

Abstract
Proteins of the SNX (sorting nexin) superfamily are characterized by the presence of a PX (Phox homology) domain and associate with PtdIns3P (phosphatidylinositol-3-monophosphate)-rich regions of the endosomal system. SNX27 is the only sorting nexin that contains a PDZ domain. In the present study, we used a proteomic approach to identify a novel interaction between SNX27 and ZO-2 [zonula occludens-2; also known as TJP2 (tight junction protein 2)], a component of the epithelial tight junction. The SNX27–ZO-2 interaction requires the PDZ domain of SNX27 and the C-terminal PDZ-binding motif of ZO-2. When tight junctions were perturbed by chelation of extracellular Ca2+, ZO-2 transiently localized to SNX27-positive early endosomes. Depletion of SNX27 in mpkCCD (mouse primary kidney cortical collecting duct) cell monolayers resulted in a decrease in the rate of ZO-2, but not ZO-1, mobility at cell–cell contact regions after photobleaching and an increase in junctional permeability to large solutes. The findings of the present study identify an important new SNX27-binding partner and suggest a role for endocytic pathways in the intracellular trafficking of ZO-2 and possibly other tight junction proteins. Our results also indicate a role for SNX27–ZO-2 interactions in tight junction maintenance and function.
Keywords: endosome, sorting nexin 27 (SNX27), tight junction, zonula occludens-2 (ZO-2)
INTRODUCTION
The SNX (sorting nexin) family of proteins is characterized by the presence of a phosphoinositide-binding PX domain, which targets proteins to phosphatidylinositol-3-monophosphate-rich membranes of the endosomal system [1–4]. In addition to a single PX domain, many SNXs contain other known protein–protein interaction domains [1,2,4].
A subset of twelve SNXs contain BAR (Bin, amphiphysin, Rvs) domains [2,4]. These SNXs recognize regions of membrane curvature and work in concert with the PX domain for optimal endosomal targeting [3,5]. Other sorting nexins contain SH3 (Src homology 3) or RGS (regulator of G-protein signalling) domains, thought to regulate SNX localization or function as a scaffold for signalling pathways [2,4]. SNX17, SNX27 and SNX31 all possess a C-terminal Ras association/FERM (4.1/ezrin/radixin/moesin)-like domain, which was recently shown to associate with the Ras GTPase [6]. The relatively large number of proteins in the SNX family, along with the diverse array of protein-interaction domains present in each, suggests that sorting nexins play important and varied roles in endosomal protein trafficking. SNX27 is the only sorting nexin that contains a PDZ domain [2,4,7].
PDZ domains are one of the largest families of protein interaction domains and are known to modulate the trafficking and subcellular distribution of cellular proteins including ion channels, receptors and adhesion molecules [8–10]. In most cases, PDZ domains of proteins function by interacting with short amino acid motifs at the C-termini of target proteins, but other modes of interaction have been reported [8,10,11]. A growing number of proteins have been identified as binding partners of the SNX27 PDZ domain, and this PDZ-mediated interaction is thought to regulate the trafficking of these proteins through the endosomal system [12–16]. In the present study, we expand on the growing list of SNX27 PDZ domain-interacting proteins by demonstrating that SNX27 interacts with ZO-2 (zonula occludens-2), a component of the TJ (tight junction).
The TJ is made up of transmembrane proteins from the claudin, and marvel (occludin and tricellulin) protein families, which are arranged in strands that encompass the apico-lateral portion of the membrane in polarized epithelia [17,18]. Canonically the TJ serves as both barrier and fence in epithelial cellular monolayers by enforcing selective permeability through the paracellular pathway and by acting as a diffusion barrier within plasma membranes, maintaining cell polarity [17,18]. However, disruption of the TJ structure was found not to interfere with apical–basal polarity while drastically increasing monolayer permeability, arguing against the fence function of the TJ [19].
Members of the MAGUK (membrane-associated guanylate kinase) family are cytosolic proteins peripheral to the plasma membrane and are important components of the TJ [20,21]. There are three MAGUK proteins, ZO-1, ZO-2 and ZO-3, all containing an enzymatically inactive GUK (guanylate kinase) domain, three PDZ domains, a U5 motif and a single SH3 domain [20,21]. Localization of ZO family members to the TJ may be mediated by direct binding to the claudins, occludin or tricellulin via these protein-interaction domains [20,21]. The first PDZ domain of all three ZO proteins interact with claudin C-terminal PDZ-binding motifs, whereas the U5 and GUK domains are responsible for occludin and tricellulin binding [20,21]. Further, multiple groups have shown that ZO protein localization is not driven by these TJ interactions; instead the interactions are necessary for TJ protein polymerization [19,20].
In addition to containing three PDZ domains, ZO-2 and ZO-3 terminate in a consensus PDZ-binding motif, suggesting that additional PDZ–protein interactions may be important for regulating the trafficking or function of these junctional proteins. In the present study, we show the ZO-2 C-terminal PDZ-binding motif is responsible for SNX27 binding. We also demonstrate a defect in ZO-2 trafficking to the TJ in cells lacking SNX27 with concomitant defects in barrier function, suggesting that SNX27 plays a role in the formation and/or maintenance of epithelial TJs.
EXPERIMENTAL
Antibodies
A polyclonal antibody against SNX27 was generated as described previously [16]. Monoclonal antibodies against fusion proteins tagged with c-Myc (clone 9E10), HA (haemagglutinin)-11 and mCherry were purchased from Abcam, Covance and Clontech respectively. ZO-2 rabbit polyclonal and mouse monoclonal antibodies were purchased from Invitrogen and Santa Cruz Biotechnology respectively. A ZO-3 polyclonal antibody (clone H-130) was purchased from Santa Cruz Biotechnology. A ZO-1 rat monoclonal antibody (clone R40.76) was a generous gift from Dr Alan Fanning (University of North Carolina, Chapel Hill, NC, U.S.A.). The Cell Light™ Early Endosome (RFP–Rab5a) marker was purchased from Invitrogen and used at a 25 MOI (multiplicity of infection). A anti-EEA1 (early endosome antigen 1) monoclonal antibody was purchased from BD Biosciences.
Molecular biology
cDNAs encoding wild-type and mutant human SNX27b were generated as described previously [16]. pBluescriptR-ZO-2 plasmid DNA (catalogue number 7492108) was purchased from A.T.C.C. (Manassas, VA, U.S.A.). To generate HA-tagged ZO-2, human ZO-2 cDNA was first excised (EcoRI-BamHI) from pBluescriptR and cloned into pCMV-HA (Clontech) (EcoRI-BglII), and an EcoRI site was then introduced to the 5′ untranslated region of ZO-2. This procedure was performed with QuikChange™ site-directed mutagenesis kit (Stratagene) using the forward primer 5′-GGGACCTGTGTCCGGAATCCCGGT-GCGAGGAGAC-3′ and the reverse primer 5′-GTCTCCTCGC-ACCGGGATTCCGGACACAGGTCCC-3′. Finally, excision by EcoRI served to remove extraneous untranslated sequence and to ensure a continuous HA–ZO-2 reading frame. Generation of pmCherry-ZO-2 was performed by EcoRI-KpnI digestion of pCMV-HA-ZO-2 and subcloning into the corresponding sites in pmCherry-c1. The BglII site in pmCherry-c1 was then destroyed by site-directed mutagenesis using the forward primer 5′-CA-AGTCCGGACTCAGTCTCGAGCTCAAGCTTCG-3′ and the reverse primer 5′-CGAAGCTTGAGCTCGAGACTGAGTCCG-GACTTG-3′ to ensure a continuous Cherry–ZO-2 reading frame. A premature termination codon in ZO-2 expression constructs (ΔDTEL) was introduced by mutagenesis using the forward primer 5′-GCCCGATACCGGTAGACAGAATTATAGATG-3′ and the reverse primer 5′-CATCTATAATTCTGTCTACCGGT-ATCGGGC-3′. All cDNA constructs were sequenced to verify the absence of undesired secondary mutations.
Cell culture and transfection
mpkCCD (mouse primary kidney cortical collecting duct) clone 3 cells were kindly provided by Dr Mark Knepper (National Heart, Lung, and Blood Institute, Bethesda, MA, U.S.A.). mpkCCD and MDCK (Madin–Darby canine kidney) cells were maintained in high glucose Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with 10 % FBS (Sigma) in humidified chambers with 5 % CO2 at 37 °C. mpkCCD cells were transfected using Lipofectamine™ LTX reagent including Plus reagent (Invitrogen) using manufacturer’s recommendations. Cells were typically used in biochemical and functional assays 24–48 h post-transfection. Cells stably expressing SNX27 silencing and non-silencing shRNAs were generated using an inducible lentiviral shRNA system (pINDUCER10) and selected with puromycin as described in Valdes et al. [16]. shRNA expression of puromycin resistant clones was induced with 1μg/ml doxycycline for 72–96 h prior to experimental analysis. SNX27 shRNA-resistant human SNX27 (SNX27r) was generated by site-directed mutagenesis, subcloned in the modified retroviral bicistronic GFP vector, MIGRI, and introduced into mpkCCD SNX27 shRNA-expressing cells as described previously [16]. Double SNX27/ZO-2-knockdown cells were generated by infection of uninduced pINDUCER10-SNX27/control shRNA cells with viral particles of pGIPZ-mouse ZO-2 shRNA (clone ID, V3lMM-488756; Thermo Scientific). ZO-2 shRNA expressing cells were sorted for GFP.
