

Abstract
Cytochrome P450 1B1 (CYP1B1) is highly expressed in human and murine ocular tissues during development. Mutations in this gene are implicated in the development of primary congenital glaucoma (PCG) in humans. Mice deficient in Cyp1b1 (Cyp1b1−/−) present developmental abnormalities similar to human primary congenital glaucoma. The present work describes the ultrastructural morphology of the iridocorneal angle of 42 eyes from 1 week to 8-month-old Cyp1b1−/− mice. Morphometric and semiquantitative analysis of the data revealed that 3-week-old Cyp1b1−/− mice present significantly (p = 0.002) decreased amount of trabecular meshwork (TM) collagen and higher TM endothelial cell and collagen lesion scores (p = 0.002) than age-matched controls. Collagen loss and lesion scores were progressively increased in older animals with 8-month old animals presenting severe atrophy of the TM. Our findings advance the understanding of the effects of CYP1B1 mutations in the TM development and primary congenital glaucoma, and suggest a link between TM morphologic alterations and increased intraocular pressure.
Keywords: Congenital Glaucoma, CYP1B1, Collagen Fibrils, Electron Microscopy, Eye, Mouse, Oxidative Stress, Trabecular Meshwork
Glaucoma is a leading cause of vision loss in the adult human population. It represents a large, diverse group of intraocular pressure (IOP)-dependent neurodegenerative disorders, that result in loss of normal function and integrity of the retinal ganglion cells and their axons in the optic nerve, ultimately causing vision loss.1 Increased IOP is considered a major risk factor for development of glaucoma and, thus, has been the main focus of treatment. Defects in the aqueous humor drainage are the main cause of elevated IOP.2 The aqueous humor outflow system is dependent primarily on the trabecular meshwork (TM), a sponge-like, three-dimensionally arranged tissue composed of connective tissue beams lined by endothelial-like cells (trabecular cells), that is circumferentially present and expended at the base of the iris and peripheral cornea (iridocorneal angle). The TM is responsible for draining the aqueous humor from the anterior chamber into the venous system via Schlemm’s canal and collector channels, maintaining homeostasis of the IOP.3
Although less prevalent than the adult forms, primary congenital glaucoma (PCG) causes a devastating loss of vision in children. The pathogenesis of the disease is associated with developmental defects of the TM, but the underlying mechanisms are not well understood.3 Genetic linkage analyses and mutation studies have identified the Cytochrome P450 family 1 subfamily B polypeptide 1 (CYP1B1) as a causative gene in PCG.4,5,6,7 CYP1B1 and other genes of the CYP450 family catalyze NADPH supported monooxygenation of diverse xenobiotics and endogenous molecules.8,9 Strong expression of CYP1B1 in the fetal human and murine eye indicates that CYP1B1 plays an important role in the ocular development and function.10,11,12 Recently our group demonstrated that Cyp1b1 deficiency significantly impaired trabecular cell function and oxidative homeostasis of the TM tissue in mice. Cyp1b1−/− mice presented increased IOP and microscopic abnormalities of the iridocorneal angle.13 Here we determined the detailed ultrastructural morphology of the postnatal iridocorneal angle of Cyp1b1−/− mice and demonstrate progressive abnormalities in the extracellular matrix of the trabecular meshwork.
Material and Methods
Animals
All experiments were carried out in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the Institutional Animal Care and Use Committee of the University of Wisconsin School of Medicine and Public Health. The generation and screening of Cyp1b1−/− mice on a C57BL/6 background were previously described.13 A total of 42 eyes, 21 from wild type and 21 from Cyp1b1−/− mice were collected at 1, 2, 3 and 6 weeks and 3, 7 and 8 months of age. Enucleated globes were immersion-fixed in 2% paraformaldehyde (PFA) and 2.5% glutaraldehyde in 0.1 M phosphate-buffered saline (PBS; pH 7.4) at 4°C overnight. Four areas of the trabecular meshwork were sampled representing the dorsal, ventral, temporal and nasal regions of the globe. Samples were processed for routine transmission electron microscopy. Briefly, fixed globes were treated with 1% OsO4 in PBS for 2 hours at room temperature followed by three 10-minute washes with 0.1 M sodium acetate buffer. Tissues were then stained with 2% uranyl acetate in sodium acetate buffer for 1 hour at room temperature, washed in buffer, dehydrated in a graded ethanol series (40%–100%), and infiltrated with propylene oxide-812 resin (1005 Embed 812; EMS, Fort Washington, PA). The samples were embedded with fresh 100% 812 resin in molds and polymerized in a 60°C oven for 36 hours. One-micron sections were stained with toluidine blue and examined under the light microscope. Ultrathin sections (90 nm) were analyzed using a JEOL 100CX electron microscope (JEOL Ltd., Tokyo, Japan).
