

Abstract
Prostate cancer is the most common malignancy in men in the United States, and one in seven men with prostate cancer dies of the disease. A major issue of prostate diagnosis is that there is no good method to reliably distinguish aggressive prostate cancer from non-aggressive prostate cancer. This leads to significant unnecessary suffering among prostate cancer patients and massive unnecessary health care expenditures. In this study, we aim to identify glycoproteins associated with aggressive prostate cancer using OCT-embedded frozen tissues obtained from patients with known clinical outcome. To eliminate the interfering of mass spectrometric analysis by the compounds in OCT and identify extracellular proteins that are likely to serve as biomarkers in body fluids, we employed the glycoproteomic analysis using solid-phase extraction of glycopeptides, which allowed the immobilization of glycopeptides to solid support and removal of OCT from sample proteins before releasing the glycopeptides from solid support for mass spectrometry analysis. Tumor tissues were cryostat microdissected from 4 cases of aggressive and 4 cases of non-aggressive prostate tumors, and glycopeptides were isolated and labeled with iTRAQ reagents before the samples were analyzed with LTQ Orbitrap Velos. From the aggressive prostate cancer tissues, we identified the overexpression of four glycoproteins involved in extracellular matrix remodeling and further verified two glycoproteins, cathepsin L and periostin, using Western blot and immunohistochemistry analyses. This is the first proteomic study to identify proteins associated with aggressive prostate cancer using OCT-embedded frozen tissues. Further study of these proteins will be needed to understand the roles of extracellular matrix proteins in cancer progression and their potential clinical utility in improving diagnosis of aggressive prostate cancer.
Keywords: prostate cancer, aggressive tumor, tumor metastasis, glycoprotein, OCT-embedded frozen tissue
Introduction
Prostate cancer is the most common malignancy in men and the second leading cause of death from cancer in the United States 1. Metastases are the major cause of death from cancer 2. . Therefore, aggressive (AG) prostate cancer leads to a higher death rate and requires more treatment than non-aggressive (NAG) prostate cancer. Since the discovery of PSA, assays that detect this serum biomarker (together with digital rectal exams) have been used for the screening of prostate cancer 3. PSA testing has resulted in early detection and intervention 4. However, the major limitation of PSA is the low specificity and high prevalence of detecting benign prostatic hyperplasia, especially in older men 5. Most important, PSA fails to distinguish aggressive prostate cancer from non-aggressive prostate cancer. Indeed, illustrating the limitations of the current PSA-based screening method, a recently published study randomly assigned 76,693 men at 10 U.S. study centers to receive either annual PSA screening (38,343 subjects) or usual care as the control (38,350 subjects); this study reported no statistical differences in prostate cancer specific mortality between the groups after 7 to 10 years of follow-up 6.
Besides preoperative PSA, clinical risk assessment tools for prostate cancer metastasis before surgery largely rely on prostate biopsy Gleason score 7, 8. However, the risk assessment based on this clinical criterion is too imprecise to be useful due to the biopsy sampling error and inter-observer grading differences. It is also unable to be used as a screening test for early detection of aggressive prostate cancer. Currently, aggressive prostate cancer is under-detected and under treated while non-aggressive prostate cancer is over-detected and over-treated. Consequences of the difficulty of distinguishing the aggressive and non-aggressive prostate cancer are that prostate cancer patients suffer from unnecessary surgeries, and health care faces massive unnecessary expenditures. Therefore, reliable biomarkers to distinguish aggressive and non-aggressive prostate cancer are badly needed to prevent patients with non-aggressive prostate cancer from overtreatment, and to allow patients with aggressive cancer to receive appropriate treatment earlier in the course of their disease.
Proteins specific to aggressive or non-aggressive cancer can potentially be identified in cancer tissues. If these proteins can be detected in body fluids, the proteins may be able to be used as molecular markers for the early detection of aggressive prostate cancer 9. This possibility has not been explored, in part, due to the challenge of identifying and isolating aggressive and non-aggressive prostate tumor tissues, which requires thorough histological characterization and patient follow up for disease progression. In addition, proteomic analysis of such tissues is challenging due to the limited amount of tissues available and the interferences from the procedures used to preserve the histology of the tissue specimens. To preserve frozen tissues for histological characterization, surgically resected tissues are usually embedded in cryopreservation medium, such as optimal cutting temperature (OCT) compound10. This can assist with the cutting of sections for histopathologic analysis by enclosing the tissue in a frozen solid matrix. OCT is a viscous aqueous solution and consists of a resin-polyvinyl alcohol, benzalonium chloride, an antifungal agent, and polyethylene glycol (PEG) to lower the freezing temperature 10. The presence of OCT tissue embedding medium in the frozen sections raises a concern regarding their usability for proteomics analysis and especially for mass spectrometric analysis, as OCT was reported to suppress ion formation in mass spectrometers 11.
