

Abstract
The asymmetric Pd-catalyzed decarboxylative allylic alkylation of differentially N-protected piperazin-2-ones allows for the synthesis of a variety of highly enantioenriched tertiary piperazine-2-ones. Deprotection and reduction affords the corresponding tertiary piperazines, which can be employed for the synthesis of medicinally important analogs. The introduction of these chiral tertiary piperazines resulted in imatinib analogs that exhibited comparable antiproliferative activity to that of their corresponding imatinib counterparts.
Keywords: allylic alkylation, enantioselective, palladium, piperazine, imatinib
Piperazine is a common structural motif in pharmaceuticals and is considered to be a privileged scaffold in medicinal chemistry.[1] Piperazine itself has been used as an anthelmintic and notable piperazine-containing small molecule pharmaceuticals include imatinib (1), a kinase-inhibiting anticancer agent;[2] ciprofloxacin (2), a potent fluoroquinolone antibiotic;[3] piribedil (3), an antiparkinsonian agent;[4] and indinavir (4), an HIV protease inhibitor (Figure 1a).[5] Common methods for the selective asymmetric preparation of substituted piperazines[6] include enantio selective hydrogenation,[7] enzyme-mediated chiral resolution,[8] α-lithiation mediated by (−)-sparteine and other chiral diamines,[9] Pd-catalyzed cyclizations,[10] or synthesis from amino acids or other members of the chiral pool.[11] One of the most straight forward methods for the synthesis of chiral piperazines is the reduction of the corresponding chiral keto- or diketopiperazine. However, methods for the asymmetric preparation of these piperazine precursors are currently limited.
Figure 1.
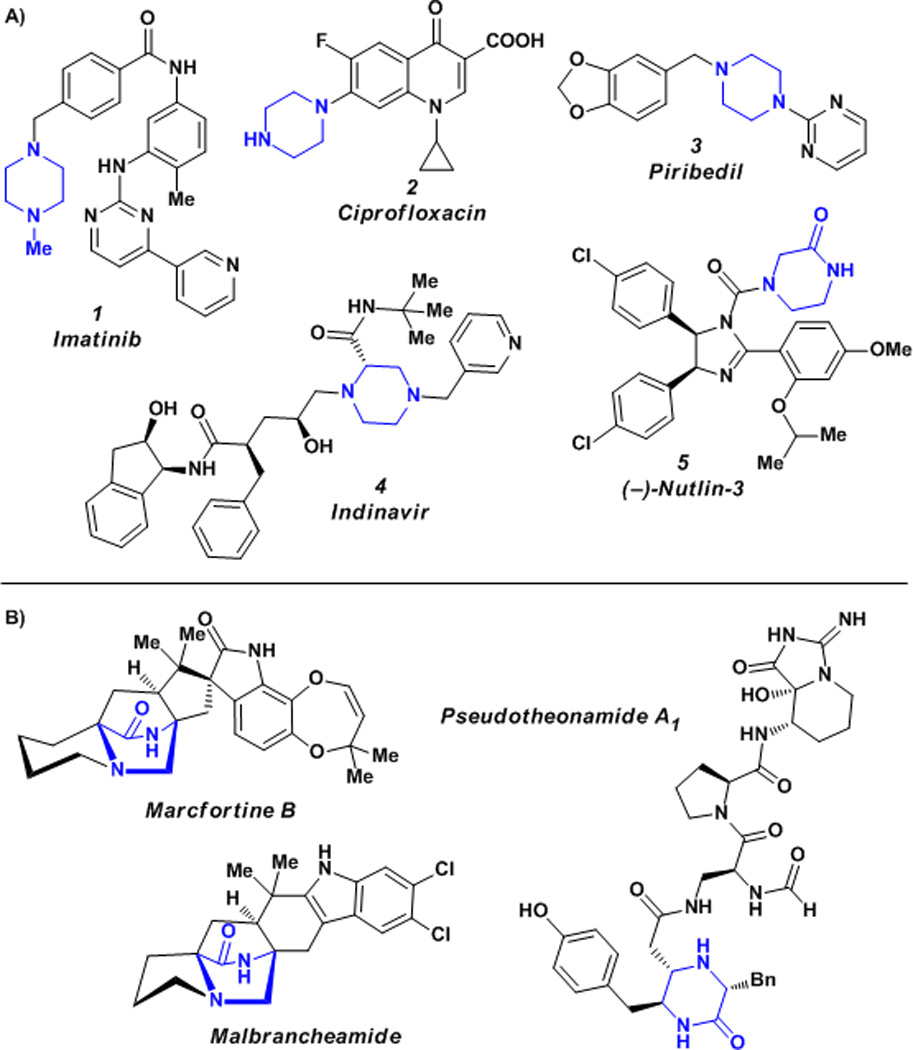
A) Representative piperazine and piperazin-2-one containing pharmaceuticals. B) Representative bioactive natural products possessing piperazin-2-one core structures.
Piperazin-2-ones possess an additional functionality (i.e. the carbonyl) that allow for the synthesis of more highly substituted piperazine-2-ones, which, upon reduction, yield substituted piperazine derivatives. Although piperazin-2-ones are employed infrequently in medicinal chemistry, they can be found in some pharmaceutical agents including the p53/MDM2 inhibitor (−)-nutlin-3 (5, Figure 1a),[12] and in several naturally occurring bioactive compounds including the marcfortines,[13] pseudotheonamides,[14] and malbrancheamides (Figure 1b).[15] Piperazin-2-ones also play a crucial role as conformationally constrained peptidomimetics. These piperazin-2-ones mimic inverse γ-turns in peptides, which play crucial roles in the secondary structures of proteins.[16] Chiral piperazin-2-ones[17] can be prepared from amino acids or other members of the chiral pool[18] or by chiral auxiliary-mediated alkylations[19] or dynamic resolutions.[20] However, most of the available methods are not capable of generating chiral α-tertiary piperazin-2-ones (6, Figure 2) and there are no previous examples that prepare this motif by catalytic enantioselective methods. Thus, there is an unaddressed absence of catalytic asymmetric synthesis strategies to these valuable compounds.