Morphometric analysis
In order to quantify the relative amount of collagen in the trabecular meshwork five nonconsecutive x15,000 TEM images of the anterior, mid and posterior TM were processed and analyzed using ImageJ software (National Institutes of Health, Bethesda, MD http://fiji.sc/Fiji). Briefly, the image’s scale bar was used to set up the system’s spatial calibration. A background subtraction was performed on the images (Process>Subtract Background option) follow by segmentation of the collagen fibers (Image>Adjust>Threshold tool option). Using the threshold tool the collagen fibers were manually selected and the amount of collagen in the image was measured (Analyze>Measure tool) and expressed as a % of the image. In order to standardize the data we avoided imaging the intra-trabecular spaces, focusing predominantly on the trabecular beams.
Semiquantitative analysis
Alterations in the ultrastructural morphology of trabecular cells and distribution of the collagen lesions in the TM were also accessed using a semiquantitative scoring system. TM collagen lesion distribution score ranged from 0 to 4 where 0 indicates no lesion; Score 1 indicates collagen fiber disarrangement affecting < 25% of the TM; Score 2, 25–50% of the TM, Score 3, 50–75% of the TM, and 4 > 75% of the TM (Table 1). Trabecular cell morphology was analyzed by accessing the presence of the following ultrastructural lesions (criteria): Irregular cell surface, loss of contact with basement membrane, cytoplasmic vacuolization, cell swelling and presence of irregular cytoplasmic material. Samples were scored from 0 to 4 where 0 indicates no lesion, 1 indicates 1 criterion present, 2 indicates 2 criteria, 3 indicates 3–4 criteria and 4 indicates all 5 criteria were present (Table 2).
Table 1.
Scoring system for the distribution of the collagen lesions in the trabecular meshwork of Cyp1b1−/− mice
| Score | % of TM collagen affected |
|---|---|
| 0 | No lesions |
| 1 | < 25 |
| 2 | 25–50 |
| 3 | 50–75 |
| 4 | >75 |
Table 2.
Scoring system for semi-quantitative analysis of the trabecular cell ultrastructural morphology of Cyp1b1−/− mice
| Ultrastructural Criteria |
|---|
| Irregular cell surface |
| Loss of contact with basement membrane |
| Cytoplasmic vacuolization |
| Cell swelling |
| Irregular cytoplasmic material |
| Score | |
|---|---|
| 0 | No lesions |
| 1 | 1 criterion |
| 2 | 2 criteria present |
| 3 | 3–4 criteria present |
| 4 | All 5 criteria present |
Statistical analysis
Statistical analyses were performed using the two-tailed unpaired Student’s t-test for collagen morphometric data and Wilcoxon–Mann–Whitney two-sample rank-sum test for the semiquantitative analyses. Collagen quantification data is reported as mean ± SEM and semiquantitative analysis data is reported as median ± range. Differences were regarded as significant when p<0.01.
Results
Histologic analysis of the iridocorneal angles of Cyp1b1−/− animals showed that all the animals exhibited an open ciliary cleft and no abnormalities on the Schlemm’s canal or aqueous veins (Fig. 1). Ultrastructural lesions were noted in the extracellular matrix of the trabecular meshwork. One and two-week-old Cyp1b1−/− animals presented a relatively normal trabecular meshwork, characterized by multiple trabecular beams composed of a core of collagen fibers interspersed with small amounts of elastic fibers covered by trabecular cells (Figs 2 and 3). Starting at 3 weeks of age Cyp1b1−/− animals presented marked disruption of the anterior and posterior trabecular meshwork characterized by multifocal atrophy of the trabecular beams with accentuation of the inter-trabecular spaces. The collagen fibers on the collagenous layer of the trabecular beams were markedly fragmented and irregularly distributed when compared to control groups. The TM cells covering the trabecular beams exhibited irregular cytoplasmic processes, loss of contact with basement membranes, variable cell swelling with cytoplasmic vacuolization and intracytoplasmic accumulation of organelle debris (Figs. 4 and 5). 6 week, 3, 7 and 8 month-old Cyp1b1−/− animals presented progressively more severe disarrangement of trabecular collagen and consequently more dramatic overall architectural changes of the TM (Figs. 6–9). At the latest time point, 8-month-old Cyp1b1−/− animals presented a complete collapsed of the TM with formation of one atrophic and irregular trabecular beam spanning the anterior and posterior TM (Figs. 10–12).
