer in the RGB image.
Fig. 2.

Comparison between axial NLSI x-z scans and H&E stained sections for normal skin, skin with preclinical hyperplasia and clinically visible tumors (top to bottom). Axial x-z scans clearly displays stratum corneum (green layer), the deeper epidermis (blue layer) and the dermis (violet layer) (2a). The RGB colors in the images were obtained by multiplying the emission spectrum from skin with the spectra for the red, green and blue sensitivities of the human eye. The RGB colors in NLSI scans correspond to the wavelengths in tissue emission spectra shown. Epidermal proliferation can be clearly observed with in vivo NLSI in 2c and 2e. The corresponding H&E stained images show normal SKH1-hr mice skin (b), hyperplasia with no atypia (d) and severe hyperplasia with atypia (part of an acanthoma) (f). The corresponding axial H&E obtained from biopsy of the imaged spot may not represent the exact position of the imaged spot. (x-z scan scale – 100 μm × 60 μm, 224 pixels × 224 pixels).
On the other hand compared to normal skin, H&E stained sections of DMBA treated mice skin with preclinical hyperplasia showed an evident increase in epidermal thickness as observed in Fig. 2d. This change was clearly seen in NLSI scan as well (Fig. 2(c)) characterized by an increased thickness of the blue colored layer (stratum spinosum and basale). In contrast, SHG from dermis was visibly diminished as compared to normal skin. In skin with clinically visible tumors, H&E stained section (Fig. 2(f)) showed a greater degree of epidermal proliferation as compared to the preclinical stage. In the corresponding NLSI, only epidermal autofluorescence was observed with no dermal SHG as seen in Fig. 2(e).
Transverse x-y spectral images in normal mice skin (Fig. 3 ) revealed corneocytes typically present in stratum corneum at a depth of 3 μm from skin surface. From 6 μm downwards, autofluorescence from epidermal cells located in stratum granulosum was seen. Autofluorescence from actively dividing epidermal cells present in stratum basale continued to dominate at 12 μm. For subsequent deeper scans in normal skin, epidermal autofluorescence gradually diminished while dermal SHG predominated. In DMBA treated mice skin with preclinical hyperplasia, epidermal autofluorescence persisted down to 24 μm deep, which is twice as much compared to normal mice skin. In addition, SHG from dermis was relatively weaker compared to normal skin and could be visualized only below 24 μm onwards. In clinically visible skin tumors, scans demonstrated that presence of a disorganized stratum corneum layer up to a depth of 6 μm. In addition, epidermal autofluorescence was observed all the way down to the deepest scan considered for analysis, with no detectable dermal SHG. In addition, the scans clearly revealed cells showing transformed cytonuclear morphology with visibly enlarged nuclei and mitotic cells (as seen in Fig. 4 ). These features were notably absent in normal skin and skin with preclinical hyperplasia
Fig. 3.

Transverse x-y NLSI obtained at depths of 3 μm, 6 μm, 12 μm, 18 μm, 24 μm and 36 μm from surface for normal skin, skin with preclinical hyperplasia and clinically visible skin tumor. Epidermal autofluorescence was present down to 24 μm for preclinical hyperplasia and persisted even deeper for clinically visible skin tumors respectively, when compared to normal mice skin. The RGB colors in the images were obtained by multiplying the emission spectrum by the spectra for the red, green and blue sensitivities of the human eye. RGB colors in NLSI scans correspond to the wavelengths in tissue emission spectra shown. (x-y scan scale – 100 μm × 100 μm, 224 pixels × 224 pixels).
Fig. 4.

Higher magnification of transverse NLSI x-y scans showing the cytonuclear morphology in (a) normal skin, (b) skin with preclinical hyperplasia and (c) clinically visible skin tumor. Yellow continuous circle denotes epidermal cells with enlarged nuclei and white dotted circle indicates dividing (mitotic) epidermal cells in skin tumor.
3.2. Tissue spectral changes and mean spectral emission intensities in different stages of DMBA induced skin carcinogenesis
The rat tail tendon reference sample gave a characteristic emission spectrum as seen in Fig. 5(a) , which was composed of a strong SHG component that peaks at 382 nm which corresponds to collagen in the rat tail and an autofluorescence component that peaks at about 450 - 480 nm. This autofluorescence component could be contributed by (i) collagen cross-links (digestible by collagenase) [42] or (ii) elastin [43] present in the rat tail tendon. Emission spectra from reference sample solutions of keratin, free NADH and FAD gave characteristic peaks at 480 nm, 460 nm and 535 nm respectively (Fig. 5(a)), which was in agreement with literature for the corresponding fluorophores [41, 44].
Fig. 5.
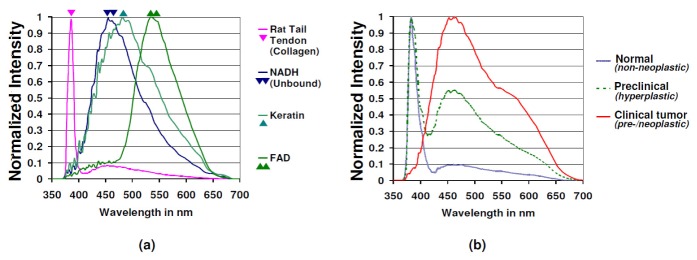
(a) Reference emission spectra (normalized) from rat tail tendon collagen, free NADH, keratin and FAD, (b) Normalized emission spectra from axial x-z scans in normal mice skin (z = 17, s = 17, n = 12), mice skin with preclinical changes (z = 8, s = 8, n = 6) and clinically visible mice skin tumors (z = 7, s = 7, n = 6). The corresponding spectrum is an average of the emission spectra obtained from ‘z’ axial x-z NLSI scans (100 μm × 60 μm) in ‘s’ regions from ‘n’ mice for each clinco-pathologic grade.
As seen in Fig. 5(b), the emission spectra of normal mice skin characteristically possessed (a) an SHG component from 370 nm to 410 nm and (b) a broadband autofluorescence component from 410 nm to 650 nm. Comparing the emission spectra from reference samples obtained ex vivo with that of normal mice skin obtained in vivo gave the following findings – (a) The SHG component obtained from normal mice skin in vivo had a similar emission peak with that of the rat tail tendon sample ex vivo, indicating that source of SHG in mice skin is obviously dermal collagen. (b) The ex vivo emission spectra of reference sample solutions indicate that the broad autofluorescence component obtained in mice skin in vivo can be attributed to contributions from fluorophores like NADH, keratin and FAD in epidermis and collagen and elastin in dermis.
