

Abstract
Chlamydia are obligate intracellular bacterial pathogens that cause a variety of diseases. Likemany Gram-negative bacteria, they employ type III secretion systems (T3SS) for invasion, establishing and maintaining their unique intracellular niche, and possibly cellular exit. Computational structure prediction indicated that ORF CT584 is homologous to other T3SS needle tip proteins. Tip proteins have been shown to be localized to the extracellular end of the T3SS needle and play a key role in controlling secretion of effector proteins. We have previously demonstrated that T3SS needle tip proteins from different bacteria share many biophysical characteristics. To support the hypothesis that CT584 is a T3SS needle tip protein, biophysical properties of CT584 were explored as a function of pH and temperature, using spectroscopic techniques. Far-UV circular dichroism, Fourier transform infrared spectroscopy, UV absorbance spectroscopy, ANS extrinsic fluorescence, turbidity, right angle static light scattering, and analytical ultracentrifugation were all employed to monitor the secondary, tertiary, quaternary, and aggregation behavior of this protein. An empirical phase diagram approach is also employed to facilitate such comparisons. These analyses demonstrate that CT584 shares many biophysical characteristics with other T3SS needle tip proteins. These data support the hypothesis that CT584 is a member of the same functional family, although future biologic analyses are required.
Chlamydia infections are common throughout the world and are associated with a range of diseases, including pneumonia, atherosclerosis, pelvic inflammatory disease, reproductive sterility, and blindness (1, 2). Chlamydia trachomatis alone is responsible for an estimated 92 million sexually transmitted infections a year (3). Despite this prevalence, much is still unknown about the pathogenesis of these obligate intracellular pathogens. Chlamydia invade eukaryotic cells as a metabolically inactive elementary body (EB) and form an inclusion which evades fusion with the late endosome/lysosome endocytic vesicles (4–7). In this compartment, EB convert into metabolically active reticulate bodies (RB) which replicate by binary fission (4). After approximately 18 h, RB begin differentiating back into EB and are eventually exocytosed to repeat the infection cycle. The pathogens are also known to exist in a persistent form inside host cells presumably to evade immune system responses (2, 8, 9).
Like many pathogenic Gram-negative bacteria, Chlamydia use type III secretion systems (T3SSs)1 to facilitate infection and exploitation of eukaryotic cells (10–14). In particular, the T3SS of Chlamydia is implicated in host cell invasion, intracellular trafficking, and maintenance of the unique intracellular niche of Chlamydia. Early evidence for the T3SS in these bacteria was based primarily on genetic information and electron microscopy where protrusions observed on the bacterial surface are presumed to be T3SS needles (13–16). The lack of a system for genetic exchange in C. trachomatis has hampered functional characterization of T3SS components. Furthermore, most of the proteins encoded by Chlamydia do not share sufficient primary sequence homology to support functional assignment, including many of the T3SS proteins. Despite this, numerous proteins have been demonstrated to be secreted in heterologous systems (e.g., Shigella) in a T3SS-dependent manner and/or are present in the cytosol (or cytosolic face of inclusion) of Chlamydia-infected cells, providing support for identification of effector molecules and utility of the T3SS in Chlamydia (17–23). Unlike most bacteria possessing T3SSs, chlamydial T3SS genes are found at multiple loci throughout the genome which has also made identification of functional components more difficult (10, 24).
T3SSs are composed of a basal body and a needle that is responsible for the translocation of effector proteins from the bacterial cytosol to the surface of a host cell or into its cytoplasm (12, 25, 26). This secretion apparatus is comprised of more than 20 distinct proteins and has a possible evolutionary relationship to bacterial flagella. One class of protein in this system of particular interest is the tip protein which is so named for its propensity to localize to the extracellular needle tip (27–30). These proteins have been shown to regulate secretion of the other effector proteins and thus play a pivotal role in the pathogenesis of the bacteria (27). Furthermore, T3SS needle tip proteins, such as LcrV from Yersinia pestis, have been demonstrated to be excellent protective vaccinogens (31–33). In addition, some tip proteins, such as IpaD from Shigella flexneri, may have additional roles, though these functions are not fully understood.
Previously, we have employed biophysical tools to understand the characteristics of T3SS needle tip proteins from proteobacteria. This work revealed protein subfamilies based on their response to pH and temperature (34–36). These distinctions were made after creation of empirical phase diagrams (EPDs). EPDs summarize data from disparate techniques using matrix mathematics and provide a global representation of results (37, 38). These diagrams are two-dimensional figures with respect to pH and temperature with different physical states expressed in terms of color. Past work has shown the utility of EPDs in highlighting structure–function similarities in proteins with little primary sequence homology (36, 39).
The identity of the T3SS needle tip protein is presently unknown for Chlamydia. As presented here, computational structure prediction and homology search indicates that a C. trachomatis ORF (CT584), a protein with no primary sequence homology to any other protein with a determined function, exhibits predicted structural homology to T3SS needle tip proteins. As such, we hypothesize that CT584 is a functional T3SS needle tip protein. To support this hypothesis, we employed biophysical analyses, including construction of an EPD, of CT584 for comparison to characteristics shared by previously analyzed T3SS needle tip proteins.
MATERIALS AND METHODS
CT584 Cloning, Expression, and Purification
CT584 was PCR amplified from C. trachomatis (L2/434/Bu) using primers 5′-GGA ATT CCA TAT GAC GAC GAA ACC CAA AAC TC and 5′-CGC CGG CCG CCA CAG ATT TCG TTA ATT CTT C (Integrated DNA technologies, Coralville, IA). Purified amplicons were digested with restriction enzymes (NdeI and EagI) and ligated into pET21b (Novagen). Isogenic transformants were selected and propagated in Escherichia coli TOP10 (Invitrogen, Carlsbad, CA). Plasmid constructs were confirmed via DNA sequencing (University of Kansas, DNA Sequencing Facility). For protein expression, plasmids containing CT584 were freshly transformed into E. coli BL21(DE3) (Bioline, Boston, MA) and grown at 37 °C in 1 L of Luria-Bertani broth with 100 μg/mL ampicillin. Cells were grown to an OD600 of 0.7 prior to 1 mM isopropyl β-D-thiogalactopyranoside being added. After induction for 4 h, cells were centrifuged and pellets frozen. Thawed cells were resuspended and lysed using sonication in a Tris buffer [40 mM Tris, 500 mM NaCl, and 5%glycerol (pH 8.0)]. Six-Histagged CT584 was purified using a cobalt metal affinity resin (Talon beads, Pierce) and gravity filtration column. Protein was eluted in Tris buffer containing 500 mM imidazole, and affinity-purified CT584 was applied to a Superdex 200 gel filtration column (GE Healthcare) employing an AKTA FPLC system (GE Healthcare) in Tris buffer. Protein-containing fractions were collected at approximately 150 kDa. Coomassie-stained sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) of the protein sample revealed the expected 21 kDa monomer at >95% purity.