Figure 1.

Iridocorneal angle; 2 week-old wild-type mice. Normal morphology of the iridocorneal angle (ICA). C= cornea, Cb= ciliary body, aTM=anterior trabecular meshwork, pTM= posterior trabecular meshwork, *= Schlemm’s canal. Transmission electron microscopy, uranyl acetate. 710x.
Figure 2.
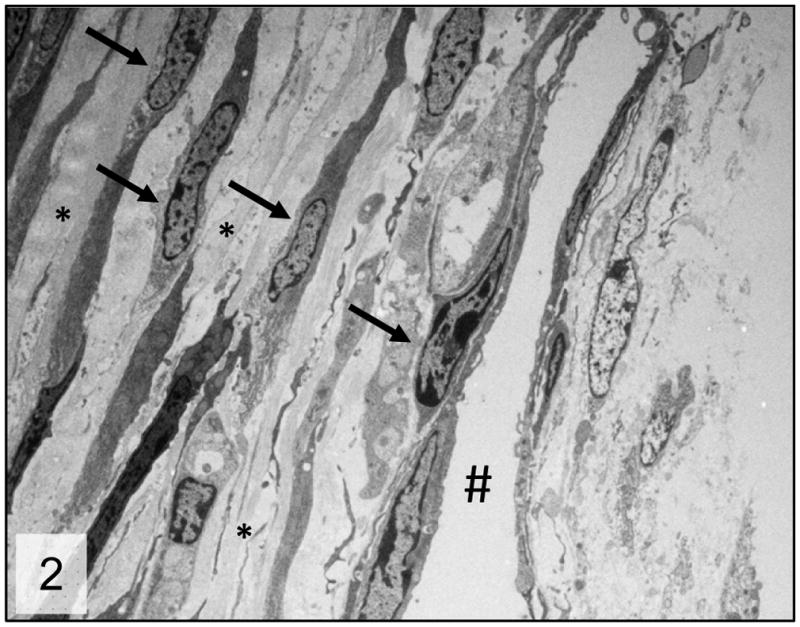
Trabecular meshwork; 2 week-old wild-type mice. Normal trabecular meshwork arranged in multiple trabecular beams composed of a core of collagen fibers (*) surrounded by trabecular cells (arrows). # = Schlemm’s canal. Transmission electron microscopy, uranyl acetate. 2650x.
Figure 3.
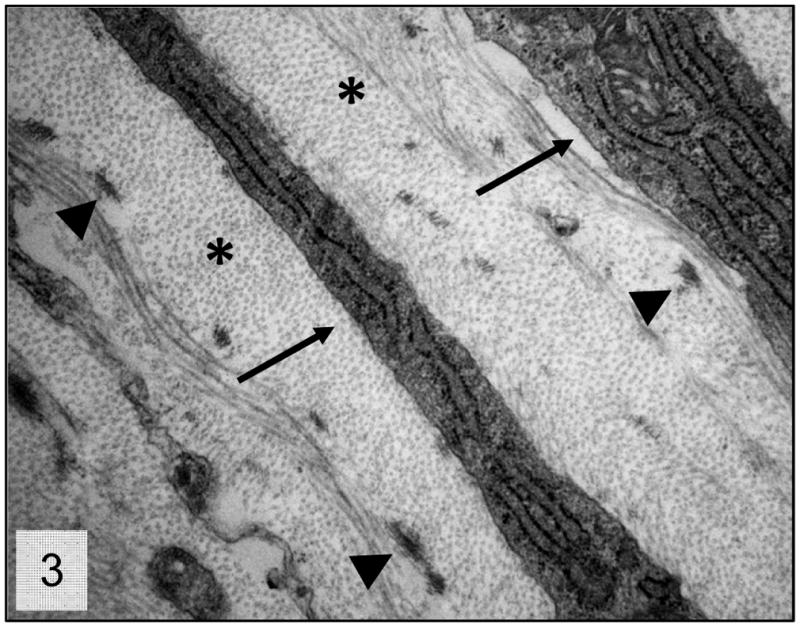
Trabecular meshwork; 2 week-old wild-type mice. Higher magnification presenting the normal relationship between the trabecular beam’s components. Collagen fibers (*), trabecular cells cytoplasmic projections (arrows) and elastic fiber aggregates (arrow heads). Transmission electron microscopy, uranyl acetate. 8800x.