Protein glycosylation is one of the most abundant protein modifications, and the altered glycosylation is known to associate with disease states. Extracellular proteins are most glycosylated and likely to enter into the body fluid to serve as potential biomarkers. Therefore, focusing on the subproteome, glycoproteome, is not only reducing the sample complexity but also targeting the information-rich subproteome. In addition, capturing glycopeptides using chemical immobilization has great potential to eliminate the OCT from the glycoproteins before the glycopeptides are released from solid support for mass spectrometry analysis.
In this study, we initially identified and quantified glycoproteins from OCT-embedded frozen tissues using glycopeptide isolation and LC-MS/MS analysis. We were able to identify glycoprotein changes related to aggressive prostate cancer and further verified the protein candidates using antibody based methods.
Experimental Procedures
Materials
Hydrazide resin and Sodium periodate were from Bio-Rad (Hercules, CA) ; Sequencing grade trypsin was purchased from Promega (Madison, WI); PNGase F was from New England Biolabs (Ipswich, MA); C18 columns were from Waters (Milford, MA); iTRAQ reagent was from Applied Biosystems (Foster City, CA); rabbit anti-human periostin antibody and mouse anti-human cathepsin L antibody for Western blot analysis were from Abcam (Cambridge, UK ); goat anti-cathepsin L antibody for immunohistochemistry was from R&D systems (Minneapolis, MN); BCA assay kit, HRP-labeled secondary antibodies and Novex ECL Chemiluminescent Substrate Reagent Kit were from Pierce (Rockford, IL); Dako LSAB+ System–AP kit (Carpinteria, CA); All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Prostate cancer tissues
Samples and clinical information were obtained with informed consent and the approval of the Institutional Review Board of the Johns Hopkins University. In this study, tissues from 4 non-aggressive and 4 aggressive primary prostate tumors were analyzed. The non-aggressive and aggressive primary cancer tissues were collected from radical prostatectomy specimens at Johns Hopkins Hospital and Johns Hopkins Bayview Medical Center and were not needed for pathologic diagnosis. Under the NCI-funded Johns Hopkins prostate cancer SPORE project, annual follow-up data is obtained from consenting subjects in this cohort, allowing clinical follow up data to be used in conjunction with pathologic data. The cancer tissues were selected from cases with known Gleason score and clinical outcome following surgery. The non-aggressive cancer tissues were microdisected from primary tissues with Gleason scores of 3+3=6 and without cancer recurrence after 11-15 years of follow-up. The aggressive cancer tissues were microdissected from primary tissues of tumors with Gleason score of 5+4=9 (3 cases) or 5+3=8 (one case) and either seminal vesicle or pelvic lymph node involvement (or both). Two of the men with aggressive prostate cancer died of prostate cancer 2 and 6 years after surgery, one died of myocardial infarction 5 years after surgery with known prostate cancer recurrence, and one is thought to have been surgically cured of aggressive prostate cancer as he shows no evidence of recurrence 15 years after surgery.
Glycopeptide isolation
OCT-embedded prostate frozen tissues were sectioned and stained with hematoxylin and eosin. The microscopic view of a histologic specimen was used for cryostat microdissection to remove the non tumor tissue. The proteins from each prostate cancer tissue were collected from one 10 μm OCT tumor section using lysis buffer (50 mM Tris, pH8.0, 150 mM NaCl, 0.1% SDS, 0.5% Na Deoxycholate, 1% Triton × 100). BCA assay was performed to determine the protein concentration. For each tissue specimen, 100 g proteins were used in the following experiment. 8M urea and 5mM TCEP as final concentration were added to the sample and incubated at 60°C for 2 hrs. 10mM iodoacetamide as final concentration was added to the sample and incubated for 30 mins at room temperature in the dark. The solution was diluted 8 fold with 100mM KH2PO4 (pH 8.0). 2μg trypsin (Promega) was added to the solution and incubated at 37°C overnight with shaking. Silver staining was used to determine whether the trypsin digestion was complete, which was indicated by disappearance of upper protein bands and appearance of lower peptide bands (<10kDa). After trypsin digestion, samples were centrifuged at 13,000 rpm for 5 minutes to remove any particulate matter. The peptides were cleaned by C18 column and followed by N-linked glycopeptides isolation using the method of solid phase extraction of glycopeptides (SPEG) 12, 13. The enriched N-linked glycopeptides were concentrated by C18 columns and dried down and resuspended in 10μl of 0.4% acetic acid.
iTRAQ labeling
Glycopeptides (10 μL) from each sample were labeled with iTRAQ 8plex (AB SCIEX) according to the manufacturer’s instruction. Peptides of four AG-prostate tumors were labeled by iTRAQ with 113, 114, 115 and 121 respectively, while peptides of four NAG-prostate tumors were labeled by iTRAQ with116, 117, 118 and 119. Labeled peptides were then mixed and cleaned by SCX column.