Figure 2.
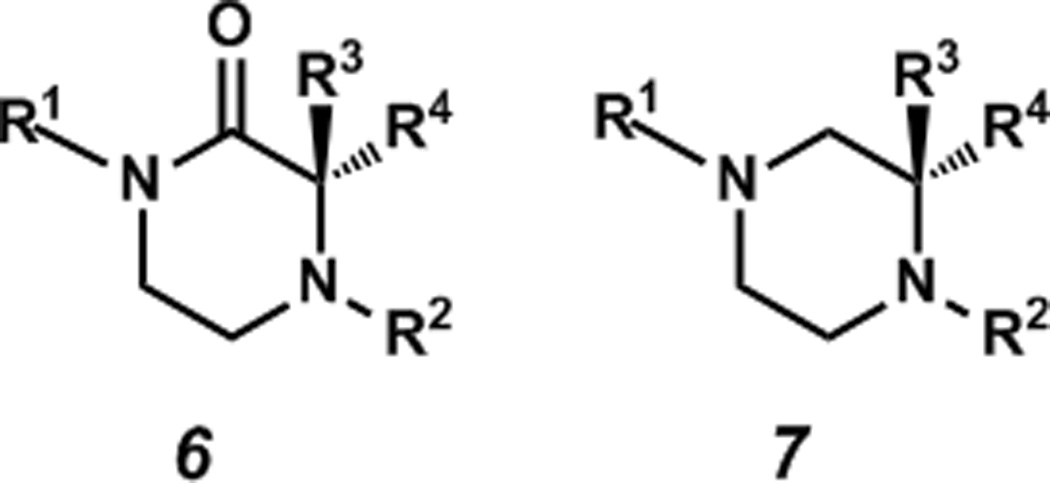
α-Tertiary piperazin-2-one and α-tertiary piperazine.
Our research group has had a longstanding interest in the construction of α-tetrasubstituted carbonyl compounds including quaternary centers using transition metal catalysis and has developed conditions for the asymmetric allylic alkylation of lactams to furnish α-quaternary lactam products.[21] Morpholin-3-ones were also identified as viable substrates under the same conditions, generating an α-tertiary stereo center.[22] We sought to extend this catalyst system to enantio selectively generate α-tertiary piperazin-2-ones (6), which, upon subsequent reduction, would generate chiral tertiary piperazines (7). α-Tertiary piperazine species are not well precedented in the literature presumably due to the difficulties associated with their preparation, and no general methods exist for their asymmetric synthesis, let alone catalytic asymmetric synthesis. A direct, catalytic asymmetric synthesis of tertiary piperazin-2-ones and their subsequent reduction to the piperazines would provide access to an invaluable scaffold, enabling the exploration of unprecedented chemical space (Figure 3).
Figure 3.
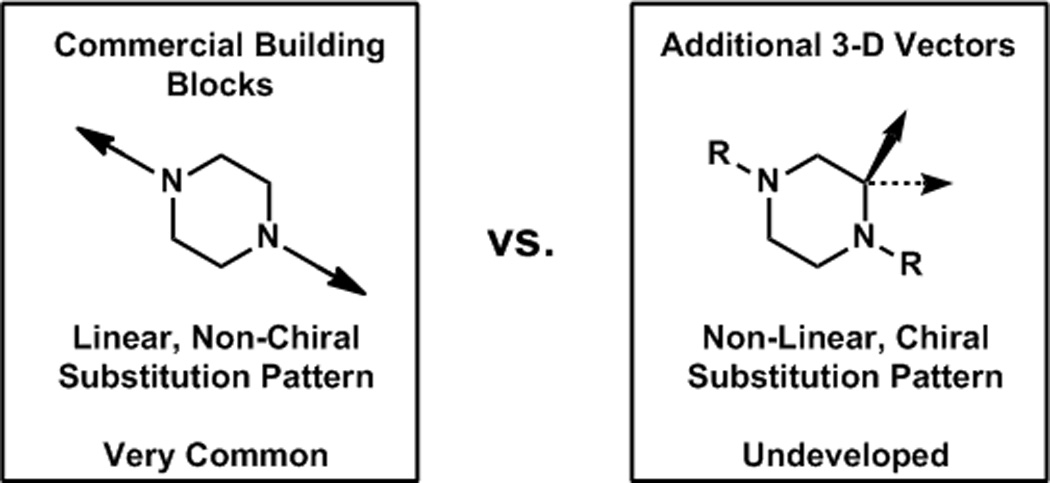
Entry into largely unexploited chemical space.
Medicinal chemistry has long utilized linearly substituted, non chiral piperazines and has also, although less frequently, utilized chiral α-secondary piperazines, as in indinavir (4).22 Access to enantiopure α-tertiary piperazines would provide a unique opportunity to explore these three dimensionally elaborated piperazines in drug discovery.
