

Abstract
Somatic mutations or deletions of TP53 and PTEN in ductal carcinoma in situ (DCIS) lesions have been implicated in progression to invasive ductal carcinomas. A recent molecular and mutational analysis of breast cancers revealed that inactivation of tumor suppressors, p53 and PTEN are strongly associated with triple negative breast cancer. In addition, these tumor suppressors play important roles in regulating self-renewal in normal and malignant stem cells. To investigate their role in breast carcinogenesis, we knocked down these genes in human mammary cells and in non-transformed MCF10A cells. p53 and PTEN knockdown synergized to activate pro-inflammatory IL6/Stat3/NF-κB signaling. This resulted in generation of highly metastatic EMT-like cancer stem cells (CSCs) resulting in tumors whose gene expression profile mimicked that found in basal/claudin-low molecular subtype within the triple negative breast tumors. Constitutive activation of this loop in transformed cells was dependent on proteolytic degradation of SOCS3 resulting in low levels of this protein in basal/claudin low cell lines and primary tumors. In non-transformed cells, transient activation of the IL6 inflammatory loop induced SOCS3 expression leading to pathway inactivation. In transformed cells, enforced expression of SOCS3 or interfering with IL6 pathway via IL6R blockade inhibited tumor growth and metastasis in mouse xenograft models. Furthermore, circulating tumor cells were significantly reduced in tumor bearing animals when treated with anti-IL6R antibodies. These studies uncover important connections between inflammation and carcinogenesis and suggest that blocking pro-inflammatory cytokines may be utilized as an attractive strategy to target triple negative breast tumors which currently lacks molecularly targeted therapies.
Introduction
The tumor suppressor genes, PTEN and TP53, are frequently inactivated in human malignancies including breast cancers (1, 2). A recent comprehensive molecular analyses demonstrated that 80% of basal breast cancers (Triple Negative breast cancer-TNBC) harbor p53 mutation (3). p53 also regulates asymmetric self-renewal and thus its inactivation expands stem cell population by increasing the proportion of symmetric self-renewal (4).
An intrinsic claudin-low subtype within the TNBC was recently identified based on low/absent expression of luminal differentiation markers, enrichment of epithelial to mesenchymal transition (EMT) and stem cell markers (5). IL6, a potent pleiotropic cytokine, is induced in response to acute and chronic inflammation. Suppressor of cytokine signaling 3 (SOCS3) is a critical negative regulator of IL6-mediated pathways (6).
We investigated the role of SOCS3 in p53 and PTEN mediated activation of inflammatory feedback loop and malignant transformation. Constitutive activation of inflammatory loop in transformed cells but not in their untransformed counterparts depends on proteolytic degradation of SOCS3. Using primary breast cancer samples, we provided evidence that loss of SOCS3 expression is associated with basal breast carcinomas. These studies reveal important differences in the regulation of inflammatory cascades in benign tissues and malignant tumors. Furthermore, they identify therapeutic strategies to target CSCs in TNBC by blocking these inflammatory loops.
Results
Knockdown of p53 and PTEN in mammary epithelial cells results in activation of an inflammatory pathway and expands stem cell population
In order to investigate the molecular changes induced by inactivation of tumor suppressors in breast cancer, we down regulated p53 (p53−) and PTEN (PTEN−) individually or in combination (p53−PTEN−) in normal human mammary epithelial cells (HNMEC) and immortalized MCF10A cells (Figures 1a and Supplementary 1a). Simultaneous knock down of p53 and PTEN in MCF10A cells results in activation of Akt/β-catenin and Stat3/NF-κB pathways as well as increased expression of Vimentin and decreased expression of E-cadherin (Figure 1a). To determine the functional relevance of pathway activation, we utilized the non-adherent sphere forming assay to assess mammary stem cell activity in vitro (7). p53 and PTEN knockdown in HNMECs and MCF10A cell line increased the sphere formation 2–10 fold compared to control or single gene deleted cells (Figure 1b and c). Utilizing the CSC markers, CD44+CD24− (8) as well as CD49f+EpCAM− which represents the mesenchymal CSC phenotype (9), we demonstrated that MCF10Ap53−PTEN− cells display a significant increase in the CD44+CD24− cell population compared to parental, p53− or PTEN− cells (Figures 1d and e). Although MCF10Ap53−PTEN− cells contained an increased EpCAM−CD49f+ population, these cells also displayed a distinct EpCAM−CD49f− population not found in parental MCF10A, MCF10A-p53− or MCF10A-PTEN− cells which are predominantly EpCAM+CD49f+ (Figures 1d and f).
Figure 1. p53 and PTEN knockdown in mammary epithelial cells activates inflammatory Stat3/NF-κB pathway expanding stem cell population.

(a) Western blot analyses of MCF10A, p53 (p53−), PTEN (PTEN−) or combined p53 and PTEN knockdown cells (p53−PTEN−) show expressions of p53, PTEN and EMT markers, Vimentin, E-cadherin as well as the activations of Stat3/NF-κB and Akt/Wnt/b-catenin pathways. (b) Sphere forming assay in control HNMEC and MCF10A cells compared to p53−, PTEN− or p53−PTEN− cells; scale bar, 100μm. (c) Number of spheres formed per 10,000 cells plated. (d) Flow cytometry analysis of p53− and PTEN− cells with CD44/CD24 and EpCAM/CD49f markers. (e, f) Quantification of CD44+CD24− cells and EpCAM/CD49f subpopulations were analyzed by flow cytometry. Means ± SD (n=3),*p≤0.05, ** p≤0.005.