LC-MS/MS analysis
iTRAQ labeled glycopeptides were separated on a C18 column (75 μm × 10 cm, 5 μm, 120Å, Magic C18, MicromBioresources, Auburn, Ca) at 750 nL/min in 15 minute loading time. The peptides were then eluted at 300 nL/min by using a gradient of 90 min at the voltage of 2.0 kV. Eluting peptides were sprayed into an LTQ Orbitrap Velos mass spectrometer (Thermo Scientific) with 1 μm emitter tip (New Objective, Woburn, MA) using 5-40% solution B (90% acetonitrile in 0.1% formic acid) gradient.
Identification of protein and glycosylation sites
MS/MS spectra were searched with MASCOT using Proteome Discoverer (version 1.0) (Thermo Fisher) against the human subdatabase of NCBI Reference Sequence (RefSeq) (version 40, released at April 16, 2010) containing 29,704 sequences. The precursor mass tolerance was set as 15 ppm while the fragment mass tolerance was set as 0.05Da. The enzyme was set to trypsin, allowing one missed cleavage, and the flexible modifications were set as deamidation (NQ) and oxidation (M). Carbamidomethylation (C) was set as fixed modification. The criterion of peptide probability score is ≥ 0.95 (Decoy target FDR, 0.05) so that low probability protein identifications can be filtered out. The N-linked glycosylation site is the Asp contained within the consensus N-linked glycosylation motif.
Statistic analysis
To determine whether there was a significant difference in the glycoproteins of the AG and NAG-prostate cancer groups, P-values of the iTRAQ tag intensities representing the different groups were calculated using two-tailed t-tests.
Western blot analysis
25μg of proteins was resolved on SDS-PAGE and transferred electrophoretically onto a 0.2μm nitrocellulose membrane. The membrane was blocked by 5% non-fat milk/0.1% TBS-Tween 20 at room temperature for 2 hours. The membrane was then probed with primary antibody (Rabbit anti-human periostin at 1:2000, mouse anti-human cathepsin L at 1:200) at 4°C overnight, followed by a three-time wash of 0.1% TBS-Tween 20. HRP conjugated secondary antibody was added at 1:2000 and incubated at room temperature for 1 hour. Three washes of 0.1% TBS-Tween 20 were performed. The signal was visualized by superSignal West Femto Maximum Sensitivity Substrate (Pierce).
Immunohistochemstry staining
Rabbit anti-human periostin and goat anti-antibody were used in a dilution of 1:500 and 1:50 to stain sections from formalin-fixed and paraffin-embedded tissue specimens and were detected using Dako LSAB+ System–AP kit according to the manufacturer’s instruction.
Results
Quantitative glycoproteomic profiling of aggressive (AG) prostate tumors and non-aggressive (NAG) prostate tumors
The scheme of present study is shown in Figure 1. Briefly, to identify glycoproteins associated with AG-prostate cancer, we quantitatively analyzed glycoproteins from OCT-embedded AG-prostate tumor and NAG-prostate tumor tissues. The candidate proteins were further verified using Western blot and immunohistochemistry.
Figure 1.

Workflow of identifying altered glycoprotein changes related to aggressive prostate carcinoma
Glycopeptides were isolated from 4 individuals of patients with AG-prostate tumors and 4 individuals of patients with NAG-prostate tumors using solid-phase extraction of glycopeptides (SPEG). To determine the relative abundance of glycoproteins in AG and NAG-prostate cancer tissues, the glycopeptides isolated from each specimen were labeled with iTRAQ prior to LC-MS/MS analysis. From mass spectrometry analysis of glycopeptides, we were able to identify 102 unique formerly N-linked glycopeptides with 95% confidence, representing 79 unique glycoproteins (Supplementary Table 1).