Given that secondary and tertiary nitrogen atoms may exhibit undesired reactivity in Pd-catalyzed reactions, it is necessary to protect both nitrogens of the ketopiperazine ring. Taking into consideration our prior results, in which N-benzoyl protected lactams reacted with high enantioselectivities in the decarboxylative asymmetric allylic alkylation,[21e] we began our studies with the 1,4-bisbenzoylated piperazin-2-one 8a (Table 1). When utilizing Pd2 (pmdba)3 [tris(4,4’-methoxydibenzylideneacetone) dipalladium (0)] at 5 mol % catalyst loading and the (S)-(CF3)3-t-BuPHOX ligand at 12.5 mol % loading in a 0.014 M solution of toluene, the bis-N-benzoylated α-allylated product 9a (Table 1) was formed in high yield but modest ee. It was reasoned that the sp2-hybridized nature of the N(4) position was detrimental to the enantioselectivity of the reaction due to its ability to stabilize the enolate intermediate.[23] Taking into consideration the need for an alkyl protecting group at the N(4) position, we next examined 4-benzylpiperazin-2-one 8b (R1=Bz, R2=Bn, R3=Me, R4=H). Under our standard conditions, the N-benzyl-protected α-allylated compound 9b was obtained in both good yield and ee. Additional N(4)-protecting groups were investigated which allow for the chemoselective deprotection of N(4). The para-methoxybenzyl protecting group, which can be cleaved by acidic or oxidative conditions, would allow for orthogonal deprotection. 4-Methoxybenzylpiperazin-2-one 9c was also obtained in good ee but with slightly lower yield than the 4-benzyl compound 9b. Given the slightly higher yield, the 4-benzyl protecting group was selected for further optimization.
Table 1.
Catalytic enantioselective piperazin-2-one decarboxylative allylic alkylation. Scope of protecting group tolerance and scope of α-substituents.
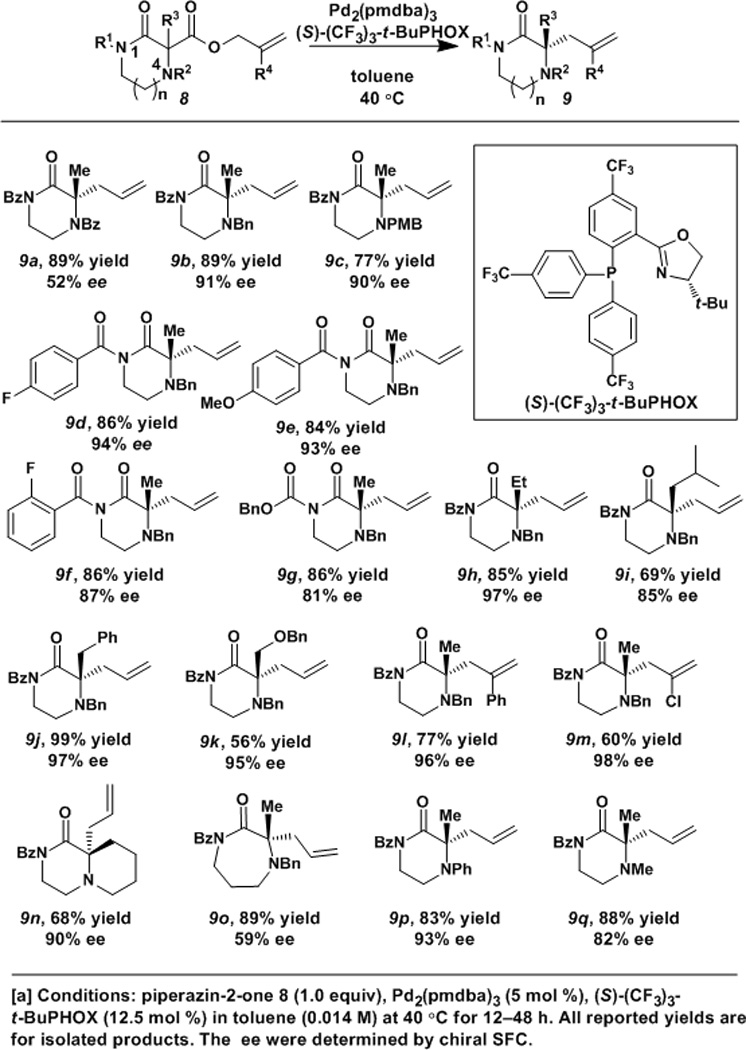 |
Conditions: piperazin-2-one 8 (1.0 equiv), Pd2(pmdba)3 (5 mol %), (S)-(CF3)3-t-BuPHOX (12.5 mol %) in toluene (0.014 M) at 40 °C for 12–48 h. All reported yields are for isolated products. The ee were determined by chiral SFC.
In efforts to increase the ee of the allylic alkylated products, additional protecting groups at the N (1) position were examined. Considering that benzoylated compounds provided the best results in the lactam case,[21e] we examined additional acyl protecting groups (Table 1). The para-fluoro benzoyl and para-methoxybenzoyl compounds 9d and 9e were obtained in nearly identical ee and just slightly lower yields, demonstrating that substantial electronic changes of the N(1)-substituent do not have a strong influence on the reaction efficiency or selectivity. However, the reaction is somewhat sensitive toward ortho substitution at the N(1)-benzoyl group as 9f was obtained in a significantly lower enantiomeric excess compared to 9d and 9e. Additionally, the1-carboxybenzyl ketopiperazine 9g was also prepared in high yield, albeit moderate ee. Given these data, the unsubstituted benzoyl group was selected as the optimal choice of N(1)-protecting group, and the benzyl group was selected as the optimal N(4)-group.
With protecting groups for both nitrogen atoms investigated, the scope of the reaction with regard to the α-substituent was examined. Piperazin-2-ones bearing alkyl (9h, 9i) and benzyl (9j) groups were prepared, as was benzyl ether 9k (Table 1), which provides an additional handle for further functionalization. Additionally, bicyclic product 9n, which is reminiscent of the marcfortine core, was obtained in good yield in the reaction (Table 1). The effect of expanding ring size was also examined. The 1,4-diazepan-2-one 9o was formed with only moderate enantiomeric excess, a result that suggests that the reaction is sensitive to ring size, contrary to the lactam examples.[21e]
Common piperazine pharmacophores include N-arylpiperazines and N-methylpiperazines,24 and we sought to determine if 4-aryl ketopiperazines and 4-methyl ketopiperazines were also competent substrates in this chemistry. The low ee observed in bis-benzoylated compound 9a suggests that an sp2-hybridized N(4) position would prove detrimental to the enantioselectivity of the reaction. Despite this, 4-phenyl compound 9p, with its partial sp2-nature of its aniline nitrogen, could be obtained in good yield and with excellent enantiomeric excess (Table 1). The 4-methylketopiperazine 9q could also be prepared in good yield but with slightly diminished ee.