Gene expression analysis of MCF10Ap53−PTEN− cells reveals a mesenchymal gene expression profile resembling basal/claudin-low breast cancer
We observed a gradual induction of mesenchymal morphology upon culture of MCF10A-p53−PTEN− cells as assessed by immunohistochemistry or light microscopy characterized by increased nuclear β-catenin staining and loss of E-cadherin expression at cell-cell junctions (Supplementary 1b and c). Molecular characterization of claudin-low breast cancers based on a distinct gene expression signature comprised of 1048 genes (5) revealed that claudin-low breast tumors displayed an EMT like stem cell phenotype characterized by increased expressions of CD44 and CD49f and lacking expressions of CD24 and EpCAM. We utilized the Affymetrix Human Genome HG-U219 Strip Arrays to characterize the gene expression profiles of MCF10A, p53−, PTEN− or p53−PTEN− cells. A high number of genes are differentially expressed between the MCF10A and p53−, PTEN− or p53−PTEN−cells. Interestingly the highest number of differentially expressed genes (560) was observed between the parental MCF10A and MCF10Ap53−PTEN− cells compared to genes (129 and 116) that are differentially expressed between the parental and MCF10Ap53− or MCF10APTEN− cells respectively (Supplementary 2a). These findings confirm an additive effect in altering gene expression by knockdown of p53 and PTEN. Furthermore 903 out of 1048 genes that distinguish the basal/claudin-low subtype were represented in MCF10Ap53−PTEN− cells (Figure 2a). In addition, we evaluated the effect of p53 and/or PTEN knockdown on the expression of EMT and stem cell related genes. Down regulation of p53 and PTEN markedly induced EMT and stem cell related genes compared to parental, p53 or PTEN knockdown cells (Figures 2b and 2c). Moreover, induction of EMT generates cells with stem cell features (10). Consistent with these findings, of EMT related genes (Vimentin, N-Cad and Snail) are upregulated and epithelial genes (EpCAM, E-Cad, Claudin, Occludin and BMPR) are down regulated in MCF10Ap53−PTEN− cells compared to parental cells (Figure 2d). The functional relevance of the EMT and stem cell gene expression profile has been suggested by the increased ability of these EMT like cells to migrate in in vitro assays. Consistent with this, MCF10Ap53−PTEN− cells demonstrate significantly higher motility compared to parental, MCF10Ap53− or MCF10APTEN− cells (Figures 1e and f) consistent with their EMT/CSC phenotype.
Figure 2. Gene expression analysis of MCF10A p53−PTEN− cells reveal a mesenchymal-like claudin-low breast cancer subtype.

(a) Heatmap and unsupervised clustering analysis of MCF10A, p53−, PTEN− or p53−PTEN− cells. (b, c) MCF10Ap53−PTEN− cells display previously described EMT and CSC gene expression signature respectively. (d) Expressions of EMT genes were validated by RT-PCR. (e, f) Boyden Chamber assay shows an increased motility of MCF10Ap53−PTEN− cells compared to MCF10A, p53− or PTEN− cells in vitro. Means ± SD (n=3),*p≤0.05, ** p≤0.005.
p53 and PTEN knockdown transforms MCF10A cells generating aggressive metastatic tumors in NOD/SCID mice
To investigate whether the molecular and phenotypic changes that were observed upon knockdown of p53 and PTEN were sufficient to transform mammary epithelial cells, we transplanted p53 and/or PTEN deleted HNMECs or MCF10A cells into humanized mouse mammary fad pads. Individual PTEN or p53 deleted NMECs characterized by increased stem cells generated atypical hyperplasia or low grade DCIS while control cells generated normal ductal structures (Figure 3a) when these cells were introduced into cleared mouse mammary fat pads. Combined deletion of p53 and PTEN in HNMECs generated a high grade DCIS in humanized mouse fat pads (Figure 3a). These lesions were characterized by complete disorganization of the myoepithelial layer with reduced expression of luminal cytokeratins (CK18) and increased expression of basal markers (CK5/6) as well as enhanced proliferation as assessed by Ki67 staining (Figure 3a).
Figure 3. p53 and PTEN knockdown transforms MCF10A cells generating tumors in NOD/SCID mice and are organized in a cancer stem cell hierarchy displaying EMT phenotype.

(a) IHC analyses of cytokines and proliferation marker demonstrated that the p53 and PTEN knockdown compared to single gene knockdown in HNMECs developed high-grade DCIS lesions when implanted into NOD/SCID mouse mammary fat pads. (b) Deletion of p53 and PTEN in MCF10A cells generated transplantable tumors while single p53 or PTEN deletion failed to do so in NOD/SCID mice. Representative macroscopic pictures of primary and secondary tumors are shown. (c) Flow cytometry sorted EpCAM/CD49f subpopulations (as abbreviated: E+49+, E−49−, E−49+) were evaluated in mouse xenografts. These indicated subpopulations of MCF10A p53−PTEN− cells were implanted into the mammary fat pad of NOD/SCID mice at serial dilutions and tumor growth was evaluated. Scale bars equal 500mm. (d) A gene expression analysis by RT-PCR revealed that EpCAM−CD49f+ and EpCAM−CD49f− subpopulations compared to EpCAM+CD49f+ overexpress EMT-related genes and lack of epithelial genes. Means ± SD (n=10), scale bars equal 100 μm.
In contrast to NMECs where p53 and PTEN knockdown generated high grade DCIS, dual knockdown of these tumor suppressors transformed MCF10A cells as was demonstrated by soft agar colony formation as well as by mouse xenotransplantation assays (Figures 3B, Supplementary 2a and b). In contrast, individual p53 or PTEN deletion failed to transform MCF10A cells (Figures 3b, Supplementary 2a and b). Tumors generated by MCF10Ap53−PTEN− cells were capable of forming tumors upon serial transplantation (Figure 3b). The mesenchymal enrichment of these tumors was confirmed by prominent Vimentin expression, lack of expression of the epithelial marker E-cadherin as well as increased rate of proliferation as assessed by Ki67 expression (Supplementary 2c). Consistent with our in vitro findings, these tumors displayed an aggressive phenotype characterized by spontaneous metastasis to lung (Supplementary 2c).