Protein-interaction analysis
The purification of GST and GST–PDZ fusion proteins was performed as described previously [16]. For GST pull-down experiments, mpkCCD cells were lysed in BB150 [binding buffer 150; 50 mM Tris (pH 7.6), 150 mM NaCl, 0.2 % CHAPS and 10 mM EDTA] plus Complete™ protease inhibitor cocktail (Roche). Protein concentration was determined using the bicinchoninic acid assay (Pierce Biotechnology). GST pull downs were typically performed using 0.5–1 mg of protein and 10–20 μg of fusion protein immobilized on glutathione beads for 2 h at 4 °C. The beads were washed three times in lysis buffer and boiled in sample buffer. For immunoprecipitation experiments, cells were lysed in BB150 containing Complete™ protease inhibitors. Immunoprecipitations were performed using 1–2 mg of protein lysate and 30 μl of anti c-Myc polyclonal antibody conjugated to Sepharose beads (Sigma). Immune complexes and proteins bound to GST-fusion proteins were analysed by Western blotting and detected using ZO-2 or SNX27 polyclonal antibodies at 1:1000 dilutions followed by secondary antibodies conjugated to IRdye 780 nm (rabbit) or IRdye 680 nm (mouse) (LI-COR) at a 1:10000 dilution. Myc tags were detected by Western blotting using the 9E10 monoclonal antibody at a 1:5000 dilution followed by a secondary antibody conjugated to horseradish peroxidase and detected by enhanced chemiluminescence (Millipore).
Peptide pull down
Wild-type (RGYYGQSARYRDTEL) and the indicated mutant ZO-2 peptides with an N-terminal linker sequence (SGSG) were synthesized at the Tufts University Peptide Synthesis core. A biotin moiety was conjugated to the N-terminal of each peptide to facilitate interaction with streptavidin. Binding assays were performed as described previously [23].
MS
To identify novel proteins bound to the PDZ domain of SNX27, samples were electrophoresed by SDS/PAGE, and protein bands visualized by staining with Coomassie Blue. The band of interest was excised and digested with trypsin at 1:50 enzyme/substrate ratio. The dried tryptic digest was analysed on an LTQ-Orbitrap XL (Thermo Fisher Scientific) interfaced with an Eksigent nano-LC 1D plus system (Eksigent Technologies) using CID (collision-induced dissociation) fragmentation. Samples were loaded on to an Agilent Zorbax 300SB-C18 trap column at a flow rate of 5 μl/min for 10 min, and then separated on a reverse-phase PicoFrit analytical column (New Objective) using a 40-min linear gradient of 2–40 % acetonitrile in 0.1 % formic acid at a flow rate of 300 nl/min. The LTQ-Orbitrap XL settings were: spray voltage, 1.5 kV; and full MS mass range, m/z 200–2000. The LTQ-Orbitrap XL was operated in a data-dependent mode; i.e. MS1 in the ion trap, scan for precursor ions followed by six data-dependent MS2 scans for precursor ions above a threshold ion count of 2000 with collision energy of 35 %. The raw file generated from the LTQ-Orbitrap XL was analysed as described previously [16].
Immunofluorescence
mpkCCD cells were plated on to glass coverslips and transfected as indicated. Cells were fixed with 4 % PFA for 10 min, permeabilized with 0.1 % Triton X-100 for 5 min and incubated in blocking buffer [1 % BSA in PBS] for 1 h at 37 °C. The fixed coverslips were incubated with primary antibodies (HA-11, ZO-1 and ZO-2 at a 1:2000 dilution; and SNX27 #14520 at a 1:100 dilution) in blocking buffer for 1 h at 4 °C. Coverslips were washed extensively with PBS and incubated with the indicated secondary antibodies (Alexa Fluor™ 488, 568, 594 or 647 nm at a 1:1000 dilution; Invitrogen/Molecular Probes) in blocking buffer for 1 h at 37 °C. Following further washing with PBS, coverslips were mounted using FluorSave™ (EMD Millipore). Samples were examined by confocal laser microscopy with a Zeiss 510 confocal system equipped with UV–visible lasers (Carl Zeiss MicroImaging). High-resolution (100 nm/pixel) images were obtained with a 63×, 1.4 numerical aperture Plan-Apochromat oil-immersion objective. Images were montaged using Illustrator CS4 software (Adobe systems). Co-localization analysis was performed with the FIJI plugin, Coloc_2 (http://fiji.sc/Colocalization_Analysis). The degree of co-localization of SNX27 (green) and ZO-2 (red) was quantified and expressed as Pearson co-localization coefficient (1, perfect correlation and – 1, no correlation). The significance of co-localization was determined by Costes’ randomization analysis [24].
FRAP
FRAP was measured using the FRAP module on a Leica SP5 confocal system (Leica Microsystems) by irreversibly photobleaching YFP–ZO-2 or YFP–ZO-1 fluorescence in a ROI (region of interest) of a cell with a high-intensity laser (488 nm). Subsequent diffusion of unbleached fluorescent proteins into the bleached region was imaged with non-bleaching attenuated laser light. Cells were placed in 35 mm coverglass bottom culture dishes (MatTek Corporation), in Iscove’s modified Dulbecco’s medium containing 20 mM Hepes at 37 °C, and images were collected via a HCX PL APO CS 63× magnification and a numerical aperture of 1.40 oil-immersion objective. A 2–4 μm strip of fluorescently tagged ZO-2 or ZO-1 was photobleached, and the whole cell was imaged while the fluorescence recovery into the photobleached ROI was monitored. The scanner was rotated to keep the ROI parallel to direction of scanning. Pre-bleach imaging conditions were established for maximal fluorescence signal without pixel saturation, and identical conditions were used for imaging during recovery (post-bleach). The bleaching conditions (laser power and the minimal number of laser iterations required for photobleaching) were empirically determined so that after photobleaching, the fluorescent signal of the photobleached ROI decreased to within background intensity levels. Average fluorescent intensities of the ROIs were collected with Fiji (FRAP analysis module, http://imagejdocu.tudor.lu/doku.php?id=plugin:analysis:frap_analysis:start), background corrected and normalized to the pre-bleach intensity. Experimental data were compared with a simulation of a double exponential plot [y = y0 + a(1 − e− bx) + c(1 − e− dx)] using Sigma Plot (Systat Software). The equation is a double exponential made up of two individual events. The first event is a(1 − e− bx) and the second event is c(1 − e− dx). y0 represents the y intercept. Of the two events, a and c control the maxima in conjunction with y0. The two events are additive (y0 + a + c= maximum). This was used to determine the mobile fraction (average of the maxima/100 %). e, b and d relate to the slope and direction of the plot. These can be used to determine the rate of recovery. Simulations were then used to calculate the mobile fraction of YFP–ZO-2 or YFP–ZO-1.
Barrier assays: FITC–dextran and TER (transepithelial electrical resistance)
TJ barrier assays were performed as described previously [21]. Briefly, to measure permeability to dextran, FITC-conjugated dextran (3 kDa) was added to the apical compartment at a concentration of 1 mg/ml. After 2 h, samples were removed from the basolateral compartment and fluorescent dextran concentrations were quantified using a Victor 3™ (PerkinElmer) and Wallac 1420 software (Amersham Pharmacia Biotech). Experimental values were determined by extrapolation from a standard curve of known fluorescent dextran concentrations. The apparent permeabilities were defined as described in [21].
Statistical analyses
All statistical analyses were performed as described in the corresponding Figure legend using GraphPad software.
RESULTS
Identification of ZO-2 as a novel SNX27 binding partner
To expand our understanding of the role played by SNX27 as an adaptor in endocytic trafficking pathways, we used a proteomic approach to identify novel interacting partners of the SNX27 PDZ domain, which is unique within the SNX family. A GST–PDZ fusion protein or GST control protein was incubated with mpkCCD cell lysates or with lysis buffer alone (Figure 1A). After washing, proteins bound to GST beads were separated by SDS/PAGE and identified by Coomassie Blue staining (Figure 1A). A protein of approximately 160 kDa was uniquely present in GST–PDZ pull downs (Figure 1A, lane iii). This band was excised and subjected to in-gel trypsin digest; its identity was revealed by MALDI-MS/MS (matrix-assisted laser desorption ionization–time-of-flight twin MS) to be ZO-2 (Figure 1B).
Figure 1. Identification of ZO-2 as a potential SNX27 PDZ binding partner.