Glycoprotein changes associated with aggressive prostate tumors
To determine the glycoprotein changes associated with aggressive prostate tumors, the relative abundance of each glycopeptide in tissues of four cases of AG prostate cancer and four cases of NAG-prostate cancer were determined by iTRAQ labeling and tandem mass spectrometry (Supplementary Table 1). Using the peak intensity of iTRAQ reporter tags which represented the relative abundance of the peptides, t-tests were performed to identify the glycopeptides and the glycoproteins associated with AG-prostate tumors. According to iTRAQ results, glycopeptides from three glycoproteins, microfibrillar-associated protein 4, periostin, and cathepsin L, showed a significant difference between the AG-prostate cancer group (iTRAQ Tag 113, 114, 115 and 121) and the NAG-prostate cancer group (iTRAQ Tag 116, 117, 118 and 119) (with p-values of less than 0.05 ) (Table 1, Supplementary Table 1, Supplementary Figure 1 and Figure 2). Two peptides, vDLEDFEnNTAYAk and fnGSVSFFR (lower case v, e, n, f, and k represent the iTRAQ labeled N-termini and Lys, lower case n in the nXT/S motif represents the formerly glycosylated Asp and delaminated after SPEG isolation), were identified from microfibrillar-associated protein 4. The quantitation of the two peptides was consistent: the average ratio of the AG-prostate tumor group vs. the reference channel (iTRAQ tag 113, labeled as one of the AG-prostate tumors) was 1.29±0.22, while the average ratio of the NAG-prostate tumor group was 0.40±0.21. In addition, collagen XII showed a ~7-fold increase in the AG group compared to the NAG group with a p-value of 0.08 (Table 1, Supplementary Figure 2E, and Figure 3E).
Table 1.
Glycoproteins overexpressed in aggressive (AG) prostate tumors
| Proteins | Peptides $ | AG-tumor # | NAG-tumor # | Ratio of AG/NAG $ |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 113 | 114 | 115 | 121 | 116 | 117 | 118 | 119 | |||
| Microfibrillar-associated protein 4 |
vDLEDFEnNTAYAk | 1.00 | 0.95 | 1.50 | 1.30 | 0.31 | 0.20 | 0.41 | 0.53 | 3.28 |
| Microfibrillar-associated protein 4 |
fnGSVSFFR | 1.00 | 1.56 | 1.29 | 1.70 | 0.14 | 0.34 | 0.38 | 0.87 | 3.22 |
| Periostin | eVnDTLLVNELk | 1.00 | 0.57 | 1.24 | 0.54 | 0.26 | 0.17 | 0.34 | 0.52 | 2.62 |
| Cathepsin L | ySVAnDTGFVDIPk | 1.00 | 0.82 | 0.96 | 1.33 | 0.38 | 0.44 | 0.78 | 0.62 | 1.85 |
| Collagen XII, alpha 1 | nLQVYnATSNSLTV k |
1.00 | 4.82 | 2.60 | 0.77 | 0.26 | 0.30 | 0.29 | 0.46 | 6.98 |
Lower case v, e, n, f, and k represent the iTRAQ labeled N-termini and Lys, lower case n in the nXT/S motif represents the formerly glycosylated Asp and delaminated after SPEG isolation
The number showed the ratio of different channel vs 113
The ratio of AG/NAG uses the average of each group
Figure 2.

Statistical analysis of protein expression in aggressive (AG) prostate tumor and non-aggressive (NAG) prostate tumor.
A. microfibrillar-associated protein 4 (peptide: vDLEDFEnNTAYAk);
B. microfibrillar-associated protein 4 (peptide: fnGSVSFFR)
C. periostin
D. cathepsin L
E. collagen XII
Verification of glycoproteomic results using Western blot
To verify the proteomic results, the 4 cases of AG-prostate tumors and 4 cases of NAG-prostate tumors were analyzed by Western blot assay. Periostin showed overexpression in all four AG-prostate tumors, while cathepsin L was elevated in 3 out of 4 AG-prostate tumors, compared to the NAG-prostate tumors (Figure 3). We were not able to identify specific bands for Collagen XII and microfibrillar-associated protein 4 by Western blot to verify their expression. IgG was detected evenly in each sample and was used as a quantitation control. These observations confirmed the glycoproteomic results for periostin and cathepsin L.
Figure 3.
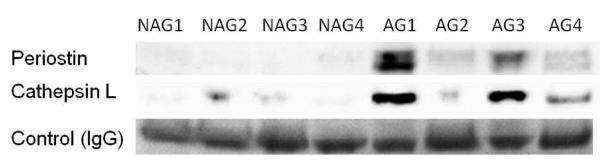
Validations for candidate proteins using Western blot analysis. Periostin showed upregulated expression in all 4 AG-prostate tumors, while cathepsin L was up-regulated in 3 out of 4 AG-prostate tumors, compared to NAG-prostate tumors. IgG was detected evenly in each sample and used as a quantitation control.
Verification of overexpressed proteins using immunohistochemistry
To verify further the overexpression of the identified proteins, we stained cathepsin L and periostin in primary prostate tumors, normal prostate tissues, and other organs metastasized with prostate cancer.
Figure 4A shows the Cathepsin L staining in primary prostate cancer tissues. Cytoplasmic staining was detected in both normal and tumor epithelial cells. However, the percentage of cells stained with cathepsin L was increased in the prostate tumor due to increased epithelial cells in tumor tissues. The staining of cathepsin L was increased in prostate cancer metastasized to different organs compared to the normal prostate tissues (Figure 4B).