MCF10Ap53−PTEN− tumors are hierarchically organized and contain tumor-initiating CSCs
To determine whether the MCF10Ap53−PTEN− cells contain a tumor-initiating cell population, we investigated the tumor initiation potential of EpCAM/CD49f subsets in mouse xenografts. Since the majority of MCF10A-p53−PTEN− cells are CD44 positive, this CSC marker is not informative in these cells. However, MCF10A-p53−PTEN− cells contained a distinct subpopulation of EpCAM−CD49f− cells compared to parental MCF10A, p53− or PTEN− cells. To examine the tumor initiating capacity of this subpopulation, we flow sorted EpCAM−CD49f−, EpCAM−CD49f+, EpCAM+CD49f+ and EpCAM+CD49f− cells and implanted serial dilutions of these cells into the mouse mammary fat pads and the tumor growth was monitored over 8 weeks. EpCAM−CD49f− and EpCAM−CD49f+ subpopulations as well as unsorted cells grew large and aggressive tumors that metastasized to the lung (Figures 3c). In contrast, the EpCAM+CD49f+ subpopulation generated significantly smaller tumors after long latency and did not generate any lung metastasis (Figures 3c) while the EpCAM+CD49f− subpopulation completely lacked tumor-initiating capacity (data not shown). Furthermore, flow cytometry analyses of CSC markers in primary tumors generated by the indicated phenotypes demonstrated predominantly EpCAM−CD49f+ (>60%) or EpCAM−CD49f− (>30%) populations (Supplementary 2d). This may be attributed to the factors within the tumor microenvironment that may preferentially induce mesenchymal phenotype. To provide a molecular explanation as to why EpCAM+CD49f− and EpCAM−CD49f− subpopulations compared to EpCAM+CD49f+ or EpCAM+CD49f− cells displayed a higher tumor initiating capacity, we examined the expression of EMT related genes in these cell populations. EpCAM+CD49f− and EpCAM−CD49f− subpopulations expressed significantly higher levels of EMT genes, Vimentin, N-cadherin and Snail, whereas the EpCAM+CD49f+ population expressed significantly higher levels of epithelial related genes, EpCAM, E-Cadherin, Claudin, BMPR2 and Occludin (Figure 3d). Moreover, IL6 expression was significantly higher in EpCAM+CD49f− and EpCAM−CD49f− subpopulations compared to EpCAM+CD49f+ population (Figure 3d).
P53 and PTEN knockdown induces inflammatory cytokine production
We next investigated the molecular mechanism of malignant transformation mediated by the simultaneous knockdown of p53 and PTEN in MCF10A cells. Since we have shown that MCF10Ap53−PTEN− cells display activation of the Stat3/NF-κB pathway, we examined whether this led to an increased production of inflammatory cytokines. Inflammatory cytokines including IL6 and TGF-β have previously been linked to malignant transformation and induction of EMT via activation of the Stat3/NF-κB signaling pathway. Consistent with these studies, we show by enzyme linked immune sorbent assay (ELISA) and RT-PCR that production of IL6 and TGF-β are significantly elevated in MCF10Ap53−PTEN− cells compared to parental, p53− or PTEN− cells (Figures 4a, b, d and e). Parental cells expressed low levels of TGF-β but there was no detectable IL6. Utilizing RT-PCR and ELISA assays, we determined that basal/claudin-low breast cancer cell lines produced significantly greater IL6 than luminal or HER2+ cell lines (Figures 4c and f), consistent with previous reports (11). It was suggested that IL6 expression in breast cancer subtypes is controlled by DNA methylation at its promoter region (12). To determine whether elevated IL6 expression was due to DNA demethylation in MCF10Ap53−PTEN− cells, we examined the implicated CpG sites by pyrosequencing. There was no detectable IL6 promoter methylation in parental, p53 and/or PTEN knockdown cells (Figure 4g). Moreover, 5-aza-2 deoxycytidine (AZA), the DNA methyltransferase (DNMT) inhibitor had no effect on DNA methylation in these cells (data not shown) suggesting that IL6 expression is not controlled by DNA methylation.
Figure 4. p53 and PTEN knockdown activates the production of inflammatory cytokines via proteolytic degradation of SOCS3.

(a, b) IL6 and TGF-β production analyzed by ELISA assay in MCF10A, p53−, PTEN−, or p53−PTEN− cells. (c) Elevated IL6 production in basal/claudin low compared to luminal and HER2 amplified breast cancer cell lines is shown by ELISA. (d, e) Expression of IL6 and TGF-β by RT-PCR in MCF10Ap53−PTEN− cells compared to the parental, p53− or PTEN− cells. (f) Expression of IL6 in basal/claudin low, luminal and HER2 amplified breast cancer cell lines. (g) DNA methylation analysis by pyrosequencing of IL6 promoter sequences (−666 to −426) in MCF10Ap53−PTEN− cells compared to the parental or single p53−or PTEN− cells show no substantial methylation. (h–i) Western blot analysis of SOCS3 expression in p53−, PTEN− or p53−PTEN− cells as well as breast cancer cell lines. (j) SOCS3 protein expression was evaluated by IHC in primary breast cancer tissue microarray samples, showing a lower level of SOCS3 expression in basal-like breast cancers. (k) Two independent gene expression data sets show a strong correlation between Il6 and SOCS3 mRNA expression (Correlation Coefficient=0.731 and 0.647 respectively). (l) SOCS3 expression levels in MCF10Ap53−PTEN− and parental MCF10A cells are evaluated by western blotting following 2 hr treatment with MG132 (10μM) or cycloheximide (10 μg/ml). (m) Polyubiquitination of SOCS3 is shown in Flag-ubiquitin transfected MCF10Ap53−PTEN− cells with or without MG132 treatment. Means ± SD (n=3),*p≤0.05, ** p≤0.005.