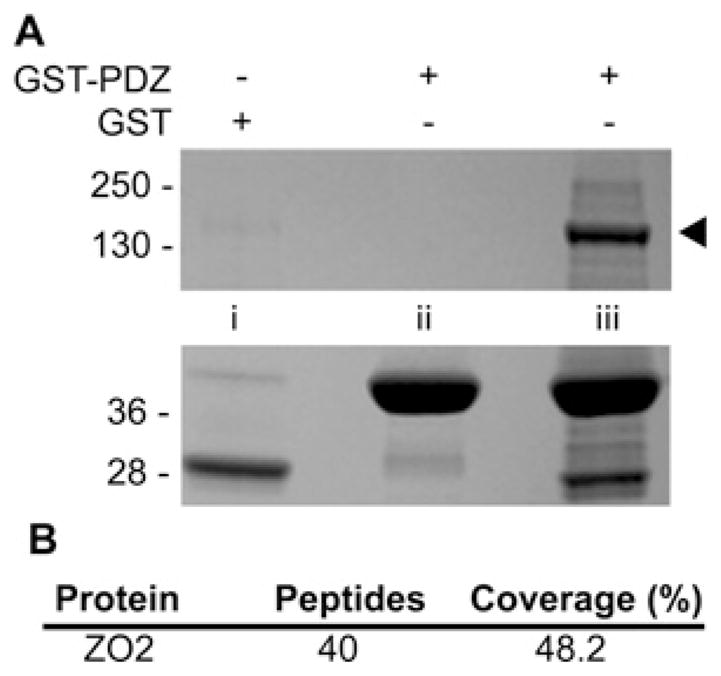
(A) mpkCCD cell extracts (lanes i and iii) or lysis buffer (lane ii) were incubated with GST–SNX27 PDZ (lanes ii and iii) or GST controls (lane i). Bound proteins were separated by SDS/PAGE and identified by Coomassie Blue staining. Prominent bands between 28 and 40 kDa are GST fusions. Band marked by arrow was excised, in-gel digested with trypsin and identified as ZO-2 by MS. Molecular mass is shown on the left-hand side in kDa. (B) ZO-2 peptide coverage statistics.
SNX27–ZO-2 binding is mediated by a type I PDZ interaction
To validate our finding that the PDZ domain of SNX27 can specifically bind ZO-2, GST pull down and immunoprecipitation experiments were performed (Figure 2). mpkCCD cell lysates were incubated with glutathione beads coated with GST alone, GST–PDZ or GST–PDZ in which the functionally critical GYGF motif is mutated to GGGG (GST–mPDZ). ZO-1, ZO-2 or ZO-3 bound to GST protein-coated beads were detected by Western blotting (Figure 2A). ZO-2 clearly associated with the wild-type PDZ domain of SNX27, but not control GST alone. Whereas a negligible amount of ZO-1 was observed in GST–PDZ pull downs, ZO-3 was not detected. No amount of ZO-1, ZO-2 or ZO-3 was detected with GST–mPDZ.
Figure 2. Validation of ZO-2 as a novel SNX27 binding partner.

(A) mpkCCD cell extracts were incubated with GST or GST-fusion proteins containing the SNX27 wild-type PDZ domain. ZO-1, ZO-2 or ZO-3 were detected by Western blotting. Equal GST loading was verified by Coomassie Blue staining. (B) mpkCCD cells were transiently transfected with empty Myc-tagged vector or plasmids containing Myc-fusion proteins of the indicated SNX27 sequences. The expressed fusion proteins were immunoprecipitated using an anti-Myc antibody. Bound ZO-2 was identified by Western blotting (top). SNX27 expression was confirmed by Western blotting for Myc (bottom). (C) mpkCCD cell extracts were incubated with wild-type (WT) (YRDTEL) or the indicated mutant C-terminal ZO-2 peptide conjugated to Streptavadin–agarose beads. Bound SNX27 was detected by Western blotting. (D) Wild-type ZO-2 C-terminal peptide can disrupt the endogenous SNX27–ZO-2 interaction. mpkCCD cell extracts (500 μg) were incubated with GST or GST-fusion proteins (20 μg) containing the SNX27 wild-type PDZ domain in the presence of increasing (0.2, 2 and 20 μg) amounts of wild-type (DTEL) or mutant (GGGG) ZO-2 C-terminal peptide. The presence of endogenous ZO-2 in GST pull downs was detected by Western blotting (upper panel). Equal GST loading was verified by Coomassie Blue staining (lower panel). (E) mpkCCD cell extracts were generated from confluent cells cultured under normal physiological calcium conditions. The extracts were incubated with an anti-SNX27 polyclonal antibody or pre-immune serum. Immune complexes were collected using protein A–Sepharose. Bound ZO-2 was detected by Western blotting using an anti-ZO-2 monoclonal antibody. Molecular mass is shown on the left-hand side in kDa. IP, immunoprecipitation.
To further verify that ZO-2 binding depends on the SNX27 PDZ domain, a series of Myc-tagged SNX27 constructs were expressed in mpkCCD cells and Myc–SNX27-interacting proteins were isolated by immunoprecipitation (Figure 2B). Although full-length Myc–SNX27 and Myc–SNX27 ΔFERM readily co-immunoprecipitated with ZO-2, both the Myc–SNX27mPDZ (a mutant SNX27 protein in which the conserved and functional GYGF sequence in the SNX27 PDZ domain was mutated to GGGG) and Myc–SNX27 ΔPDZ mutants were defective for ZO-2 binding. Thus we conclude that the PDZ domain of SNX27 is necessary and sufficient for ZO-2 binding [9].
Given that the PDZ domain of SNX27 was found to specifically bind ZO-2, we postulated that the C-terminus of ZO-2 mediates the SNX27 interaction. However, since ZO-2 contains three PDZ domains [20], we could not rule out the possibility of a PDZ–PDZ domain interaction between the two proteins. Secondly, we proposed that ZO-2 conforms to the type I PDZ-binding motif [9–11]. To test both hypotheses, biotinylated ZO-2 peptides corresponding to the wild-type (YRDTEL) or the indicated mutant ZO-2 C-terminus were synthesized. We incubated mpkCCD cell lysates with the immobilized peptides and visualized interacting proteins following SDS/PAGE and Western blot analysis (Figure 2C). SNX27 was readily detected in complexes containing the ZO-2-wild-type peptide and peptides where the residue at the – 1 position (glutamic acid) is substituted with glycine (Figure 2C). In contrast, SNX27 binding to peptides substituted with glycine at residues 0 or – 2 was abolished (Figure 2C). Amino acids closer to the N-terminus have been proposed to have a less essential role in PDZ domain binding [10,11]. In agreement with this, binding of ZO-2 peptide mutated at position – 3 or – 5 led to diminished SNX27 binding (Figure 2C). Previous reports have demonstrated a PDZ–PDZ domain interaction between different ZO proteins in the same family [25]. To rule out the possibility of a ZO-2–SNX27 PDZ–PDZ domain interaction we asked if a C-terminal ZO-2 peptide could compete with endogenous ZO-2 for interaction with GST–SNX27 (Figure 2D). When increased amounts of ZO-2 wild-type peptide (0.2–20 μg) were added to pools of GST–SNX27 and mpkCCD cell lysate, decreasing amounts of ZO-2 was found in GST–SNX27 PDZ-bound complexes (Figure 2D). In contrast, ZO-2 bound to the GST–SNX27 PDZ complexes was unaffected by the presence of increasing amounts of ZO-2 mutant C-terminal peptide (Figure 2D). In conclusion, the C-terminus of ZO-2 does conform to a canonical type-1 PDZ-binding motif and is the only motif or domain responsible for binding SNX27.
We also asked whether the C-terminus of ZO-2 was capable of binding the ZO-2 PDZ domains. However, we failed to detect any interaction between the ZO-2 wild-type C-terminus peptide and full-length ZO-2 (results not shown).
To determine whether the SNX27–ZO-2 interaction occurs at normal physiological levels, endogenous SNX7 was immuno-precipitated from mpkCCD cells at confluence under conditions of normal physiological calcium. Endogenous ZO-2 was detected in the SNX27 immune complex, but not the pre-immune control (Figure 2E). Thus we conclude that SNX27 associates with ZO-2 under normal physiological conditions via the PDZ domain of SNX27 and the type I PDZ-binding motif of ZO-2.
ZO-2 co-localizes with SNX27 on early endosomes
Many reports have localized SNX27 to the early endosome in a variety of cell types [14,15,26], whereas ZO-2 accumulates at the TJ of confluent cells and in the nucleus of subconfluent cells [27]. ZO-1 and other TJ proteins have also been detected in the endosomal network when TJs are disrupted by pharmacological or physiological perturbations [28,29].