From the normalized tissue spectral plots of axial x-z scans in normal skin, the epidermal autofluorescence intensity was found to be only one tenth of the dermal SHG intensity (as plotted in continuous blue line in Fig. 5(b). In contrast, the emission spectra of DMBA treated mice skin with preclinical hyperplasia (plotted in dashed green line in Fig. 5(b) showed the epidermal autofluorescence peaked at around 450 nm – 460 nm and its intensity was roughly as high as half the dermal SHG intensity. This suggested that relative contribution of the epidermal autofluorescence component in the spectral plot increased by about 5 times in skin with preclinical hyperplasia when compared to normal mice skin. Unlike normal mice skin and preclinical hyperplastic skin, the tissue emission spectra from axial x-z scans obtained from clinically visible skin tumors (plotted in continuous red line in Fig. 5(b)) showed the epidermal autofluorescence component (emission peak about 450 nm – 460 nm) to be the sole contributor to the spectral graph, while the relative contribution of dermal SHG was negligible.
Upon evaluating the variation in emission spectral plots for normal skin, skin with preclinical hyperplasia and clinically visible skin tumors at different skin depth (3 μm – 36 μm), it was seen that each clinico-pathologic grade had its own distinctive pattern. As seen in Fig. 6(a) , at a depth of 3 μm, all the three grades of skin regions had a common emission peak at around 480 nm that corresponds to keratin. Interestingly there was another notable peak at 540 nm – 550 nm for normal skin and skin with preclinical hyperplasia, while clinically visible skin tumors had a unique peak near 570 nm. The dermal SHG component was absent for all three grades at this depth. As the scans approached a depth of 6 μm, the emission peak shifted to 450 nm – 460 nm for all three grades (Fig. 6(b)), suggesting a dominant presence of NADH from this point onwards. While the peak at 540 nm – 550 nm begins to clearly diminish for normal mice skin and skin with preclinical hyperplasia, the unique emission peak at 570 nm is quite evident for neoplastic skin at this depth.
Fig. 6.

Normalized emission spectra for normal mice skin (y = 17, s = 17, n = 12), skin with preclinical hyperplasia (y = 8, s = 8, n = 6) and clinically visible skin tumors (y = 7, s = 7, n = 6) at depths of (a) 3 μm, (b) 6 μm, (c) 12 μm, (d) 18 μm, (e) 24 μm and (f) 36 μm from skin surface. The depicted spectrum is an average of the emission spectra obtained at the corresponding depth from ‘y’ transverse x-y NLSI scans (100 μm × 100 μm) in ‘s’ regions from ‘n’ mice for each clinico-pathologic grade.
From the depth of 12 μm downwards, the dermal SHG was the dominant component of the spectral plot for normal mice skin, while the epidermal autofluorescence decreased relatively and remained at its lowest level from 24 μm downwards as displayed in Fig. 6(c) – Fig. 6(f). However in skin with preclinical hyperplasia, the spectral contribution from epidermal autofluorescence remained high up to a depth of 24 μm, following which it gradually decreased. Despite this decrease, the relative contribution of epidermal autofluorescence to tissue emission spectra in mice skin with preclinical hyperplasia remained higher than that of normal mice skin even at 36 μm (Fig. 6(f)). In contrast, the dermal SHG spectral component in mice skin with preclinical hyperplasia kept rising with increasing skin depth, albeit at a relatively slower rate than normal skin. Clinically visible skin tumors on the other hand, exhibited a consistent spectral profile with no alterations from 12 μm to 36 μm, characterized by the predominant epidermal autofluorescence peak at 450 – 460 nm and absence of dermal SHG. Another notable point was that the spectral peaks present at 540 nm – 550 nm for normal and skin with preclinical hyperplasia and at 570 nm for clinically visible skin tumors were not observed anymore at depths below 12 μm.
The mean total autofluorescence and SHG intensity quantified from the area under the spectral plots for each clinico-pathologic grade of mice skin is depicted in Fig. 7 . There was no significant difference in the mean total autofluorescence intensity between normal mice skin, mice skin with preclinical hyperplasia or clinically visible skin tumors. On the other hand, the mean total SHG intensity was significantly decreased in skin with preclinical hyperplasia by about a factor of 3.77 compared to normal skin (p-value < 0.05). The mean total SHG intensity measured from visible skin tumors was almost negligible.
Fig. 7.
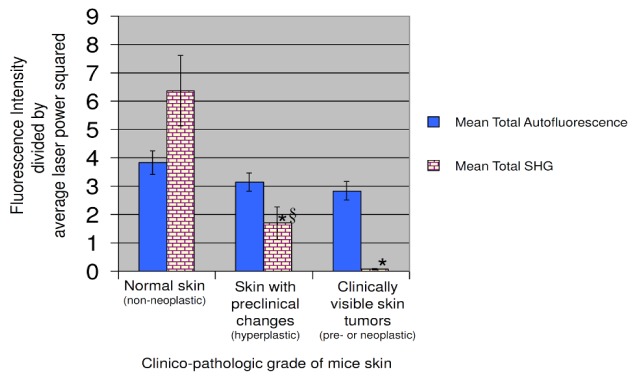
Comparison of mean total autofluorescence intensity (410 nm – 650 nm) and mean total SHG intensity (370 nm – 410 nm) for normal mice skin (z = 17, s = 17, n = 12), skin with preclinical hyperplasia (z = 8,s = 8, n = 6) and clinically visible skin tumors (z = 7,s = 7, n = 6). The intensities were calculated and averaged from ‘z’ axial x-z NLSI scans (100 μm × 60 μm) in ‘s’ regions from ‘n’ mice for each clinico-pathologic grade. Y-bars stand for standard error of the mean (* - p-value < 0.05 with respect to normal skin, § - p-value < 0.05 for skin with preclinical hyperplasia with respect to clinically visible skin tumors).
The ratio formulated as AUC450nm-460nm/AUC525nm-535nm was observed to be the lowest for normal mice skin as seen in Fig. 8 . On the other hand, this ratio was found to be increased by about 12% for skin with preclinical hyperplasia and 9% for clinically visible skin tumors (p-value < 0.05). There was no significant difference in the ratio obtained from skin with preclinical hyperplasia and clinically visible skin tumors.
Fig. 8.