Down-regulation of SOCS3 protein in MCF10Ap53−PTEN− cells and breast cancer cell lines correlates with increased levels of inflammatory cytokines
The mechanism of IL6 regulation is well studied in immune cells where it has been demonstrated that SOCS3 is a negative regulator of IL6 mediated immune responses (6). To determine whether a similar mechanism regulates IL6 production in normal and malignant breast cells, we examined the expression of SOCS3 by western blotting in parental MCF10A as well as in p53−, PTEN− and p53−PTEN− cells. While MCF10A cells expressed abundant SOCS3 protein, single p53 or PTEN knockdown cells expressed decreased SOCS3 protein, whereas there was no detectable SOCS3 protein in MCF10Ap53−PTEN− cells (Figure 4h). We further examined SOCS3 expression in number of breast cancer cell lines and demonstrated that when compared to normal, luminal and HER2-positive breast cancer cell lines, basal/claudin low Sum159 and MDA-M231 cells that have activated IL6 feedback loop expressed low or undetectable SOCS3 protein (Figure 4i). We have recently shown that trastuzumab resistance is mediated by activation of an IL6 feedback loop (13). Interestingly, decreased SOCS3 expression was found in trastuzumab resistant BT474-shPTEN-LTT compared to parental BT474 cells (Figure 4i). To determine whether the lack of SOCS3 protein expression correlates with SOCS3 mRNA level, we examined the SOCS3 transcript levels in these cells utilizing a quantitative RT-PCR. Surprisingly, we found increased levels of SOCS3 transcripts in MCF10Ap53−PTEN− cells compared to parental MCF10A or p53 knockdown cells (Supplementary 3a). Increased levels of SOCS3 mRNA were also detected in breast cancer cell lines representing the basal/claudin subtype (Supplementary 3b) which lack SOCS3 protein expression. To investigate whether similar discordance between SOCS3 mRNA and protein expression was also present in primary human breast tumors, we compared published SOCS3 mRNA levels in primary human breast tumors with SOCS3 protein expression as determined by immunochemistry utilizing a primary breast cancer tissue microarray. Basal-like breast cancers expressed significantly lower levels of SOCS3 protein compared to normal, luminal and HER2+ subtypes (Figure 4j). In contrast to the loss of SOCS3 protein expression in the basal/claudin low subtype, SOCS3 mRNA was more than two fold higher in basal/claudin low breast cancer compared to the luminal and HER2+ subtypes (Supplementary 3c). These findings suggest that SOCS3 protein expression is controlled at the translational/post translational level.
SOCS3 protein expression is silenced via proteasomal degradation not by DNA methylation in MCF10Ap53−PTEN− cells
Although SOCS3 silencing was shown to be regulated by IL6-mediated hypermethylation of SOCS3 promoter in colorectal and hepatocellular carcinoma(14, 15), we found no evidence of methylation at the CpG sites of SOCS3 promoter in parental MCF10A, p53−, PTEN− or p53−PTEN− cells or in breast cancer cell lines (MCF7, BT474, MDA-MB-231) representing luminal, HER2-positive and basal/claudin-low subtypes (Supplementary 4a). Furthermore, IL6 or AZA treatment failed to alter methylation of the SOCS3 promoter in any of the cell lines tested (Supplementary 4a). These findings suggested that constitutively activated Stat3/NF-κB signaling in MCF10Ap53−PTEN− cells may regulate SOCS3 post-transcriptionally. We reasoned that if the proteasome degradation is controlled by IL6 or TGF-β mediated Stat3/NF-κB signaling, then inhibiting these pathways may restore SOCS3 protein expression. We treated MCF10Ap53−PTEN− cells with inhibitors of IL6R (Tocilizumab), TGF-β (A831), Stat3 (Stat3 Inh) or NF-κB (Bay11) for 2 or 72 hours. Inhibition of these pathways in MCF10Ap53−PTEN− cells resulted in restoration of SOCS3 protein expression (Supplementary 4b). Consistent with these we also determined that steady state levels of SOCS3 transcription were up-regulated by the IL6/Stat3/NF-κB pathway in parental MCF10A cells (Supplementary 4c). However, higher levels of SOCS3 mRNA in MCF10Ap53−PTEN− cells did not translate into higher protein levels as demonstrated by western blotting (Figure 4h) suggesting increased protein turnover in these cells. We also utilized two different data set and demonstrated a strong positive correlation between IL6 and SOCS3 mRNA expressions (Figure 4k). Although SOCS3 phosphorylation was previously proposed for its rapid degradation (16), we found no evidence of SOCS3 phosphorylation in MCF10A-p53-PTEN- cells (data not shown). Treatment of MCF10Ap53−PTEN− cells with proteasome inhibitor, MG132 for 2 hours restored the SOCS3 protein expression while inhibition of protein biosynthesis with cyclohezimide (CHX) completely eliminated the SOCS3 protein expression (Figure 4l, left panel). In contrast, SOCS3 protein levels in parental MCF10A cells were not affected by the MCG132 or CHX treatment (Figure 4l, right panel) suggesting a longer protein half-life in these cells. We next transfected MCF10A-p53-PTEN- cells with SOCS3-GFP expression vector and confirmed that ectopic expression of SOCS3 is also targeted by proteasomal degradation. While treatment of SOCS3-GFP expressing cells with proteasome inhibitors (MG132 or PS-341) significantly increased the level of SOCS3-GFP protein, CHX treatment eliminated the accumulation of this protein (Supplementary 4d). In order to confirm the polyubiquitination of SOCS3 protein in MCF10Ap53−PTEN− cells, we transfected these cells with flag-Ubiquitin and demonstrated SOCS3 binding to ubiquitin in these cells as demonstrated by immunoprecipitation with anti-flag antibody and western blotting with SOCS3 antibody (Figure 4m).
