

Abstract
Perivascular adipose tissue (PVAT) is implicated in the development of vascular diseases; however, the roles of PVAT on OPN expression in diabetic vasculature remain to be determined. This study investigated the role of adipokines derived from diabetic PVAT in regulating the vascular expression of OPN and explored the mechanisms involved. Aortic sections of ob/ob and high-fat diet (HFD)-induced obese (DIO) mice showed an increased expression of OPN, which was paralleled by increased amounts of PVAT characterized by enlargement of adipocytes. In the earlier phase of HFD feeding, macrophage infiltration was mainly localized to the area of PVAT at which adipocytes were enlarged, suggesting a potential link of activated adipocytes to macrophage infiltration. PVAT sections of ob/ob and DIO mice revealed a significantly greater number of macrophages with increased expression of adipokines, including resistin and visfatin. The distribution of resistin in PVAT mostly co-localized with macrophages, while visfatin was expressed in macrophages and other cells. In in vitro studies, OPN expression in vascular smooth muscle cells (VSMCs) co-cultured with PVAT of DIO mice was significantly increased, which was attenuated by a resistin-neutralizing antibody. Likewise, resistin up-regulated expression of OPN mRNA and protein in cultured VSMCs and the pivotal role of AP-1 in resistin-induced OPN transcription was demonstrated. Resistin produced by PVAT plays a pivotal role in the up-regulated expression of OPN in the diabetic vasculature via a signalling pathway that involves activation of AP-1. © 2013 The Authors. Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: resistin, PVAT, macrophage, OPN, obesity
Introduction
Perivascular adipose tissue (PVAT) is a highly active endocrine organ, producing and releasing a range of bioactive substances 1. PVAT include adipocytes, endothelial cells and inflammatory cells. These cell populations change with age, nutritional status and environmental conditions. In diabetic mice and human individuals, the peri-aortic PVAT is characterized by inflammatory cell infiltration and enhanced cytokine expression 2,3. PVAT is a modulator of vascular function 4 and altered PVAT function is involved in the development of vascular diseases, including hypertension 5,6 and atherosclerosis 7,8
Vascular smooth muscle cell (VSMC) migration and proliferation are critical to the pathophysiological processes of proliferative vascular diseases including atherosclerosis. Atherosclerotic vascular disease is a major complication in diabetic patients, and osteopontin (OPN) expression is increased in the media of diabetic arteries 9. Many studies have demonstrated that OPN promotes migration, extracellular matrix invasion and proliferation of VSMCs 10,11. OPN expression is increased in atherosclerotic plaques and neointima after experimental angioplasty 10,12 and has been implicated in atherosclerosis and vascular injury response 13. Moreover, angiotensin II-induced inflammatory activation in VSMCs was reduced by down-regulating OPN expression 4. Therefore, OPN is not only a marker of the proliferative VSMCs phenotype but also an active player in the progression of atherosclerosis and restenosis 15.
In obese and type 2 diabetes, PVAT may regulate vascular function via the endocrine and paracrine effects of adipokines through cross-talk among inflammatory cells, adipocytes and vascular cells 15. Inflammatory PVAT may in turn secrete proatherogenic cytokines 16 and causes local endothelial dysfunction 17, thus contributing to the progression of systemic and local vascular disease. Bioactive substances of PVAT induce VSMCs proliferation through activation of mitogen-activated protein kinase pathways 18,19. In the pathophysiological processes of vascular remodelling, phenotypic changes in vascular cells correlate with up-regulation of proinflammatory adipokines, which may contribute to vascular dysfunction 20,21. However, although VSMCs are in closer proximity to PVAT, little attention has been devoted to the role of PVAT and PVAT-derived bioactive substances in the regulation of the VSMC phenotype. Thus, mapping the signalling pathway that mediates OPN expression in diabetic vasculature may reveal important pharmacological targets for the prevention of vascular diseases.
PVAT-derived adipokines may provide a molecular link between diabetes and cardiovascular disease. Among a variety of adipokines, resistin and visfatin have been reported as major proinflammatory mediators 22,23; however, the cellular source and the precise roles of these adipokines on OPN production in diabetic vasculature remain unknown. To investigate whether PVAT has a paracrine role in the development of diabetes-associated vascular diseases, we determined the production of resistin and visfatin in PVAT of diabetic mice, examined the effects of these adipokines on OPN expression in VSMCs and elucidated the mechanisms involved.
Materials and methods
Animals
Male C57BL/6JHamSlc-lepob/lepob diabetic (ob/ob) mice and their non-diabetic littermates (ob/+) were purchased from Japan SCL Inc. (Hamamatsu, Japan) at age 8 weeks. Male C57BL/6 mice (aged 8 weeks) were obtained from Samtako Inc. (Osan, Gyunggi-do, Korea). All mice were routinely fed a low-fat standard rodent diet (11% kcal from fat), with water freely available. To induce diet-induced obesity (DIO), C57BL/6 mice (aged 8 weeks) were fed a high-fat diet (HFD; 45% kcal from fat, Harland Tekland, Madison, WI, USA) for 15 weeks. Blood glucose levels were determined using a standard glucometer (Accucheck, Seoul, Korea). Serum total cholesterol and triglycerides were measured with specific determination kits (Asan, Seoul, Korea). All experiments involving animals conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 2011). All experimental procedures were conducted in accordance with the Animal Care Guidelines of Pusan National University Institutional Animal Care and Use Committee.