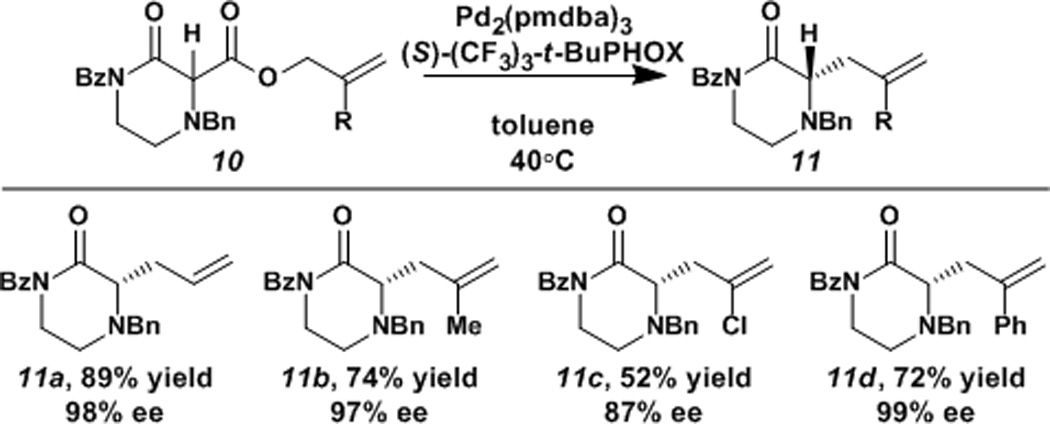
Contrary to results with the piperdinone substrates, we were delighted to find that even the unsubstituted α-secondary ketopiperazine 11a could be obtained in excellent yield and enantioselectivity (Table 2). Previous attempts to generate trisubstituted stereo centers via our asymmetric allylic alkylation of lactam and ketone substrates were unsuccessful. Such experiments have generally resulted in mixtures of mono-, di-, and unallylated products, and the desired trisubstituted product was formed in poor yield and with only moderate ee. We were delighted to find that in the case at hand, unsubstituted α-secondary ketopiperazine 11a could be obtained with no detectable amounts of di- or unallylated byproducts. It is likely that the low acidity of the α-hydrogen of the monosubstituted piperazin-2-one substrate and product is key to obtaining a high yield of the monoallylated product. Given this exciting result, additional allyl substrates were tested (Table 2). Numerous allyl groups are compatible, including methallyl 11b, chloroallyl 11c, and phenylallyl 11d which were all obtained in fair to excellent yield and high enantioselectivity.
Table 2.
Scope of allyl substituents for α-secondary piperazin-2-ones.
 |
The ketopiperazine products can be converted to the related piperazines in two steps, hydrolysis of the benzoyl group to piperazine-2-one 12 followed by reduction of the amide to piperazine 13 (Scheme 1A). The deprotected N(1) position can be further alkylated to form for instance, di-allyl piperazin-2-one 15 (Scheme 1B). Cross-metathesis can also be performed (Scheme 1C). Additionally, the 4-methoxybenzylgroupcan be selectively cleaved under oxidative conditions to form piperazine-2-one 17 (Scheme 1D).
Scheme 1.
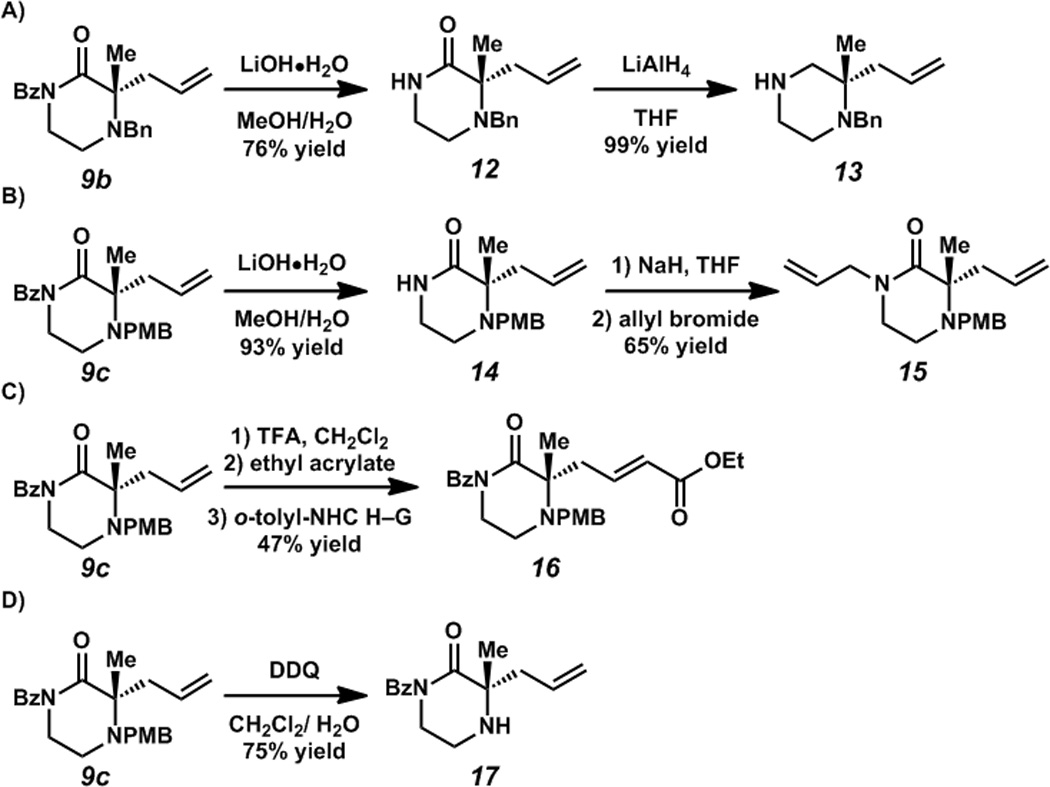
A) Protecting group cleavage and reduction to yield piperazine 13. B) Protecting group cleavage and alkylation to yield piperazin-2-one 15. C) Cross-metathesis with ethyl acrylate. D) Oxidative cleavage of PMB protecting group.