Figures 4, 6 and 8.
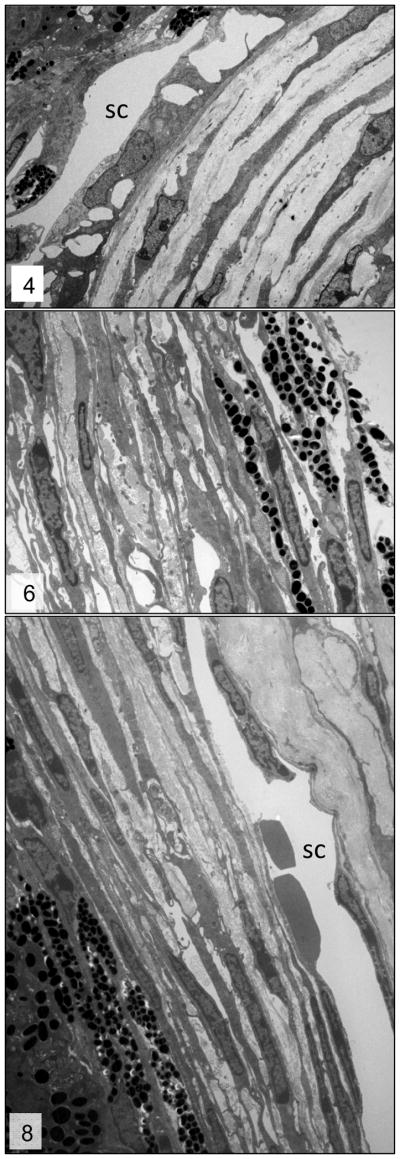
Trabecular meshwork; 3–week-, 6-week- and 3-month-old wild-type mice, respectively. Normal trabecular meshwork. Please note the relationship between the collagenous core (*) of the trabecular beams and trabecular cells (arrows). sc = Schlemm’s canal. Transmission electron microscopy, uranyl acetate. 2650x.
Figures 5, 7 and 9.
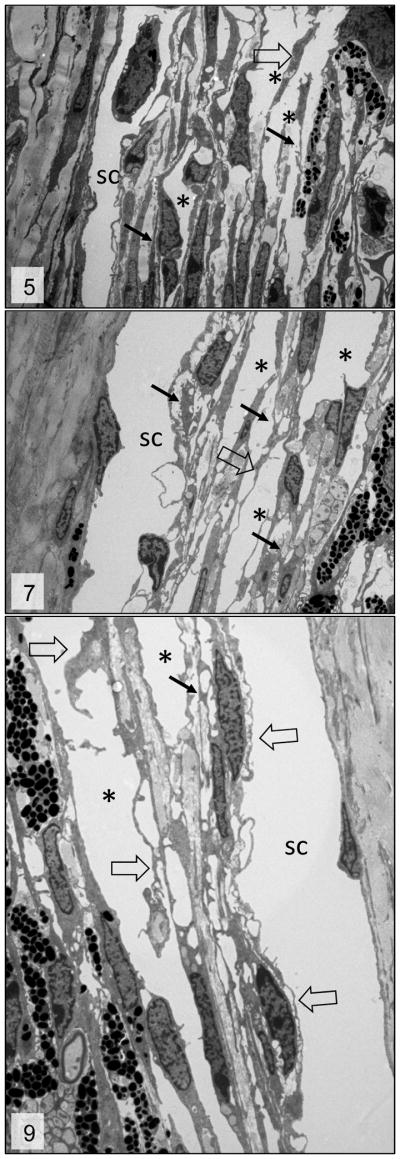
Trabecular meshwork; 3 week, 6week and 3 month-old Cyp1b1−/− mice, respectively. Irregular trabecular beams with accentuation of the inter-trabecular spaces (*), fragmentation of the collagen fibers (arrows) and irregular and reactive trabecular cells (open arrows). sc = Schlemm’s canal. Transmission electron microscopy, uranyl acetate. 2650x.
Figure 10.

Iridocorneal angle; 8-month-old Cyp1b1−/− mice. Complete collapsed of the trabecular meshwork (TM) with formation of one atrophic and irregular trabecular beam spanning the anterior and posterior TM. ICA= Iridocorneal angle, C= cornea, Cb= ciliary body, ir= iris, *= Schlemm’s canal. Transmission electron microscopy, uranyl acetate. 710x.