Addition of recombinant IL6 or TGF-β transiently increase the stem cell population but fails to transform MCF10A cells
We next assessed the ability of recombinant IL6 or TGF-β to activate a positive feedback loop and transform MCF10A cells. Iliopoulos et al., previously reported that transient IL6 exposure transforms MCF10A cells by expanding the CD44+CD24− stem cell population via activation of an epigenetic switch involving IL6, Lin28, Let7 and NF-κB signaling (17). To replicate these findings, we treated MCF10A, p53−, PTEN− and p53−PTEN− cells with recombinant IL6 for three days and assessed malignant transformation of these cells by in vitro soft agar assay as well as by their tumorigenic capacity in mouse xenografts. Contrary to the findings of Iliopoulos et al., we found no evidence of transformation in any of the cell lines treated with IL6 (Supplementary 5a and b). As shown in figure 3b, while individual knockdown of p53 or PTEN tumor suppressors failed to transform MCF10A cells, knockdown of both tumor suppressors generated tumors in mouse xenograft models. Stimulation of MCF10A, p53 and/or PTEN knockdown cells with recombinant IL6 or TGF-β significantly increased the CD44+CD24− population (Figures 5a, Supplementary 5c) as well as the EpCAM−CD49f+ population within 3–5 days (Supplementary 5d and e). However, this effect was transient and reversible. In the absence of exogenous IL6 addition, the CD44+CD24− population returned to baseline by 14 days (Figure 5b) when IL6 or TGF-β secretion also returned baseline levels (Figure 5c and d). In contrast, MCF10Ap53−PTEN− cells maintain high levels of IL6 and TGF-β secretion (Figure 6c and 6d). Together these findings demonstrate sustained activation of an inflammatory loop maintained by IL6 occurs in fully transformed MCF10-p53−PTEN− but not in parental or single gene deleted cells.
Figure 5. IL6 transiently increase stem cell population but does not transform MCF10A cells and ectopic SOCS3 expression prevents IL6 feedback loop in MCF10Ap53−PTEN− cells.

(a) Percentage of CD44+CD24− cells assessed by flow cytometry following 3 day treatment of MCF10A, MCF10Ap53−, MCF10APTEN− or MCF10Ap53−PTEN− cells with recombinant IL6 (50ng/ml) or TGF-β (5 ng/ml) for 2h. (b) Transiently increased CD44+CD24− subpopulation in MCF10A cells is shown by flow cytometry following 3 days treatment with recombinant IL6 (50 ng/ml), then IL6 was removed from the culture and cells were analyzed at day 7 and 14. (c, d) IL6 and TGF-β production analyzed by ELISA 7 days after recombinant IL6 (50ng/ml) or TGF-β (5 ng/ml) treatment in MCF10A, p53−, PTEN− and p53−PTEN− cells. (e) SOCS3 expression is analyzed by western blot in MCF10A cells. Cells are treated with recombinant IL6 (50ng/ml) or TGF-β (5 ng/ml) for the indicated time points. (f, g) IL6 production was analyzed by ELISA after stimulation of MCF10A, and MCF10A , p53−, PTEN− and p53−PTEN− cells with recombinant IL6 or TGF-β for the indicated time points. Means ± SD (n=3),*p≤0.05, ** p≤0.005.
Figure 6. SOCS3 expression or IL6 blockade inhibits tumor growth and reduces circulating tumor cells in mice bearing MCF10Ap53−PTEN− xenografts.

(a) Expressions of SOCS3, phosphorylated NF-κB and phosphorylated Stat3 are analyzed by western blotting in MCF10A, MCF10Ap53−PTEN− and SOCS3 overexpressing MCF10Ap53−PTEN− cells. (b, c) Expressions of IL6 and SOCS3 are analyzed by RT-PCR in p53−PTEN− and p53−PTEN−SOCS3+ cells. (d) Production of IL6 is analyzed by ELISA in p53−PTEN− and p53−PTEN−SOCS3+ cells. (e) MCF10Ap53−PTEN− and MCF10Ap53−PTEN−SOCS3+ implanted in the mammary fat pad of NOD/SCID mice and monitored over 8 weeks. (f) MCF10Ap53−PTEN− and MCF10Ap53−PTEN−SOCS3+ tumor weights are presented in bar graphs and representative macroscopic tumor pictures are in insert. (g) Human IL6 levels in mice bearing MCF10Ap53−PTEN− or MCF10Ap53−PTEN−SOCS3+ tumors as well as in mice without tumor were measured by ELISA. (h) Mice with MCF10Ap53−PTEN− tumors were treated with tocilizumab and docetaxel and tumor growth was monitored during the course of 8 weeks.
SOCS3 expression in MCF10A cells negatively controls the inflammatory feedback loop
In normal macrophages, SOCS3 expression is induced by IL6 mediated Stat3/NF-κB signaling and serves as a negative regulator of this pathway (18). We therefore examined the effects of IL6 or TGF-β on SOCS3 protein expression in MCF10A cells. IL6 or TGF-β stimulation increased the SOCS3 protein expression in MCF10A cells within 30 minutes to 12 hours but these levels of SOCS3 returned to below baseline after 24 hours (Figure 5e). However, SOCS3 expression returned to baseline levels following 3 days of IL6 or TGF-β stimulation (Figure 6e). Interestingly, as determined by ELISA, IL6 or TGF-β mediated changes in SOCS3 protein expression were inversely correlated with IL6 secretion in MCF10A cells (Figures 5f and 5g). Higher SOCS3 protein expression at the indicated time points coincided with reduced IL6 production while reduced SOCS3 expression coincided with increased IL6 production. These findings provide a molecular explanation for the transient phenotypic changes in MCF10A cells characterized by increases in the CD44+CD24− population upon IL6 or TGF-β stimulation at day 3. However, following the restoration of SOCS3 protein expression after 5 days, the inflammatory loop is down regulated and the CD44+CD24− population returns to baseline.