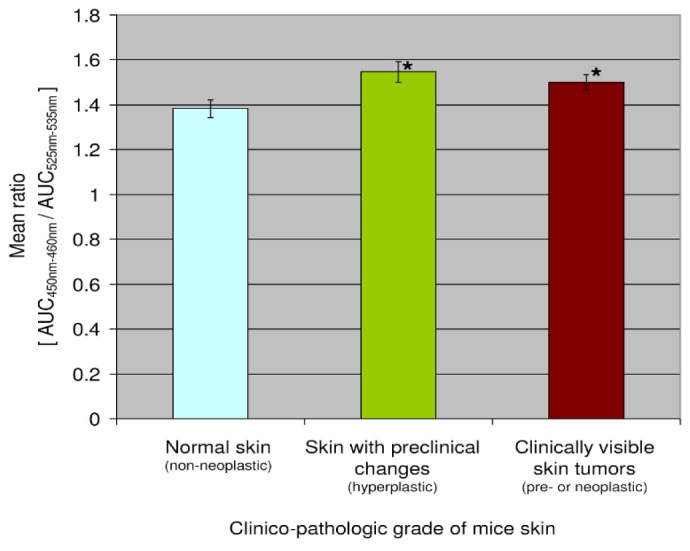
Comparison of mean ratio AUC450nm-460nm/AUC525nm-535nm (AUC- Area under curve) between normal mice skin (y = 49, s = 17, n = 12), skin with preclinical hyperplasia (y = 42, s = 8, n = 6) and clinically visible skin tumors (y = 63, s = 7, n = 6). The ratio was calculated and averaged from ‘y’ transverse x-y NLSI scans (100 μm × 100 μm) in ‘s’ regions from ‘n’ mice for each clinico-pathologic grade. Y-bars stand for standard error of the mean (* - p-value < 0.05 with respect to normal mice skin).
4. Discussion
4.1 Spectroscopic changes monitored by in vivo nonlinear spectral imaging during DMBA induced skin carcinogenesis
This study attempted to determine whether in vivo NLSI based on TPEF and SHG could monitor morphological, spectroscopic and metabolic changes in mice skin at different stages of DMBA induced carcinogenesis. When in vivo NLSI was applied on various clinico-pathologic grades of mice skin, distinct microscopic changes could be observed. These observations mainly included DMBA treated mice skin region with preclinical changes displaying a clear increase in the area with epidermal autofluorescence when compared to control normal mice skin. This increase in area with epidermal autofluorescence could be due to a) increased keratin deposition and elevated corneocyte turn-over that lead to thickened stratum corneum (hyperkeratosis) and b) increase in area with intracellular NADH fluorescence due to epidermal proliferation. The degree of epidermal proliferation was even more severe in clinically visible skin tumors such that the dermal SHG could not be visualized anymore. These findings was readily confirmed in the corresponding transverse x-y scans, where epidermal autofluorescence persisted up to a depth of 24 μm for skin with preclinical hyperplasia and down to the deepest scan taken for clinically visible skin tumors. In contrast, epidermal autofluorescence in normal mice skin diminished below a depth of 12 μm. In addition, the transverse scans in clinically visible skin tumors displayed an increased presence of cells with enlarged nuclei and actively dividing cells. These features are indicative of altered nuclear-cytoplasmic ratio and elevated mitotic activity that is characteristic of epithelial dysplasia. Thus, these findings reveal that NLSI can clearly visualize cytonuclear details that might aid the pathologists to determine and monitor epithelial dysplasia in the imaged organs real time and in vivo.
In conjunction with the microscopic observations, NLSI was able to generate additional spectral details that gave an insight of the biochemical changes during different stages of skin carcinogenesis. The acquired emission spectra varied distinctively for each clinico-pathologic grade of mice skin. At 3 μm, the tissue emission spectra for all three grades of mice skin shared a common peak at 480 nm that corresponded to keratin present in stratum corneum. There were additional peaks at 540 nm for normal mice skin and DMBA treated skin with preclinical hyperplasia which is probably due to high concentration of lipids in stratum corneum, especially phospholipids that have a similar emission peak [41, 45]. On the other hand, clinically visible skin tumors exhibited a unique peak at 570 nm which may be due to lipofuscin or lipofuscin-like pigments which have emission around the same range [46]. Possibly the increased generation of lipofuscin in the non-dividing post-mitotic cells is due to lipid oxidation with time and ageing [47]. Due to rapid proliferation in these tumors, these post-mitotic cells are rapidly extruded towards the skin surface leading to lipofuscin accumulation in these layers. This hypothesis is however based on the emission peaks of lipofuscin obtained from literature [46]. Since reference samples for lipofuscin were not commercially available to evaluate the corresponding emission spectra, this lipofuscin hypothesis needs further validation in future studies.
As the x-y scans were performed deeper to the stratum corneum, there was a clear blue-shift in the emission spectra towards 450 nm – 460 nm for all three clinico-pathologic grades. This is probably attributed to intracellular NADH present in metabolically active epidermal cells (stratum spinosum and basale) that lie below the metabolically inert stratum corneum. However beyond a depth of 12 um, NADH associated fluorescence diminished in normal mice skin. At the depth below 12 um, dermal SHG dominated in the spectra for normal skin. This spectral transition from epidermis to dermis was comparable with the findings in the in vivo study by Palero et al. on normal hairless mice skin [18, 45]. Interestingly, this spectral pattern was altered for DMBA treated skin with preclinical hyperplasia and clinically visible skin tumors with NADH associated fluorescence being present as deep as 24 μm and 36 μm respectively. This was clearly indicative of actively dividing epidermal cells extending deeper and thence resulting in a thicker epidermis.
Another characteristic feature observed was the reduction of dermal SHG signal for mice skin with preclinical hyperplasia and its complete absence in clinically visible skin tumors. This finding was confirmed upon quantifying the total SHG intensity from the spectral plots which also showed a similar trend for skin with preclinical hyperplasia and visible skin tumors. This is obviously due to downward displacement of dermis caused by epidermal proliferation. This in turn leads to reduced detection of SHG signal from skin surface due to signal attenuation. Another possible cause for diminished SHG signal with advancing stages of carcinogenesis could be dermal collagen degradation or lysis during epidermal invasion into dermis as observed by Xiong et al. [26]. However this possibility seems improbable in the present study for the following reasons – a) advanced skin tumors such as SCC and carcinoma–in–situ had been excluded from the present data set and b) no dermal invasion was observed histopathologically in the corresponding H&E stained sections of the assessed skin with preclinical hyperplasia and clinically visible early skin tumors, which rules out epidermal invasion into dermis.
Our findings distinctly showed that NLSI could effectively visualize microscopic and spectroscopic differences between normal skin and skin with preclinical hyperplasia, both of which would appear clinically normal to the dermatologist. The remarkable aspect of these findings was that – a) NLSI was able detect these early cancer related changes with out any exogenous labels and b) this was achieved in real-time, in vivo and in a non-invasive manner. This property would also be very useful for surgeons to distinguish between normal and high risk tissue around the margins of a cancerous lesion during its excision, such as during Moh’s surgery [48]. Thus the findings of this study demonstrates the immense potential of in vivo NLSI to monitor structural and biochemical changes during carcinogenesis in skin and possibly in other organs that have high incidence of cancer such as lungs, breast and colon.