To test whether ZO family members might specifically be recruited to the endosomal system in a SNX27-dependent manner we overexpressed Myc–SNX27 and fluorescently labelled ZO-2 or ZO-1 in confluent mpkCCD cells. Upon imaging fixed cells, we saw striking recruitment of fluorescently tagged ZO-2 to SNX27-positive Rab5 early endosomes (Figure 3, upper panels). In contrast, ZO-1 was exclusively visible at TJs (Figure 3, lower panels). Hence, the specific biochemical interaction between SNX27 and ZO-2 may direct trafficking of ZO-2 to the endosomal system in epithelial cells.
Figure 3. Recruitment of ZO-2, but not ZO-1, to the early endosome by SNX27.
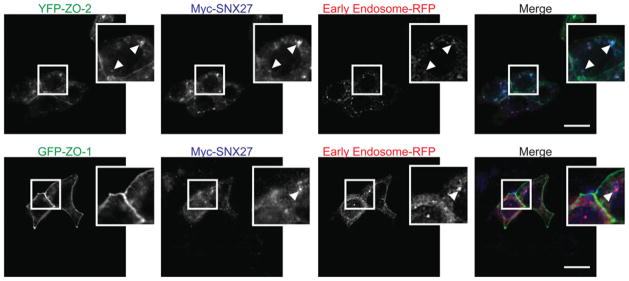
mpkCCD cells were infected with RFP–Rab5a (early endosome marker) and co-transfected with Myc–SNX27 and either YFP–ZO-2 (upper panels) or YFP–ZO-1 (lower panels) infected with RFP–Rab5a and grown to confluence. At this point, the cells were fixed/permeabilized and stained with an anti-Myc monoclonal antibody and visualized using Alexa Fluor® anti-rat 647 nm. Arrows indicate co-localized protein (scale bar = 15 μm).
We hypothesized that the PDZ domain of SNX27 might be responsible for the recruitment of ZO-2 to the early endosome. To test this idea, GFP–SNX27 and mCherry–ZO-2 fusion proteins were co-expressed in mpkCCD cells (Figure 4 and Supplementary Figure S1 at http://www.biochemj.org/bj/455/bj4550095add.htm). At 48 h after transfection the cells were at confluence and subjected to the ‘calcium-switch’ protocol for the indicated times. The cells were subsequently fixed and examined by confocal microscopy (Figure 4). As expected, pre-treatment mCherry–ZO-2 was found to localize at points of cell–cell contact (Figures 4i and 4iii, top row). Surprisingly, we also observed a portion of GFP–SNX27 at contact sites at an identical confocal plane (Figure 4iii, top row). After 1 h of calcium depletion, complete loss of mCherry–ZO-2 from the plasma membrane was observed. At this point, all mCherry–ZO-2 exhibited a punctate cytoplasmic distribution where co-localization with GFP–SNX27 was commonly observed (Figure 4iii, middle row). After a 1 h recovery, both mCherry–ZO-2 and GFP–SNX27 were recruited to the initial sites of cell contact (Figure 4iii, bottom row). When co-expressed with mCherry–ZO-2, GFP was not observed at the sites of cell–cell contacts (Figure 4i). However, when co-expressed with mCherry alone, a small amount of GFP–SNX27 was detected at the cell–cell contact sites (Figures 4ii and 4iii). Under these conditions, GFP–SNX27 may be recruiting endogenous ZO-2 or another interacting protein to cell–cell contact sites. Finally, we examined the role of the PDZ domain of SNX27 in the recruitment of mCherry–ZO-2 to endosomal sites. Whereas mCherry–ZO-2 was readily detected at points of cell–cell contact, GFP–SNX27 ΔPDZ was completely absent (Figure 4iv, top row). As expected, calcium depletion resulted in the internalization of mCherry–ZO-2 from contact sites, but co-localization with GFP-SNX27 ΔPDZ was not observed (Figure 4iv, middle row). Following restoration of calcium for 1 h, many GFP–SNX27 ΔPDZ-expressing cells exhibited mCherry–ZO-2 at sites of new cell–cell contacts (Figure 4iv, bottom row). Taken together, these results suggest that the SNX27 PDZ–ZO-2 interaction might play an important role in the trafficking of ZO-2 at sites of nascent cell–cell contacts.
Figure 4. Co-localization of ZO-2 and SNX27 is dependent on the PDZ domain of SNX27.

mpkCCD cells were transiently transfected with the indicated GFP and mCherry constructs. The cells were grown to confluence, depleted of calcium using 2 mM EGTA and then returned to normal growth medium. Cells were fixed at indicated time points and visualized with confocal microscopy (scale bar = 10 μm).
We next examined the distribution of endogenous SNX27 and ZO-2 during the restructuring of cell–cell contacts (Figure 5). The localization of endogenous SNX27 and ZO-2 in mpkCCD cells was visualized using specific polyclonal and monoclonal antibodies respectively (Figure 5A) and co-localization analysis was performed using Costes’ randomization and Pearson’s correlation [24]. At cell confluence, ZO-2 was present at the TJ, whereas SNX27 exhibited a largely punctate cytoplasmic distribution (Figure 5Ai). Whereas Costes’ randomization analysis shows that the Pearson’s correlation between the two images is significantly higher than would be if the image was randomized [%(Robs>Rrand) = 100], the Pearson’s correlation is still low (R = 0.058; Figure 5Bi). After 15, 30 and 60 min of calcium depletion using the chelating agent EGTA, most endogenous ZO-2 was internalized and points of co-localization between SNX27 and ZO-2 were visually apparent (Figures 5Aii, 5Aiii and 5Aiv). Further, co-localization analysis showed increased Pearson’s correlations compared with confluent cells. (R = 0.202, 0.160 and 0.443; Figures 5Bii, 5Biii and 5Biv). The co-localization of endogenous SNX27 and ZO-2 following TJ disruption suggests that SNX27 may mediate the internalization of ZO-2 to cytoplasmic vesicles. Co-localization between the two proteins visually diminished from the 60 min of calcium depletion upon restoration of normal physiological calcium for 30 and 60 min, although ZO-2 remained largely cytoplasmic (Figures 5Av and 5Avi). Again, co-localization analysis of the representative images supported our visual analysis (R = 0.091 and 0.20; Figures 5Av and 5Avi). After 60 min of calcium restoration, most endogenous ZO-2 was observed at nascent cell–cell contact sites, but SNX27 was not detected at this location, suggesting that ZO-2 returns to the TJ on SNX27-independent vesicles (Figure 5Avi). To support the trends observed in the representative images of Figures 5(A) and 5(B), co-localization analysis was performed on multiple images from the indicated time points. Pearson’s correlations from individual images were quantified, averaged and compared across time points (Figure 5C). A one-way ANOVA shows that Pearson’s correlations of images from the 30 and 60 min time points are significantly higher than those from the 0 min time point (P < 0.001).
Figure 5. Co-localization of SNX27 and ZO-2 during TJ disruption.

(A) mpkCCD cells were grown to confluence, depleted of calcium using 2 mM EGTA and then returned to normal growth medium. During this process cells were fixed and permeabilized at the indicated time points and stained with an anti-SNX27 polyclonal or anti-ZO-2 monoclonal antibodies (scale bar = 10 μm). (B) For the corresponding representative images in (A) Costes’ randomization analysis was performed to determine co-localization. Box plot represents Pearson’s correlations for the ZO-2 channel compared with 200 randomized SNX27 channels. The red lines and values represent Pearson’s correlation of the corresponding image [%(R obs >R rand) = 100%]. (C) Average Pearson’s correlation of SNX27 and ZO-2 channels for images corresponding to indicated time points (0 min n = 11, 15 min n = 2, 30 min n = 7, 60 min n = 6, 30 min recovery n = 10 and 60 min recovery n = 7). *ANOVA P < 0.001. Error bars represent the S.D.
Other PDZ proteins may play a role in ZO-2 trafficking
If the SNX27–ZO-2 interaction is responsible for the phenotypes we observed in the present study, we hypothesized that a mutant ZO-2 lacking the SNX27-binding site (ZO-2 ΔDTEL) would produce similar deficiencies in trafficking and function. Therefore we examined a mutant ZO-2 ΔDTEL by transiently transfecting mpkCCD cells with N-terminal tagged HA–ZO-2 and HA–ZO-2 ΔDTEL (Figure 6). Figure 6(A) shows comparable expression levels and complete translation of the two fusion proteins. The intracellular distribution of wild-type ZO-2 and ZO-2 ΔDTEL in confluent mpkCCD cells was compared. In confluent monolayers, HA–ZO-2 was predominantly located at the cell periphery, overlapping the distribution of ZO-1 (Figure 6B). In contrast, we noted a striking nuclear localization of HA–ZO-2 ΔDTEL (Figure 6B). When we quantified the percentage of cells exhibiting nuclear HA–ZO-2 in three independent experiments (Figure 6C), we found that nuclear HA–ZO-2 was detected in approximately 1 % of cells, whereas nuclear HA–ZO-2 ΔDTEL was clearly visible in almost 80 %of transfected cells (P < 0.0001). A similar phenotype was observed in MDCK cell monolayers (results not shown). Hence, the C-terminal PDZ-binding motif plays a critical role in the targeting of ZO-2 in epithelial cells.