Tissue preparation and histological assessment
Isolated aortas with PVAT were fixed for 1 day in 10% formalin, transferred to 70% ethanol and embedded in paraffin blocks. Blocks of 5 µm thickness were cut and stained with haematoxylin and eosin (H&E) and with anti-perilipin A/B (Sigma, St. Louis, MO, USA) to detect adipocytes. For immunohistochemical studies, paraffin-embedded tissue samples were deparaffinized before blocking endogenous peroxidase with 0.3% H2O2 for 30 min. Non-specific protein binding was blocked with CAS-BLOCK (Zymed Laboratories, San Francisco, CA, USA) and then stained for macrophages, OPN, resistin and visfatin with anti-CD68 (Santa Cruz Biotechnology, CA, USA), anti-OPN (Abcam, Cambridge, MA, USA), anti-resistin (R&D Systems, Minneapolis, MN, USA) and anti-visfatin antibodies (Abcam). These sections were incubated with biotinylated anti-rabbit antibody (Vector, Burlingame, CA, USA), and they were visualized using a Vectastain ABC kit (Vector) with a solution of 3,3′-diaminobenzidine tetrahydrochloride (Diaminobenzidine substrate kit, Vector). In double-immunofluorescence experiments, the incubating solution contained goat anti-CD68, goat anti-resistin (Santa Cruz) antibodies and mouse anti-visfatin (Santa Cruz). After washing, the sections were incubated with secondary antibody solution containing fluorescence 594-conjugated anti-rabbit IgG (Life Technologies, Carlsbad, CA, USA) and fluorescence 488-conjugated anti-mouse IgG (Life Technologies) for 1 h at room temperature, washed and mounted using Vectashield mounting medium (Vector). Fluorescence was detected by microscopy (Olympus Corp., Tokyo, Japan).
Western blot analysis
Protein samples (30 µg each) were loaded into a 10% SDS–polyacrylamide gel and transferred to a nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ, USA). The blocked membrane was then incubated with antibodies for OPN (Abcam), resistin and visfatin (Santa Cruz). Immunoreactive bands were visualized with the chemiluminescent reagent from the Supersignal West Dura Extended Duration Substrate Kit (Pierce Chemical, Rockford, IL, USA). Signals were quantified using a GS-710 calibrated imaging densitometer (Bio-Rad, Hercules, CA, USA). The results were expressed as relative density.
Co-culture of VSMCs with isolated PVAT
Primary cultures of VSMCs were grown by the explant technique from the aortae of Sprague–Dawley rats, as previously described 14. Cells were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco BRL, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS). Experiments were performed with cells from passages 4–9. PVATs were collected from mice fed ND (15 weeks) and HFD (15 weeks) above an ice-cooled plate. The PVATs were rinsed three times with DMEM supplemented with 10% FBS, and 10 mg PVAT samples were placed in the culture wells and co-cultured with VSMCs for 24 h.
RT–PCR analysis
To assess the effects of adipokines on the expression of OPN mRNA, cells were stimulated with recombinant resistin (Phoenix Pharmaceuticals, Belmont, CA, USA). Total RNA was extracted using TRIzol reagent (Invitrogen, San Diego, CA, USA). PCR primers for amplification of OPN were: 5′-AGA CTG GCA GTG GTT TGC TT-3′ and 5′-ATG GCT TTC ATT GGA GTT GC-3′ (antisense). Equal amounts of RT–PCR products were separated on a 1% agarose gel and stained with ethidium bromide.
Preparation of promoter constructs
A series of constructs of the OPN promoter in a luciferase expression vector pGL3 basic (Promega, Madison, WI, USA) was prepared. The Rattus OPN promoter was amplified from Rattus genomic DNA with PCR primers (forward 5′-AGTGTAGGAAGCAGTCAGTCCTGTCAG-3′; reverse 5′-TACCTTGGCTGGCTTCTCGAGCATGCT-3′) and cloned into pGL3-Basic to generate the construct pLuc–OPN-2286. Additional deletion constructs lacking distal promoter sequences (denoted pLuc–OPN-538 and pLuc–OPN-234) were prepared by digestion of pLuc–OPN-2286 with the restriction enzymes NheI, Sac1 or Xho1. The OPN promoter sequence was analysed for transcription factor binding sites within the 5′-upstream region, using the sequence motif search of GenomeNet.
Transient transfection, luciferase and transcription factor assays
All plasmids were prepared using QIAprep spin kit. Cells were transfected with plasmids using FuGENE6 Transfection Reagent (Roche Applied Science, Indianapolis, IN, USA), following the manufacturer's guidelines. Cell lysates were prepared using a passive lysis buffer from a Promega assay system (Promega, WI, USA) and used to measure the luciferase activity according to the manufacturer's instructions for the dual luciferase reporter assay system (Promega). The luciferase assays were carried out using a Glomax™ 20/20 luminometer (Promega). Cell lysates were extracted using a nuclear extraction kit (Chemicon International, Temecula, CA, USA) according to the manufacturer's protocol. The DNA-binding activity of AP-1 and C/EBP were measured using colorimetric AP-1 and C/EBP transcription factor assay kits (Active Motif, Carlsbad, CA, USA).
Statistical analysis
All data are expressed as mean ± standard error of the mean (SEM). The change in variable parameters between untreated control and treated groups was analysed by one-way analysis of variance (ANOVA), followed by Tukey's multiple comparison tests as a post hoc comparison. Differences were considered statistically significant at p < 0.05.
Results
Characteristics of genetic and diet-induced obese mice
At age 8 weeks, body weight and plasma levels of glucose and total cholesterol in ob/ob mice (n = 6) were significantly higher than those in ob/+ (n = 6) and control mice (n = 7), while the plasma levels of triglyceride were similar among these three groups (Table1). To produce HFD-induced obese (DIO) mice, male C57BL/6 mice (aged 8 weeks) were fed a control chow or HFD for 5, 10 and 15 weeks to produce differing durations and magnitudes of obesity. Body weight was gradually increased with the duration of HFD. At 10 and 15 weeks under HFD mice showed increased adiposity, characterized by statistically significant differences in terms of body weight, plasma levels of glucose and total cholesterol (Table2). At 15 weeks under HFD, mice also showed an increased level of insulin and glucose intolerance (data not shown).
Table 1.
Changes in body weight and plasma lipid profiles in WT, ob/+ and ob/ob mice
| WT | ob/+ | ob/ob | |
|---|---|---|---|
| Body weight (g) | 22.24 ± 0.74 | 23.91 ± 0.88 | 46.63 ± 0.75** |
| Glucose (mg/dl) | 176.80 ± 10.41 | 154.50 ± 8.44 | 519.33 ± 7.44** |
| Cholesterol (mg/dl) | 74.66 ± 2.16 | 66.67 ± 6.89 | 128.92 ± 12.29** |
| Triglyceride (mg/dl) | 68.75 ± 5.19 | 57.71 ± 3.62 | 67.81 ± 2.55 |
Data are expressed as mean ± SEM from mice at 8 weeks (n = 6 or 7 mice/group).
p < 0.01 versus wild-type (WT) mice.