4.2 Metabolic changes monitored during DMBA induced skin carcinogenesis by in vivo NLSI
Cellular metabolism has often been assessed quantitatively by calculating the redox ratio calculated on the basis of the concentrations of endogenous metabolic fluorophores such NADH and FAD, derived from their fluorescence intensities. The redox ratio was originally formulated to study oxidation-reduction changes in brain and kidney in rats under anoxia by Chance et al., who found that the redox ratio was affected by vascular oxygen supply and cellular metabolism rate [29, 49–51]. The recent discovery that cancer progression maybe associated with tumor hypoxia [52], led to a newer perspective on the redox ratio as being a potential indicator of ongoing carcinogenesis. Skala et al. had observed the redox ratio (formulated as FAD/FAD + NADH) to decrease in precancerous stages in an oral cancer model of hamster cheek in vivo [53]. Similarly Varone et al. [54] demonstrated fluctuation in the metabolic redox ratio between engineered normal and precancerous squamous epithelial tissues ex vivo. Numerous ex vivo or in vivo studies that measured the redox ratio either found a decrease in the redox ratio or an increase in the inversed redox ratio (formulated as NADH/FAD) in neoplastic samples, suggestive of hypoxia in cancerous cells [9, 35]. Most cancer based redox studies generally relied on fluorescence lifetime measurements to assess the redox ratio, with the exception of a few studies where the redox ratio was assessed from the acquired emission spectra [28, 36, 37, 55, 56]. However till date, there have been no in vivo studies that utilized NLSI to assess metabolic activity in epidermis of skin during ongoing carcinogenesis in an animal model.
In the present study, we utilized simulated band-pass filters at 450 nm – 460 nm and 525 nm – 535 nm to indirectly estimate NADH and FAD respectively from the epidermis. To minimize the contribution of other fluorophores (keratin, lipids, lipofuscin, elastin and collagen) in the mentioned wavelength ranges, the analysis was performed in a spatially discriminatory manner exclusively on epidermal cells in the deeper layers. As a result, autofluorescence from keratin (from stratum corneum and hair follicle), lipids (from stratum corneum), lipofuscin (from ageing cells in the superficial layers), elastin and collagen (from dermis) were minimized. The ratio AUC450nm-460nm/AUC525nm-535nm can therefore be considered an indirect approximation to the ratio NADH/FAD that had been utilized in earlier studies. Although it should be noted that calculating AUC at 450 nm – 460 nm and 525 nm – 535 nm only provides a relative measure of NADH and FAD respectively in the epidermis and is not an absolute measure of NADH and FAD concentration in the epidermis. It is difficult to assess the absolute concentration of fluorophores like NADH and FAD in an in vivo scenario, as it is not possible to have an ‘in vivo’ reference of known concentration. In contrast, ex vivo quantification of fluorophores is feasible as demonstrated in the study of Keyes et al. [57], but this approach is not applicable for an in vivo study. Moreover, for absolute quantitative measurement of fluorophores in tissues ‘in vivo’, factors like attenuation of laser power needs to be corrected. This can be problematic, as rates of laser power absorption and scattering could differ between normal and cancerous tissue. By expressing the parameters AUC450nm-460nm and AUC525nm-535nm in a ratiometric form as described above, the effects of laser power attenuation can therefore be eliminated.
The ratio formulated as AUC450nm-460nm/AUC525nm-535nm was found to be increased for mice skin with preclinical hyperplasia and clinically visible skin tumors. If this ratio is considered analogous to the NADH/FAD ratio used in earlier studies, the findings suggest that there might be an elevated epidermal metabolism in these early stages of skin carcinogenesis. A possible explanation may be that NADH production could be raised because of increased glycolysis in the proliferative epidermal cells due to either the diminishing Pasteur effect (reduced oxygen) or the Warburg effect (in presence of oxygen) [32, 33]. However it should be noted that despite the difference being statistically significant (p < 0.05), the margin of difference was relatively low for the three clinico-pathologic grades of mice skin, when compared to the redox ratio difference observed in cancerous and normal samples of other organs analyzed in earlier studies [10, 28, 56]. One possible explanation is that epidermal cells during skin carcinogenesis may not be affected by hypoxia as much as compared to other visceral organs, because of ambient oxygen that can easily diffuse into skin from the atmosphere. In the study by Stücker et al. [58], it was estimated that total thickness of human skin supplied by external atmospheric oxygen was about as deep as 266 – 375 μm. Another reason could be that the AUC450nm-460nm/AUC525nm-535nm ratio assessments were primarily performed during the early stages of skin carcinogenesis – preclinical hyperplastic and clinically visible early skin tumors (< 2mm diameter). It would have been informative to assess the ratio in advanced stages of DMBA-induced skin carcinogenesis as well such as keratoacanthomas, squamous cell carcinoma in situ and squamous cell carcinoma in skin. Unfortunately since all these lesions were > 4 mm in diameter and had very thick layers of keratin above, it was not possible to image and assess the ratio from the deeply located proliferative and metabolically active zone of these lesions using the current nonlinear imaging setup. However, it must be noted that in vivo NLSI was sensitive to even small alterations in the ratio during early stages of mice skin carcinogenesis.
4.3 Limitations
Due to decreased signal to noise ratio with increasing depth, it was challenging to obtain high resolution spectral images at depths lower than 48 μm using the described NLSI setup. In addition, increased scattering of the laser signal by highly keratinized layers of dead cells made it impossible to perform spectral imaging in the deeper proliferative layers in advanced lesions. Therefore these types of lesions were excluded from further data analysis. In addition, it was not possible to determine with spectral imaging if skin tumors like papillomas and acanthomas was non-invasive or turning invasive. These kind of microscopic changes happen only at the epidermo-dermal junction that could be located as deep as 200 μm – 500 μm. However, the use of in vivo NLSI is unnecessary for advanced skin tumors, as the dermatologist would excise these types of lesions for therapeutic reasons and send it for conventional histopathology anyway. So far, in vivo imaging in human skin has been performed as deep as 320 μm by Koenig et al. [59]. Imaging to such depth was probably attained due to two reasons: a) Koenig et al. used an objective of high numerical aperture (NA) of 1.3 which is more efficient in collecting emission signal at increasing depths when compared the objective used in this study which had an NA of only 0.8, and b) Koenig et al. used an oil immersion objective which has the added advantage of being able to collect more autofluorescence signal at increasing depths due to lower refraction index mismatch induced spherical aberrations, when compared to the water immersion objective used in this study. In addition, high resolution deeper imaging can be obtained by further increasing the laser power and scan time. Although it should be borne in mind, that use of excessively high laser power and prolonged scan time during imaging can also increase formation of cyclobutane pyrimidine dimer mutations in DNA and thus raise the risk of cSCC from femtosecond laser irradiation [60]. Therefore an optimized imaging protocol is needed to simultaneously improve imaging efficiency and minimize laser radiation associated hazard.