Ultraviolet Absorption Spectroscopy
Spectra for each protein were analyzed from 200 to 400 nm with an Agilent 8453 diode-array spectrophotometer equipped with a Peltier temperature controller. The experimental spectral resolution was 1 nm, and the integration time was set to 25 s. Spectra were recorded from 10 to 90 °C in 2.5 °C increments with a 5 min equilibration time at each temperature. The resultant absorbance data were analyzed using Agilent UV–visible ChemStation software by fitting spectra to a fifth-degree Savitzky–Golay polynomial followed by a second-derivative calculation. This derivative spectrum was subjected to a 99-point interpolation which produces an effective resolution of 0.01 nm.
Far-UV Circular Dichroism
Circular dichroism (CD) spectra were recorded from 190 to 260 nm with a 1 nm resolution at 20 °C. Spectra were recorded with a Jasco (Tokyo, Japan) J-720 spectrometer equipped with a Peltier-controlled sample holder. For thermal stress experiments, CD data were acquired at 222 nmfrom10 to85 °C at a rate of 15 °C/h. This wavelength was chosen to monitor changes in the protein’s α-helical content. CD signals were converted to molar ellipticity using the Jasco software.
Fluorescence Spectroscopy
A PTI QM-1 spectrophotometer was used to collect fluorescence data. For intrinsic Tyr fluorescence, the excitation wavelength was 275 nm and emission was recorded from 280 to 380 nm in 1 nm increments with a 0.5 s integration time. In the case of ANS fluorescence, samples were excited at 372 nm with emission spectra recorded from 400 to 600 nm in 2 nm intervals. ANS was added from a 2 mg/mL ethanol solution to CT584 to yield a 10:1 molar ANS: protein ratio. In both cases, emission was recorded by a photomultiplier tube (PMT) 90° from the angle of incident light. Approximately 1 mL of the samples was pipetted into a 1 cm path length quartz cuvette, and data were collected from 10 to 85 °C in 2.5 °C increments. Light scattering was recorded simultaneously via intrinsic fluorescence acquisition with a second photomultiplier tube located 180° to the emission acquisition at 275 nm. Blank solutions containing all of the chemical components except protein were also measured, and the resultant spectra were subtracted from the protein emission spectra. Peak positions were calculated in Origin by fitting the data to a secondary polynomial curve followed by a first-derivative calculation.
Empirical Phase Diagrams
Data from all the techniques described above were combined to create empirical phase diagrams (EPDs). The underlying theory and mathematics employed have been described in detail previously (37). In brief, data from the biophysical studies were formed into vectors with each vector representing a distinct pH–temperature setting. A density matrix is formed by assembling these vectors which can be manipulated to calculate eigenvalues. The three largest eigenvalues are selected and employed to create new eigenvectors that expand a truncated version of the data set in three dimensions. Each dimension is assigned a color (red, green, and blue), and the data are expressed in an EPD by plotting these color assignments. Thus, color assignments are arbitrary. No equilibria are implied across “apparent” phase boundaries, and these are therefore not thermodynamic phase diagrams but are empirical in nature.
Fourier Transform Infrared Spectroscopy
A Bruker (Billerica, MA) Tensor 27 FTIR spectrometer equipped with a ZnSe attenuated total reflectance (ATR) plate (Pike Technologies, Madison, WI) was used to collect Fourier transform infrared (FTIR) spectra at room temperature (25 °C). Data were collected over 256 composite scans with a resolution of 4 cm−1. The samples were analyzed in 20 mM citrate/phosphate buffer at 1.8 mg/mL. Water spectra were subtracted with the OPUS spectroscopy software associated with the spectrometer, and data were further analyzed using GRAMS/AI (Galactic, Inc.). The baseline was corrected between 1800 and 2400 cm−1, and the spectra were then smoothed using a five-point Savitsky–Golay function. Following Fourier self-deconvolution, the amide I/II bands of the spectrum (1500–1700 cm−1) were decomposed using a peaks fitting tool which employs Gaussian band profiles. The second-derivative spectra from 1500 to 1700 cm−1 provided the initial peak positions for fitting.
Analytical Ultracentrifugation
Protein molecular mass and oligomerization state were assessed using a Beckman Coulter (Palo Alto, CA) XL-I analytical ultracentrifuge in both sedimentation velocity and equilibrium modes. For sedimentation velocity, CT584 was diluted to 0.2 and 0.5 mg/mL in citrate phosphate buffer and injected into two-channel, 12 mm optical path length cells. A four-hole An60Ti rotor at 10 °C housed the cells, and samples were run in triplicate at 36000 rpm for 8 h. Sedimentation velocity analysis was conducted using SEDFIT (version 11.71) which was developed at the National Institutes of Health by P. Schuck. Continuous c(s) distribution analysis was employed, and both the Simplex and Marquardt–Levenberg models were tested to ensure the final fit was in a global minimum. Both time and radial invariant noise were removed. In equilibrium mode, the same settings were used as before except six-channel centerpieces were included in the assembly and the rotor speed was 17000 rpm, with five measurements taken hourly after a 12 h delay. Data were analyzed using SEDPHAT (version 6.21) with a monomer–n-mer self-association model giving the best fit both for global reduced χ2 and for local root-mean-square deviation (rmsd) values.
RESULTS
Computational Structure Prediction and Homology Comparison
Identifying candidate T3SS needle tip proteins using primary sequence homology has not proven to be useful in Chlamydia. To identify potential T3SS needle tip candidates, computational structure prediction followed by a structural homology search was employed. I-TASSER is a hierarchal protein structure, molecular modeling program that incorporates a secondary structure-enhanced Profile–Profile Alignment and iterative implementation of the Threading Assembly Refinement program (40). This computational modeling program has proven to be the most effective program at accurately predicting protein three-dimensional structure blindly from primary sequence (41, 42). Using this program, predicted structures of proteins with unknown function can then be used for comparison to structures of proteins for which the function has previously been determined. Similar to primary sequence homology BLAST analysis, this approach can provide candidates for subsequent functional analysis.