Table 2.
Changes in body weight and plasma lipid profiles in mice fed HFD
| Control | 5 weeks | 10 weeks | 15 weeks | |
|---|---|---|---|---|
| Body weight (g) | 22.47 ± 0.19 | 26.17 ± 0.43* | 37.46 ± 0.80** | 50.34 ± 0.45** |
| Glucose (mg/dl) | 172.50 ± 3.73 | 239.25 ± 8.67* | 302.25 ± 10.98** | 486.75 ± 16.48** |
| Cholesterol (mg/dl) | 54.93 ± 4.57 | 90.32 ± 30.74 | 161.62 ± 2.20* | 171.13 ± 7.71* |
| Triglyceride (mg/dl) | 69.70 ± 4.87 | 78.06 ± 9.47 | 116.75 ± 11.86** | 137.97 ± 11.10** |
Data are expressed as mean ± SEM from five or six mice fed a high-fat diet (HFD) for 0 (control), 5, 10, and 15 weeks.
p < 0.05 and
p < 0.01 versus control.
OPN expression in diabetic vasculature
OPN is known to be an active player in the progression of atherosclerosis and restenosis 15. To investigate the potential role of OPN in diabetic vascular diseases, we examined OPN expression in diabetic vasculature. As shown in Figure 1A, immunohistochemical studies showed an enhanced expression of OPN in the medial layer of aortic sections from ob/ob mice compared with those in ob/+ and wild-type control mice. In another series of experiments to analyse whether the levels of OPN expression change during DIO, male C57BL/6 mice (aged 8 weeks) were maintained on either a normal diet (ND) or a high-fat diet (HFD) for 5, 10 and 15 weeks. As shown in Figure 1B, OPN was minimally expressed in the abdominal aortas of control or mice fed ND for 15 weeks. In the aorta of HFD-fed mice, OPN was not typically present in the aortae of mice fed HFD for 5 weeks, but its expression started to increase at 10 weeks of HFD and markedly increased at 15 weeks of HFD (Figure 1B), suggesting a potential link of obesity to increased OPN expression in the vasculature.
Figure 1.
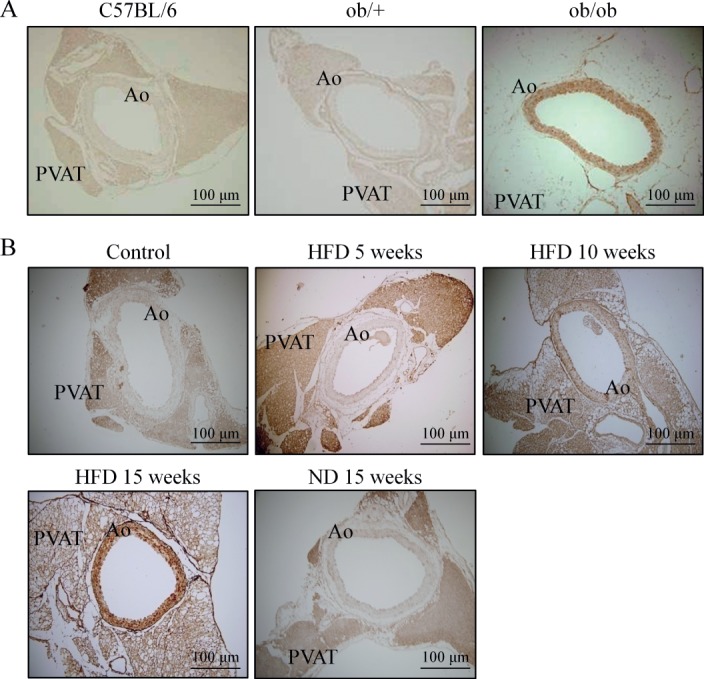
The expression of OPN in diabetic vasculature. (A) Immunohistochemical staining for OPN in aortic sections of control (C57BL/6), ob/+ and ob/ob mice. (B) Time course of OPN expression in aortic sections of mice fed a normal diet (ND) and HFD for 5, 10 and 15 weeks. Representative images are from five to seven independent experiments: Ao, abdominal aorta; PVAT, perivascular adipose tissue.
Characteristics of PVAT in diabetic mice
The total amount of PVAT surrounding the aorta was significantly increased in ob/ob mice compared to ob/+ and control mice. In H&E-stained sections of the abdominal aorta surrounded by PVAT, the area of PVAT in ob/ob mice was significantly larger than that in control mice (data not shown). Moreover, the size of adipocytes in PVAT was markedly increased in ob/ob mice (Figure 2A). In parallel, peri-aortic adipose tissue surrounding the thoracic and abdominal aorta showed a similar gross appearance in colour, and the total amount of PVAT surrounding the aorta was significantly increased in mice fed HFD for 15 weeks compared to those of control or ND-fed mice. An increase in PVAT area in DIO mice was also demonstrated in H&E-stained sections of the abdominal aorta (data not shown). The size of adipocytes in PVAT of DIO mice was markedly increased (Figure 2B) and the abdominal aorta was surrounded by adipocytes in close proximity to aortic adventitia, suggesting a potential paracrine role of PVAT in modulating vascular function.
Figure 2.
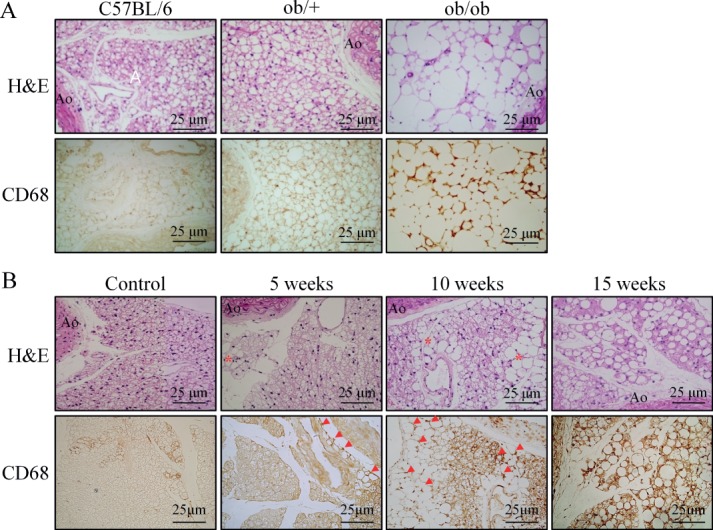
Characteristics of PVAT in diabetic and HFD-fed obese mice. (A) H&E-stained sections of PVAT surrounding the abdominal aorta of control (C57BL/6), ob/+ and ob/ob mice. (B) Time course changes of PVAT morphology in mice fed HFD for 5, 10 and 15 weeks. Representative images are from five or six independent experiments: Ao, abdominal aorta. Bottom in each group: (A) immunohistochemical staining for macrophages (CD68+) in PVAT of control (C57BL/6), ob/+ and ob/ob mice. (B) Time course of macrophage infiltration in PVAT of HFD-fed mice. Representative images are from five or six independent experiments: *area where adipocytes are enlarged; arrows, macrophage staining.