NLSI performed in this study could clearly detect changes in skin layer organization and features of epithelial dysplasia such as altered nuclear-cytoplasmic ratio and mitotic cells during different stages of skin carcinogenesis. However intranuclear details such as nuclear hyperchromaticity could not be visualized by NLSI. These intranuclear details are often assessed by pathologists to identify the grade of dysplasia. This is highly crucial for the clinician too as the dysplasia grade play a pivotal role in determining the prognosis and treatment modality for the patient. However since NLSI is capable of identifying changes in tissue layer organization and features of epithelial dysplasia, in vivo NLSI will be extremely useful to determine the necessity for an invasive biopsy in a suspect region. This will help in reducing the number of unnecessary invasive biopsies considerably.
In regard to metabolic assessment of precancerous or cancerous tissues, earlier studies calculated the metabolic redox ratio by performed imaging on tissues with two different excitation wavelengths to minimize spectral overlap between NADH and FAD and obtain separate emission intensities [28, 36, 55]. In this study we performed imaging with a single wavelength and attempted to monitor the metabolic status of the epidermis by evaluating the ratio AUC450nm-460nm/AUC525nm-535nm which may serve as a ballpark estimate of the NADH/FAD ratio. This approach has its advantages as it employed a single wavelength excitation that allowed a) quicker image acquisition per spot and therefore shorter imaging duration per mouse and b) minimizing motion artifacts that arise during the wavelength switch. However this technique has its limitations, as the spectral overlap between NADH and FAD is higher when using a single near infrared (NIR) wavelength of 765 nm as compared to use of two different imaging NIR wavelengths. For the sake of measuring the ratio AUC450nm-460nm/AUC525nm-535nm in a simple manner, the spectral overlap between NADH and FAD was not taken into consideration. In order to compensate for this spectral overlap, it should be noted that spectral overlap depends on (a) fluorophore concentration and (b) the imaging wavelength. When the imaging wavelength is kept constant and the fluorophore concentration is known, the amount of spectral overlap and hence the overlap correction factor can be determined ex vivo as in the case of the reference NADH and FAD solutions used in this study. As mentioned earlier, the spectral overlap between NADH and FAD measured ex vivo in reference solutions was 11% at the simulated band-pass filter of 450 nm – 460 nm, while it was 29% at 525 nm – 535 nm. The corresponding spectral overlap correction factor for ratio AUC450nm-460nm/AUC525nm-535nm would therefore be 1.25 (i.e. 1 – 0.11/ 1 – 0.29 = 1.25). If this correction factor was to be applied to the ratios obtained from in vivo measurements, the revised results would still show elevated AUC450nm-460nm/AUC525nm-535nm values for early preclinical and clinical stages of carcinogenesis. This is because a correction factor of 1.25 would only further increase the existing difference shown presently in the results. However it would be inaccurate to extrapolate this spectral overlap correction factor from ex vivo reference samples of known fluorophore concentration to an in vivo environment with unknown fluorophore concentration.
Another factor to be noted is that in order for AUC450nm-460nm and AUC525nm-535nm to provide a reliable estimate of NADH and FAD in epidermis, the spectral contribution from other fluorophores such as keratin, lipids, lipofuscin, elastin and collagen needed to be minimized. This was achieved by performing the ratio analysis in a spatially discriminatory and depth specific manner exclusively in epidermal cells, thus excluding superficial dead layers such as stratum corneum, hair follicles and dermis. However, the mentioned fluorophores still exist in epidermal cells as well, albeit in significantly lesser amounts than NADH and FAD. Keratinocytes contain intracellular keratin intermediate filaments that may affect reliable estimation of NADH in epidermis from AUC450nm-460nm, while lipofuscin and lipids present due to ageing or waste product accumulation [61] could similarly interfere with FAD estimation from AUC525nm-535nm. Future studies that use single wavelength imaging for NLSI, should therefore try to unmix the contribution of all fluorophores in the epidermis by adopting more complex algorithms such as classical linear unmixing, principal component analysis (PCA), parallel factor analysis (PARAFAC), non negative matrix factorization (NMF) and spectral phasor analysis (SPA) [62]. The ratio calculated after unmixing with the aforementioned methods would be a more reliable indicator of the metabolic status in the epidermal cells.
5. Conclusion
From this study, it can be seen that in vivo NLSI could readily detect spectroscopic alterations that were present in the preclinical stages of skin carcinogenesis. The acquired spectral RGB images clearly revealed the changes in structural layers of skin during early carcinogenesis, which were confirmed by histopathology. The emission spectra revealed distinctive spectral shapes for normal skin, skin with preclinical hyperplasia and clinically visible skin tumors. This distinction between the spectral profiles of the three clinico-pathologic grades was observed the best at depth of 12 – 36 μm from mice skin surface. The results also showed the mean total SHG intensity decreased as the clinico-pathologic grade progressed from normal to preclinical hyperplasia and eventually clinically visible tumors. Furthermore, the ratio AUC450nm-460nm/AUC525nm-535nm which is an indirect approximation of NADH/FAD in epidermal cells was found to be higher in skin with preclinical hyperplasia and clinically visible early skin tumors, when compared to normal skin. This suggests that the epidermal cells may have elevated cellular metabolism in the early preclinical stage of hyperplasia itself. Therefore based on all the results obtained in this study, it can be concluded that in vivo NLSI would be a useful tool to screen for early preclinical stages of cSCC.
Acknowledgment
We are extremely indebted to Dr. Pablo Secades and Dr. Norman Sachs for their insight in developing the carcinogenesis protocol. We would like to thank Bastiaan Tuk for his invaluable guidance for the histology procedures. We acknowledge Dr. Henriette de Bruijn, Angelique van der Ploeg, Dr. Dominique Kuiper and Dr. Mathieu Sommers for their help in the animal experiment design. We appreciate Dr. Daniela Salvatori, Dr. Raoul Kuiper and Dr. Senada Koljenović for their initial input on pathology assessment of the skin biopsies. We are grateful to Dr. Gerhard Blab who helped design the ImageJ plug-in needed for data analysis. We also thank Dave van den Heuvel for making the necessary arrangements during the imaging protocol. This study was supported by the Dutch Technology Foundation (STW), which is part of the Netherlands Organization for Scientific Research (NWO), and which is partly funded by the Ministry of Economic Affairs (Grant # UU 10322).