One of the C. trachomatis proteins that exhibited predicted structural similarity to other T3SS needle tip proteins was ORF CT584. The I-TASSER-predicted structure of CT584 (Figure 1) was compared pairwise (DaliLite) to each of the three determined T3SS needle tip protein structures known: IpaD (PDB entry 2J0O), BipD (PDB entry 2IXR), and LcrV (PDB entry 1R6F) (43). This pairwise analysis will generate a Z score indicative of the measure of quality for the alignment, with scores between 8 and 20 supporting an intermediate level of confidence that two proteins are homologous. For reference, comparison of IpaD and BipD structures returns a relatively high Z score of 17.3 with 220 amino acids aligned despite only 27% sequence identity. In contrast, IpaD or BipD compared to LcrV results in a very low Z score of <3.9, reflecting the structural differences between these functionally similar proteins. Comparison of the predicted structure of CT584 to the structure of IpaD revealed an intermediate Z score of 8.4. Only 108 amino acids of the predicted CT584 structure (of 184 total) were aligned to IpaD, although the aligned region is structured very similarly (Figure 1). Given the structural homology between IpaD and BipD, it was expected that a pairwise comparison of CT584 with BipD should exhibit some homology. This analysis resulted in a Z score of 6.9 and a region of structural similarity of 119 amino acids. In contrast to the results observed for IpaD and BipD, the Z score for the LcrV comparison to the predicted structure of CT584 was 2.1, close to a no confidence score (<2.0).
Figure 1.
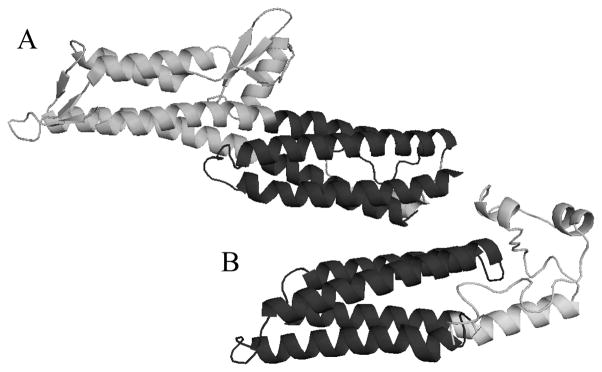
Protein ribbon structure of IpaD and I-TASSER-predicted structure of C. trachomatis CT584. (A) IpaD from S. flexneri (PDB entry 2J0O) and (B) I-TASSER structural prediction of CT584. The region of shared structural similarity is colored dark gray.
Overall, structural prediction of CT584 and comparison to known T3SS needle tip protein structures provided an intermediate level of confidence that CT584 is structurally similar to T3SS needle proteins, specifically, IpaD and BipD. The predicted structure of CT584 forms two α-helical coiled-coil domains (Figure 1). While this is a widely distributed structural feature in biology, all of the needle tip proteins contain this structure (29, 44, 45). Additionally, unique N- and C-terminal domains flank the coiled-coil segment of needle tip proteins. The predicted structure of CT584 also contains an N- and C-terminal region that flanks the coiled-coil domain.
Size exclusion chromatography indicated that CT584 is forming a higher-order complex (~150 kDa), possibly a hexamer or heptamer. This molecular mass and assembly of oligomers are very similar to those observed for natively purified T3SS needle tip proteins, including IpaD (29, 46). To provide further support for the hypothesis that CT584 is a Chlamydia T3SS needle tip protein, biophysical characterization of this protein was performed and the results were compared to prior studies that revealed protein subfamilies on the basis of their response to pH and temperature (34–36).
Far-UV Circular Dichroism
To characterize the secondary structure of CT584, circular dichroism (CD) and FTIR spectroscopy were employed. Both of these techniques permit quantitation of α-helical and β-sheet content in addition to turns and disordered structural elements (47, 48). Circular dichroism spectra from 190 to 260 nm at 10 °C in pH 7 buffer indicate that, under physiologic conditions, the protein has primarily α-helical structure (Figure 2A). This conclusion is based on the presence of two negative peaks located at 208 and 222 nm. Secondary structure analysis using Dichroweb estimates that the protein is 55%α-helical. CD spectra were measured frompH3 to 8, and the strongest minima were recorded at pH 3, suggesting that maximum secondary structure exists under more acidic conditions. In contrast, pH 6 possessed the least α-helical content, while pH 8 had minima nearly as strong as those of pH 3 (not illustrated).
Figure 2.

Far-UV circular dichroism. Spectra from 190 to 260 nm expressed in terms of molar ellipticity (A) and pH vs temperature trends at 222 nm monitored from 10 to 85 °C (B). Individual data points are the average of three trials, and the standard deviation for each sample was less than 8%ofmeasured values: (■) pH3, (○) 4, (▲) 5, (▽) 6, (◆) 7, and (left-pointing triangles) 8.
In addition to acquiring full-length spectra at 10 °C, we monitored the intensity of the minimum at 222 nm as a function of temperature from10 to 85 °C in 2.5 °C increments (Figure 2B). By thermally stressing the protein in this manner, we can ascertain the relative stability of the secondary structure. CT584 shows only small changes in secondary structure at pH 3 and 6 compared to other conditions. Transitions from pH 4 to 8 show increasing transition onsets (T0), with T0 occurring near 20 °C at pH4 and at approximately 42 °C in the pH8 environment. The most distinctive trend is at pH 5 where there appears to be a biphasic transition initiated at 27 °C. This suggests the possible existence of at least two independently folding domains.
FTIR
Fourier transform infrared spectroscopy provides an orthogonal method with which to measure protein secondary structure and was performed here at pH7 and 25 °C. We can estimate secondary structural elements by deconvoluting the amide I band (Figure 3) (48). Decomposition of the amide I and II bands was accomplished using Fourier self-deconvolution with the second-derivative trace of the spectra guiding initial peak position estimates. From this analysis, six peaks were clearly identified at 1695.5, 1683.7, 1674.2, 1652.9, 1635.9, and 1625.5 cm−1. The first two peaks are generally associated with β-turns, while peaks at 1674.7 cm−1 and the two below 1650 cm−1 are attributed to β-sheets (48). The dominant peak, located at 1652.9 cm−1, can be assigned to α-helical content. On the basis of the relative areas, helix comprises 60% of the protein’s secondary structure which is within 5% of estimates from CD spectra. The β-sheet contribution to the amide I peak is estimated to be approximately 30%which compares with only 17%in CD analysis.
Figure 3.

Deconvoluted amide I and II bands in solution measured by ATR-FTIR at 1.8 mg/mL and pH 7. The Fourier self-deconvolution bandwidth and enhancement parameters were 21 and 2.5, respectively. Six peaks were identified as secondary structure elements at 1695.5, 1683.7, 1674.2, 1652.9, 1635.9, and 1625.5 cm−1.