Figure 6. C-terminal PDZ-binding motif of ZO-2 is important for localization.
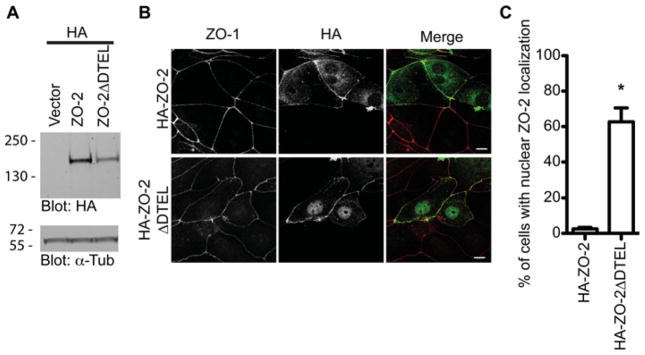
(A) Western blot of indicated HA-tagged ZO-2 constructs in mpkCCD cells. Equal loading was confirmed by Western blotting using an anti-α-tubulin antibody. (B) Localization of HA–ZO-2 fusion proteins was assessed in mpkCCD cells by staining with an anti-HA monoclonal antibody and anti-ZO-1 polyclonal antibody (scale bar = 10 μm). (C) The proportion of cells expressing nuclear HA–ZO-2 in mpkCCD cells was quantified. The histogram is representative of three independent experiments (Student’s t test, *P < 0.005, n>100; error bars represent the S.D.).
We postulated that if the SNX27–ZO-2 interaction was important for ZO-2 targeting in epithelial cells, the nuclear HA–ZO-2 ΔDTEL phenotype could be recapitulated in mpkCCD cells lacking SNX27. To this end we used an inducible lentiviral-mediated shRNA expression system to suppress the endogenous levels of SNX27 in mpkCCD cells (Figure 7A). Puromycin-resistant clones were treated with doxycycline to induce shRNA and RFP expression. After 72 h of treatment, SNX27 levels were suppressed by over 90 %(Figure 7A), yet ZO-2 was easily detected at the sites of cell–cell contacts (Figure 7B) in both control and SNX27-depleted mpkCCD cells. Surprisingly, SNX27-knockdown cells did not present an endogenous nuclear ZO-2 phenotype to parallel the nuclear localization of HA–ZO-2 ΔDTEL. Hence, other PDZ domain-containing proteins may mediate wild-type ZO-2 nuclear exit or negatively regulate ZO-2 nuclear targeting.
Figure 7. Depletion of SNX27 from mpkCCD cells at confluence has no effect of ZO-2 localization at the TJ.
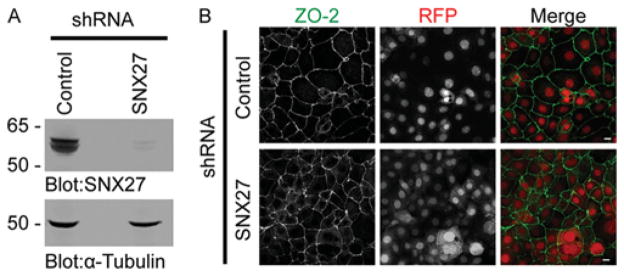
(A) The protein levels of SNX27 (upper panel) and α-tubulin (lower panel) in control and SNX27 shRNA-treated cells were assessed by Western blotting. (B) ZO-2 localization of control and shRNA-expressing cells was detected by immunostaining using an anti-ZO-2 polyclonal antibody. shRNA-expressing cells are marked by RFP (scale bar10 μm).
Depletion of SNX27 decreases ZO-2 mobility at the TJ
Since SNX27 and ZO-2 co-localized upon calcium depletion, we next hypothesized that SNX27 may be important for ZO-2 internalization upon TJ disruption. To test this we performed a ‘calcium-switch’ assay on SNX27-depleted cells. However, fixed and ZO-2 stained cells did not show a significant difference in the rate of TJ disruption nor a change in localization at all time points (Supplementary Figure S2 at http://www.biochemj.org/bj/455/bj4550095add.htm).
Previously, it has been shown that the ZO family of proteins is highly dynamic at the TJ [30]. We hypothesized that if the interaction between SNX27 and ZO-2 was in part responsible for ZO-2 trafficking we would be able to identify a difference in ZO-2 TJ dynamics upon depletion of SNX27. Whereas these differences may present as indistinguishable subtleties in our previous fixed cell assays, we proposed that a live-cell kinetic assay would make this difference clear.
Therefore FRAP was performed to compare the mobility of ZO-2 or ZO-1 in control and SNX27-depleted cells (Figure 8). YFP–ZO-2 (Figure 8Ai) or YFP–ZO-1 (Figure 8Aii) was transfected into control or SNX27-knockdown cells. Expressed YFP-tagged ZO-2/ZO-1 at cell–cell contacts was photobleached and the rate of fluorescence recovery was assessed for up to 6 min (Figure 8A). YFP–ZO-2 fluorescence returned the bleached regions to a lesser extent in SNX27-depleted cells (Figure 8Bi), whereas the extent to which YFP–ZO-1 fluorescence returned to bleached regions was independent of SNX27 (Figure 8Bii). The recovery of YFP–ZO-2/YFP–ZO-1 to the bleached region was analysed and fitted to a double-exponential curve (Figure 8B). The curve was used to quantify the mobile fraction of YFP–ZO-2 under each condition, revealing a significantly greater mobile fraction in the control cells (Figure 8Ci; P < 0.05).
Figure 8. Decreased rate of recovery of YFP–ZO-2 fluorescence at the TJ in mpkCCD cells lacking SNX27.

Control or SNX27-knockdown cell monolayers were transiently transfected with YFP–ZO-2 or YFP–ZO-1. Points of cell–cell contact in both YFP- and RFP-expressing cells were photobleached and the rate of recovery of ZO-2 or ZO-1 at the TJ at the indicated times was monitored by YFP (A). Cells expressing shRNA and were identified through RFP fluorescence. High magnification images of TJ sections at the indicated times are shown. Box represents the bleached region of interest (scale bar = 10 μm). (B) Quantitative FRAP analysis of the bleached area shown in (Ai) and (Bi). Curves were fit to the data using a double-exponential equation (Bi, replicates = 3, control n = 8 and SNX27 n = 18; and Bii, replicates = 3, control n = 8 and SNX27 n = 12). (C) Average mobile fractions of YFP–ZO-2 and YFP–ZO-1 in wild-type and SNX27-knockdown mpkCCD cells. Double exponential fit curves were used to calculate the mobile fraction of YFP–ZO-2 under the indicated conditions (Ci, Student’s t test, *P < 0.05, control n = 18 and SNX27 n = 18; Cii, Student’s t test, P > 0.05, control n = 8 and SNX27 n = 12; error bars represent the S.D.).
To ensure that our analysis was not skewed by differences in YFP–ZO-2 expression levels between cells, the initial YFP fluorescence intensity of each image was compared with the mobile fraction (Supplementary Figure S3A at http://www.biochemj.org/bj/455/bj4550095add.htm). A linear regression was performed to analyse the relationship (R = 0.53 and slope = 0.0048; Supplementary Figure 3A). Whereas the positive slope denotes a relationship between the two, the grade of the slope and small R value means the relationship is not very important. Further, the average and distribution of YFP–ZO-2 fluorescence intensities is similar between control shRNA images and SNX27 shRNA images (Supplementary Figure S3B).
To determine if the change in kinetics was due to lateral diffusion of ZO-2 through the TJ or mobility from the cytoplasm to the junction, we compared the relative fluorescence at the edge (blue) of the bleached area with the levels in the middle (magenta) over time in both control and SNX27 knockdown cells (Supplementary Figure S4 at http://www.biochemj.org/bj/455/bj4550095add.htm). The recovery of fluorescence occurred at a similar rate in all bleached regions. At the same time, the mobility of ZO-2 at a junctional site adjacent to the bleach area was monitored. Here, we observed a stable fluorescence signal through time in both control and SNX27-knockdown cells (Supplementary Figure S4, black arrows and lines). Taken together, these data suggest that the ZO-2 at the TJ does not diffuse laterally within the junction in either control or SNX27-knockdown cells. Therefore we reason that the majority of ZO-2 kinetic change at the TJ upon SNX27 knockdown was due to an exchange of ZO-2 between the junction and cytoplasm.
Ultimately, in the absence of functional SNX27, turnover of ZO-2 at the sites of cell–cell contact is hindered, but not abolished. Therefore SNX27 regulates the dynamics of ZO-2 at the TJ.