References and links
- 1.Gallagher R. P., Hill G. B., Bajdik C. D., Coldman A. J., Fincham S., McLean D. I., Threlfall W. J., “Sunlight exposure, pigmentation factors, and risk of nonmelanocytic skin cancer. II. Squamous cell carcinoma,” Arch. Dermatol. 131(2), 164–169 (1995). 10.1001/archderm.1995.01690140048007 [DOI] [PubMed] [Google Scholar]
- 2.Veierød M. B., Couto E., Lund E., Adami H.-O., Weiderpass E., “Host characteristics, sun exposure, indoor tanning and risk of squamous cell carcinoma of the skin,” Int. J. Cancer 135(2), 413–422 (2014). 10.1002/ijc.28657 [DOI] [PubMed] [Google Scholar]
- 3.Fargnoli M. C., Kostaki D., Piccioni A., Micantonio T., Peris K., “Dermoscopy in the diagnosis and management of non-melanoma skin cancers,” Eur. J. Dermatol. 22(4), 456–463 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Göppert-Mayer M., “Über Elementarakte mit zwei Quantensprüngen,” Annalen der Physik 401(3), 273–294 (1931). 10.1002/andp.19314010303 [DOI] [Google Scholar]
- 5.Bloembergen N., Pershan P. S., “Light Waves at the Boundary of Nonlinear Media,” Phys. Rev. 128(2), 606–622 (1962). 10.1103/PhysRev.128.606 [DOI] [Google Scholar]
- 6.Wang B. G., König K., Halbhuber K. J., “Two-photon microscopy of deep intravital tissues and its merits in clinical research,” J. Microsc. 238(1), 1–20 (2010). 10.1111/j.1365-2818.2009.03330.x [DOI] [PubMed] [Google Scholar]
- 7.König K., “Multiphoton microscopy in life sciences,” J. Microsc. 200(2), 83–104 (2000). 10.1046/j.1365-2818.2000.00738.x [DOI] [PubMed] [Google Scholar]
- 8.König K., “Clinical multiphoton tomography,” J Biophotonics 1(1), 13–23 (2008). 10.1002/jbio.200710022 [DOI] [PubMed] [Google Scholar]
- 9.Perry S. W., Burke R. M., Brown E. B., “Two-Photon and Second Harmonic Microscopy in Clinical and Translational Cancer Research,” Ann. Biomed. Eng. 40(2), 277–291 (2012). 10.1007/s10439-012-0512-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen G., Wang L., Lu J., Zhu W., Zhang H., Chen J., Zhuo S., Yan J., “Optical diagnosis for lung cancer using multiphoton imaging,” Scanning 35(6), 362–365 (2013). 10.1002/sca.21076 [DOI] [PubMed] [Google Scholar]
- 11.Adur J., Pelegati V. B., de Thomaz A. A., Baratti M. O., Andrade L. A. L. A., Carvalho H. F., Bottcher-Luiz F., Cesar C. L., “Second harmonic generation microscopy as a powerful diagnostic imaging modality for human ovarian cancer,” J Biophotonics 7(1-2), 37–48 (2014). 10.1002/jbio.201200108 [DOI] [PubMed] [Google Scholar]
- 12.Liu N. R., Chen G. N., Wu S. S., Chen R., “Distinguishing human normal or cancerous esophagus tissue ex vivo using multiphoton microscopy,” J. Opt. 16(2), 025301 (2014). 10.1088/2040-8978/16/2/025301 [DOI] [Google Scholar]
- 13.Zhuo S., Chen J., Luo T., Jiang X., Xie S., Chen R., “Two-layered multiphoton microscopic imaging of cervical tissue,” Lasers Med. Sci. 24(3), 359–363 (2009). 10.1007/s10103-008-0570-2 [DOI] [PubMed] [Google Scholar]
- 14.Kolanjiappan K., Ramachandran C. R., Manoharan S., “Biochemical changes in tumor tissues of oral cancer patients,” Clin. Biochem. 36(1), 61–65 (2003). 10.1016/S0009-9120(02)00421-6 [DOI] [PubMed] [Google Scholar]
- 15.Chen Y. J., Cheng Y. D., Liu H. Y., Lin P. Y., Wang C. S., “Observation of biochemical imaging changes in human pancreatic cancer tissue using Fourier-transform infrared microspectroscopy,” Chang Gung Med. J. 29(5), 518–527 (2006). [PubMed] [Google Scholar]
- 16.Calabrese C., Pisi A., Di Febo G., Liguori G., Filippini G., Cervellera M., Righi V., Lucchi P., Mucci A., Schenetti L., Tonini V., Tosi M. R., Tugnoli V., “Biochemical Alterations from Normal Mucosa to Gastric Cancer by Ex vivo Magnetic Resonance Spectroscopy,” Cancer Epidemiol. Biomarkers Prev. 17(6), 1386–1395 (2008). 10.1158/1055-9965.EPI-07-2676 [DOI] [PubMed] [Google Scholar]
- 17.Bader A. N., Pena A.-M., Johan van Voskuilen C., Palero J. A., Leroy F., Colonna A., Gerritsen H. C., “Fast nonlinear spectral microscopy of in vivo human skin,” Biomed. Opt. Express 2(2), 365–373 (2011). 10.1364/BOE.2.000365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palero J. A., de Bruijn H. S., van der Ploeg-van den Heuvel A., Sterenborg H. J. C. M., Gerritsen H. C., “In vivo nonlinear spectral imaging in mouse skin,” Opt. Express 14(10), 4395–4402 (2006). 10.1364/OE.14.004395 [DOI] [PubMed] [Google Scholar]
- 19.Palero J. A., de Bruijn H. S., van der Ploeg van den Heuvel A., Sterenborg H. J., Gerritsen H. C., “Spectrally resolved multiphoton imaging of in vivo and excised mouse skin tissues,” Biophys. J. 93(3), 992–1007 (2007). 10.1529/biophysj.106.099457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen J., Zhuo S., Chen R., Jiang X., Xie S., Zou Q., “Depth-resolved spectral imaging of rabbit oesophageal tissue based on two-photon excited fluorescence and second-harmonic generation,” New J. Phys. 9(7), 212 (2007). 10.1088/1367-2630/9/7/212 [DOI] [Google Scholar]
- 21.Zhuo S., Chen J., Jiang X., Xie S., Chen R., Cao N., Zou Q., Xiong S., “The layered-resolved microstructure and spectroscopy of mouse oral mucosa using multiphoton microscopy,” Phys. Med. Biol. 52(16), 4967–4980 (2007). 10.1088/0031-9155/52/16/017 [DOI] [PubMed] [Google Scholar]
- 22.Dimitrow E., Ziemer M., Koehler M. J., Norgauer J., König K., Elsner P., Kaatz M., “Sensitivity and Specificity of Multiphoton Laser Tomography for In Vivo and Ex Vivo Diagnosis of Malignant Melanoma,” J. Invest. Dermatol. 129(7), 1752–1758 (2009). 10.1038/jid.2008.439 [DOI] [PubMed] [Google Scholar]