Second-Derivative UV Absorption Spectroscopy
Tertiary structure can be monitored through a variety of spectroscopic techniques, including intrinsic fluorescence, near-UV CD, and second-derivative UV absorption spectroscopy. Intrinsic fluorescence is not useful here because of the insensitivity of Tyr residues to changes in environment. Second-derivative UV absorption spectroscopy provides a fairly global picture of tertiary structure changes since all three aromatic amino acid residues can be simultaneously monitored (49, 50). CT584 has 13 Phe, one Tyr, and no Trp residues. Representative second derivative negative peaks are shown in Figure 4 near 265 and 269 nm, corresponding to Phe and Tyr, respectively. The shallow slope seen at lower temperatures in all pH environments is not due to alterations in tertiary structure but rather to dynamic aspects of the protein’s behavior and suggests burial and immobilization of the Phe residues. This phenomenon is discussed in detail elsewhere (in publication). Initial peak positions are nearly identical under all pH conditions and are slightly longer (~0.5–1 nm) than those observed in the free amino acid, indicating that on average the aromatic residues are buried (37, 49). The Phe negative peak (Figure 4A) exhibits red shifts at pH 7 and 8 which manifest a high error associated with aggregation. Confirmation of this protein association is observed in light scattering and turbidity data (Figure 5C, D). At pH5 and 6, initial blue shifts lead to erratic data with a large error which again is probably the result of protein aggregation. The two most acidic pH environments examined display broad transitions to longer wavelengths. The Tyr peak centered around 269 nm displays little discernible transition at pH 3 and 4, but distinct blue shifts accompanied by a larger error are present at pH 5 and 6. These shifts to a shorter wavelength indicate a more solvent-exposed state for the Tyr residue. Aggregation, which coincides with this transition, again accounts for the increased variability. Red shifts appear at pH 7 and 8 with greater variability observed at pH 7.
Figure 4.

Second-derivative UV absorption spectroscopy. Absorption peak position minima centered around 253 nm(Phe, A), 259 nm (Phe, B), 265 nm (Phe, C), and 269 nm (Tyr, D) are presented as a function of temperature from10 to 85 °C. Error bars are the standard deviations from at least three trials: (■) pH3, (○) 4, (▲) 5, (▽) 6, (◆) 7, and (left-pointing triangles) 8.
Figure 5.

IpaD ANS and light scattering studies. ANS fluorescence wavelength peak position (A) and intensity (B) monitored from10 to 85 °C. Light scattering intensities at 275 nm (C) and optical densities at 350 nm were measured under the same conditions. Error bars are the standard deviations from at least three trials: (■) pH3, (○) 4, (▲) 5, (▽) 6, (◆) 7, and (left-pointing triangles) 8.
Extrinsic Fluorescence via ANS
Extrinsic fluorescence using ANS as a probe was performed to monitor the appearance of apolar binding sites inCT584 with respect to both temperature and wavelength. Initial peak positions are nearly identical at pH 4–6 but occur at shorter wavelengths under more acidic or basic conditions (Figure 5A). Results at pH 7 and 8 display low-temperature red shifts which begin at~15 °C and are followed by transitions to shorter wavelengths at midrange temperatures. Trends in the middle pH range (pH 4–6) manifest less pronounced blue shifts at lower temperatures. Peak position wavelength trends are less revealing at high temperatures where error, particularly at pH 5 and 6, precludes definitive conclusions. In contrast, at pH 3 the general trend reveals a biphasic red shift suggesting the existence of two independent domains.
Structural changes in CT584 can also be observed through examination of the fluorescence intensity of the ANS molecule. ANS interacts strongly with apolar regions in proteins. As the structure of the protein begins to loosen, the dye partitions into the apolar core, causing an increase in its fluorescence quantum yield (51). Because of the negative charge of ANS in solution, there may also be some ionic interactions that might be more expected under acidic circumstances (52). Only broad, comparatively small transitions can be observed below pH 5. From pH 5 to 8, however, transitions occur at increasing temperatures with respect to alkalinity (Figure 5B). T0 in the pH 8 environment begins at approximately 50 °C, which is the highest transition temperature observed. The initial fluorescence signal is strongest at pH5, indicating amore disrupted three-dimensional structure. In contrast, the fluorescence intensity at pH 8 is nearly 1 order of magnitude lower than that under any other condition. This lower quantum yield may be due in part to the pI of the protein (5.61) which would cause it to be more negatively charged in an alkaline environment. Specifically, this pH is sufficiently far from the pKa of the four His residues and sufficiently close to the pKa of the three Cys residues to expect a significant alteration in charge state compared to that even at pH 7. Even with such a change in ionization state, however, it is likely that the tertiary structure at pH 8 is adequately compact to preclude the level of ANS binding observed under more acidic conditions.
Aggregation
In addition to secondary and tertiary structure changes, the stability of proteins can also be measured in terms of their aggregation behavior. Light scattering provides insights into the formation of higher-order aggregates and is more sensitive than turbidity measurements. Alterations in static light scattering intensity may in some cases be the result of variation in the refractive index of scatterers. Increases in scattering are, however, most often a consequence of a change in association behavior. In this case, transition temperatures in light scattering data increase with respect to pH with the exception of those at pH 3 and 4 (Figure 5C). At pH 3, the intensity of the initial light scattering signal is less than half that observed under other conditions and remains smaller than the decrease in intensity seen at ~40 °C at higher pH. The initial decrease in intensity observed at pH 4–8 is probably due to changes in the oligomerization behavior discussed in Analytical Ultracentrifugation (Figure 6). Optical density data, recorded at 350 nm during the UV absorption experiments, also indicate an increasing transition temperature occurring with higher pH (Figure 5D). The exception again is environments below pH 5. The OD transition onsets for all pH conditions begin at higher temperatures compared to the right angle light scattering results. This phenomenon is attributed to the fact that light scattering is a more sensitive measure of aggregation.
Figure 6.
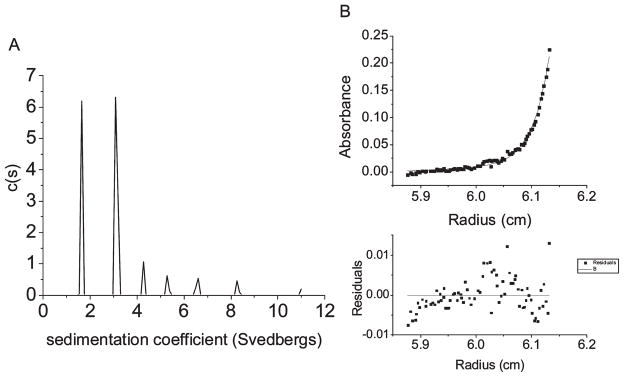
Analytical ultracentrifugation. Sedimentation velocity analysis for CT584 at 0.5 mg/mL and 36000 rpm at 10 °C using a continuous c(s) distribution model from SEDFIT (A). Sedimentation equilibrium results at 10 °C and 18000 rpm using a monomer to pentamer self-association model in SEDPHAT (B).