Figure 4.



Target validation by immunohistochemistry.
4A. Immunohistochemical analysis of prostate primary tumor and its adjacent normal tissue with antibody specific to cathepsin L. Cathepsin L was expressed in epithelial cells. Arrows indicate the epithelial cell staining of antibody against Cathespin L.
4B. Immunohistochemical staining revealed a strong overexpression of cathepsin L in organs containing metastastic prostate cancer.
4C. Immunohistochemical analysis of prostate primary tumor and its adjacent normal tissue with antibody specific to periostin. Staining exhibited low background in the normal prostate samples but revealed an overexpression in peritumoral stroma of gleason 3 tumors and strong overexpression in peritumoral stroma of gleason 4 tumors. Arrows point the peritumoral stroma staining of antibody against periostin.
Figure 4C shows the immunohistochemstry staining of periostin in primary prostate cancer tissues. Staining exhibited low background in the normal prostate samples but revealed an overexpression in peritumoral stroma of gleason 3 tumors and strong overexpression in peritumoral stroma of gleason 4 tumors. This indicates that periostin expression may correlate with aggressive prostate cancer.
Discussion
Several glycoproteins were identified in this study and showed significant altered expression in aggressive prostate tumors compared to non-aggressive prostate tumors. This is the first glycoproteomic study to elucidate the differentially expressed proteins associated with aggressive prostate cancer, proteins that are potentially useful for diagnosis of aggressive prostate cancer. Furthermore, though OCT-embedded tissues have been used in proteomic analyses14, our study was the first to demonstrate the use of OCT-embedded frozen tissues in glycoproteomic analysis. Because glycoproteomics has several advantages in identifying extracellular proteins as potential biomarkers, our finding may facilitate the usage of OCT-embedded frozen tissues in biomarker discovery.
Tumor invasion and metastasis is a complicated and multifaceted process which may include intravasation, survival in the circulating system, arrest and extravasation into a new tissue, initiation and maintenance of tumor cell growth, and reactivation of angiogenesis 15. In this multistep process, interactions between cancer cells and stromal cells as well as between cancer cells and the extracellular matrix (ECM) are required. Therefore, component changes in the ECM within the tumor microevironment could play a fundamental impact on the metastatic process 16.
Cathepsin L acts as endopeptidase, which can degrade many intracellular and extracellular proteins 17 and thereby modify their function. Cathepsin L has been reported to be upregulated in a variety of malignancies including breast, lung, colon, gastric, head and neck carcinomas, melanomas, and gliomas 18-20. Furthermore, the overexpression of cathepsin L correlates positively with the malignancy states 21. Several studies have revealed that cathepsin L plays an important role in the processes of invasion and migration. The data show that cell-cell adhesion is diminished and degradation of ECM is increased when extracellular activity of cathepsin L increases 18, 19, 22, 23, which suggests that cathepsin L can increase metastatic tumor development. Studies have also found that inhibition of cathepsin L mRNA decreased tumor growth of murine myeloma24 and that anti-cathepsin L ScFv (single chain variable fragment) inhibited the tumorigenic and metastatic phenotype of human melanoma25.
Periostin is a unique and important ECM protein involved in cell development and adhesion26. Periostin has been shown to interact with many other ECM proteins, including fibronectin, collagen V, tenascin-C and periostin itself 27, 28. The epithelial-mesenchymal transition (EMT), which gives epithelial cancer cells invasive and metastatic potential, is one of the critical steps of tumor metastasis 29-32, and it has been shown that periostin can facilitate cell migration and differentiation during EMT 33. Periostin has also been found to be overexpressed in various types of human cancer, such as breast cancer, colon cancer, lung cancer, pancreatic cancer, prostate cancer, and ovarian cancer 15, 34-37. Most recently, periostin was found to be overexpressed in prostate cancer tissue compared to benign prostate hyperplasia 38. However, our study is the first to discover the overexpression of periostin in aggressive prostate cancer using quantitative glycoproteonics and mass spectrometry. Consistent with our glycoproteomics findings, periostin was found to promote tumor metastasis in colon cancer15, melanoma 39, head and neck squamous cell carcinoma, gastric cancer and lymph node metastases 40. For example, the average expression level of periostin is not increased in primary tumors of melanoma, whereas periostin overexpression is found in around 60% of metastatic melanoma tumors in the liver or lymph nodes 39.