- 23.Seidenari S., “Multiphoton Laser Microscopy and Fluorescence Lifetime Imaging for the Assessment of the Skin,” in Non Invasive Diagnostic Techniques in Clinical Dermatology, Berardesca E., Maibach H. I., Wilhelm K.-P., eds. (Springer Berlin Heidelberg, 2014), pp. 89–100. [Google Scholar]
- 24.Manfredini M., Arginelli F., Dunsby C., French P., Talbot C., König K., Pellacani G., Ponti G., Seidenari S., “High-resolution imaging of basal cell carcinoma: a comparison between multiphoton microscopy with fluorescence lifetime imaging and reflectance confocal microscopy,” Skin Res. Technol. 19(1), e433–e443 (2013). 10.1111/j.1600-0846.2012.00661.x [DOI] [PubMed] [Google Scholar]
- 25.De Giorgi V., Massi D., Sestini S., Cicchi R., Pavone F. S., Lotti T., “Combined non-linear laser imaging (two-photon excitation fluorescence microscopy, fluorescence lifetime imaging microscopy, multispectral multiphoton microscopy) in cutaneous tumours: first experiences,” J. Eur. Acad. Dermatol. Venereol. 23(3), 314–316 (2009). 10.1111/j.1468-3083.2008.03045.x [DOI] [PubMed] [Google Scholar]
- 26.Xiong S. Y., Yang J. G., Zhuang J., “Nonlinear spectral imaging of human normal skin, basal cell carcinoma and squamous cell carcinoma based on two-photon excited fluorescence and second-harmonic generation,” Laser Phys. 21(10), 1844–1849 (2011). 10.1134/S1054660X11170269 [DOI] [Google Scholar]
- 27.Leupold D., Scholz M., Stankovic G., Reda J., Buder S., Eichhorn R., Wessler G., Stücker M., Hoffmann K., Bauer J., Garbe C., “The stepwise two-photon excited melanin fluorescence is a unique diagnostic tool for the detection of malignant transformation in melanocytes,” Pigment Cell Melanoma Res 24(3), 438–445 (2011). 10.1111/j.1755-148X.2011.00853.x [DOI] [PubMed] [Google Scholar]
- 28.Edward K., Qiu S., Resto V., McCammon S., Vargas G., “In vivo layer-resolved characterization of oral dysplasia via nonlinear optical micro-spectroscopy,” Biomed. Opt. Express 3(7), 1579–1593 (2012). 10.1364/BOE.3.001579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chance B., Cohen P., Jobsis F., Schoener B., “Intracellular oxidation-reduction states in vivo,” Science 137(3529), 499–508 (1962). 10.1126/science.137.3529.499 [DOI] [PubMed] [Google Scholar]
- 30.Ozawa K., Yamamoto M., Shimahara Y., Kishida A., Tabata R., Takahashi M., Terada Y., Iwata S., Kobayashi T., “The redox theory in evolution,” J. Hep. Bil. Pancr. Surg. 2(3), 205–214 (1995). 10.1007/BF02350900 [DOI] [Google Scholar]
- 31.Gulledge C. J., Dewhirst M. W., “Tumor oxygenation: a matter of supply and demand,” Anticancer Res. 16(2), 741–749 (1996). [PubMed] [Google Scholar]
- 32.Warburg O., “On the origin of cancer cells,” Science 123(3191), 309–314 (1956). 10.1126/science.123.3191.309 [DOI] [PubMed] [Google Scholar]
- 33.Diaz-Ruiz R., Rigoulet M., Devin A., “The Warburg and Crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression,” Biochimica et Biophysica Acta (BBA) - Bioenergetics 1807(6), 568–576 (2011). 10.1016/j.bbabio.2010.08.010 [DOI] [PubMed] [Google Scholar]
- 34.Skala M. C., Riching K. M., Gendron-Fitzpatrick A., Eickhoff J., Eliceiri K. W., White J. G., Ramanujam N., “In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia,” Proc. Natl. Acad. Sci. U.S.A. 104(49), 19494–19499 (2007). 10.1073/pnas.0708425104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walsh A. J., Cook R. S., Manning H. C., Hicks D. J., Lafontant A., Arteaga C. L., Skala M. C., “Optical Metabolic Imaging Identifies Glycolytic Levels, Subtypes, and Early-Treatment Response in Breast Cancer,” Cancer Res. 73(20), 6164–6174 (2013). 10.1158/0008-5472.CAN-13-0527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cicchi R., Sturiale A., Nesi G., Kapsokalyvas D., Alemanno G., Tonelli F., Pavone F. S., “Multiphoton morpho-functional imaging of healthy colon mucosa, adenomatous polyp and adenocarcinoma,” Biomed. Opt. Express 4(7), 1204–1213 (2013). 10.1364/BOE.4.001204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirkpatrick N. D., Brewer M. A., Utzinger U., “Endogenous Optical Biomarkers of Ovarian Cancer Evaluated with Multiphoton Microscopy,” Cancer Epidemiol. Biomarkers Prev. 16(10), 2048–2057 (2007). 10.1158/1055-9965.EPI-07-0009 [DOI] [PubMed] [Google Scholar]
- 38.Weagle G., Paterson P. E., Kennedy J., Pottier R., “The nature of the chromophore responsible for naturally occurring fluorescence in mouse skin,” J. Photochem. Photobiol. B 2(3), 313–320 (1988). 10.1016/1011-1344(88)85051-6 [DOI] [PubMed] [Google Scholar]
- 39.Sachs N., Secades P., van Hulst L., Kreft M., Song J.-Y., Sonnenberg A., “Loss of integrin α3 prevents skin tumor formation by promoting epidermal turnover and depletion of slow-cycling cells,” Proc. Natl. Acad. Sci. U.S.A. 109(52), 21468–21473 (2012). 10.1073/pnas.1204614110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palero J. A., de Bruijn H. S., van der Ploeg van den Heuvel A., Sterenborg H. J. C. M., Gerritsen H. C., “Spectrally Resolved Multiphoton Imaging of In Vivo and Excised Mouse Skin Tissues,” Biophys. J. 93(3), 992–1007 (2007). 10.1529/biophysj.106.099457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramanujam N., “Fluorescence Spectroscopy In Vivo,” in Encyclopedia of Analytical Chemistry (John Wiley & Sons, Ltd, 2006). [Google Scholar]
- 42.Gillies R., Zonios G., Anderson R. R., Kollias N., “Fluorescence Excitation Spectroscopy Provides Information About Human Skin In Vivo,” J. Invest. Dermatol. 115(4), 704–707 (2000). 10.1046/j.1523-1747.2000.00091.x [DOI] [PubMed] [Google Scholar]