Finally, we have demonstrated that these tertiary piperazines can successfully be incorporated into known piperazine-containing pharmaceuticals, leading to novel analogs with comparable bioactivities in preliminary testing. Substitution on the piperazine ring has been shown to modulate bioactivity and, in some cases, result in more specific and stronger enzyme binding.[25] We considered imatinib (1), an antiproliferative agent developed for the treatment of several cancers, notably including Philadelphia chromosome-positive chronic myelogenous leukemia (CML),[2] to be an ideal candidate for proof of concept. Imatinib targets the Abl tyrosine kinase domain of the Bcr-Abl fusion protein, and it is known that genetic point mutations can render imatinib ineffective because it is no longer able to bind to the enzyme.[26] The piperazine moiety of the molecule forms two key hydrogen bonds to two amino acid residues, and thus plays a crucial role in binding.[27]
Given that the piperazine is so crucial to the binding of imatinib, we wanted to explore whether highly substituted and congested piperazine analogs would disrupt these interactions. Enantiomerically pure, benzylated analog 18 (Scheme 2) was assessed for its antiproliferative activity against the human K562 CML cell line.
Scheme 2.
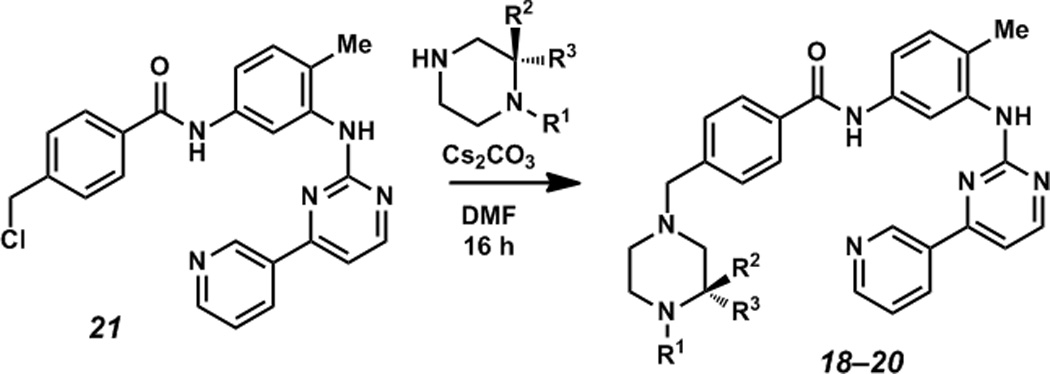
Synthesis of imatinib analogs from precursor 19.
N-substituted analog 18 was found to have significantly less antiproliferative activity than imatinib (1, Table 3) signifying that too much bulk around N(4) might perturb key interactions. We also synthesized two novel des-methyl tertiary piperazine-containing imatinib analogs (Scheme 2). These analogs, (S)-20 and (R)-20, were assayed for their antiproliferative activity and were found to have activities slightly greater than that of N-desmethyl imatinib (CGP 74588) 19, the main bioactive metabolite of imatinib. The (R)-enantiomer is slightly more potent ((R)-20, Table 3). These results point to the potential utility of stereochemically-rich piperazines as important building blocks for medicinal chemistry. Additionally, these novel substructures will likely open up new chemical space around a privileged scaffold in drug discovery.
Table 3.
Antiproliferative activity of imatinib and imatinib analogs
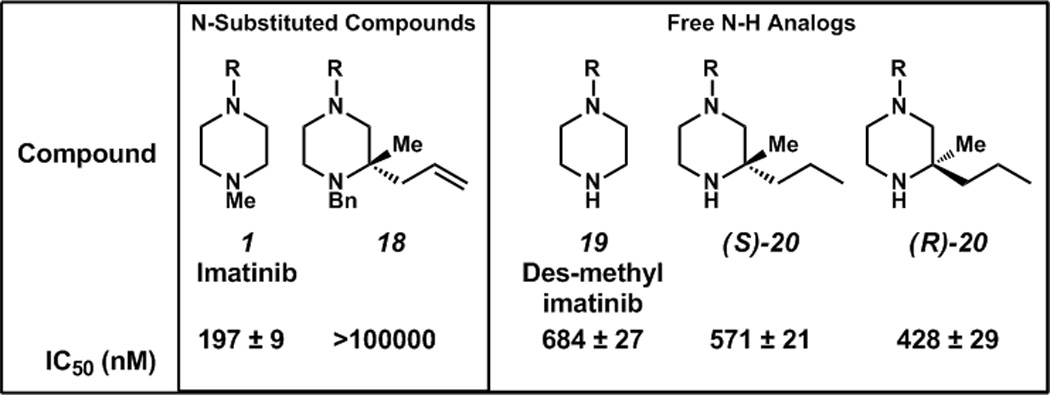 |
IC50 values are against K562 CML cells and are reported ± standard deviation
In summary, we have developed the first catalytic enantioselective synthesis of α-tertiary piperazin-2-ones. These important molecules can be easily converted to novel α-tertiary piperazines. This method utilizes palladium catalysts derived from Pd2(pmdba)3 and electron-deficient PHOX ligands to deliver α-tertiary piperazin-2-ones in good to excellent yields and enantioselectivities. This method also allows for the synthesis of α-secondary piperazin-2-ones in modest to excellent yields and good to excellent enantioselectivities. In addition to being tolerant of a variety of N-substitutions, this reaction is also tolerant of substitution at the stereocenter including fused bicycles such as those found in piperazine-2-one-containing natural products. We have further demonstrated that these chiral piperazin-2-ones can be reduced to the corresponding chiral piperazines, and these chiral α-tertiary piperazines can successfully be incorporated into known piperazine-containing pharmaceuticals. Specifically, these piperazines can be incorporated into the imatinib framework without major perturbation of the drug’s antiproliferative activity against the human K562 CML cell line, thus indicating the enormous potential that these novel three-dimensionally elaborated chiral piperazines have in drug discovery.