Figure 12.
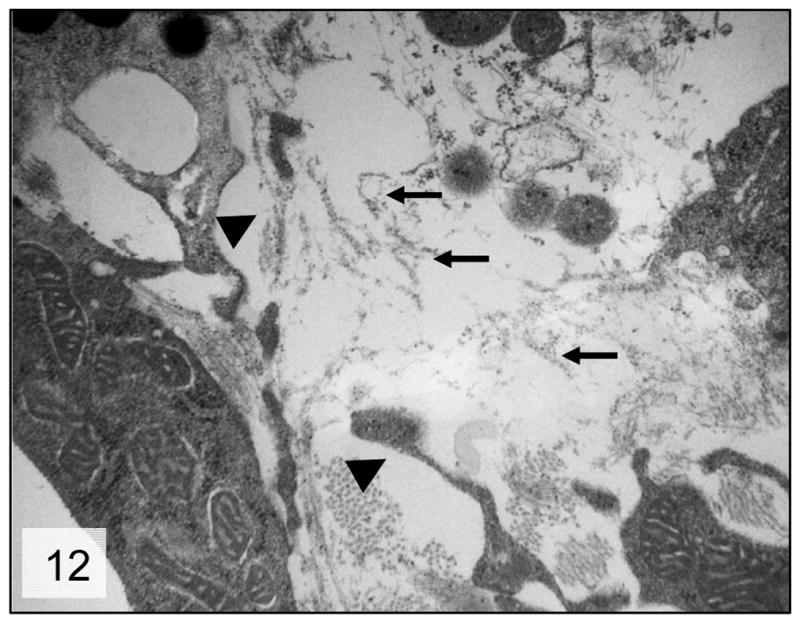
Trabecular meshwork; 8-month-old Cyp1b1−/− mice. Higher magnification of the trabecular beam presenting marked degeneration of the collagen fibers (arrows) and irregular trabecular cell projections (arrow heads); Transmission electron microscopy, uranyl acetate. 8800x.
The morphometric quantification of the relative amount of collagen occupying the trabecular meshwork is represented in Figure 13. Starting at 3 weeks of age Cyp1b1−/− mice presented significantly (p < 0.002) less collagen in the trabecular beams (11.1% ± 3.8) than age-matched wild type (WT) controls (35.2% ± 6.7) as well as younger 2-week-old WT (44.9% ± 4.3) and Cyp1b1−/− (39% ± 8.4) mice. The relative amount of TM collagen in affected animals progressively decreased with age and Cyp1b1−/− mice were had significantly less TM collagen than age-match WT animals from six weeks of age through the course of the study (Fig. 13). The semiquantitative analysis of the trabecular cell and TM collagen lesions presented a similar spatial distribution pattern between the ages. Trabecular cell and collagen median scores of Cyp1b1−/− were significantly (p=0.002) more severe than respective controls in all ages (Figs. 14 and 15). There was a statistically significant increase in the median collagen score between 2-week (1 ± 1) and 3-week-old (3 ± 1) Cyp1b1−/− animals and 6-week (2.5 ± 0.5) and 3-month-old (4 ± 1) Cyp1b1−/− animals and a statistically significant increase in median trabecular cell score between 2 (1 ± 1) and 3-week-old (3 ± 1) Cyp1b1−/− animals.
Figure 13.

Morphometric quantification of the relative amounts of collagen in the trabecular meshwork of Cyp1b1−/− mice. 3-week-old Cyp1b1−/− mice presented significantly less collagen in the trabecular meshwork than wild-type, and younger control and mutant mice. TM collagen loss progressed with age. Means with different letters are significantly different (n=42 [3/group/age], Mean ± SD, Student’s t-test, p = 0.0004). w= week, m= month, WT = wild type.
Figure 14.

Semiquantitative scoring analysis of the distribution of the collagen lesions in the trabecular meshwork of Cyp1b1−/− mice. Cyp1b1−/− mice presented significantly higher median scores than wild-type mice in all ages. There was a significant increase in median score between 2- and 3-week-old and 6-week and 3-month-old Cyp1b1−/− mice. (n=42 [3/group/age], Wilcoxon–Mann–Whitney, Median ± range, *differences between Cyp1b1−/− and wild-type, p=0.002; ** differences between Cyp1b1−/− mice, p=0.005). w= week, m= month, WT = wild-type.
Figure 15.