Ectopic SOCS3 expression in MCF10Ap53−PTEN− cells blocks the IL6 feedback loop by inhibiting the Stat3/NF-κB signaling
Our findings suggested that a reciprocal relationship exists between IL6 and SOCS3 in MCF10A cells. However efficient silencing of this protein via proteolytic degradation in MCF10A-p53−PTEN− cells resulted in constitutive activation of an IL6 inflammatory feedback loop. We determined whether ectopic expression of SOCS3 can block the IL6 feedback loop in transformed cells. We generated a MCF10Ap53−PTEN− cell line stably expressing SOCS3-GFP fusion protein (MCF10Ap53−PTEN−SOCS3+). SOCS3-GFP protein is detected at the expected size about ~55kDa. Expression of SOCS3-GFP in MCF10Ap53−PTEN− cells significantly decreased Stat3 and NF-κB activity (Figure 6a). Increased SOCS3 mRNA expression in transfected cells was confirmed by RT-PCR assay (Figure 6b). Furthermore ectopic SOCS3 expression markedly reduced IL6 expression and secretion as determined by RT-PCR and ELISA assays respectively (Figures 6c and d). Although, strong GFP and DsRed expression in these cells precluded the examination of changes in CD44+CD24− phenotype by flow, we show that CD44 mRNA expression is reversed by ectopic SOCS3 expression (Supplementary 4e) as well as investigated tumor-initiating capacity of these cells in mouse xenografts.
SOCS3 expression or IL6 blockade inhibits tumor growth and reduces circulating tumor cells in mouse xenografts generated by MCF10Ap53−PTEN− cells
Loss of SOCS3 expression is associated with increased risk of recurrent disease while higher levels are correlated with earlier tumor stage and better clinical outcome (19, 20). Consistent with these findings, our in vitro studies suggested that SOCS3 may function as a tumor suppressor by inhibiting the IL6/NF-κB inflammatory loop which generates EMT like CSCs. To determine the functional relevance of SOCS3 in mouse xenografts, we implanted SOCS3 overexpressing and control MCF10Ap53−PTEN− cells into mouse mammary fat pads and monitored the tumor growth over 8 weeks. As expected, control MCF10Ap53−PTEN− cells generated extremely large tumors (> 1cm) within 8 weeks. In contrast, SOCS3 expression reduced tumor growth by approximately 70% as well as increasing tumor latency (Figures 6e and f). We reasoned that reduced tumor growth by SOCS3 expressing cells may be due to reduced IL6 secretion from these tumors. Consistent with our in vitro assays, circulating human IL6 levels in mice bearing SOCS3 expressing tumors were markedly diminished compared to mice with control tumors (Figure 6g).
In order to demonstrate direct role of the IL6 feedback loop in tumor growth, we utilized the IL6R antibody (tocilizumab) (21). Although docetaxel treatment failed to inhibit tumor growth, tocilizumab treatment significantly reduced the tumor growth (Figure 6h) suggesting that these tumors depend on an IL6 feedback loop.
Discussion
Despite frequent inactivation of the tumor suppressors p53 and PTEN in breast tumors (1–3) and increased risk of breast cancer development in patients with germline mutations of p53 and PTEN, their mechanistic role in tumor development and progression has been poorly understood. As we demonstrated p53 and PTEN knockdown in MCF10A cells resulted in transformation of these cells and further expansion of the stem cell population compared to single gene knockdown as demonstrated by CD44+CD24− and Epcam−CD49f+ stem cell markers. Consistent with the findings of Mani et al., (10), MCF10A-p53−PTEN− cells also displayed an increased EMT phenotype characterized by expression of EMT related genes such as Vimentin, Snail and N-Cadherin.
Utilizing Affymetrix gene expression analyses, we confirmed that malignant MCF10Ap53−PTEN− cells displayed a gene expression pattern which closely resembled that of the basal/claudin-low breast cancer subtype. This subtype is characterized by expression of EMT and stem cell genes (5, 22).
The Inflammatory cytokines IL6 and TGF-β have been found to regulate stem cells and EMT (23–26). Consistent with these data, we found that MCF10A-p53−PTEN− cells secreted significantly higher levels of IL6 (>1000 fold) and TGF-β (>2 fold) compared to parental or single gene deleted cells. When we analyzed different subsets of MCF10A-p53−PTEN− cells, we found that EpCAM−CD49f− and EpCAM−CD49f+ subpopulations secreted significantly higher levels of IL6 and TGF-β and were more tumorigenic than other subpopulations. In addition, these subsets of cells displayed a prominent EMT phenotype.
It was previously reported that addition of exogenous IL6 transforms MCF10A cells by activating an irreversible inflammatory feedback loop involving IL6/Stat3/NF-κB (17). However, we found no evidence of transformation of MCF10A cells upon addition of recombinant IL6. Although IL6 or TGF-β treatment of MCF10A cells increased the stem cell population, these effects were transient and activation of the IL6-NF-κB inflammatory loop was not sustained following IL6 withdrawal. Furthermore the proportion of CSCs returned to baseline over time. As a consequence, parental or single gene deleted cells were not transformed by addition of IL6. We demonstrated that SOCS3 is abundantly expressed in MCF10A cells where it negatively regulates the IL6 feedback loop via Stat3/NF-κB pathway (6, 27). Unexpectedly, we found that SOCS3 protein was undetectable in MCF10A-p53−PTEN− cells. Basal/claudin-low breast cancer cell lines that display an activated IL6 feedback loop also had low SOCS3 protein levels. Together these findings suggest that loss of SOCS3 protein expression allows for maintenance of the IL6 mediated inflammatory feedback loop. Despite previous reports (14, 15), we found no evidence of methylation at the implicated CpG sites of SOCS3 promoter. In contrast, SOCS3 protein expression in MCF10A-p53−PTEN− cells is increased upon proteasome inhibition. Although SOCS3 Tyr phosphorylation was proposed to be mediating this degradation (16), there was no detectable phospho-SOCS3 in MCF10A-p53−PTEN− cells. Proteolytic degradation of SOCS3 protein in turn is controlled by constitutive activation of the IL6 mediated Stat3/NF-κB pathway supporting a reciprocal relationship between Stat3/NF-κB signaling and SOCS3 expression (28). SOCS3 inhibits IL6 mediated Stat3 signaling by binding to the Jak2 kinase domain and IL6 receptor β-chain (29). SOCS3 homozygous deletion in the pancreas accelerates development of pancreatic intraepithelial neoplasia (PanIN) and pancreatic cancer by activating Stat3/NF-κB signaling in mouse models (30). In addition deletion of SOCS3 in liver cells promotes hepatocellular carcinoma (31). Furthermore, loss of SOCS3 expression is strongly associated with increased risk of recurrent disease in breast cancer patients (19). In line with these findings, we demonstrated that loss of SOCS3 protein expression in MCF10A-p53−PTEN− cells plays crucial role in tumor development and progression via activating IL6 mediated Stat3/NF-κB pathway. In summary, our studies reveal important differences between the regulation of inflammatory loops in normal tissues and cancer, differences which are depicted in figure 7. In non-transformed cells, activation of an inflammatory loop involving IL6/Stat3/NF-κB enhances the expression of SOCS3 which in turn shuts down the pathway through inhibition of IL6/Stat3. This accounts for the transient reversible activation of inflammation resulting from tissue damage. In contrast, transformation of normal mammary cells via inactivation of p53 and PTEN is accompanied by proteolytic degradation of SOCS3 leading to constitutive activation of the IL6/Stat3/NF-κB inflammatory loop. Molecular link between the tumor suppressors and SOCS3 as well as increased activity of proteolytic enzymes targeting SOCS3 will need further investigation.