Depletion of SNX27 destabilizes the barrier for large molecules
Given that ZO-2 trafficking to, or stability at, the TJ may be regulated by SNX27, we next investigated the functional role of SNX27 at the TJ by measuring permeability of solutes >4 Å (1 Å = 0.1 nm) using fluorescein-conjugated dextran flux assays (Figure 9B). These assays were performed in control or SNX27-depleted polarized mpkCCD cells. To rule out off-target SNX27 shRNA effects, a human SNX27 cDNA resistant to the targeting sequence (SNX27r) was expressed in SNX27 shRNA knockdown cells (Figure 9A). After 72 h of SNX27 shRNA induction, endogenous SNX27 levels had been depleted over 90 % (Figure 9B). In contrast, expression of SNX27r in SNX27-knockdown cells restored the protein levels of SNX27 close to the level of endogenous SNX27 present in control shRNA-expressing cells (Figure 9B).
Figure 9. Loss of SNX27 alters TJ permeability characteristics.
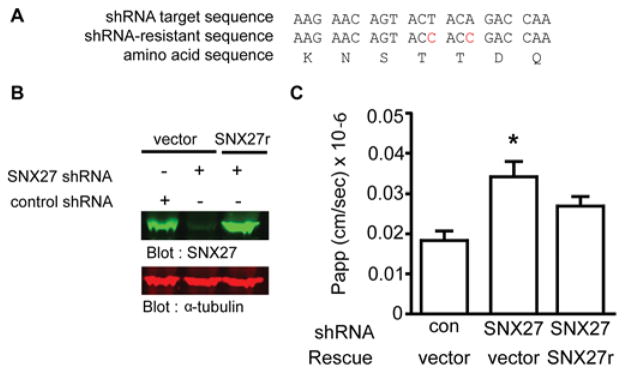
(A) SNX27 shRNA-resistant rescue strategy. (B) Resistance of SNX27r to SNX27 shRNA as revealed by Western blotting for SNX27 (upper panel). Equal loading confirmed by Western blotting for α-tubulin. (C) Flux of 3 kDa fluorescein-conjugated dextran was measured in steady-state control, SNX27 shRNA or SNX27 shRNA/SNX27r expressed mpkCCD cells. Flux of six cells per experiment were quantified (one-way ANOVA, P < 0.05; Dunnett’s multiple comparison post test *P < 0.05; error bars represent the S.D.). Con, control.
SNX27 depletion caused a significant increase in permeability of 3 kDa FITC–dextran (Figure 9C). Exogenous expression of SNX27r restored barrier function close to that of cells expressing control shRNA. Clearly, SNX27 contributes to TJ permeability, an effect we propose to be mediated by the PDZ-dependent interaction with ZO-2.
Finally, to address this hypothesis we depleted both SNX27 and ZO-2 from mpkCCD cells and measured 3 kDa dextran flux (Figure 10). Western blot analysis revealed an over 90 % decrease in SNX27 and ZO-2 protein levels in cells expressing their respective shRNAs (Figure 10A). Consistent with previous results, increased TJ permeability to 3 kDa dextran was observed in cells lacking SNX27. In both control and SNX27-knockdown cells also lacking ZO-2, total dextran permeability was not significantly different to that in cells lacking SNX27 alone. However, both double-knockdown cells had increased permeability relative to the control cells (Figure 10B). These data support the hypothesis that the contribution of SNX27 to the permeability to large solutes in the TJ is due to the specific delivery of ZO-2 to the TJ.
Figure 10. Effect of SNX27 on TJ permeability is ZO-2 dependent.

mpkCCD cells were infected with lentivirus expressing either inducible SNX27 or control shRNA in combination with constitutive ZO-2 or control shRNA (con). ZO-2 shRNA cells were selected for GFP by flow cytometry. SNX27 shRNA expression was induced by doxycycline (1 μg/ml). (A) Protein expression was assessed post-assay by Western blotting. (B) Permeability of the indicated mpkCCD cells to 3 kDa FITC–dextran was measured. The data represent the mean of two assays from four independent experiments (one-way ANOVA, P < 0.05; Dunnett’s multiple comparison post test, *P < 0.05 and ** P < 0.01; error bars represent the S.D.).
DISCUSSION
Using proteomic approaches, we identified ZO-2 as a novel binding partner of SNX27 and characterized the role of SNX27 in TJ function and the intracellular trafficking of ZO-2. Using GST pull down and co-immunoprecipitation assays, we showed that the C-terminal PDZ-binding motif of ZO-2 (DTEL) interacts with the PDZ domain of SNX27. The two other members of the ZO family, ZO-1 and ZO-3, were not identified in our proteomic screens. We did not detect ZO-3 and detected ZO-1 only very weakly in GST–SNX27 PDZ pull downs by Western blotting. This is not surprising since the C-terminus of ZO-1 does not terminate in a consensus PDZ-binding motif. Further, a recent proteomic screen looking for ZO-1-interacting proteins did not identify SNX27 while identifying other SNX family members [31]. The C-terminus of ZO-3 does terminate in a consensus PDZ-binding motif [9]. The detection of ZO-1 in PDZ pull downs may be indirect and due to the known interaction of ZO-2 with ZO-1 [25].
In confluent monolayers, ZO-2 is localized at the TJ. However, when junctions are disassembled, such as during cell division or non-physiologically by calcium depletion, junctional components may reside on cytosolic vesicular structures [28,29,32]. Given that SNX27 is known to localize to early endosomes, we hypothesized that ZO-2 may transiently interact with SNX27 during intracellular trafficking as nascent cell–cell contacts form [12,15]. Using tagged SNX27 and ZO-2 proteins as well as antibody staining of native proteins, we show overlap of the two proteins on the early endosome. Whereas other TJ proteins have been reported at these sites [28,29,32,33], this is the first time that ZO-2 has been identified on endosomes in epithelial cells. Using a ‘calcium-switch’ strategy, Ivanov et al. [28] identified a clathrin-dependent mechanism for trafficking of occludin, claudin 1, claudin 4 and ZO-1. Thus TJ proteins may traffic to and from the TJ in a multimeric complex mediated by PDZ protein interactions involving the MAGUKs and SNX27. A careful analysis of all proteins in the SNX27–ZO-2 complex and an examination of various endosomal pools of TJ proteins will be required to fully understand the role of SNX27 in the formation and maintenance of TJs.
We postulated that a SNX27-binding-defective mutant ZO-2 would exhibit intracellular trafficking defects. We found the majority of ZO-2 ΔDTEL mutant to localize to the nucleus, even when cells were plated at high density, a condition where ZO-2 is known to selectively accumulate at the cell periphery. In contrast, in cells lacking SNX27 ZO-2 was readily found at the TJ. These data strongly suggest that other PDZ proteins modulate the nuclear trafficking of ZO-2, and it will be critical to identify these unknown regulators of ZO-2 localization. To date, only the mammalian homologue of Drosophila Scribble, hScrib, has been proposed to regulate ZO-2 trafficking by binding ZO-2-DTEL [34]. However, a function for the Scrib–ZO-2 interaction has yet to be elucidated.
ZO-2 contains several predicted nuclear import and export signals [35–37]. Deletion of any of the four canine ZO-2 nuclear export signals leads to nuclear ZO-2 accumulation [36]. However, if all nuclear import signals are removed, nuclear ZO-2 is still apparent [36,37]. To address these observations, the authors discussed the possibility that ZO-2 may use a ‘piggyback’ mechanism to enter the nucleus [37]. Previous data indicate that protein kinase Cε-dependent phosphorylation of a serine residue in the NES (nuclear export sequence) is essential for ZO-2 nuclear export [38]. The authors demonstrated that ZO-2, once synthesized, is translocated to the nucleus and later relocated to the plasma membrane [38]. Although the majority of ZO-2 ΔDTEL is found in the nucleus, some of this mutant protein is detected at sites of cell–cell contact. This suggests that the ZO-2 mutant readily escapes the nucleus, but is unstable at the plasma membrane. It remains to be tested whether the C-terminal PDZ-binding motif is required for nuclear export, stability at the TJ or a combination of both. Knowing the mechanism of ZO-2 nuclear localization to be SNX27-independent, we postulated that SNX27 might regulate the function of ZO-2 at the TJ. By immunofluorescence, the localization of ZO-2 at cell–cell contacts in mpkCCD cells lacking SNX27 was unaltered. Given that TJs are highly dynamic, only when we used FRAP technology to study ZO-2 dynamics were deficiencies in ZO-2 trafficking in SNX27-depleted cells apparent. Shen et al. [30] demonstrated that ZO-1 was highly dynamic at the TJ with approximately 70 % of ZO-1 present in the mobile fraction. We noted a similar amount of mobile ZO-2 (80.6 ± 15.2 %) and ZO-1 (79.3 ± 15.4 %) in the present study. In cells lacking SNX27, the fraction of mobile ZO-2 at the TJ after photobleaching was approximately 10 % lower than in control cells. This relatively small, but significant, decrease is comparable with the decrease in occludin mobility when casein kinase is inhibited [39]. Hence, we propose that the interaction between the PDZ-binding motif of ZO-2 and PDZ domain of SNX27 is important in ZO-2 dynamics at the TJ.