Increased macrophage infiltration in diabetic PVAT
Representative immunohistochemical staining of macrophages (CD68+) distribution in PVAT showed a higher percentage of macrophages in PVAT from diabetic mice than from control groups, and the cells appeared to be randomly distributed along the septa (Figure 2A). Quantitative analysis showed a significantly higher percentage of CD68+ macrophages than trabecular cells in PVAT compared with the other groups (data not shown). Interestingly, in the early phase of HFD feeding in mice, enlarged adipocytes were localized around the PVAT. In the PVAT of mice fed HFD for 10 and 15 weeks, almost all adipocytes in the PVAT surrounding the abdominal aorta were enlarged (Figure 2B). Interestingly, macrophage infiltration in the earlier phase of HFD feeding was mainly localized to the PVAT at which adipocytes were enlarged. At 10 or 15 weeks of HFD feeding, immunohistochemical staining of macrophage (CD68+) distribution showed a higher percentage of macrophages in PVAT from DIO mice than from control groups, and the cells appeared to be randomly distributed along the septa (Figure 2B). These results suggest a potential link between macrophage infiltration and adipocyte activation during obesity progression.
Adipokine expression in diabetic PVAT
An elevated expression of resistin and visfatin in diabetic PVAT was demonstrated compared with those in corresponding control mice (Figure 3A). In addition, lysates of PVAT also showed elevated protein levels of resistin and visfatin in ob/ob mice compared to those in control mice (Figure 3B). Similar to PVAT from ob/ob mice, an elevated expression of resistin and visfatin in PVAT of DIO mice was demonstrated compared with those in corresponding control mice. Similar to macrophage infiltration in PVAT in DIO mice, the expression of these adipokines was mainly observed in the localized area of PVAT at which macrophages were infiltrated (Figure 3C). Confocal microscopy analysis also showed enhanced signals for macrophages (CD68+) as well as signals for elevated expression of resistin and visfatin in PVAT of ob/ob and DIO mice compared with those in corresponding control mice. The signals for CD68, resistin and visfatin were almost exclusively demonstrated along the septa. The signals for resistin on PVAT mostly co-localized with macrophages, and the signal was higher in the septal area. However, although the distribution of visfatin on PVAT was observed mainly co-localized with CD68+ cells, a slight signal could also be detected in the interstitial spaces and in other cell types (Figure 4A, B).
Figure 3.
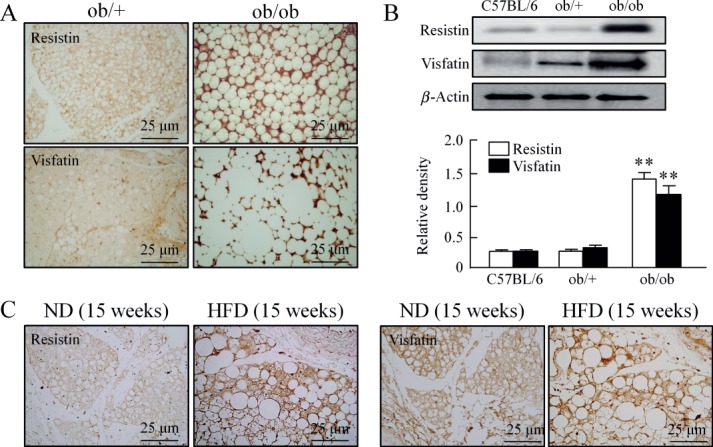
The expression of resistin and visfatin in diabetic PVAT. (A) Immunohistochemical staining for resistin and visfatin in PVAT of ob/+ and ob/ob mice (n = 5). (B) Immunoblotting of resistin and visfatin in PVAT of control, ob/+ and ob/ob mice. Quantitative data are expressed as mean ± SEM of three to five experiments; **p < 0.01 versus corresponding control. (C) Representative immunohistochemical stain for resistin (n = 5) and visfatin (n = 5) in PVAT of mice fed ND or HFD for 15 weeks.
Figure 4.

Cellular localization of resistin and visfatin in PVAT. Representative immunofluorescence for macrophages, resistin and visfatin in PVAT surrounding the abdominal aorta of ob/ob (A, n = 5) and HFD-fed mice (B, n = 6). Macrophage fluorescence (CD68, red) was merged with those of resistin or visfatin (green), showing that resistin exclusively co-localized with CD68; arrows, non-co-localized areas of CD68 and visfatin.
Role of PVAT-derived resistin on OPN expression in VSMCs
Since the average serum concentration of resistin in healthy control subjects was 30.7 ng/ml and was significantly elevated in patients with diabetes (49.7 ng/ml) 24, we used 1–100 ng/ml human recombinant resistin to evaluate the direct effect of resistin on OPN expression in VSMCs. As shown in Figure 5A, B, the expression levels of OPN protein and mRNA were increased in a concentration (1–30 ng/ml)-dependent manner by resistin but not by visfatin. These results suggest a pivotal role of resistin in the up-regulation of OPN expression at the transcriptional level. In another series of experiments, we isolated PVAT from DIO mice showing an increased expression of resistin compared to control (Figure 6A). To evaluate the functional role of resistin produced in PVAT of DIO mice, VSMCs were co-cultured with PVAT for 48 h and then the VSMCs were collected for western blot analysis. Compared to OPN expression in VSMCs co-cultured with control PVAT, OPN expression in VSMCs co-cultured with PVAT of DIO mice was significantly increased, which was attenuated by pretreatment with anti-resistin antibody (Figure 6B). These results indicate a functional role of PVAT-derived resistin on OPN expression in the vasculature of obese mice.