Analytical Ultracentrifugation
To gain a better understanding of the presence or absence of self-association events that may be occurring in CT584, the protein was analyzed using analytical ultracentrifugation (AUC) at 10 °C. Some tip proteins are known to oligomerize in their native state and are reported to form pentamers or hexamers when localized to the needle tip (29, 46, 53, 54). Samples of CT584 were examined in both sedimentation velocity and equilibrium modes using interference and absorption optical systems. Samples were run at 36000 rpm at 0.5 and 1.0mg/mL in velocity runs. In Figure 6A, the results from a continuous c(s) distribution analysis are shown at 0.5 mg/mL. The peak at 1.6 S corresponds to approximately 32 kDa, while the peak at 3.1 S is just over 88 kDa. When the concentration was doubled to 1mg/mL, the size of the first peak fell to 29 kDa while the sizes of the higher-order species were also altered. Since the size of monomeric CT584 is just over 21.1 kDa and there is a change in the apparent size of species when concentration is varied, the oligomerization kinetics must be fast on the time scale of sedimentation (55). Fast kinetics skews the apparent sizes of the species of interest in velocity analysis. For this reason, we chose to analyze CT584 using sedimentation equilibrium as well.
Equilibrium runs were conducted at 0.5mg/mL and 10 °C with a rotor speed of 17000 rpm. The data were fit to multiple models. Despite the complex self-association phenomenon observed, the best fits occurred with the monomer–n-mer self-association model (Figure 6B). A larger nonparticipating species set at the size of the smallest higher-order aggregate (4.3 S) was included in this analysis to improve the fit. The result shown in Figure 6B is monomer to pentamer with the residuals shown below. The data were fit to four different equilibrium conditions, and the global reduced χ2 was 1.4. The pentameric and hexameric models both gave good fits. On the basis of the size of the second peak in the sedimentation velocity analysis, however, it is most likely that the correct model is monomer in equilibrium with pentamer with small amounts of higher-order aggregation occurring.
Empirical Phase Diagrams
Empirical phase diagrams (EPDs) provide an effective way to summarize data from multiple techniques and enable one to gain amore global picture of the effect of temperature and pH on protein structure (37–39, 56). In addition, proteins with similar function but little sequence homology generally have similar EPDs which highlights the utility of this technique in considering structure–function relationships (36, 39). In the case of CT584, even at low temperatures (i.e., 10 °C), there are conformational differences from pH 6 to 8 (Figure 7C). The difference between pH 7 and 8 is primarily due to ANS intensity data, while pH 6 differs because of the CD results. The changes at lower temperatures can be ascribed to these data sets because the differences in results from these techniques are significantly larger than those from the other parent data sets. A small transition occurs at pH7 near 15 °C that arises from a red shift in ANS wavelength peak position. If this smaller transition is ignored, the trend of increasing stability with respect to pH above pH 4 is in agreement among the multiple techniques. Acidic environments (pH3 and 4) display more subtle changes in the empirical phase diagram. At pH 3, for example, the greater amount of secondary structure and the more buried aromatic residues seen in UV absorption data suggest that pH 3 may be more stable than any other condition. The dramatic changes in color seen at high temperatures near the protein pI are generally indicative of aggregation and are in agreement with the parent data.
Figure 7.

Empirical phase diagrams. The empirical phase diagram is a way of summarizing data from different techniques and was created using all of the biophysical techniques for CT584 (C). For the sake of comparison, previously reported (36) empirical phase diagrams for IpaD (A) and LcrV (B) are also presented.
DISCUSSION
We conducted this biophysical characterization of CT584 to explore the possibility that it functions as the tip protein in the T3SS of C. trachomatis. Tip proteins from other T3SSs have been thoroughly examined with biological assays (27, 28, 30, 57–64), crystallographic methods (29, 44, 45), biophysical characterization, and a number of other techniques (34–36). This work, however, represents the first biophysical exploration of CT584 and one of the first studies of any kind of this protein. As such, the data presented above should be compared to those of CT584 putative functional homologues.
Previous work with EPDs has highlighted their utility in revealing structure–function relationships between proteins with minimal sequence homology (36, 39). The tip proteins IpaD (S. flexneri), BipD (Burkholderia pseudomallei), SipD (Salmonella spp.), LcrV (Yersinia spp.), and PcrV (Pseudomonas aeruginosa) were previously compared and were shown to fall into two distinct subfamilies (36). The first subfamily (IpaD, BipD, and SipD) produced EPDs that manifested different structures at pH 6 and7 even at 10 °C (see the example in Figure 7A). The physical states of the proteins were the same at pH ≥ 7 and, in general, were disrupted under acidic conditions (pH <5). The second subfamily’s hallmark was nearly identical conformations at low temperatures from pH 5 to 8 with stability increasing in response to higher pH (see the example in Figure 7B). EPDs created so far for proteins that are not T3SS tip proteins do not display trends that closely resemble that of either subfamily. CT584’s phase diagram is similar to those of all of the tip proteins mentioned above in that there is a dramatic difference in protein structure (conformation) above and below pH 5. This difference was associated with a disrupted physical structure in acidic media with the other T3SS proteins but may be a region of greater stability for CT584. Defining which subfamily the Chlamydia protein most resembles is more challenging since it appears to possess one or more of the defining characteristics of each group. Like the IpaD subfamily, CT584 displays conformational differences at low temperatures between pH 6 and 7 with the EPD color shift arising from changes in secondary structure. The observed increasing stability as a function of increasing pH, however, would place it in the LcrV subfamily. It is tempting to speculate on the basis of these two observations that, since C. trachomatis belongs to a different phylum than the other bacteria mentioned, similarities to both subfamilies trace it back to a common ancestor prior to the split in the protein family.
While EPD similarities to known tip proteins are not sufficient evidence to establish CT584’s function, the protein has other properties described here that strengthen the possibility. Many of the T3SS tip proteins have been shown to oligomerize in vitro and in vivo, and it is believed that, on the needle tip, they are pentamers or hexamers (29, 46, 53, 54). AUC sedimentation velocity and equilibrium analysis demonstrate that in aqueous solution, CT584 oligomerizes most probably to a pentamer with some higher-order aggregates present. If this self-association occurs in more biologically relevant situations, it suggests a role for the protein similar to those of other tip proteins. Light scattering displayed an initial drop in intensity from 10 to 37 °C at pH 7, and additional work is needed to ascertain if this change is due to an alteration in oligomerization properties (perhaps due to the formation of a less dense particle).
Biphasic transitions are visible under various conditions as a function of temperature for CT584. Circular dichroism traces at pH 5 and 6 as well as ANS peak position transitions in several environments display this behavior which suggests the presence of two independent folding domains. This response to temperature is significant because IpaD and SipD have similar biphasic transitions and have been shown to possess N-terminal and C-terminal domains which exhibit differences in thermal stability (34–36).