There are two pathways by which solutes may pass through the TJ. It is known that ions smaller than approximately 4 Å pass through claudin-based pores (pore pathway). ZO-1 and occludin have been implicated in the pathway by which larger molecules pass through the junction (leak pathway) [40]. We looked at the effect of SNX27 on both pathways by generating multiple mpkCCD cell lines expressing SNX27 shRNA. In some, an increased resistance of the barrier to ions (measured by TER) was observed, but these effects were not rescued upon re-expression of a shRNA-resistant SNX27 cDNA expression construct (results not shown). Furthermore, knockdown of ZO-2 decreased TER in both control and SNX27 shRNA background cells (results not shown). Hence, we concluded that the effects of SNX27 knockdown on TER are likely to be non-specific. In contrast, all mpkCCD SNX27-knockdown cell lines exhibited an increased permeability to larger solutes, a phenotype partially rescued by re-expression of SNX27. A double shRNA knockdown combination of both SNX27 and ZO-2 or control and ZO-2 exhibited similar solute permeability to cells having a single knockdown of SNX27. This suggested that the effect of SNX27 on mpkCCD permeability to solutes was mediated by defective trafficking of ZO-2 to the TJ.
In summary, we identified a novel interaction between the PDZ domain of SNX27 and the C-terminal PDZ-binding motif of ZO-2. There may be several mechanisms by which ZO-2 traffics through the cell. We propose that one mechanism of ZO-2 removal from the junction is via interaction with SNX27-positive vesicles. ZO-2 is subsequently recycled to the TJ by exchange to an unidentified protein. In the absence of SNX27, the dynamics of ZO-2 exchange at the TJ are slowed. We propose the change in dynamics is due to the deficient removal of ZO-2 from the junction by SNX-27 positive vesicles and proper targeting ZO-2 to the protein responsible for recycling. Further, in the absence of SNX27, permeability of the TJ to large solutes is increased. We postulate that the slowed exchange ZO-2 at the TJ is not sufficient to maintain the integrity of the TJ structure.
Thus, ZO-2 joins the expanding list of SNX27-interacting proteins, which includes cell adhesion markers, receptors and ion channels, and highlights the complexity of the contribution of SNX27 to regulating trafficking of proteins to multiple sites of function within the cell.
Supplementary Material
Figure S1 Additional images for Figure 4 of the main text of mpkCCD cells transiently transfected with the indicated GFP and mCherry constructs
The cells were grown to confluence and subjected to the calcium-switch procedure for the indicated times. Cells were fixed and visualized with confocal microscopy.
Figure S2 SNX27 knockdown does not alter ZO-2 localization during calcium switch
Control or SNX27-knockdown cells were subjected to calcium switch and fixed at specified time points. Cells were stained using a polyclonal antibody against ZO-2 and visualized by confocal microscopy.
Figure S3 YFP–ZO-2 expression levels does not significantly alter the mobile fraction
(A) Average fluorescence intensity of YFP–ZO-2 compared with the mobile fraction (linear regression analysis, R = 0.53 and slope = 0.0048). (B) Average initial YFP–ZO-2 fluorescence intensity of control and SNX27-knockdown mpkCCD cells analysed by FRAP (error bars represent the S.D.).
Figure S4 Pertubation of ZO-2 kinetics in SNX27-knockdown cells is due to hindrance of cytoplasmic/TJ exchange
Control or SNX27-knockdown cell monolayers were transiently transfected with YFP–ZO-2 (A and C). Points of cell–cell contact in both YFP- and RFP-expressing cells were photobleached and the rate of fluorescence recovery of ZO-2 at the TJ for the indicated times and regions of interest (arrows) was monitored by YFP and quantified (B and D). The colour of lines in (B) and (C) represent the co-ordinated region of interest monitored in (A) and (C). Cells expressing shRNA and YFP–ZO-2 were identified through RFP and YFP fluorescence. High magnification images of TJ sections at the indicated times are shown (error bars represent the S.D.).
Acknowledgments
We thank Angel Aponte (NHLBI Proteomics core) for MS expertise; Andrew Udofa for experimental contributions at the initial stages of the project; Dr Mark Knepper for the kind donation of mpkCCD cells; Dr Pat Sokolove, Kendall Masada and Dr Steven Gee for critically reading the paper prior to submission; and Dr Alan Fanning (University of North Carolina, Chapel Hill, NC, U.S.A.) and Dr Christina van Itallie (NIH, Bethesday, MD, U.S.A.) for generous donation of antibodies, YFP-ZO-1 and useful discussions.
FUNDING
This work was supported by the NIH intramural research program.
Abbreviations used
- BB150
binding buffer 150
- FERM
4.1/ezrin/radixin/moesin
- GUK
guanylate kinase
- HA
haemagglutinin
- MAGUK
membrane-associated guanylate kinase
- MDCK
Madin–Darby canine kidney
- mpkCCD
mouse primary kidney cortical collecting duct
- ROI
region of interest
- SH3
Src homology 3
- SNX
sorting nexin
- TER
transepithelial electrical resistance
- TJ
tight junction
- ZO
zonula occludens
Footnotes
AUTHOR CONTRIBUTION
All experiments were designed and performed by Seth Zimmerman, Christina Hueschen and Martin Playford. Daniela Malide performed the FRAP procedures. Seth Zimmerman, Sharon Milgram and Martin Playford wrote the paper. Sharon Milgram managed and advised the project.
References
- 1.Carlton JG, Cullen PJ. Sorting nexins. Curr Biol. 2005;15:R819–R820. doi: 10.1016/j.cub.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 2.Cullen PJ. Endosomal sorting and signalling: an emerging role for sorting nexins. Nat Rev Mol Cell Biol. 2008;9:574–582. doi: 10.1038/nrm2427. [DOI] [PubMed] [Google Scholar]
- 3.van Weering JR, Verkade P, Cullen PJ. SNX-BAR proteins in phosphoinositide-mediated, tubular-based endosomal sorting. Semin Cell Dev Biol. 2010;21:371–380. doi: 10.1016/j.semcdb.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Worby CA, Dixon JE. Sorting out the cellular functions of sorting nexins. Nat Rev Mol Cell Biol. 2002;3:919–931. doi: 10.1038/nrm974. [DOI] [PubMed] [Google Scholar]
- 5.Teasdale RD, Loci D, Houghton F, Karlsson L, Gleeson PA. A large family of endosome-localized proteins related to sorting nexin 1. Biochem J. 2001;358:7–16. doi: 10.1042/0264-6021:3580007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghai R, Mobli M, Norwood SJ, Bugarcic A, Teasdale RD, King GF, Collins BM. Phox homology band 4.1/ezrin/radixin/moesin-like proteins function as molecular scaffolds that interact with cargo receptors and Ras GTPases. Proc Natl Acad Sci USA. 2011;108:7763–7768. doi: 10.1073/pnas.1017110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kajii Y, Muraoka S, Hiraoka S, Fujiyama K, Umino A, Nishikawa T. A developmentally regulated and psychostimulant-inducible novel rat gene mrt1 encoding PDZ-PX proteins isolated in the neocortex. Mol Psychiatry. 2003;8:434–444. doi: 10.1038/sj.mp.4001258. [DOI] [PubMed] [Google Scholar]
- 8.Brone B, Eggermont J. PDZ proteins retain and regulate membrane transporters in polarized epithelial cell membranes. Am J Physiol. 2005;288:C20–C29. doi: 10.1152/ajpcell.00368.2004. [DOI] [PubMed] [Google Scholar]
- 9.Jelen F, Oleksy A, Smietana K, Otlewski J. PDZ domains: common players in the cell signaling. Acta Biochim Pol. 2003;50:985–1017. [PubMed] [Google Scholar]
- 10.Nourry C, Grant SG, Borg JP. PDZ domain proteins: plug and play! Sci STKE. 2003:RE7. doi: 10.1126/stke.2003.179.re7. [DOI] [PubMed] [Google Scholar]
- 11.Lee HJ, Zheng JJ. PDZ domains and their binding partners: structure, specificity, and modification. Cell Commun Signaling. 2010;8:8. doi: 10.1186/1478-811X-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cai L, Loo LS, Atlashkin V, Hanson BJ, Hong W. Deficiency of sorting nexin 27 (SNX27) leads to growth retardation and elevated levels of N-methyl-D-aspartate receptor 2C (NR2C) Mol Cell Biol. 2011;31:1734–1747. doi: 10.1128/MCB.01044-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joubert L, Hanson B, Barthet G, Sebben M, Claeysen S, Hong W, Marin P, Dumuis A, Bockaert J. New sorting nexin (SNX27) and NHERF specifically interact with the 5-HT4a receptor splice variant: roles in receptor targeting. J Cell Sci. 2004;117:5367–5379. doi: 10.1242/jcs.01379. [DOI] [PubMed] [Google Scholar]