Figure 5.
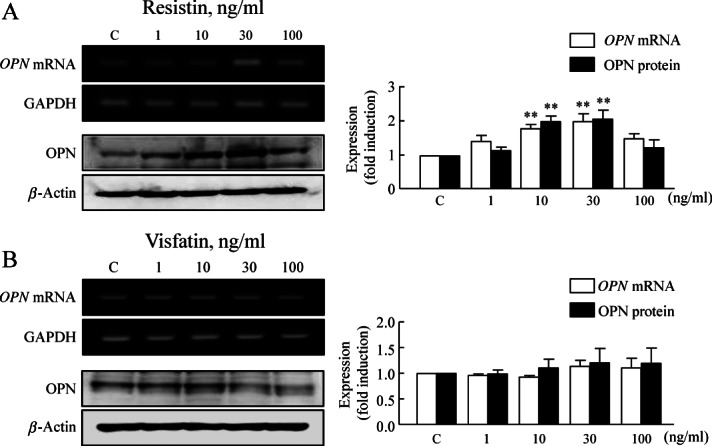
The effects of resisitin and visfatin on OPN expression in VSMCs. Representative blots show the expression of OPN mRNA and protein in cultured VSMCs stimulated with resistin (A) or visfatin (B) for 24 h at the indicated doses. Quantitative data are expressed as mean ± SEM of four to six experiments; **p < 0.01 versus corresponding control.
Figure 6.
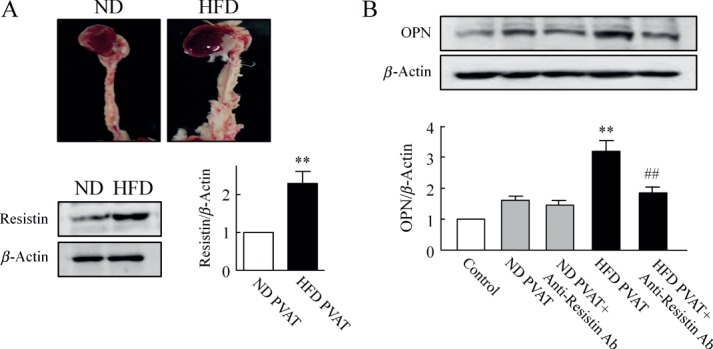
Functional role of PVAT-derived resistin on OPN expression in VSMCs. (A) Representative photographs of PVAT surrounding aorta of mice fed ND or HFD for 15 weeks. Representative blots show the expression of resistin in PVAT from ND- and HFD-fed mice. Quantitative data are expressed as the mean ± SEM of five experiments; **p < 0.01 versus ND PVAT. (B) Immunoblots for OPN expression in VSMCs co-cultured with PVAT explants for 48 h. PVAT was isolated from mice fed ND or HFD for 15 weeks and then co-cultured with VSMCs in the presence or absence of anti-resistin Ab (10 µg/ml). Quantitative results are expressed as mean ± SEM of five experiments; **p < 0.01 versus control; ##p < 0.01 versus HFD PVAT.
Transcriptional regulation of resistin-induced OPN expression in VSMCs
To locate the regions responsible for resistin-induced OPN transcription within the 2268 nt OPN promoter, three constructs with different promoter sizes were prepared and transfected transiently into VSMCs, and then luciferase activity was measured. As shown in Figure 7A, the luciferase reporter activities of pLuc–OPN-2268 and pLuc–OPN-538 after resistin stimulation were 4.45- and 8.11-fold higher, respectively, than that in control. However, the luciferase reporter activities of pLuc–OPN-234 was not affected by resistin, suggesting that the region nt 538–234 is responsible for resistin-enhanced OPN promoter activity in VSMCs. Sequence analysis within the region nt 538–234 demonstrated the presence of consensus elements for transcription factors including AP-1 and C/EBP (Figure 7B). In chemiluminescent transcription factor assays, resistin enhanced AP-1 activity but not the activity of C/EBP α/β in VSMCs, indicating a central role of AP-1 in OPN transcription induced by resistin.
Figure 7.

The role of resistin in the transcriptional regulation of OPN expression in VSMCs. (A) Cells were co-transfected with various promoter constructs and an empty luciferase vector pRL CMV for 24 h and treated with resistin (30 ng/ml) for 4 h, then luciferase activity was determined. Results are expressed as the mean ± SEM of five experiments; **p < 0.01 versus corresponding control. (B) Nucleotide sequence of the promoter region of the OPN gene. The 538 bp sequence of the 5′-flanking region of OPN is shown. The underlined sequences are the possible transcription factor binding sites as predicted by Genome Net. (C) Direct interaction of AP-1 and C/EBP α/β with the OPN promoter. ELISA-based transcription factor activation assay was performed in a plate with an oligonucleotide containing TPA-responsive element (AP-1) or a C/EBP α/β consensus binding site. Nuclear extract from cells treated with resistin (1–100 ng/ml) for 1 h was incubated. Results are expressed as mean ± SEM of four experiments; **p < 0.01 versus control (C).
Discussion
OPN expression in the vasculature of both genetic (ob/ob) and diet-induced obese (DIO) mice was paralleled by an increased accumulation of PVAT characterized by enlargement of adipocytes, an increased macrophage infiltration and up-regulated production of resistin. OPN expression in VSMCs co-cultured with PVAT of DIO mice was significantly increased compared to control, which was attenuated by immunodepletion of resistin. Here we also demonstrated a novel in vitro function of recombinant resistin in OPN expression in VSMCs, suggesting a potential pathogenic mechanism linking PVAT-derived resistin to vascular complications in diabetes.
OPN is not typically present in normal blood vessels, but its expression is increased in atherosclerotic plaques 12,25. OPN transgenic mice exhibit exaggerated atherosclerosis and neointimal formation 26,27, while OPN deficiency attenuates atherosclerosis 28,29. Therefore, OPN is considered to be an active player in the progression of atherosclerosis and restenosis 30. Consistent with previous reports 9 in which OPN expression was increased in diabetic vasculature, OPN expression in the medial layer of the vasculature was markedly increased in aortae from ob/ob and DIO mice. Thus, it is suggested that OPN is involved in the development of vascular complications in diabetes. Although OPN is considered to be an important mediator in a remarkable range of pathological responses 31, little is known about the role of PVAT in the enhanced expression of OPN in diabetic vasculature.