Supplementary Material
Acknowledgments
The authors wish to thank NIH-NIGMS (R01GM080269-01) for financial support. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1144469 (fellowship to K.M.K.).The authors also wish to thank the Deutsche Forschungsgemeinschaft (DFG postdoctoral fellowship to C.E.), Amgen, Abbott, Boehringer Ingelheim, and Caltech for financial support. Corey Reeves is acknowledged for providing allyl cyanoformate and for insightful discussions. Douglas Duquette is acknowledged for providing allyl 1H-imidazole-1-carboxylate reagents and for insightful discussions. Scott Virgil is acknowledged for assistance with instrumentation and insightful discussions.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org
Contributor Information
Katerina M. Korch, The Warren and Katharine Schlinger Laboratory for Chemistry and Chemical Engineering, Division of Chemistry and Chemical Engineering, California Institute of Technology 1200 E. California Blvd, MC 101-20, Pasadena, CA 91125 (USA)
Christian Eidamshaus, The Warren and Katharine Schlinger Laboratory for Chemistry and Chemical Engineering, Division of Chemistry and Chemical Engineering, California Institute of Technology 1200 E. California Blvd, MC 101-20, Pasadena, CA 91125 (USA).
Douglas C. Behenna, The Warren and Katharine Schlinger Laboratory for Chemistry and Chemical Engineering, Division of Chemistry and Chemical Engineering, California Institute of Technology 1200 E. California Blvd, MC 101-20, Pasadena, CA 91125 (USA)
Sangkil Nam, Molecular Medicine, Beckman Research Institute, City of Hope Comprehensive Cancer Center 1500 East Duarte Road, Duarte, CA 91010 (USA).
David Horne, Molecular Medicine, Beckman Research Institute, City of Hope Comprehensive Cancer Center 1500 East Duarte Road, Duarte, CA 91010 (USA).
Brian M. Stoltz, Email: stoltz@caltech.edu, The Warren and Katharine Schlinger Laboratory for Chemistry and Chemical Engineering, Division of Chemistry and Chemical Engineering, California Institute of Technology 1200 E. California Blvd, MC 101-20, Pasadena, CA 91125 (USA).
References
- 1.a) Welsch ME, Snyder SA, Stockwell BR. Curr. Opin. Chem. Biol. 2010;14:1–15. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) DeSimone RW, Currie KS, Mitchell SA, Darrow JW, Pippin DA. Comb. Chem. High Throughput Screening. 2004;7:473–493. doi: 10.2174/1386207043328544. [DOI] [PubMed] [Google Scholar]
- 2.Capdeville R, Buchdunger E, Zimmermann J, Matter A. Nat. Rev. Drug Discovery. 2002;1:493–502. doi: 10.1038/nrd839. [DOI] [PubMed] [Google Scholar]
- 3.Sharma PC, Jain A, Jain S, Pahwa R, Yar MS. J. Enzyme Inhib. Med. Chem. 2010;25:577–589. doi: 10.3109/14756360903373350. [DOI] [PubMed] [Google Scholar]
- 4.Mittur A. Curr. Drug Ther. 2011;6:17–34. [Google Scholar]
- 5.Dorsey BD, Vacca JP. Infect. Dis. Ther. 2002;25:65–83. [Google Scholar]
- 6.For a review see: Dinsmore CJ, Beshore DC. Org. Prep. Proced. Int. 2009;34:367–404.