- 14.Lauffer BE, Melero C, Temkin P, Lei C, Hong W, Kortemme T, von Zastrow M. SNX27 mediates PDZ-directed sorting from endosomes to the plasma membrane. J Cell Biol. 2010;190:565–574. doi: 10.1083/jcb.201004060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lunn ML, Nassirpour R, Arrabit C, Tan J, McLeod I, Arias CM, Sawchenko PE, Yates JR, 3rd, Slesinger PA. A unique sorting nexin regulates trafficking of potassium channels via a PDZ domain interaction. Nat Neurosci. 2007;10:1249–1259. doi: 10.1038/nn1953. [DOI] [PubMed] [Google Scholar]
- 16.Valdes JL, Tang J, McDermott MI, Kuo JC, Zimmerman SP, Wincovitch SM, Waterman CM, Milgram SL, Playford MP. Sorting nexin 27 protein regulates trafficking of a p21-activated kinase (PAK) interacting exchange factor (beta-Pix)-G protein-coupled receptor kinase interacting protein (GIT) complex via a PDZ domain interaction. J Biol Chem. 2011;286:39403–39416. doi: 10.1074/jbc.M111.260802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harbor Perspect Biol. 2009;1:a002584. doi: 10.1101/cshperspect.a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furuse M. Molecular basis of the core structure of tight junctions. Cold Spring Harbor Perspect Biol. 2010;2:a002907. doi: 10.1101/cshperspect.a002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Umeda K, Ikenouchi J, Katahira-Tayama S, Furuse K, Sasaki H, Nakayama M, Matsui T, Tsukita S, Furuse M. ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell. 2006;126:741–754. doi: 10.1016/j.cell.2006.06.043. [DOI] [PubMed] [Google Scholar]
- 20.Fanning AS, Anderson JM. Zonula occludens-1 and -2 are cytosolic scaffolds that regulate the assembly of cellular junctions. Ann NY Acad Sci. 2009;1165:113–120. doi: 10.1111/j.1749-6632.2009.04440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Itallie CM, Fanning AS, Bridges A, Anderson JM. ZO-1 stabilizes the tight junction solute barrier through coupling to the perijunctional cytoskeleton. Mol Biol Cell. 2009;20:3930–3940. doi: 10.1091/mbc.E09-04-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, Pendergast AM, Bronson R, Aster JC, Scott ML, Baltimore D. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood. 1998;92:3780–3792. [PubMed] [Google Scholar]
- 23.Thelin WR, Kesimer M, Tarran R, Kreda SM, Grubb BR, Sheehan JK, Stutts MJ, Milgram SL. The cystic fibrosis transmembrane conductance regulator is regulated by a direct interaction with the protein phosphatase 2A. J Biol Chem. 2005;280:41512–41520. doi: 10.1074/jbc.M507308200. [DOI] [PubMed] [Google Scholar]
- 24.Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein–protein colocalization in live cells. Biophys J. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wittchen ES, Haskins J, Stevenson BR. Protein interactions at the tight junction. Actin has multiple binding partners, and ZO-1 forms independent complexes with ZO-2 and ZO-3. J Biol Chem. 1999;274:35179–35185. doi: 10.1074/jbc.274.49.35179. [DOI] [PubMed] [Google Scholar]
- 26.MacNeil AJ, Mansour M, Pohajdak B. Sorting nexin 27 interacts with the cytohesin associated scaffolding protein (CASP) in lymphocytes. Biochem Biophys Res Commun. 2007;359:848–853. doi: 10.1016/j.bbrc.2007.05.162. [DOI] [PubMed] [Google Scholar]
- 27.Islas S, Vega J, Ponce L, Gonzalez-Mariscal L. Nuclear localization of the tight junction protein ZO-2 in epithelial cells. Exp Cell Res. 2002;274:138–148. doi: 10.1006/excr.2001.5457. [DOI] [PubMed] [Google Scholar]
- 28.Ivanov AI, Nusrat A, Parkos CA. Endocytosis of epithelial apical junctional proteins by a clathrin-mediated pathway into a unique storage compartment. Mol Biol Cell. 2004;15:176–188. doi: 10.1091/mbc.E03-05-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsuda M, Kubo A, Furuse M, Tsukita S. A peculiar internalization of claudins, tight junction-specific adhesion molecules, during the intercellular movement of epithelial cells. J Cell Sci. 2004;117:1247–1257. doi: 10.1242/jcs.00972. [DOI] [PubMed] [Google Scholar]
- 30.Shen L, Weber CR, Turner JR. The tight junction protein complex undergoes rapid and continuous molecular remodeling at steady state. J Cell Biol. 2008;181:683–695. doi: 10.1083/jcb.200711165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Itallie CM, Aponte A, Tietgens AJ, Gucek M, Fredriksson K, Anderson JM. The N and C termini of ZO-1 are surrounded by distinct proteins and functional protein networks. J Biol Chem. 2013;288:13775–13788. doi: 10.1074/jbc.M113.466193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu D, Turner JR. Stimulus-induced reorganization of tight junction structure: the role of membrane traffic. Biochim Biophys Acta. 2008;1778:709–716. doi: 10.1016/j.bbamem.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marchiando AM, Shen L, Graham WV, Weber CR, Schwarz BT, Austin JR, 2nd, Raleigh DR, Guan Y, Watson AJ, Montrose MH, Turner JR. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol. 2010;189:111–126. doi: 10.1083/jcb.200902153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Metais JY, Navarro C, Santoni MJ, Audebert S, Borg JP. hScrib interacts with ZO-2 at the cell–cell junctions of epithelial cells. FEBS Lett. 2005;579:3725–3730. doi: 10.1016/j.febslet.2005.05.062. [DOI] [PubMed] [Google Scholar]
- 35.Gonzalez-Mariscal L, Betanzos A, Avila-Flores A. MAGUK proteins: structure and role in the tight junction. Semin Cell Dev Biol. 2000;11:315–324. doi: 10.1006/scdb.2000.0178. [DOI] [PubMed] [Google Scholar]
- 36.Gonzalez-Mariscal L, Ponce A, Alarcon L, Jaramillo BE. The tight junction protein ZO-2 has several functional nuclear export signals. Exp Cell Res. 2006;312:3323–3335. doi: 10.1016/j.yexcr.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 37.Jaramillo BE, Ponce A, Moreno J, Betanzos A, Huerta M, Lopez-Bayghen E, Gonzalez-Mariscal L. Characterization of the tight junction protein ZO-2 localized at the nucleus of epithelial cells. Exp Cell Res. 2004;297:247–258. doi: 10.1016/j.yexcr.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 38.Chamorro D, Alarcon L, Ponce A, Tapia R, Gonzalez-Aguilar H, Robles-Flores M, Mejia-Castillo T, Segovia J, Bandala Y, Juaristi E, Gonzalez-Mariscal L. Phosphorylation of zona occludens-2 by protein kinase C epsilon regulates its nuclear exportation. Mol Biol Cell. 2009;20:4120–4129. doi: 10.1091/mbc.E08-11-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raleigh DR, Boe DM, Yu D, Weber CR, Marchiando AM, Bradford EM, Wang Y, Wu L, Schneeberger EE, Shen L, Turner JR. Occludin S408 phosphorylation regulates tight junction protein interactions and barrier function. J Cell Biol. 2011;193:565–582. doi: 10.1083/jcb.201010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen L, Weber CR, Raleigh DR, Yu D, Turner JR. Tight junction pore and leak pathways: a dynamic duo. Annu Rev Physiol. 2011;73:283–309. doi: 10.1146/annurev-physiol-012110-142150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Additional images for Figure 4 of the main text of mpkCCD cells transiently transfected with the indicated GFP and mCherry constructs
The cells were grown to confluence and subjected to the calcium-switch procedure for the indicated times. Cells were fixed and visualized with confocal microscopy.
Figure S2 SNX27 knockdown does not alter ZO-2 localization during calcium switch
Control or SNX27-knockdown cells were subjected to calcium switch and fixed at specified time points. Cells were stained using a polyclonal antibody against ZO-2 and visualized by confocal microscopy.
Figure S3 YFP–ZO-2 expression levels does not significantly alter the mobile fraction
(A) Average fluorescence intensity of YFP–ZO-2 compared with the mobile fraction (linear regression analysis, R = 0.53 and slope = 0.0048). (B) Average initial YFP–ZO-2 fluorescence intensity of control and SNX27-knockdown mpkCCD cells analysed by FRAP (error bars represent the S.D.).
Figure S4 Pertubation of ZO-2 kinetics in SNX27-knockdown cells is due to hindrance of cytoplasmic/TJ exchange
Control or SNX27-knockdown cell monolayers were transiently transfected with YFP–ZO-2 (A and C). Points of cell–cell contact in both YFP- and RFP-expressing cells were photobleached and the rate of fluorescence recovery of ZO-2 at the TJ for the indicated times and regions of interest (arrows) was monitored by YFP and quantified (B and D). The colour of lines in (B) and (C) represent the co-ordinated region of interest monitored in (A) and (C). Cells expressing shRNA and YFP–ZO-2 were identified through RFP and YFP fluorescence. High magnification images of TJ sections at the indicated times are shown (error bars represent the S.D.).