PVAT may regulate vascular function via the endocrine and paracrine effects of adipokines produced by cross-talk between inflammatory cells, adipocytes and vascular cells 20. Perivascular adipocytes exhibit an enhanced pro-inflammatory state, with reduced adipocytic differentiation in obese or diabetic subjects 32. These phenotypic changes in PVAT were also enhanced in DIO mice 20,33. Consistent with these reports, the total amount of PVAT surrounding the abdominal aorta and the size of adipocytes in PVAT were markedly increased in DIO mice. In addition, macrophage infiltration in PVAT was increased with duration of HFD feeding in mice. At 5 weeks of HFD feeding, enlarged adipocytes were localized to PVAT. In PVAT surrounding abdominal aorta from mice fed HFD for 10 and 15 weeks, the enlarged adipocytes were observed throughout the PVAT. Interestingly, macrophage infiltration in the early phase of HFD feeding was mainly localized to the PVAT area in which adipocytes were enlarged. In addition, OPN expression in the vasculature paralleled the process of inflammation in PVAT during the development of obesity, suggesting that vascular expression of OPN is closely linked to macrophage infiltration in PVAT.
PVAT expresses many kinds of adipokine, and PVAT-derived bioactive substances stimulate VSMCs proliferation 34. Thus, it appears likely that PVAT-derived adipokines might increase OPN expression in the vasculature of diabetic mice. Among a variety of adipokines, resistin and visfatin were proposed as important pro-inflammatory mediators 35, considered to be important regulators for VSMCs proliferation 36. Thus, we determined the expression of resistin and visfatin in PVAT of ob/ob and DIO mice. In PVAT of DIO mice, an increased macrophage infiltration was accompanied by the increased expression of resistin and visfatin expression, almost exclusively along the septa. Interestingly, although it is known that resistin is mainly expressed in adipose tissues in rodents 37, our present study showed that resistin in PVAT was mostly co-localized with macrophages. Similar to other reports in which macrophages, rather than adipocytes, appear to be the most important source of resistin in human subjects 38,39, our data suggested that resistin was largely produced by macrophages in PVAT of DIO mice. However, visfatin was detected in macrophages and in the interstitial space or in other cell types, suggesting that visfatin was produced by macrophages as well as by other cells, including adipocytes.
The average serum level of resistin in patients with diabetes (49.7 ng/ml) was markedly increased compared to that in healthy control subjects (30.7 ng/ml) 24. Plasma resistin levels correlate with markers of inflammation and coronary atherosclerosis 40 and play a pivotal role in the pathogenesis of neointimal thickening after mechanical injury 41. Moreover, hyper-resistinaemia was associated with vascular complications in patients with diabetes 42, suggesting its critical role in the development of diabetic vascular disease. Thus, in an in vitro study, VSMCs were co-cultured with PVAT to investigate the potential link between PVAT-derived adipokines and enhanced expression of OPN in diabetic vasculature. OPN expression in VSMCs co-cultured with diabetic PVAT was significantly increased compared to control PVAT, which was attenuated by immunodepletion of resistin with neutralizing antibody. Likewise, the OPN expression in VSMCs was up-regulated by exogenous resistin, indicating a pivotal role of resistin on the up-regulation of OPN expression. However, in contrast to other reports in which PVAT-derived visfatin was found to be a VSMC growth factor 33,43, we observed no effect of visfatin on the expression of OPN in VSMCs. Considering the fact that intracellular visfatin converts VSMCs from a proliferative to a non-proliferative state 42, and that the action of intracellular visfatin on VSMCs proliferation differs from that of extracellular visfatin 33, further studies are needed to explain these discrepancies.
In the in vitro study, resistin increased the expression of OPN mRNA in VSMCs in a time- and dose-dependent manner. The regulation of OPN transcription has been suggested to require the concerted action of various transcription factors in VSMCs. Previously, the NFAT-binding site has been demonstrated in the proximal region, where both activator protein (AP)-1 and upstream transcription factor have been shown to bind 44, and were involved in OPN expression in VSMCs exposed to hyperglycaemia. Although the possible synergisms between NFAT and AP-1 have not been examined, it is possible that all these proteins interact to overcome stereological constraints and enable stable assembly within this limited DNA region. Further, it has also been shown that resistin is linked to activation of AP-1 involved in the induction of adhesion molecules, chemokines and reactive oxygen species in endothelial 45 and smooth muscle cells 46. Interestingly, in the present experiments using luciferase-reporter plasmids containing different lengths of the 5′ region of the mouse OPN promoter, basal activity of the OPN promoter was greatly reduced when the regions harbouring the AP-1 and C/EBP were deleted. In addition, AP-1 activity, but not C/EBP activity, was markedly increased by exogenous resistin in a dose-dependent manner, indicating an important role of signalling pathways that activate AP-1 in resistin-induced OPN expression in VSMCs. Although signalling pathways involved in OPN expression might be diverse and dependent on a variety of different stimuli, the results of our present study indicated a potential role of resistin derived from macrophages in PVAT in the increased expression of OPN in diabetic vasculature.
We demonstrated that DIO was associated with an increased accumulation of PVAT characterized by an increased production of resistin. Resistin in PVAT was mostly derived from macrophages and played a pivotal role in the up-regulation of OPN expression at the transcriptional level in VSMCs via a signalling pathway that involves activation of AP-1. Our findings suggest a potential role of resistin in PVAT in diabetic vascular complications through phenotypic conversion of VSMCs of diabetic vasculature. Thus, strategies targeting macrophage recruitment and resistin could be promising options for the prevention of diabetes-associated vascular diseases.
Acknowledgments
This study was supported by the medical research centre programme of the Ministry of Education, Science and Technology/Korea Science and Engineering Foundation (Grant No. 2005–0049416).
Author contributions
SYP, KHK and KWS conceived and carried out experiments; JUB collected and analysed data; SJL and WSL were involved in the literature search and generation of figures; YHK carried out additional experiments to make the revised version of the manuscript; and CDK was involved in study design and data interpretation. All authors were involved in writing the paper and had final approval of the submitted and published versions.