Microfibrillar-associated protein-4 (MFAP4) is an ECM protein that is upregulated in aggressive prostate cancer. MFAP4 binds to collagen and contains a C-terminal fibrinogen-like domain and an N-terminal integrin-binding motif 41. The fibrinogen-like domain is responsible for the carbohydrate binding activity. The N-terminal part of MFAP4 includes one cysteine-residue and a ligand motif Arg-Gly-Asp (RGD) for cell surface integrins 41, 42. The function of MFAP4 is not yet clear. Human recombinant MFAP4 was reported to bind the collagen domain of surfactant protein A (SP-A), which suggests that MFAP4 may be involved in inflammatory processes 41. In a study of MFAP4 in catfish, Niu et al reported that MFAP4 may play a novel role in teleost immune responses43.
Type XII collagen is a homotrimer found in association with type I collagen. It is thought to modify the interactions between collagen I fibrils and the surrounding matrix 44.
In this study, the co-upregulation of cathepsin L and several ECM proteins, such as periostin, microfibrillar-associated protein-4, and collagen XII, in aggressive prostate tumor may indicate that there are some interactions among ECM proteins to facilitate the process of prostate tumor metastasis. Further studies will facilitate the understanding of the mechanisms involved in the function, regulation, and biological activities of cathepsin L and ECM proteins in tumor metastasis.
In addition, to avoid the tissue heterogeneity of using the whole tissue for comparative studies, we used the cell captured by laser microdissection from OCT-embedded tissue. However, there is a concern about using OCT-embedded frozen tissue in mass spectrometry analysis. OCT contains several chemicals to lower the freezing temperature, such as PEG, which may suppress the ionization and cause the contamination of mass spectrometry analysis. Somiari and coworkers presented the first high-throughput proteomic analysis of human breast infiltrating ductal carcinoma using OCT-embedded biopsies, using two dimensional gel electrophoresis (2-D DIGE) technology 14. Asomugha et al used the same technologies for identification of crystalline modifications in the human lens cortex and nucleus 45 . 2-D DIGE could separate the proteins from the OCT chemicals so as to avoid contaminating the mass spectrometers. However, to the best of our knowledge, directly analyzing OCT samples with mass spectrometers has not been reported. Our strategy of using solid phase extraction of N-glycopeptide overcomes this difficulty by chemical immobolization of glycopeptides and subsequentially removing OCT in the washing step so that the glycopeptides isolated from OCT-embedded tissues could be isotopically labeled and directly injected to mass spectrometry instruments for protein identification and quantification. In our study, glycopeptides were readily identified from the OCT-embedded frozen tissues and no contamination was detected in the spectrum of mass spec analysis. These results demonstrate: 1) the protein glycosylation is preserved in the OCT-embedded frozen tissues; 2) the OCT in the sample can be removed during the procedure of glycopeptide capture to eliminate the interference of OCT to the mass spectrometry analysis. Therefore, OCT-embedded frozen tissues can be used for glycoproteomic analysis and can provide invaluable specimen sources for identification of glycoprotein biomarkers.
Supplementary Material
Acknowledgment
This work was supported by federal funds from the National Institutes of Health, National Cancer Institute, the Early Detection Research Network (NIH/NCI/EDRN) grant U01CA152813. We also gratefully acknowledge the support of Dr. Robert Cole and Robert O’Meally from Johns Hopkins University for their assistance in mass spectrometry analysis and data processing. We thank Xiaer Sun from Johns Hopkins University for technical assistance, Dr. Dylan Back from Johns Hopkins University for evaluating the immunohistochemistry results, and Lauren Hurwitz for proof reading the manuscript.