- 7.a) Kukula P, Prins R. J. Catal. 2002;208:404–411. [Google Scholar]; b) uwano RK, Ito Y. J. Org. Chem. 1999;64:1232–1237. [Google Scholar]; c) ossen KR, Pye PJ, DiMichele LM, Volante RP, Reider PJ. Tetrahedron Lett. 1998;39:6823–6826. [Google Scholar]
- 8.a) Komeda H, Harada H, Washika S, Sakamoto T, Ueda M, Asano Y. Eur. J. Biochem. 2004;271:1580–1590. doi: 10.1111/j.1432-1033.2004.04069.x. [DOI] [PubMed] [Google Scholar]; b) Eichhorn E, Roduit J-P, Shaw N, Heinzmann K, Kiener A. Tetrahedron: Asymmetry. 1997;8:2533–2536. [Google Scholar]
- 9.a) McDermott BP, Campbell AD, Ertan A. Synlett. 2008:875–879. [Google Scholar]; b) erkheij MB, van der Sluis L, Sewing C, den Boer DJ, Terpstra JW, Hiemstra H, Bakker WII, van den Hoogenband A, van Maarseveen JH. Tetrahedron Lett. 2005;46:2369–2371. [Google Scholar]; c) Robinson SP, Sheikh NS, Baxter CA, Coldham I. Tetrahedron Lett. 2010;51:3642–3644. [Google Scholar]
- 10.a) Cochran BM, Michael FE. Org. Lett. 2008;10:329–332. doi: 10.1021/ol702891p. [DOI] [PubMed] [Google Scholar]; b) Nakhla JS, Wolfe JP. Org. Lett. 2007;9:3279–3282. doi: 10.1021/ol071241f. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) akano HN, Yokoyama J, Fujita R, Hongo H. Tetrahedron Lett. 2002;43:7761–7764. [Google Scholar]; d) to KI, Imahayashi Y, Kuroda T, Eno S, Saito B, Katsuki T. Tetrahedron Lett. 2004;45:7277–7281. [Google Scholar]; e) Uozumi Y, Tanahashi A, Hayashi T. J. Org. Chem. 1993;58:6826–6832. [Google Scholar]
- 11.a) Kwon SH, Lee SM, Byun SM, Chin J, Kim BM. Org. Lett. 2012;14:3664–3667. doi: 10.1021/ol301506b. [DOI] [PubMed] [Google Scholar]; b) Dekeukeleire S, D’hooghe M, Vanwalleghem M, Van Brabandt W, De Kimpe N. Tetrahedron. 2012;68:10827–10834. [Google Scholar]; c) Mickelson JW, Belonga KL, Jacobsen EJ. J. Org. Chem. 1995;60:4177–4183. [Google Scholar]; d) Ruider SA, Müller S, Carreira EM. Angew. Chem. Int. Ed. 2013;52:11908–11911. doi: 10.1002/anie.201306563. Angew. Chem.2013, 125, 12125–12128. [DOI] [PubMed] [Google Scholar]; e) Yar M, McGarrigle EM, Aggarwal VK. Angew. Chem. Int. Ed. 2008;47:3784–3786. doi: 10.1002/anie.200800373. Angew. Chem.2008, 120, 3844–3846. [DOI] [PubMed] [Google Scholar]; f) Santes V, Gómez E, Zárate V, Santillan R, Farfán N, Rojas-Lima S. Tetrahedron: Asymmetry. 2001;12:241–247. [Google Scholar]; g) Warshawsky AM, Patel MV, Chen T-M. J. Org. Chem. 1997;62:6439–6440. [Google Scholar]; h) Schanen V, Riche C, Chiaroni A, Quirion J-C, Husson H-P. Tetrahedron Lett. 1994;35:2533–2536. [Google Scholar]; i) Nordstrøm LU, Madsen R. Chem. Commun. 2007:5034–5036. doi: 10.1039/b712685a. [DOI] [PubMed] [Google Scholar]; j) Crestey F, Witt M, Jaroszewski JW, Franzyk H. J. Org. Chem. 2009;74:5652–5655. doi: 10.1021/jo900441s. [DOI] [PubMed] [Google Scholar]
- 12.Secchiero P, Bosco R, Celeghini C, Zauli G. Curr. Pharm. Des. 2011;17:569–577. doi: 10.2174/138161211795222586. [DOI] [PubMed] [Google Scholar]
- 13.a) Polonsky J, Merrien M-A, Prangé T, Pascard C, Moreau S. J. Chem. Soc., Chem. Commun. 1980:601–602. [Google Scholar]; b) Prangé T, Billion M-A, Vuilhorgne M, Pascard C, Polonsky J, Moreau S. Tetrahedron Lett. 1981;22:1977–1980. [Google Scholar]
- 14.Nakao Y, Masuda A, Matsunaga S, Fusetani N. J. Am. Chem. Soc. 1999;121:2425–2431. [Google Scholar]
- 15.Martínez-Luis S, Rodríguez R, Acevedo L, González MC, Lira-Rocha A, Mata R. Tetrahedron. 2006;62:1817–1822. [Google Scholar]
- 16.a) Rübsam F, Mazitschek R, Giannis A. Tetrahedron. 2000;56:8481–8487. [Google Scholar]; b) Herrero S, García-López MT, Latorre M, Cenarruzabeitia E, Del Rio J, Herranz R. J. Org. Chem. 2002;67:3866–3873. doi: 10.1021/jo0256336. [DOI] [PubMed] [Google Scholar]; c) Limbach M, Lygin AV, Korotkov VS, Es-Sayed M, de Meijere A. Org. Biomol. Chem. 2009;7:3338–3342. doi: 10.1039/b908548c. [DOI] [PubMed] [Google Scholar]; d) Suwal S, Kodadek T. Org. Biomol. Chem. 2013;11:2088–2092. doi: 10.1039/c3ob27476d. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Chen Z, Kende AS, Colson A-O, Mendezandino JL, Ebetino FH, Bush RD, Hu XE. Synth. Commun. 2006;36:473–479. [Google Scholar]
- 17.For a review see: De Risi C, Pelà M, Pollini GP, Trapella C, Zanirato V. Tetrahedron: Asymmetry. 2010;21:255–274.