Semi-quantitative scoring analysis of the trabecular cell ultrastructural morphology of Cyp1b1−/− mice. Cyp1b1−/− mice presented significantly higher median scores than wild-type mice in all ages (*, p=0.002). There was a significant increase in median score between 2- and 3-week-old Cyp1b1−/− animals (p=0.005). (n=42 [3/group/age], Wilcoxon–Mann–Whitney, Median ± range,). w= week, m= month, WT = wild-type.
Discussion
The Cyp1b1−/− mice presented progressive ultrastructural abnormalities in the trabecular meshwork starting at 3-weeks of age that ultimately led to architectural collapse of the tissue. These developmental abnormalities resemble those reported in human patients with primary congenital glaucoma14,15.
Libby et al. described severe iridocorneal angle abnormalities in albino Cyp1b1−/− mice characterized by absence of Schlemm’s canal and trabecular meshwork and areas of anterior synechia of the iris16. The same authors reported that the lesions on Cyp1b1−/− pigmented (129X1/SvJ X B6) mice were less severe, with animals at 13 months of age presenting an open ciliary cleft, attenuated endothelium of the Schlemm’s canal and reduction of the TM to a singular trabecular beam. Our data presents similar findings on even younger animals. The presence of severe atrophy and collapse of the TM into a single trabecular beam was observed in 8-month old animals (Figs. 10 and 11), and based on the similar appearance of the lesion at 13 months of age, it appears to represent a final stage of TM atrophy.
Figure 11.
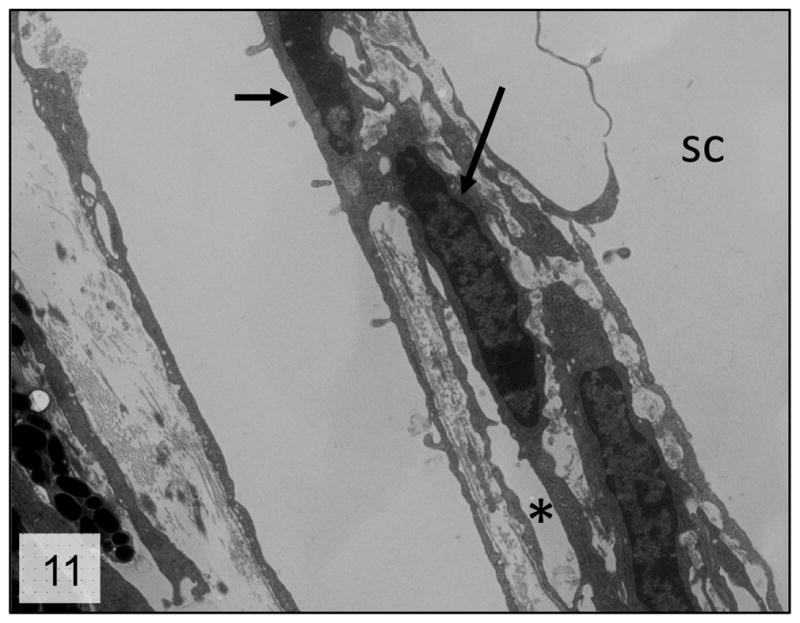
Trabecular meshwork; 8-month-old Cyp1b1−/− mice. Higher magnification of the collapsed trabecular tissue presenting irregular trabecular cells (arrows) in close proximity with scant extracellular matrix (*). sc = Schlemm’s canal; Transmission electron microscopy, uranyl acetate. 2650x.
The quantitative assessment of the extracellular matrix and trabecular endothelial cells morphology demonstrated that the loss and disruption of the collagen fibers and degeneration of TM cells were increased with time. The severity and temporal distribution of the lesions also correlated with the postnatal development period of the TM in mice. At birth the mouse TM still presents a primitive appearance, only reaching full maturity at P35 to P42. The major developmental changes on the TM occur by P18, with subsequent maturation primarily involving final enlargement of spaces in the posterior TM17. Our data showed that the major losses in TM collagen and most significant increases in severity of the TM endothelial cells and collagen morphology score occurred between 2 and 3 weeks of age, a period roughly analogous to the final stages of TM development in the mouse (P10 to P18).
These finding along with the near normal morphology of the trabecular meshwork of 1 and 2-week-old Cyp1b1−/− mice suggest that the lesions observed are more likely a failure of ECM remodeling than a failure of ECM deposition in the TM. The molecular mechanisms responsible for normal or abnormal development of the iridocorneal angle are not well defined17. The embryological development of the iridocorneal angle starts with cells of the periocular mesenchyme migrating into the developing eye to form the primitive TM18. As TM development proceeds the tissue differentiates, organizes and develops channels to produce the mature meshwork with its trabecular beams separated by intertrabecular spaces through which the aqueous percolates19. Since there were no morphological differences between 1 and 2 week-old Cyp1b1−/− mice and controls, a failure of cell migration during embryogenesis as a cause of the lesions is unlikely.