References
- (1).Lankelma JM, Voorend DM, Barwari T, Koetsveld J, Van der Spek AH, De Porto AP, Van Rooijen G, Van Noorden CJ. Life Sci. 2010;86:225–233. doi: 10.1016/j.lfs.2009.11.016. [DOI] [PubMed] [Google Scholar]
- (2).Lang K, Drell T. L. t., Zaenker KS, Entschladen F. Recent Pat Anticancer Drug Discov. 2006;1:69–80. doi: 10.2174/157489206775246511. [DOI] [PubMed] [Google Scholar]
- (3).Chan DW, Bruzek DJ, Oesterling JE, Rock RC, Walsh PC. Clin Chem. 1987;33:1916–1920. [PubMed] [Google Scholar]
- (4).Presti JC., Jr. Nat Clin Pract Urol. 2007;4:505–511. doi: 10.1038/ncpuro0887. [DOI] [PubMed] [Google Scholar]
- (5).Keetch DW, Catalona WJ, Smith DS. J Urol. 1994;151:1571–1574. doi: 10.1016/s0022-5347(17)35304-1. [DOI] [PubMed] [Google Scholar]
- (6).Andriole GL, Crawford ED, Grubb RL, 3rd, Buys SS, Chia D, Church TR, Fouad MN, Gelmann EP, Kvale PA, Reding DJ, Weissfeld JL, Yokochi LA, O’Brien B, Clapp JD, Rathmell JM, Riley TL, Hayes RB, Kramer BS, Izmirlian G, Miller AB, Pinsky PF, Prorok PC, Gohagan JK, Berg CD. N Engl J Med. 2009;360:1310–1319. doi: 10.1056/NEJMoa0810696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Partin AW, Kattan MW, Subong EN, Walsh PC, Wojno KJ, Oesterling JE, Scardino PT, Pearson JD. JAMA. 1997;277:1445–1451. [PubMed] [Google Scholar]
- (8).Makarov DV, Trock BJ, Humphreys EB, Mangold LA, Walsh PC, Epstein JI, Partin AW. Urology. 2007;69:1095–1101. doi: 10.1016/j.urology.2007.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zhang H, Chan DW. Cancer Epidemiol Biomarkers Prev. 2007;16:1915–1917. doi: 10.1158/1055-9965.EPI-07-0420. [DOI] [PubMed] [Google Scholar]
- (10).Turbett GR, Sellner LN. Diagn Mol Pathol. 1997;6:298–303. doi: 10.1097/00019606-199710000-00009. [DOI] [PubMed] [Google Scholar]
- (11).Schwartz SA, Reyzer ML, Caprioli RM. J Mass Spectrom. 2003;38:699–708. doi: 10.1002/jms.505. [DOI] [PubMed] [Google Scholar]
- (12).Zhang H, Li XJ, Martin DB, Aebersold R. Nat Biotechnol. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- (13).Tian Y, Zhou Y, Elliott S, Aebersold R, Zhang H. Nat Protoc. 2007;2:334–339. doi: 10.1038/nprot.2007.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Somiari RI, Sullivan A, Russell S, Somiari S, Hu H, Jordan R, George A, Katenhusen R, Buchowiecka A, Arciero C, Brzeski H, Hooke J, Shriver C. Proteomics. 2003;3:1863–1873. doi: 10.1002/pmic.200300560. [DOI] [PubMed] [Google Scholar]
- (15).Bao S, Ouyang G, Bai X, Huang Z, Ma C, Liu M, Shao R, Anderson RM, Rich JN, Wang XF. Cancer Cell. 2004;5:329–339. doi: 10.1016/s1535-6108(04)00081-9. [DOI] [PubMed] [Google Scholar]
- (16).Ma C, Rong Y, Radiloff DR, Datto MB, Centeno B, Bao S, Cheng AW, Lin F, Jiang S, Yeatman TJ, Wang XF. Genes Dev. 2008;22:308–321. doi: 10.1101/gad.1632008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gal S, Gottesman MM. Biochem Biophys Res Commun. 1986;139:156–162. doi: 10.1016/s0006-291x(86)80093-6. [DOI] [PubMed] [Google Scholar]
- (18).Gocheva V, Joyce JA. Cell Cycle. 2007;6:60–64. doi: 10.4161/cc.6.1.3669. [DOI] [PubMed] [Google Scholar]
- (19).Jedeszko C, Sloane BF. Biol Chem. 2004;385:1017–1027. doi: 10.1515/BC.2004.132. [DOI] [PubMed] [Google Scholar]
- (20).Kos J, Stabuc B, Schweiger A, Krasovec M, Cimerman N, Kopitar-Jerala N, Vrhovec I. Clin Cancer Res. 1997;3:1815–1822. [PubMed] [Google Scholar]
- (21).Skrzydlewska E, Sulkowska M, Koda M, Sulkowski S. World J Gastroenterol. 2005;11:1251–1266. doi: 10.3748/wjg.v11.i9.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Mohamed MM, Sloane BF. Nat Rev Cancer. 2006;6:764–775. doi: 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- (23).Strojnik T, Kavalar R, Trinkaus M, Lah TT. Cancer Detect Prev. 2005;29:448–455. doi: 10.1016/j.cdp.2005.07.006. [DOI] [PubMed] [Google Scholar]
- (24).Kirschke H, Eerola R, Hopsu-Havu VK, Bromme D, Vuorio E. Eur J Cancer. 2000;36:787–795. doi: 10.1016/s0959-8049(00)00014-9. [DOI] [PubMed] [Google Scholar]