- 18.a) Chen JJ, Nguyen T, D’Amico DC, Qian W, Human J, Aya T, Biswas K, Fotsch C, Han N, Liu Q, Nishimura N, Peterkin TAN, Yang K, Zhu J, Riahi BB, Hungate RW, Andersen NG, Colyer JT, Faul MM, Kamassah A, Wang J, Jona J, Kumar G, Johnson E, Askew BC. Bioorg. Med. Chem. Lett. 2011;21:3384–3389. doi: 10.1016/j.bmcl.2011.03.115. [DOI] [PubMed] [Google Scholar]; b) Gurjar MK, Karmakar S, Mohapatra DK, Phalgune UD. Tetrahedron Lett. 2002;43:1897–1900. [Google Scholar]; c) Pollini GP, Baricordi N, Benetti S, De Risi C, Zanirato V. Tetrahedron Lett. 2005;46:3699–3701. [Google Scholar]; d) Rychnovsky SD, Beauchamp T, Vaidyanathan R, Kwan T. J. Org. Chem. 1998;63:6363–6374. doi: 10.1021/jo9808831. [DOI] [PubMed] [Google Scholar]; e) Powell NA, Ciske FL, Clay EC, Cody WL, Downing DM, Blazecka PG, Holsworth DD, Edmunds JJ. Org. Lett. 2004;6:4069–4072. doi: 10.1021/ol048235t. [DOI] [PubMed] [Google Scholar]; f) Hicks F, Hou Y, Langston M, McCarron A, O’Brien E, Ito T, Ma C, Matthews C, O’Bryan C, Provencal D, Zhao Y, Huang J, Yang Q, Heyang L, Johnson M, Sitang Y, Yuqiang L. Org. Process Res. Dev. 2013;17:829–837. [Google Scholar]; g) Fu Y-Q, Ding L-N, Wang L-G, Tao J-C. Synth. Commun. 2008;38:2672–2683. [Google Scholar]; h) Sudhakar G, Bayya S, Reddy KJ, Sridhar B, Sharma K, Bathula SR. Eur. J. Org. Chem. 2014:1253–1265. [Google Scholar]; i) Mata L, Avenoza A, Busto JH, Peregrina JM. Chem. - Eur. J. 2013;19:6831–6839. doi: 10.1002/chem.201204392. [DOI] [PubMed] [Google Scholar]
- 19.Lencina CL, Dassonville-Klimpt A, Sonnet P. Tetrahedron: Asymmetry. 2008;19:1689–1697. [Google Scholar]
- 20.Baek J, Jang JI, Park YS. Bull. Korean Chem. Soc. 2011;32:4067–4070. [Google Scholar]
- 21.a) Behenna DC, Stoltz BM. J. Am. Chem. Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]; b) Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew. Chem. Int. Ed. 2005;44:6924–6927. doi: 10.1002/anie.200502018. Angew. Chem.2005, 117, 7084–7087; [DOI] [PubMed] [Google Scholar]; c) Seto M, Roizen JL, Stoltz BM. Angew. Chem. Int. Ed. 2008;47:6873–6876. doi: 10.1002/anie.200801424. Angew. Chem. 2008, 120, 6979–6982; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Streuff J, White DE, Virgil SC, Stoltz BM. Nat. Chem. 2010;2:192–196. doi: 10.1038/nchem.518. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Behenna DC, Liu Y, Yurino T, Kim J, White DE, Virgil SC, Stoltz BM. Nat. Chem. 2012;4:130–133. doi: 10.1038/nchem.1222. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Reeves CM, Eidamshaus C, Kim J, Stoltz BM. Angew. Chem. Int. Ed. 2013;52:6718–6721. doi: 10.1002/anie.201301815. Angew. Chem. 2013, 125, 6850–6853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dömling A, Huang Y. Synthesis. 2010:2859–2883. [Google Scholar]
- 23.We have found that stabilization of the intermediate enolate likely erodes ee by forming a solvent separated ion pair leading to an outer sphere, non-stereo selective reaction mechanism, see: Sherden NH, Behenna DC, Virgil SC, Stoltz BM. Angew. Chem. Int. Ed. 2009;48:6840–6843. doi: 10.1002/anie.200902575. Angew. Chem. 2009, 121, 6972–6975; Behenna DC, Mohr JT, Sherden NH, Marinescu SC, Harned AM, Tani K, Seto M, Ma S, Novák Z, Krout MR, McFadden RM, Roizen JL, Enquist JA, Jr, White DE, Levine SR, Petrova KV, Iwashita A, Virgil SC, Stoltz BM. Chem.–Eur. J. 2011;17:14199–14223. doi: 10.1002/chem.201003383.
- 24.Horton DA, Bourne GT, Smythe ML. Chem. Rev. 2003;103:893–930. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]
- 25.a) Rivkin A, Ahearn SP, Chichetti SM, Kim YR, Li C, Rosenau A, Kattar SD, Jung J, Shah S, Hughes BL, Crispino JL, Middleton RE, Szewczak AA, Munoz B, Shearman MS. Bioorg. Med. Chem. Lett. 2010;20:1269–1271. doi: 10.1016/j.bmcl.2009.11.101. [DOI] [PubMed] [Google Scholar]; b) Hirokawa Y, Kinoshita H, Tanaka T, Nakamura T, Fujimoto K, Kahimoto S, Kojima T, Kato S. Bioorg. Med. Chem. Lett. 2009;19:175–179. doi: 10.1016/j.bmcl.2008.10.127. [DOI] [PubMed] [Google Scholar]; c) Crawford JJ, Kenny PW, Bowyer J, Cook CR, Finlayson JE, Heyes C, Highton AJ, Hudson JA, Jestel A, Krapp S, Martin S, Macfaul PA, McDermott BP, McGuire TM, Morley AD, Morris JJ, Page KM, Ribeiro LR, Sawney H, Steinbacher S, Smith C, Dossetter AG. J. Med. Chem. 2012;55:8827–8837. doi: 10.1021/jm301119s. [DOI] [PubMed] [Google Scholar]; d) Jimenez J-M, Davis C, Boyall D, Fraysse D, Knegtel R, Settimo L, Young S, Bolton C, Chiu P, Curnock A, Rasmussen R, Tanner A, Ager I. Bioorg. Med. Chem. Lett. 2012;22:4645–4649. doi: 10.1016/j.bmcl.2012.05.114. [DOI] [PubMed] [Google Scholar]
- 26.Weisberg E, Manley PW, Cowan-Jacob SW, Hochhaus A, Griffin JD. Nat. Rev. Cancer. 2007;7:345–358. doi: 10.1038/nrc2126. [DOI] [PubMed] [Google Scholar]
- 27.Nagar B, Bornmann WG, Pellicena P, Schindler T, Veach DR, Miller WT, Clarkson B, Kuriyan J. Cancer Res. 2002;62:4236–4243. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.