Fourier transform infrared spectroscopy deconvolution results agree with the general conclusion from circular dichroism spectra that the secondary structure of CT584 is primarily α-helical. FTIR secondary structural analysis, however, of most of the tip proteins from proteobacteria listed above revealed three predominate bands of nearly equal intensity which is indicative of coiled-coil structure (34, 35, 65, 66). This structure has been confirmed by crystallographic studies of IpaD, BipD, and LcrV (29, 44, 45). In the case ofCT584, however, there is little evidence from spectroscopic data of a coiled-coil domain. This does not, of course, completely preclude the possibility that some other intramolecular interaction is interfering with its detection. For example, BipD’s trimeric coiled coil alters its FTIR spectrum such that the bands are not longer equal in intensity.
This study provides the first detailed biophysical study of the C. trachomatis protein, CT584. While many biological analyses are required to support the hypothesis that CT584 is a T3SS needle tip protein, this study represents a first step in ascertaining the function of this previously undescribed protein. While CT584 does not share homology with proteins from other bacterial genera, proteins with a high degree of homology to C. trachomatis CT584 are encoded by Chlamydia spp., including (but not limited to) Chlamydia pneumoniae, Chlamydia abortis, and Chlamydia caviae. Additionally, the protein sequence for CT584 is 100% identical within human C. trachomatis serovars (serovars A, D, and LGV) or C. pneumoniae strains. The sequence for CT584 is also highly conserved (83%identical and 91%similar) between C. pneumoniae and C. trachomatis. I-TASSER modeling using the C. pneumoniae sequence resulted in an identical predicted three-dimensional structure (data not shown). On the basis of the sequence and modeling conservation between and within species, it is expected that the biophysical characteristics described for C. trachomatis CT584 are probably conserved for the other Chlamydia homologues.
Acknowledgments
We acknowledge Dr. William D. Picking for his critical reading of and suggestions for the manuscript.
Footnotes
Abbreviations: T3SS, type III secretion system; TTSA, type III secretion apparatus; ORF, open reading frame; Tm, transition midpoint; CD, circular dichroism; UV, ultraviolet; ANS, 8-anilino-1-naphthalenesulfonate; ME, molar ellipticity; EPD, empirical phase diagram; PDB, Protein Data Bank.
References
- 1.Schachter J. Infection and disease epidemiology. In: Stephens RS, editor. Chlamydia: Intracellular biology, pathogenesis and immunity. American Society for Microbiology; Washington, DC: 1999. pp. 139–169. [Google Scholar]
- 2.Brunham RC, Rey-Ladino J. Immunology of Chlamydia infection: Implications for a Chlamydia trachomatis vaccine. Nat Rev Immunol. 2005;5:149–161. doi: 10.1038/nri1551. [DOI] [PubMed] [Google Scholar]
- 3.World Health Organization. Global Prevalence and Incidence of Selected Curable Sexually Transmitted Infections: Overview and Estimates. World Health Organization; Geneva: 2001. [Google Scholar]
- 4.Bedson SP, Bland JOW. A morphological study of psittacosis virus with the description of a developmental cycle. Br J Exp Pathol. 1932;13:461–466. [Google Scholar]
- 5.Byrne GI, Moulder JW. Parasite-specified phagocytosis of Chlamydia psittaci and Chlamydia trachomatis by L and HeLa cells. Infect Immun. 1978;19:598–606. doi: 10.1128/iai.19.2.598-606.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hackstadt T, Rockey DD, Heinzen RA, Scidmore MA. Chlamydia trachomatis interrupts an exocytic pathway to acquire endogenously synthesized sphingomyelin in transit from the Golgi apparatus to the plasma membrane. EMBO J. 1996;15:964–977. [PMC free article] [PubMed] [Google Scholar]
- 7.Scidmore MA, Rockey DD, Fischer ER, Heinzen RA, Hackstadt T. Vesicular interactions of the Chlamydia trachomatis inclusion are determined by chlamydial early protein synthesis rather than route of entry. Infect Immun. 1996;64:5366–5372. doi: 10.1128/iai.64.12.5366-5372.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beatty WL, Morrison RP, Byrne GI. Persistent chlamydiae: From cell culture to a paradigm for chlamydial pathogenesis. Microbiol Rev. 1994;58:686–699. doi: 10.1128/mr.58.4.686-699.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hogan RJ, Mathews SA, Mukhopadhyay S, Summersgill JT, Timms P. Chlamydial persistence: Beyond the biphasic paradigm. Infect Immun. 2004;72:1843–1855. doi: 10.1128/IAI.72.4.1843-1855.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hueck CJ. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev. 1998;62:379–433. doi: 10.1128/mmbr.62.2.379-433.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galan JE, Collmer A. Type III secretion machines: Bacterial devices for protein delivery into host cells. Science. 1999;284:1322–1328. doi: 10.1126/science.284.5418.1322. [DOI] [PubMed] [Google Scholar]
- 12.Galan JE, Wolf-Watz H. Protein delivery into eukaryotic cells by type III secretion machines. Nature. 2006;444:567–573. doi: 10.1038/nature05272. [DOI] [PubMed] [Google Scholar]
- 13.Hsia RC, Pannekoek Y, Ingerowski E, Bavoil PM. Type III secretion genes identify a putative virulence locus of Chlamydia. Mol Microbiol. 1997;25:351–359. doi: 10.1046/j.1365-2958.1997.4701834.x. [DOI] [PubMed] [Google Scholar]
- 14.Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q, Koonin EV, Davis RW. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science. 1998;282:754–759. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
- 15.Matsumoto A. Isolation and electron microscopic observations of intracytoplasmic inclusions containing Chlamydia psittaci. J Bacteriol. 1981;145:605–612. doi: 10.1128/jb.145.1.605-612.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsumoto A. Surface projections of Chlamydia psittaci elementary bodies as revealed by freeze-deep-etching. J Bacteriol. 1982;151:1040–1042. doi: 10.1128/jb.151.2.1040-1042.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clifton DR, Fields KA, Grieshaber SS, Dooley CA, Fischer ER, Mead DJ, Carabeo RA, Hackstadt T. A chlamydial type III translocated protein is tyrosine-phosphorylated at the site of entry and associated with recruitment of actin. Proc Natl Acad Sci USA. 2004;101:10166–10171. doi: 10.1073/pnas.0402829101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fields KA, Hackstadt T. Evidence for the secretion of Chlamydia trachomatis CopN by a type III secretion mechanism. Mol Microbiol. 2000;38:1048–1060. doi: 10.1046/j.1365-2958.2000.02212.x. [DOI] [PubMed] [Google Scholar]