References
- 1.Rajsheker S, Manka D, Blomkalns AL, et al. Crosstalk between perivascular adipose tissue and blood vessels. Curr Opin Pharmacol. 2010;10::191–196. doi: 10.1016/j.coph.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6::772–783. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- 3.Skilton MR, Sérusclat A, Sethu AH, et al. Noninvasive measurement of carotid extra-media thickness: associations with cardiovascular risk factors and intima–media thickness. JACC Cardiovasc Imaging. 2009;2::176–182. doi: 10.1016/j.jcmg.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 4.Gao YJ, Lu C, Su LY, et al. Modulation of vascular function by perivascular adipose tissue: the role of endothelium and hydrogen peroxide. Br J Pharmacol. 2007;151::323–331. doi: 10.1038/sj.bjp.0707228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao YJ, Holloway AC, Su LY, et al. Effects of fetal and neonatal exposure to nicotine on blood pressure and perivascular adipose tissue function in adult life. Eur J Pharmacol. 2008;590::264–268. doi: 10.1016/j.ejphar.2008.05.044. [DOI] [PubMed] [Google Scholar]
- 6.Lu C, Su LY, Lee RM, et al. Alterations in perivascular adipose tissue structure and function in hypertension. Eur J Pharmacol. 2011;656::68–73. doi: 10.1016/j.ejphar.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 7.Greif M, Becker A, von Ziegler F, et al. Pericardial adipose tissue determined by dual source CT is a risk factor for coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 2009;29::781–786. doi: 10.1161/ATVBAHA.108.180653. [DOI] [PubMed] [Google Scholar]
- 8.Payne GA, Borbouse L, Kumar S, et al. Epicardial perivascular adipose-derived leptin exacerbates coronary endothelial dysfunction in metabolic syndrome via a protein kinase C-β pathway. Arterioscler Thromb Vasc Biol. 2010;30::1711–1717. doi: 10.1161/ATVBAHA.110.210070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takemoto M, Yokote K, Nishimura M, et al. Enhanced expression of osteopontin in human diabetic artery and analysis of its functional role in accelerated atherogenesis. Arterioscler Thromb Vasc Biol. 2000;20::624–628. doi: 10.1161/01.atv.20.3.624. [DOI] [PubMed] [Google Scholar]
- 10.Panda D, Kundu GC, Lee BI, et al. Potential roles of osteopontin and αVβ3 integrin in the development of coronary artery restenosis after angioplasty. Proc Natl Acad Sci USA. 1997;94::9308–9313. doi: 10.1073/pnas.94.17.9308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li JJ, Zhu CG, Yu B, et al. The role of inflammation in coronary artery calcification. Ageing Res Rev. 2007;6::263–270. doi: 10.1016/j.arr.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 12.Shimizu H, Takahashi M, Takeda S, et al. Mycophenolate mofetil prevents transplant arteriosclerosis by direct inhibition of vascular smooth muscle cell proliferation. Transplantation. 2004;77::1661–1667. doi: 10.1097/01.tp.0000127592.13707.b6. [DOI] [PubMed] [Google Scholar]
- 13.Li G, Oparil S, Kelpke SS, et al. Fibroblast growth factor receptor-1 signaling induces osteopontin expression and vascular smooth muscle cell-dependent adventitial fibroblast migration in vitro. Circulation. 2002;106::854–859. doi: 10.1161/01.cir.0000024113.26985.cc. [DOI] [PubMed] [Google Scholar]
- 14.Yin BL, Hao H, Wang YY, et al. Down-regulating osteopontin reduces angiotensin II-induced inflammatory activation in vascular smooth muscle cells. Inflamm Res. 2009;58::67–73. doi: 10.1007/s00011-009-8034-0. [DOI] [PubMed] [Google Scholar]
- 15.Scatena M, Liaw L, Giachelli CM. Osteopontin: a multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler Thromb Vasc Biol. 2007;27::2302–2309. doi: 10.1161/ATVBAHA.107.144824. [DOI] [PubMed] [Google Scholar]
- 16.Henrichot E, Juge-Aubry CE, Pernin A, et al. Production of chemokines by perivascular adipose tissue: a role in the pathogenesis of atherosclerosis? Arterioscler Thromb Vasc Biol. 2005;25::2594–2599. doi: 10.1161/01.ATV.0000188508.40052.35. [DOI] [PubMed] [Google Scholar]
- 17.Greenstein AS, Khavandi K, Withers SB, et al. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation. 2009;119::1661–1670. doi: 10.1161/CIRCULATIONAHA.108.821181. [DOI] [PubMed] [Google Scholar]
- 18.Bruemmer D, Collins AR, Noh G, et al. Angiotensin II-accelerated atherosclerosis and aneurysm formation is attenuated in osteopontin-deficient mice. J Clin Invest. 2003;112::1318–1331. doi: 10.1172/JCI18141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langheim S, Dreas L, Veschini L, et al. Increased expression and secretion of resistin in epicardial adipose tissue of patients with acute coronary syndrome. Am J Physiol Heart Circ Physiol. 2010;298::746–753. doi: 10.1152/ajpheart.00617.2009. [DOI] [PubMed] [Google Scholar]
- 20.Chatterjee TK, Stoll LL, Denning GM, et al. Proinflammatory phenotype of perivascular adipocytes: influence of high-fat feeding. Circ Res. 2009;104::541–549. doi: 10.1161/CIRCRESAHA.108.182998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takaoka M, Suzuki H, Shioda S, et al. Endovascular injury induces rapid phenotypic changes in perivascular adipose tissue. Arterioscler Thromb Vasc Biol. 2010;30::1576–1582. doi: 10.1161/ATVBAHA.110.207175. [DOI] [PubMed] [Google Scholar]
- 22.Stofkova A. Resistin and visfatin: regulators of insulin sensitivity, inflammation and immunity. Endocr Regul. 2010;44::25–36. doi: 10.4149/endo_2010_01_25. [DOI] [PubMed] [Google Scholar]
- 23.Bokarewa M, Nagaev I, Dahlberg L, et al. Resistin, an adipokine with potent proinflammatory properties. J Immunol. 2005;174::5789–5795. doi: 10.4049/jimmunol.174.9.5789. [DOI] [PubMed] [Google Scholar]