Figure 7. Illustration of SOCS3-mediated IL6 signaling pathway in normal and basal/claudin-low breast cancer.
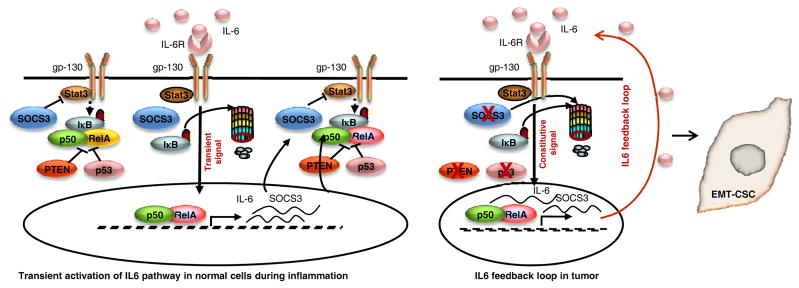
A model of IL6 signaling controlled by SOCS3 in normal cells, inflammation and tumor development is illustrated.
SOC3 mediated activation of inflammatory feedback loop generates EMT like CSCs which drive tumor growth and metastasis. The clinical relevance of this pathway is supported by our findings of reduced SOCS3 protein in the face of increased levels of SOCS3 mRNA in primary basal/claudin low breast cancers. Furthermore, we demonstrate that enforced SOCS3 expression reduced IL6 secretion by more than 90% via inhibiting activity of Stat3/NF-κB signaling as assessed by western blotting resulted in inhibition of tumor growth and metastasis.
Our findings that IL6/Stat3/NF-κB inflammatory loop is activated in basal/claudin-low but not in luminal or HER2+ breast cancer cell lines further support the importance of this loop in this subtype. We demonstrate that interfering with this loop through IL6R blockade repress the CSC population, reducing the tumor growth and metastasis in mouse xenografts. These studies provide a strong rationale for development of IL6 pathway-targeting agents for the treatment of basal/claudin-low tumors, an aggressive disease that currently lacks molecularly targeted therapeutics.
Materials and Methods
Animal experiments
All mice procedures were conducted in accordance with the University Committee on the Use and Care of Animals at the University of Michigan. Humanization of mice fat pads were performed as previously described (32). Cells infected with DsRed, p53 shRNA, PTEN shRNA or both p53 and PTEN shRNA lentiviral vectors were implanted in humanized mammary fat pads. Detailed animal procedures were provided in the supplementary material.
Normal mammary epithelial cells from reduction mammoplasties
Mammary tissues from reduction mammoplasties were dissociated as previously described (33). Tissue collection protocol was approved by the Internal Review Board (IRB). The procured tissues were confirmed to be normal by pathological examination.
Lentiviral Production and Infection of HNMECs and MCF10A Cell Line
Lentiviral vectors were constructed in house and lentiviral production was performed by the University of Michigan Vector Core facility. Details of the lentiviral constructions were provided in supplementary material.
Procedures of immunoblotting, Immunohistochemical stainings and ELISA were provided in supplementary material.
RNA extraction and real-time RT-PCR
Total RNA was extracted using RNeasy Mini or Micro kit (QIAGEN) and 1 μg of RNA was used for making cDNA using Reverse Transcription System (Promega). Each 20 μg of cDNA was analyzed in triplicate using real-time quantitative reverse transcription–PCR (qRT-PCR) assays in an ABI PRISM 7900HT sequence detection system with 384-well block module and automation accessory (Applied Biosystems).
Statistical Analyses
Statistical differences for indicated assays were determined using Student’s t-test. When P value was less than 0.05, it was considered statistically significant.
Supplementary Material
Footnotes
Conflict of interest: MSW has financial holdings and is a scientific advisor for OncoMed Pharmaceuticals, is a scientific advisor for Verastem, Paganini and MedImmune and receives research support from Dompe Pharmaceuticals and MedImmune.