- (25).Rousselet N, Mills L, Jean D, Tellez C, Bar-Eli M, Frade R. Cancer Res. 2004;64:146–151. doi: 10.1158/0008-5472.can-03-1717. [DOI] [PubMed] [Google Scholar]
- (26).Ruan K, Bao S, Ouyang G. Cell Mol Life Sci. 2009;66:2219–2230. doi: 10.1007/s00018-009-0013-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Norris RA, Damon B, Mironov V, Kasyanov V, Ramamurthi A, Moreno-Rodriguez R, Trusk T, Potts JD, Goodwin RL, Davis J, Hoffman S, Wen X, Sugi Y, Kern CB, Mjaatvedt CH, Turner DK, Oka T, Conway SJ, Molkentin JD, Forgacs G, Markwald RR. J Cell Biochem. 2007;101:695–711. doi: 10.1002/jcb.21224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Takayama G, Arima K, Kanaji T, Toda S, Tanaka H, Shoji S, McKenzie AN, Nagai H, Hotokebuchi T, Izuhara K. J Allergy Clin Immunol. 2006;118:98–104. doi: 10.1016/j.jaci.2006.02.046. [DOI] [PubMed] [Google Scholar]
- (29).Peinado H, Olmeda D, Cano A. Nat Rev Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- (30).Thiery JP, Sleeman JP. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- (31).Thiery JP. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- (32).Huber MA, Kraut N, Beug H. Curr Opin Cell Biol. 2005;17:548–558. doi: 10.1016/j.ceb.2005.08.001. [DOI] [PubMed] [Google Scholar]
- (33).Lindsley A, Snider P, Zhou H, Rogers R, Wang J, Olaopa M, Kruzynska-Frejtag A, Koushik SV, Lilly B, Burch JB, Firulli AB, Conway SJ. Dev Biol. 2007;307:340–355. doi: 10.1016/j.ydbio.2007.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kudo Y, Siriwardena BS, Hatano H, Ogawa I, Takata T. Histol Histopathol. 2007;22:1167–1174. doi: 10.14670/HH-22.1167. [DOI] [PubMed] [Google Scholar]
- (35).Puglisi F, Puppin C, Pegolo E, Andreetta C, Pascoletti G, D’Aurizio F, Pandolfi M, Fasola G, Piga A, Damante G, Di Loreto C. J Clin Pathol. 2008;61:494–498. doi: 10.1136/jcp.2007.052506. [DOI] [PubMed] [Google Scholar]
- (36).Tischler V, Fritzsche FR, Wild PJ, Stephan C, Seifert HH, Riener MO, Hermanns T, Mortezavi A, Gerhardt J, Schraml P, Jung K, Moch H, Soltermann A, Kristiansen G. BMC Cancer. 2010;10:273. doi: 10.1186/1471-2407-10-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Tsunoda T, Furusato B, Takashima Y, Ravulapalli S, Dobi A, Srivastava S, McLeod DG, Sesterhenn IA, Ornstein DK, Shirasawa S. Prostate. 2009;69:1398–1403. doi: 10.1002/pros.20988. [DOI] [PubMed] [Google Scholar]
- (38).Sun C, Song C, Ma Z, Xu K, Zhang Y, Jin H, Tong S, Ding W, Xia G, Ding Q. Proteome Sci. 2011;9:22. doi: 10.1186/1477-5956-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Tilman G, Mattiussi M, Brasseur F, van Baren N, Decottignies A. Mol Cancer. 2007;6:80. doi: 10.1186/1476-4598-6-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Li JS, Sun GW, Wei XY, Tang WH. World J Gastroenterol. 2007;13:5261–5266. doi: 10.3748/wjg.v13.i39.5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Schlosser A, Thomsen T, Shipley JM, Hein PW, Brasch F, Tornoe I, Nielsen O, Skjodt K, Palaniyar N, Steinhilber W, McCormack FX, Holmskov U. Scand J Immunol. 2006;64:104–116. doi: 10.1111/j.1365-3083.2006.01778.x. [DOI] [PubMed] [Google Scholar]
- (42).Lausen M, Lynch N, Schlosser A, Tornoe I, Saekmose SG, Teisner B, Willis AC, Crouch E, Schwaeble W, Holmskov U. J Biol Chem. 1999;274:32234–32240. doi: 10.1074/jbc.274.45.32234. [DOI] [PubMed] [Google Scholar]
- (43).Niu D, Peatman E, Liu H, Lu J, Kucuktas H, Liu S, Sun F, Zhang H, Feng T, Zhou Z, Terhune J, Waldbieser G, Li J, Liu Z. Dev Comp Immunol. 2011;35:568–579. doi: 10.1016/j.dci.2011.01.002. [DOI] [PubMed] [Google Scholar]
- (44).Veit G, Hansen U, Keene DR, Bruckner P, Chiquet-Ehrismann R, Chiquet M, Koch M. J Biol Chem. 2006;281:27461–27470. doi: 10.1074/jbc.M603147200. [DOI] [PubMed] [Google Scholar]
- (45).Asomugha CO, Gupta R, Srivastava OP. Mol Vis. 2010;16:476–494. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.