

Abstract
Dysfunction of the mucosal immune system plays an important role in inflammatory bowel disease (IBD) pathogenesis. Dendritic cells are emerging as central players based on both our increasing understanding of how genetic susceptibility impacts the mucosal immune system and the key role of dendritic cells in regulating response to gut microflora. We discuss areas of therapeutic opportunity in this evolving landscape. © 2013 The Authors. Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: inflammatory bowel disease, dendritic cell, mucosal immunity, colitis, Crohn's disease, inflammation
Introduction
The inflammatory bowel diseases (IBDs) are a heterogeneous group of disorders which have two major phenotypic forms, Crohn's disease and ulcerative colitis, characterized by chronic relapsing and remitting intestinal inflammation 1–4. Ulcerative colitis is the most common of the IBDs 5 and is characterized by a continuous pattern of inflammation beginning in the rectum and extending progressively further into the colon with increasing disease severity. By contrast, Crohn's disease patients frequently develop a discontinuous pattern of inflammation which can occur anywhere in the gastrointestinal tract, although the distal small intestine and colon are the most common locations 6.
In ulcerative colitis, inflammatory infiltrates are confined to the mucosa and consist primarily of lymphocytes and plasma cells plus granulocytes in crypt abscesses and present in the mucosa during disease flares. Ulceration is common during active disease and colonic epithelial cell changes such as goblet cell depletion, distorted crypt architecture, and epithelial dysplasia are associated with chronic disease 7. Transmural inflammation is a characteristic feature of Crohn's disease and this deeper tissue involvement in the inflammatory process appears to be responsible for many of the serious complications associated with Crohn's disease such as fibrostenotic disease, abscesses, and fistula formation 8,9. Epithelioid granulomas are also associated with Crohn's disease and provide diagnostic differentiation from ulcerative colitis when identified histologically. Epithelioid cells, so named for a resemblance to epithelial cells, are activated histiocytes with homogeneous eosinophilic cytoplasm, and a working definition of an epithelioid granuloma is a collection of at least five epithelioid cells with or without accompanying multinucleate giant cells 10. However, these granulomas are found in only about 20–40% of biopsies and 60% of surgical resection specimens 11 and, when present, must be differentiated from those associated with infectious diseases such as tuberculosis.
When Crohn's disease inflammation is confined to the mucosa and no granulomatous lesions are present, it cannot be definitively differentiated from ulcerative colitis by histological evaluation. A subset of patients present with clinical and histological features of disease which do not clearly segregate into one of the established phenotypic forms. These patients are often diagnosed as having indeterminate colitis 12,13. Some of these patients will eventually develop lesions more characteristic of one of the major forms of IBD.
Accurately determining the prevalence of inflammatory bowel disease is a challenging proposition given that IBD is not a reportable disease and that patients can have a multi-decade disease course. Prevalence estimates have been drawn based on either in-depth regional data 14 or administrative data from health plan databases 15,16. Based on these data, there are between 1 and 1.5 million individuals with ulcerative colitis or Crohn's disease in the United States alone. Clinical manifestations of disease develop during childhood in up to 25% of patients 2, and disease prevalence in children under 20 years of age is 43 per 100 000 for Crohn's disease and 28 per 100 000 for ulcerative colitis 16. As expected with a chronic disease, the prevalence increases with age to about 201 per 100 000 for Crohn's disease and 238 per 100 000 for ulcerative colitis. The incidence of Crohn's disease and ulcerative colitis in the United States increased after 1940; however, those rates appear to have stabilized over the past few decades 14. Despite some uncertainty about the total number of IBD patients, it is clear that large numbers of patients exist and that many of these patients can anticipate the need for treatment over a span of decades. While the advent of biological therapies has improved the clinical situation, IBD remains a major unmet medical need.
Current models hypothesize that IBD arises from and is sustained by interactions between genetically susceptible hosts and the gut microflora as well as, potentially, other environmental triggers. While the pathogenesis of IBD remains incompletely understood, it is clear that dysfunction of the mucosal immune system plays an important role. Dendritic cells are a key player in the mucosal immune system, serving as a bridge between the innate and the adaptive immune response 17. In this review, we will discuss the evidence for dendritic cells in IBD pathogenesis with emphasis on genetics and microbial interactions. The innate immune system is emerging as a potentially attractive therapeutic target in IBD and we will review some of the current information in this area.
Genetics of IBD
It is well established that host genetic susceptibility plays an important role in IBD pathogenesis. The first Crohn's disease susceptibility gene, nucleotide oligomerization domain receptor 2 (NOD2), which is encoded by the CARD15 gene, was identified in 2001 18,19. In the intervening years, tremendous progress has been made using genome-wide linkage and association studies to reveal additional genetic polymorphisms associated with susceptibility to ulcerative colitis or Crohn's disease. Currently there are more than 160 IBD susceptibility genes or loci recognized 20. A number of susceptibility genes are shared between ulcerative colitis and Crohn's disease 21,22, suggesting the presence of shared or interlinking pathways in inflammatory bowel disease pathogenesis. While the functional role of many loci or specific single nucleotide polymorphisms (SNPs) is incompletely understood, many of the genes are associated with aspects of mucosal immunity including innate immune response to microbial pathogens 20 (Figure 1).