References
- 1.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997 Apr;15(4):356–62. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 2.Hollstein M, Rice K, Greenblatt MS, Soussi T, Fuchs R, Sorlie T, et al. Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1994 Sep;22(17):3551–5. [PMC free article] [PubMed] [Google Scholar]
- 3.Comprehensive molecular portraits of human breast tumours. Nature. 2012 Oct 4;490(7418):61–70. doi: 10.1038/nature11412. Epub 2012/09/25. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cicalese A, Bonizzi G, Pasi CE, Faretta M, Ronzoni S, Giulini B, et al. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009 Sep 18;138(6):1083–95. doi: 10.1016/j.cell.2009.06.048. Epub 2009/09/22. eng. [DOI] [PubMed] [Google Scholar]
- 5.Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12(5):R68. doi: 10.1186/bcr2635. Epub 2010/09/04. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol. 2003 Jun;4(6):540–5. doi: 10.1038/ni931. Epub 2003/05/20. eng. [DOI] [PubMed] [Google Scholar]
- 7.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003 May 15;17(10):1253–70. doi: 10.1101/gad.1061803. Epub 2003/05/21. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003 Apr 1;100(7):3983–8. doi: 10.1073/pnas.0530291100. Epub 2003/03/12. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keller PJ, Lin AF, Arendt LM, Klebba I, Jones AD, Rudnick JA, et al. Mapping the cellular and molecular heterogeneity of normal and malignant breast tissues and cultured cell lines. Breast Cancer Res. 2010 Oct 21;12(5):R87. doi: 10.1186/bcr2755. Epub 2010/10/23. Eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008 May 16;133(4):704–15. doi: 10.1016/j.cell.2008.03.027. Epub 2008/05/20. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011 Jul;121(7):2723–35. doi: 10.1172/JCI44745. Epub 2011/06/03. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dandrea M, Donadelli M, Costanzo C, Scarpa A, Palmieri M. MeCP2/H3meK9 are involved in IL-6 gene silencing in pancreatic adenocarcinoma cell lines. Nucleic Acids Res. 2009 Nov;37(20):6681–90. doi: 10.1093/nar/gkp723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell. 2012 Aug 24;47(4):570–84. doi: 10.1016/j.molcel.2012.06.014. Epub 2012/07/24. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y, Deuring J, Peppelenbosch MP, Kuipers EJ, de Haar C, van der Woude CJ. IL-6-induced DNMT1 activity mediates SOCS3 promoter hypermethylation in ulcerative colitis-related colorectal cancer. Carcinogenesis. 2012 Oct;33(10):1889–96. doi: 10.1093/carcin/bgs214. Epub 2012/06/29. eng. [DOI] [PubMed] [Google Scholar]
- 15.Niwa Y, Kanda H, Shikauchi Y, Saiura A, Matsubara K, Kitagawa T, et al. Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene. 2005 Sep 22;24(42):6406–17. doi: 10.1038/sj.onc.1208788. [DOI] [PubMed] [Google Scholar]
- 16.Sommer U, Schmid C, Sobota RM, Lehmann U, Stevenson NJ, Johnston JA, et al. Mechanisms of SOCS3 phosphorylation upon interleukin-6 stimulation. Contributions of Src- and receptor-tyrosine kinases. J Biol Chem. 2005 Sep 9;280(36):31478–88. doi: 10.1074/jbc.M506008200. [DOI] [PubMed] [Google Scholar]
- 17.Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009 Nov 13;139(4):693–706. doi: 10.1016/j.cell.2009.10.014. Epub 2009/11/03. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, et al. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol. 2003 Jun;4(6):551–6. doi: 10.1038/ni938. Epub 2003/05/20. eng. [DOI] [PubMed] [Google Scholar]
- 19.Ying M, Li D, Yang L, Wang M, Wang N, Chen Y, et al. Loss of SOCS3 expression is associated with an increased risk of recurrent disease in breast carcinoma. J Cancer Res Clin Oncol. 2010 Oct;136(10):1617–26. doi: 10.1007/s00432-010-0819-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sasi W, Jiang WG, Sharma A, Mokbel K. Higher expression levels of SOCS 1,3,4,7 are associated with earlier tumour stage and better clinical outcome in human breast cancer. BMC Cancer. 2010;10:178. doi: 10.1186/1471-2407-10-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohsugi Y, Kishimoto T. The recombinant humanized anti-IL-6 receptor antibody tocilizumab, an innovative drug for the treatment of rheumatoid arthritis. Expert Opin Biol Ther. 2008 May;8(5):669–81. doi: 10.1517/14712598.8.5.669. Epub 2008/04/15. eng. [DOI] [PubMed] [Google Scholar]
- 22.Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009 Aug 18;106(33):13820–5. doi: 10.1073/pnas.0905718106. Epub 2009/08/12. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. 2011 Jun 10;145(6):926–40. doi: 10.1016/j.cell.2011.04.029. Epub 2011/06/15. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yao Z, Fenoglio S, Gao DC, Camiolo M, Stiles B, Lindsted T, et al. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc Natl Acad Sci U S A. 2010 Aug 31;107(35):15535–40. doi: 10.1073/pnas.1009472107. Epub 2010/08/18. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, Taffurelli M, et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J Clin Invest. 2007 Dec;117(12):3988–4002. doi: 10.1172/JCI32533. Epub 2007/12/07. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A. 2011 Jan 25;108(4):1397–402. doi: 10.1073/pnas.1018898108. Epub 2011/01/12. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997 Apr 10;336(15):1066–71. doi: 10.1056/NEJM199704103361506. Epub 1997/04/10. eng. [DOI] [PubMed] [Google Scholar]
- 28.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007 Jun;7(6):454–65. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 29.Babon JJ, Kershaw NJ, Murphy JM, Varghese LN, Laktyushin A, Young SN, et al. Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity. 2012 Feb 24;36(2):239–50. doi: 10.1016/j.immuni.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Kloppel G, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011 Apr 12;19(4):456–69. doi: 10.1016/j.ccr.2011.03.009. Epub 2011/04/13. eng. [DOI] [PubMed] [Google Scholar]
- 31.Ogata H, Kobayashi T, Chinen T, Takaki H, Sanada T, Minoda Y, et al. Deletion of the SOCS3 gene in liver parenchymal cells promotes hepatitis-induced hepatocarcinogenesis. Gastroenterology. 2006 Jul;131(1):179–93. doi: 10.1053/j.gastro.2006.04.025. [DOI] [PubMed] [Google Scholar]
- 32.Korkaya H, Paulson A, Iovino F, Wicha MS. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008 Oct 16;27(47):6120–30. doi: 10.1038/onc.2008.207. Epub 2008/07/02. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Korkaya H, Paulson A, Charafe-Jauffret E, Ginestier C, Brown M, Dutcher J, et al. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta-catenin signaling. PLoS Biol. 2009 Jun 2;7(6):e1000121. doi: 10.1371/journal.pbio.1000121. Epub 2009/06/06. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.