- 19.Ho TD, Starnbach MN. The Salmonella enterica serovar typhimurium-encoded type III secretion systems can translocate Chlamydia trachomatis proteins into the cytosol of host cells. Infect Immun. 2005;73:905–911. doi: 10.1128/IAI.73.2.905-911.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hower S, Wolf K, Fields KA. Evidence that CT694 is a novel Chlamydia trachomatis T3S substrate capable of functioning during invasion or early cycle development. Mol Microbiol. 2009;72:1423–1437. doi: 10.1111/j.1365-2958.2009.06732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kleba B, Stephens RS. Chlamydial effector proteins localized to the host cell cytoplasmic compartment. Infect Immun. 2008;76:4842–4850. doi: 10.1128/IAI.00715-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peters J, Wilson DP, Myers G, Timms P, Bavoil PM. Type III secretion a la Chlamydia. Trends Microbiol. 2007;15:241–251. doi: 10.1016/j.tim.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 23.Subtil A, Delevoye C, Balana ME, Tastevin L, Perrinet S, Dautry-Varsat A. A directed screen for chlamydial proteins secreted by a type III mechanism identifies a translocated protein and numerous other new candidates. Mol Microbiol. 2005;56:1636–1647. doi: 10.1111/j.1365-2958.2005.04647.x. [DOI] [PubMed] [Google Scholar]
- 24.Hefty PS, Stephens RS. Chlamydial type III secretion system is encoded on ten operons preceded by sigma 70-like promoter elements. J Bacteriol. 2007;189:198–206. doi: 10.1128/JB.01034-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cossart P, Sansonetti PJ. Bacterial invasion: The paradigms of enteroinvasive pathogens. Science. 2004;304:242–248. doi: 10.1126/science.1090124. [DOI] [PubMed] [Google Scholar]
- 26.He SY, Nomura K, Whittam TS. Type III protein secretion mechanism in mammalian and plant pathogens. Biochim Biophys Acta. 2004;1694:181–206. doi: 10.1016/j.bbamcr.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 27.Espina M, Olive AJ, Kenjale R, Moore DS, Ausar SF, Kaminski RW, Oaks EV, Middaugh CR, Picking WD, Picking WL. IpaD Localizes to the Tip of the Type III Secretion System Needle of Shigella flexneri. Infect Immun. 2006;74:4391–4400. doi: 10.1128/IAI.00440-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goure J, Broz P, Attree O, Cornelis GR, Attree I. Protective anti-V antibodies inhibit Pseudomonas and Yersinia translocon assembly within host membranes. J Infect Dis. 2005;192:218–225. doi: 10.1086/430932. [DOI] [PubMed] [Google Scholar]
- 29.Johnson S, Roversi P, Espina M, Olive A, Deane JE, Birket S, Field T, Picking WD, Blocker AJ, Galyov EE, Picking WL, Lea SM. Self-chaperoning of the type III secretion system needle tip proteins IpaD and BipD. J Biol Chem. 2007;282:4035–4044. doi: 10.1074/jbc.M607945200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mueller CA, Broz P, Mueller SA, Ringler P, Erne-Brand F, Sorg I, Kuhn M, Engel A, Cornelis GR. The VAntigen of Yersinia Forms a Distinct Structure at the Tip of Injectisome Needles. Science. 2005;310:674–676. doi: 10.1126/science.1118476. [DOI] [PubMed] [Google Scholar]
- 31.Frank DW, Vallis A, Wiener-Kronish JP, Roy-Burman A, Spack EG, Mullaney BP, Megdoud M, Marks JD, Fritz R, Sawa T. Generation and characterization of a protective monoclonal antibody to Pseudomonas aeruginosa PcrV. J Infect Dis. 2002;186:64–73. doi: 10.1086/341069. [DOI] [PubMed] [Google Scholar]
- 32.Holder IA, Neely AN, Frank DW. PcrV immunization enhances survival of burned Pseudomonas aeruginosa-infected mice. Infect Immun. 2001;69:5908–5910. doi: 10.1128/IAI.69.9.5908-5910.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Une T, Brubaker RR. Roles of V antigen in promoting virulence and immunity in yersiniae. J Immunol. 1984;133:2226–2230. [PubMed] [Google Scholar]
- 34.Espina M, Ausar SF, Middaugh CR, Baxter MA, Picking WD, Picking WD. Conformational stability and differential structural analysis of LcrV, PcrV, BipD, and SipD from type III secretion systems. Protein Sci. 2007;16:704–714. doi: 10.1110/ps.062645007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Espina M, Ausar SF, Middaugh CR, Picking WD, Picking WL. Spectroscopic and calorimetric analyses of invasion plasmid antigen D (IpaD) from Shigella flexneri reveal the presence of two structural domains. Biochemistry. 2006;45:9219–9227. doi: 10.1021/bi060625v. [DOI] [PubMed] [Google Scholar]
- 36.Markham AP, Birket SE, Picking WD, Picking WL, Middaugh CR. pH sensitivity of type III secretion system tip proteins. Proteins. 2008;71:1830–1842. doi: 10.1002/prot.21864. [DOI] [PubMed] [Google Scholar]
- 37.Kueltzo LA, Ersoy B, Ralston JP, Middaugh CR. Derivative absorbance spectroscopy and protein phase diagrams as tools for comprehensive protein characterization: A bGCSF case study. J Pharm Sci. 2003;92:1805–1820. doi: 10.1002/jps.10439. [DOI] [PubMed] [Google Scholar]
- 38.Fan H, Ralston J, Dibiase M, Faulkner E, Middaugh CR. Solution behavior of IFN-β-1a: An empirical phase diagram based approach. J Pharm Sci. 2005;94:1893–1911. doi: 10.1002/jps.20410. [DOI] [PubMed] [Google Scholar]
- 39.Fan H, Kashi RS, Middaugh CR. Conformational lability of two molecular chaperones Hsc70 and gp96: Effects of pH and temperature. Arch Biochem Biophys. 2006;447:34–45. doi: 10.1016/j.abb.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinf. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Battey JN, Kopp J, Bordoli L, Read RJ, Clarke ND, Schwede T. Automated server predictions in CASP7. Proteins. 2007;69(Suppl 8):68–82. doi: 10.1002/prot.21761. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 2007;69(Suppl 8):108–117. doi: 10.1002/prot.21702. [DOI] [PubMed] [Google Scholar]