The trabecular meshwork forms most of the resistance to aqueous humor outflow needed for maintenance of a pressure gradient between intraocular and venous pressure.20 The aqueous outflow facility through the drainage structures of the iridocorneal angle is dependent, among others things, on the normal spatial arrangement of the TM tissue. On this regard the ECM of the trabecular beams is of special importance, providing tensile strength and tissue flexibility to the TM.21,22 The maintenance of these mechanical properties relies on the composition of the trabecular ECM where type I and III collagen fibers provide structural support in tension, and elastin fibers enable a mechanism for reversible deformation in response to cyclic changes in IOP.23 The ECM changes present on the TM of Cyp1b1−/− mice reported here can alter the mechanical properties of the tissue and affect its ability to respond to injury. The disruption in collagen and elastic fibers can decrease tissue elasticity, alter the shape and cytoskeleton organization of TM endothelial cells due to lack of support and affect the proper expansion of the trabecular spaces leading to compromised aqueous humor outflow and increase in IOP. These findings complement previous data published by our group demonstrating increase oxidative stress and trabecular meshwork endothelial cell abnormalities in Cyp1b1−/− mice. Cyp1b1 modulates the expression of periostin, an extracellular matrix protein essential to collagen fibrillogenesis, by suppression of oxidative stress.13 As evidenced by our results the suppression of Cyp1b1 activity, and consequentially periostin expression, cause marked ultrastructural abnormalities on the ECM of the TM. Since the architectural organization of the TM is essential to maintain normal aqueous outflow, we believe these abnormalities are responsible for the increase in IOP present in these animals.
In summary, our results demonstrated progressive ultrastructural ECM abnormalities on the TM of Cyp1b1−/− mice that are likely associated with impairment of aqueous outflow and initiation of glaucoma. CYP1B1 mutations are associated with multiple forms of human glaucoma6. The Cyp1b1−/− mice present morphologic lesions similar to human cases of primary congenital glaucoma and can be a valuable tool in understanding the mechanisms by which CYP1B1 mutations affect the TM function and development, leading to possible preventive approaches for glaucoma patients with such mutation.
Acknowledgments
This work was supported by grants EY018179, EY016995, P30-EY016665, and P30 CA014520 UW Paul P. Carbone Cancer Center Support Grant from the National Institutes of Health and an unrestricted departmental award from Research to Prevent Blindness. NS is a recipient of a Research Award from American Diabetes Association, 1-10-BS-160. NS and CMS are supported by Retina Research Foundation.
References
- 1.Teixeira LBC, Dubielzig RR. The eye. In: Haschek WM, Rousseaux CG, Walling MA, editors. Haschek and Rousseaux’s Handbook of Toxicologic Pathology. 3. London, UK: Academic press; 2013. pp. 2095–2185. [Google Scholar]
- 2.Weinreb RN, Khaw PT. Primary open-angle glaucoma. Lancet. 2004;363:1711–1720. doi: 10.1016/S0140-6736(04)16257-0. [DOI] [PubMed] [Google Scholar]
- 3.Tamm ER. The trabecular meshwork outflow pathways: structural and functional aspects. Exp Eye Res. 2009;88:648–655. doi: 10.1016/j.exer.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 4.Panicker SG, Reddy AB, Mandal AK, et al. Identification of novel mutations causing familial primary congenital glaucoma in Indian pedigrees. Invest Ophthalmol Vis Sci. 2002;43:1358–1366. [PubMed] [Google Scholar]
- 5.Soley GC, Bosse KA, Flikier D, et al. Primary congenital glaucoma: a novel single-nucleotide deletion and varying phenotypic expression for the 1,546-1,555dup mutation in the GLC3A (CYP1B1) gene in 2 families of different ethnic origin. J Glaucoma. 2003;12:27–30. doi: 10.1097/00061198-200302000-00005. [DOI] [PubMed] [Google Scholar]