- 24.Shalev A, Patterson NB, Hirshberg B, et al. Resistin serum levels in type 1 diabetes pre- and post-islet transplantation. Metabolism. 2004;53::403–404. doi: 10.1016/j.metabol.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 25.Giachelli CM, Bae N, Almeida M, et al. Osteopontin is elevated during neointima formation in rat arteries and is a novel component of human atherosclerotic plaques. J Clin Invest. 1993;92::1686–1696. doi: 10.1172/JCI116755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Isoda K, Nishikawa K, Kamezawa Y, et al. Osteopontin plays an important role in the development of medial thickening and neointimal formation. Circ Res. 2002;91::77–82. doi: 10.1161/01.res.0000025268.10302.0c. [DOI] [PubMed] [Google Scholar]
- 27.Isoda K, Kamezawa Y, Ayaori M, et al. Osteopontin transgenic mice fed a high-cholesterol diet develop early fatty-streak lesions. Circulation. 2003;107::679–681. doi: 10.1161/01.cir.0000055739.13639.d7. [DOI] [PubMed] [Google Scholar]
- 28.Bruemmer D, Collins AR, Noh G, et al. Angiotensin II-accelerated atherosclerosis and aneurysm formation is attenuated in osteopontin-deficient mice. J Clin Invest. 2003;112::1318–1331. doi: 10.1172/JCI18141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsui Y, Rittling SR, Okamoto H, et al. Osteopontin deficiency attenuates atherosclerosis in female apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23::1029–1034. doi: 10.1161/01.ATV.0000074878.29805.D0. [DOI] [PubMed] [Google Scholar]
- 30.Scatena M, Liaw L, Giachelli CM. Osteopontin: a multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler Thromb Vasc Biol. 2007;27::2302–2309. doi: 10.1161/ATVBAHA.107.144824. [DOI] [PubMed] [Google Scholar]
- 31.Nomiyama T, Perez-Tilve D, Ogawa D, et al. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J Clin Invest. 117::2877–2828. doi: 10.1172/JCI31986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Police SB, Thatcher SE, Charnigo R, et al. Obesity promotes inflammation in periaortic adipose tissue and angiotensin II-induced abdominal aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2007;29::1458–1464. doi: 10.1161/ATVBAHA.109.192658. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takaoka M, Suzuki H, Shioda S, et al. Endovascular injury induces rapid phenotypic changes in perivascular adipose tissue. Arterioscler Thromb Vasc Biol. 2010;30::1576–1582. doi: 10.1161/ATVBAHA.110.207175. [DOI] [PubMed] [Google Scholar]
- 34.Miao CY, Li ZY. The role of perivascular adipose tissue in vascular smooth muscle cell growth. Br J Pharmacol. 2012;165::643–658. doi: 10.1111/j.1476-5381.2011.01404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stofkova A. Resistin and visfatin: regulators of insulin sensitivity, inflammation and immunity. Endocr Regul. 2010;44::25–36. doi: 10.4149/endo_2010_01_25. [DOI] [PubMed] [Google Scholar]
- 36.Calabro P, Samudio I, Willerson JT, et al. Resistin promotes smooth muscle cell proliferation through activation of extracellular signal-regulated kinase 1/2 and phosphatidylinositol 3-kinase pathways. Circulation. 2004;110::3335–3340. doi: 10.1161/01.CIR.0000147825.97879.E7. [DOI] [PubMed] [Google Scholar]
- 37.Steppan CM, Lazar MA. Resistin and obesity-associated insulin resistance. Trends Endocrinol Metab. 2002;13::18–23. doi: 10.1016/s1043-2760(01)00522-7. [DOI] [PubMed] [Google Scholar]
- 38.Rea R, Donnelly R. Resistin: an adipocyte-derived hormone. Has it a role in diabetes and obesity? Diabetes Obes Metab. 2004;6::163–170. doi: 10.1111/j.1462-8902.2004.00334.x. [DOI] [PubMed] [Google Scholar]
- 39.Patel L. Objective assessment of pancreatic function in all patients with cystic fibrosis. J Pediatr. 2005;147::127–128. doi: 10.1016/j.jpeds.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 40.Shyu KG, Lien LM, Wang BW, et al. Resistin contributes to neointimal formation via oxidative stress after vascular injury. Clin Sci (Lond) 2011;120::121–129. doi: 10.1042/CS20100226. [DOI] [PubMed] [Google Scholar]
- 41.Shin HJ, Park S, Yoon SJ, et al. Association between serum resistin and carotid intima media thickness in hypertension patients. Int J Cardiol. 2008;125::79–84. doi: 10.1016/j.ijcard.2007.02.028. [DOI] [PubMed] [Google Scholar]
- 42.Miao CY, Li ZY. The role of perivascular adipose tissue in vascular smooth muscle cell growth. Br J Pharmacol. 2012;165::643–658. doi: 10.1111/j.1476-5381.2011.01404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nilsson-Berglund LM, Zetterqvist AV, Nilsson-Ohman J, et al. Nuclear factor of activated T cells regulates osteopontin expression in arterial smooth muscle in response to diabetes-induced hyperglycemia. Arterioscler Thromb Vasc Biol. 2010;30::218–224. doi: 10.1161/ATVBAHA.109.199299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun J, Xu Y, Dai Z, et al. Intermittent high glucose enhances proliferation of vascular smooth muscle cells by upregulating osteopontin. Mol Cell Endocrinol. 2009;313::64–69. doi: 10.1016/j.mce.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 45.Gan AM, Butoi ED, Manea A, et al. Inflammatory effects of resistin on human smooth muscle cells: up-regulation of fractalkine and its receptor, CX3CR1, expression by TLR4 and Gi-protein pathways. Cell Tissue Res. 2013;351::161–174. doi: 10.1007/s00441-012-1510-9. [DOI] [PubMed] [Google Scholar]
- 46.Verma S, Li SH, Wang CH, et al. Resistin promotes endothelial cell activation: further evidence of adipokine–endothelial interaction. Circulation. 2003;108::736–740. doi: 10.1161/01.CIR.0000084503.91330.49. [DOI] [PubMed] [Google Scholar]