- 43.Laiho L. H., Pelet S., Hancewicz T. M., Kaplan P. D., So P. T., “Two-photon 3-D mapping of ex vivo human skin endogenous fluorescence species based on fluorescence emission spectra,” J. Biomed. Opt. 10(2), 024016 (2005). 10.1117/1.1891370 [DOI] [PubMed] [Google Scholar]
- 44.Roberts M. S., Dancik Y., Prow T. W., Thorling C. A., Lin L. L., Grice J. E., Robertson T. A., König K., Becker W., “Non-invasive imaging of skin physiology and percutaneous penetration using fluorescence spectral and lifetime imaging with multiphoton and confocal microscopy,” Eur. J. Pharm. Biopharm. 77(3), 469–488 (2011). 10.1016/j.ejpb.2010.12.023 [DOI] [PubMed] [Google Scholar]
- 45.Palero J. A., de Bruijn H. S., van der Ploeg van den Heuvel A., Sterenborg H. J. C. M., Gerritsen H. C., “Spectrally Resolved Multiphoton Imaging of In Vivo and Excised Mouse Skin Tissues,” Biophys. J. 93(3), 992–1007 (2007). 10.1529/biophysj.106.099457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mochizuki Y., Park M. K., Mori T., Kawashima S., “The difference in autofluorescence features of lipofuscin between brain and adrenal,” Zoolog. Sci. 12(3), 283–288 (1995). 10.2108/zsj.12.283 [DOI] [PubMed] [Google Scholar]
- 47.Terman A., Brunk U. T., “Lipofuscin: mechanisms of formation and increase with age,” APMIS 106(2), 265–276 (1998). 10.1111/j.1699-0463.1998.tb01346.x [DOI] [PubMed] [Google Scholar]
- 48.Mohs F. E., “Chemosurgery: microscopically controlled surgery for skin cancer--past, present and future,” J. Dermatol. Surg. Oncol. 4(1), 41–54 (1978). 10.1111/j.1524-4725.1978.tb00379.x [DOI] [PubMed] [Google Scholar]
- 49.Chance B., Schoener B., Oshino R., Itshak F., Nakase Y., “Oxidation-reduction ratio studies of mitochondria in freeze-trapped samples. NADH and flavoprotein fluorescence signals,” J. Biol. Chem. 254(11), 4764–4771 (1979). [PubMed] [Google Scholar]
- 50.Chance B., Schoener B., “Correlation of oxidation-reduction changes of intracellular reduced pyridine nucleotide and changes in electroencephalogram of the rat in anoxia,” Nature 195(4845), 956–958 (1962). 10.1038/195956a0 [DOI] [PubMed] [Google Scholar]
- 51.Chance B., “Metabolic heterogeneities in rapidly metabolizing tissues,” J. Appl. Cardiol. 4, 207–221 (1989). [Google Scholar]
- 52.Zhou J., Schmid T., Schnitzer S., Brüne B., “Tumor hypoxia and cancer progression,” Cancer Lett. 237(1), 10–21 (2006). 10.1016/j.canlet.2005.05.028 [DOI] [PubMed] [Google Scholar]
- 53.Skala M. C., Riching K. M., Bird D. K., Gendron-Fitzpatrick A., Eickhoff J., Eliceiri K. W., Keely P. J., Ramanujam N., “In vivo multiphoton fluorescence lifetime imaging of protein-bound and free nicotinamide adenine dinucleotide in normal and precancerous epithelia,” J. Biomed. Opt. 12(2), 024014 (2007). 10.1117/1.2717503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Varone A., Xylas J., Quinn K. P., Pouli D., Sridharan G., McLaughlin-Drubin M. E., Alonzo C., Lee K., Münger K., Georgakoudi I., “Endogenous Two-Photon Fluorescence Imaging Elucidates Metabolic Changes Related to Enhanced Glycolysis and Glutamine Consumption in Precancerous Epithelial Tissues,” Cancer Res. 74(11), 3067–3075 (2014). 10.1158/0008-5472.CAN-13-2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cicchi R., Crisci A., Cosci A., Nesi G., Kapsokalyvas D., Giancane S., Carini M., Pavone F. S., “Time- and Spectral-resolved two-photon imaging of healthy bladder mucosa and carcinoma in situ,” Opt. Express 18(4), 3840–3849 (2010). 10.1364/OE.18.003840 [DOI] [PubMed] [Google Scholar]
- 56.Zhuo S., Yan J., Chen G., Chen J., Liu Y., Lu J., Zhu X., Jiang X., Xie S., “Label-free monitoring of colonic cancer progression using multiphoton microscopy,” Biomed. Opt. Express 2(3), 615–619 (2011). 10.1364/BOE.2.000615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Keyes J. T., Simon B. R., Vande Geest J. P., “Location-Dependent Coronary Artery Diffusive and Convective Mass Transport Properties of a Lipophilic Drug Surrogate Measured Using Nonlinear Microscopy,” Pharm. Res. 30(4), 1147–1160 (2013). 10.1007/s11095-012-0950-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stücker M., Struk A., Altmeyer P., Herde M., Baumgärtl H., Lübbers D. W., “The cutaneous uptake of atmospheric oxygen contributes significantly to the oxygen supply of human dermis and epidermis,” J. Physiol. 538(3), 985–994 (2002). 10.1113/jphysiol.2001.013067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koenig K., “Hybrid multiphoton multimodal tomography of in vivo human skin,” Intravital 1(1), 11–26 (2012). 10.4161/intv.21938 [DOI] [Google Scholar]
- 60.Thomas G., Nadiarnykh O., van Voskuilen J., Hoy C. L., Gerritsen H. C., Sterenborg H. J. C. M., “Estimating the risk of squamous cell cancer induction in skin following nonlinear optical imaging,” J Biophotonics 7(7), 492–505 (2014). 10.1002/jbio.201200207 [DOI] [PubMed] [Google Scholar]
- 61.Watson J. M., Marion S. L., Rice P. F., Utzinger U., Brewer M. A., Hoyer P. B., Barton J. K., “Two-photon excited fluorescence imaging of endogenous contrast in a mouse model of ovarian cancer,” Lasers Surg. Med. 45(3), 155–166 (2013). 10.1002/lsm.22115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thomas G., van Voskuilen J., Gerritsen H. C., Sterenborg H. J. C. M., “Advances and challenges in label-free nonlinear optical imaging using two-photon excitation fluorescence and second harmonic generation for cancer research,” J. Photochem. Photobiol. B 141, 128–138 (2014). [DOI] [PubMed] [Google Scholar]