- 6.Vincent AL, Billingsley G, Buys Y, et al. Digenic inheritance of early-onset glaucoma: CYP1B1, a potential modifier gene. Am J Hum Genet. 2002;70:448–460. doi: 10.1086/338709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stoilov I, Akarsu AN, Sarfarazi M. Identification of three different truncating mutations in cytochrome P4501B1 (CYP1B1) as the principal cause of primary congenital glaucoma (buphthalmos) in families linked to the GLC3A locus on chromosome 2p21. Hum Mol Genet. 1997;6:641–647. doi: 10.1093/hmg/6.4.641. [DOI] [PubMed] [Google Scholar]
- 8.Savas U, Bhattacharyya KK, Christou M, et al. Mouse cytochrome P-450EF, representative of a new 1B subfamily of cytochrome P-450s. Cloning, sequence determination, and tissue expression. J Biol Chem. 1994;269:14905–14911. [PubMed] [Google Scholar]
- 9.Sutter TR, Tang YM, Hayes CL, et al. Complete cDNA sequence of a human dioxininducible mRNA identifies a new gene subfamily of cytochrome P450 that maps to chromosome 2. J Biol Chem. 1994;269:13092–13099. [PubMed] [Google Scholar]
- 10.Choudhary D, Jansson I, Rezaul K, et al. Cyp1b1 protein in the mouse eye during development: an immunohistochemical study. Drug Metab Dispos. 2007;35:987–994. doi: 10.1124/dmd.106.014282. [DOI] [PubMed] [Google Scholar]
- 11.Hakkola J, Pasanen M, Pelkonen O, et al. Expression of CYP1B1 in human adult and fetal tissues and differential inducibility of CYP1B1 and CYP1A1 by Ah receptor ligands in human placenta and cultured cells. Carcinogenesis. 1997;18:391–397. doi: 10.1093/carcin/18.2.391. [DOI] [PubMed] [Google Scholar]
- 12.Stoilov I, Rezaie T, Jansson I, et al. Expression of cytochrome P4501b1 (Cyp1b1) during early murine development. Mol Vis. 2004;10:629–636. [PubMed] [Google Scholar]
- 13.Zhao Y, Wang S, Sorensen CM, et al. Cyp1b1 Mediates periostin regulation of trabecular meshwork development by suppression of oxidative stress. Mol cell Bio. 2013;33:4225–4240. doi: 10.1128/MCB.00856-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allen L, Burian HM, Braley AE. The anterior border ring of Schwalbe and the pectinate ligament; embryologic and comparative anatomic studies. AMA Arch Ophthalmol. 1955;53:799–806. doi: 10.1001/archopht.1955.00930010807003. [DOI] [PubMed] [Google Scholar]
- 15.Maumene AE. The Pathogenesis of Congenital Glaucoma: A New Theory. Trans Am Ophthalmol Soc. 1958;56:507–570. [PMC free article] [PubMed] [Google Scholar]
- 16.Libby RT, Smith RS, Savinova OV, et al. Modification of ocular defects in mouse developmental glaucoma models by tyrosinase. Science. 2003;299:1578–81. doi: 10.1126/science.1080095. [DOI] [PubMed] [Google Scholar]
- 17.Smith RS, Zabaleta A, Savinova OV, et al. The mouse anterior chamber angle and trabecular meshwork develop without cell death. BMC Dev Biol. 2001;1:3. doi: 10.1186/1471-213X-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mann IC. The Development of the Human Eye. 1. Cambridge, UK: Cambridge University Press; 1928. [Google Scholar]
- 19.Gong H, Tripathi RC, Tripathi BJ. Morphology of the aqueous outflow pathway. Microsc Res Tech. 1996;33:336–367. doi: 10.1002/(SICI)1097-0029(19960301)33:4<336::AID-JEMT4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 20.Lütjen-Drecoll E. Functional morphology of the trabecular meshwork in primate eyes. Prog Retin Eye Res. 1999;18:91–119. doi: 10.1016/s1350-9462(98)00011-1. [DOI] [PubMed] [Google Scholar]
- 21.Borras T, Comes N. Evidence for a calcification process in the trabecular meshwork. Exp Eye Res. 2009;88:738–746. doi: 10.1016/j.exer.2008.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gabelt BT, Kaufman PL. Changes in aqueous humor dynamics with age and glaucoma. Prog Retin Eye Res. 2005;24:612–637. doi: 10.1016/j.preteyeres.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 23.Hernandez MR, Gong H. Extracellular matrix of trabecular meshwork and optic nerve head. In: Ritch R, Shields MB, Krupin T, editors. The Glaucomas. St. Louis, MO: Mosby; 1996. pp. 213–249. [Google Scholar]