- 43.Holm L, Park J. DaliLite workbench for protein structure comparison. Bioinformatics. 2000;16:566–567. doi: 10.1093/bioinformatics/16.6.566. [DOI] [PubMed] [Google Scholar]
- 44.Derewenda U, Mateja A, Devedjiev Y, Routzahn KM, Evdokimov AG, Derewenda ZS, Waugh DS. The structure of Yersinia pestis V-antigen, an essential virulence factor and mediator of immunity against plague. Structure (Cambridge, MA, US) 2004;12:301–306. doi: 10.1016/j.str.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 45.Erskine PT, Knight MJ, Ruaux A, Mikolajek H, Sang NW, Withers J, Gill R, Wood SP, Wood M, Fox GC, Cooper JB. High Resolution Structure of BipD: An Invasion Protein Associated with the Type III Secretion System of Burkholderia pseudomallei. J Mol Biol. 2006;363:125–136. doi: 10.1016/j.jmb.2006.07.069. [DOI] [PubMed] [Google Scholar]
- 46.Caroline G, Eric F, Bohn YS, Sylvie E, Attree I. Oligomerization of PcrV and LcrV, protective antigens of Pseudomonas aeruginosa and Yersinia pestis. J Biol Chem. 2008;283:23940–23949. doi: 10.1074/jbc.M803146200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hennessey JP, Jr, Johnson WC., Jr Information content in the circular dichroism of proteins. Biochemistry. 1981;20:1085–1094. doi: 10.1021/bi00508a007. [DOI] [PubMed] [Google Scholar]
- 48.Byler DM, Susi H. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolymers. 1986;25:469–487. doi: 10.1002/bip.360250307. [DOI] [PubMed] [Google Scholar]
- 49.Mach H, Middaugh CR. Simultaneous monitoring of the environment of tryptophan, tyrosine, and phenylalanine residues in proteins by near-ultraviolet second-derivative spectroscopy. Anal Biochem. 1994;222:323–331. doi: 10.1006/abio.1994.1499. [DOI] [PubMed] [Google Scholar]
- 50.Mach H, Thomson JA, Middaugh CR, Lewis RV. Examination of phenylalanine microenvironments in proteins by second-derivative absorption spectroscopy. Arch Biochem Biophys. 1991;287:33–40. doi: 10.1016/0003-9861(91)90384-u. [DOI] [PubMed] [Google Scholar]
- 51.Semisotnov GV, Rodionova NA, Razgulyaev OI, Uversky VN, Gripas AF, Gilmanshin RI. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers. 1991;31:119–128. doi: 10.1002/bip.360310111. [DOI] [PubMed] [Google Scholar]
- 52.Matulis D, Lovrien R. 1-Anilino-8-naphthalene sulfonate anion-protein binding depends primarily on ion pair formation. Biophys J. 1998;74:422–429. doi: 10.1016/S0006-3495(98)77799-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Broz P, Mueller CA, Muller SA, Philippsen A, Sorg I, Engel A, Cornelis GR. Function and molecular architecture of the Yersinia injectisome tip complex. Mol Microbiol. 2007;65:1311–1320. doi: 10.1111/j.1365-2958.2007.05871.x. [DOI] [PubMed] [Google Scholar]
- 54.Veenendaal AK, Hodgkinson JL, Schwarzer L, Stabat D, Zenk SF, Blocker AJ. The type III secretion system needle tip complex mediates host cell sensing and translocon insertion. Mol Microbiol. 2007;63:1719–1730. doi: 10.1111/j.1365-2958.2007.05620.x. [DOI] [PubMed] [Google Scholar]
- 55.Dam J, Velikovsky CA, Mariuzza RA, Urbanke C, Schuck P. Sedimentation velocity analysis of heterogeneous protein-protein interactions: Lamm equation modeling and sedimentation coefficient distributions c(s) Biophys J. 2005;89:619–634. doi: 10.1529/biophysj.105.059568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fan H, Li H, Zhang M, Middaugh CR. Effects of solutes on empirical phase diagrams of human fibroblast growth factor 1. J Pharm Sci. 2006;96:1490–1503. doi: 10.1002/jps.20796. [DOI] [PubMed] [Google Scholar]
- 57.De Geyter C, Wattiez R, Sansonetti P, Falmagne P, Ruysschaert JM, Parsot C, Cabiaux V. Characterization of the interaction of IpaB and IpaD, proteins required for entry of Shigella flexneri into epithelial cells, with a lipid membrane. Eur J Biochem. 2000;267:5769–5776. doi: 10.1046/j.1432-1327.2000.01649.x. [DOI] [PubMed] [Google Scholar]
- 58.Fields KA, Nilles ML, Cowan C, Straley SC. Virulence role of V antigen of Yersinia pestis at the bacterial surface. Infect Immun. 1999;67:5395–5408. doi: 10.1128/iai.67.10.5395-5408.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hermant D, Menard R, Arricau N, Parsot C, Popoff MY. Functional conservation of the Salmonella and Shigella effectors of entry into epithelial cells. Mol Microbiol. 1995;17:781–789. doi: 10.1111/j.1365-2958.1995.mmi_17040781.x. [DOI] [PubMed] [Google Scholar]
- 60.Lawton DG, Longstaff C, Wallace BA, Hill J, Leary SE, Titball RW, Brown KA. Interactions of the type III secretion pathway proteins LcrV and LcrG from Yersinia pestis are mediated by coiled-coil domains. J Biol Chem. 2002;277:38714–38722. doi: 10.1074/jbc.M203632200. [DOI] [PubMed] [Google Scholar]
- 61.Marquart ME, Picking WL, Picking WD. Structural analysis of invasion plasmid antigen D (IpaD) from Shigella flexneri. Biochem Biophys Res Commun. 1995;214:963–970. doi: 10.1006/bbrc.1995.2380. [DOI] [PubMed] [Google Scholar]
- 62.Nanao M, Ricard-Blum S, Di Guilmi AM, Lemaire D, Lascoux D, Chabert J, Attree I, Dessen A. Type III secretion proteins PcrV and PcrG from Pseudomonas aeruginosa form a 1:1 complex through high affinity interactions. BMC Microbiol. 2003;3:21. doi: 10.1186/1471-2180-3-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stevens MP, Wood MW, Taylor LA, Monaghan P, Hawes P, Jones PW, Wallis TS, Galyov EE. An Inv/Mxi-Spa-like type III protein secretion system in Burkholderia pseudomallei modulates intracellular behaviour of the pathogen. Mol Microbiol. 2002;46:649–659. doi: 10.1046/j.1365-2958.2002.03190.x. [DOI] [PubMed] [Google Scholar]
- 64.Venkatesan MM, Buysse JM, Kopecko DJ. Characterization of invasion plasmid antigen genes (ipaBCD) from Shigella flexneri. Proc Natl Acad Sci USA. 1988;85:9317–9321. doi: 10.1073/pnas.85.23.9317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heimburg T, Schuenemann J, Weber K, Geisler N. Specific recognition of coiled coils by infrared spectroscopy: Analysis of the three structural domains of type III intermediate filament proteins. Biochemistry. 1996;35:1375–1382. doi: 10.1021/bi9515883. [DOI] [PubMed] [Google Scholar]
- 66.Heimburg T, Schunemann J, Weber K, Geisler N. FTIR spectroscopy of multistranded coiled coil proteins. Biochemistry. 1999;38:12727–12734. doi: 10.1021/bi983079h. [DOI] [PubMed] [Google Scholar]