Figure 1.
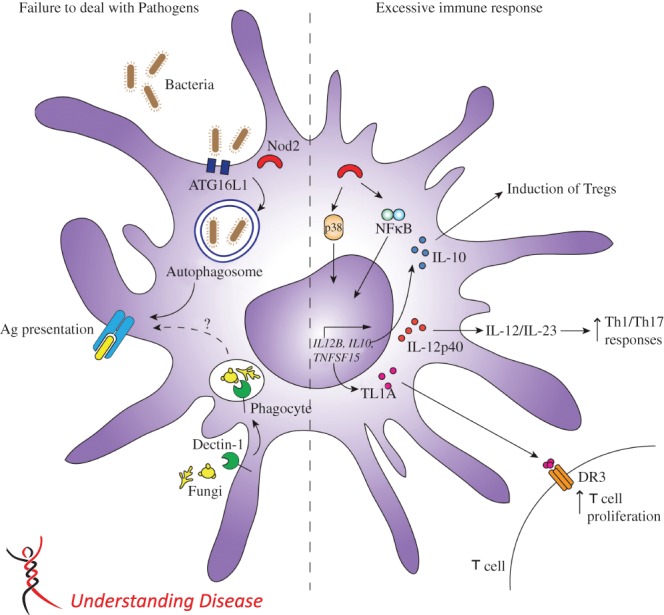
Impact of IBD genetic polymorphisms on dendritic cell function. Polymorphisms in IBD susceptibility genes in dendritic cells can be broadly categorized as either inhibiting the ability to effectively clear pathogens or contributing to excessive immune response. Dendritic cells play an important role in autophagy and presentation of bacterial antigens. SNPs associated with this pathway, including Nod2 and ATG16L1, can contribute to failure to deal with pathogens. Additionally, dectin-1 is important for the clearance of fungal pathogens by enabling dendritic cell recognition of β-1,3-glucans in fungal cell walls. A number of IBD susceptibility genes are linked to the excessive immune response which is characteristic of both Crohn's disease and ulcerative colitis. Nod2 in dendritic cells is upstream of MAPK and NF-κB, which are important regulators of pro-inflammatory cytokines. Impaired regulation of the inflammatory response caused by polymorphisms in Nod2, IL12B or TNFSF15 may result in excessive and prolonged pro-inflammatory T-cell responses. In addition, dendritic cells regulate the immune response through production of the anti-inflammatory cytokine IL-10. SNPs in IL-10 may result in loss of regulatory T cells, leading to excessive immune response.
The importance of microbial recognition and the response of the innate immune system is underscored by the association between Crohn's disease and NOD2, which is a member of the NLR (NOD, leucine-rich repeat-containing protein) family of intracellular pathogen-associated molecular recognition receptors (PRRs) 23,24. This association includes three NOD2 polymorphisms which occur with greatest frequency in individuals of European descent but are not found in Asian populations 25. Approximately 30% of patients of European ancestry will have one or more of these mutations and these patients are at increased risk for ileal involvement and fibrostenotic disease 26. Individuals heterozygous for a NOD2 polymorphism have an increased Crohn's disease risk of 2.4-fold, while homozygous individuals have a 17.1-fold increase 25. These are the highest relative risks associated with any IBD-risk gene.
NOD2 is expressed by a variety of immune and non-immune cell types including dendritic cells and is upstream of the nuclear factor-κB (NF-κB) and mitogen-activated protein (MAP) kinase signalling pathways which drive pro-inflammatory cytokine production 27. It encodes an intracellular sensor of peptidoglycan, a component of bacterial cell walls, and Crohn's disease-associated mutations are mainly located in the leucine-rich repeat region which interacts with the peptidoglycan muramyl dipeptide (MDP) motif, leading to altered bacterial recognition 28,29. How NOD2 polymorphisms predispose to Crohn's disease development remains incompletely understood, despite significant research effort in this field. Because the intestinal tract has continuous bacterial exposure, chronic activation of NOD2 signalling should result in immune cell hyporesponsiveness to subsequent NOD2 or Toll-like receptor (TLR) ligand stimulation 30,31. While the NOD2-mediated mechanisms which down-regulate pro-inflammatory cytokines during exposure to commensal bacteria are not fully characterized, it is clear that the process is defective in patients with Crohn's disease-associated NOD polymorphisms 31. One hypothesis is that NOD2 normally acts to attenuate TLR signalling, resulting in reduced activation of NF-κB, and thereby prevents excessive activation of dendritic cells and subsequent pathogenic T-cell response 32.
There is increasing evidence that PRR signals intersect with other pathways that coordinate bacterial responses. For example, NOD2 interaction with autophagy pathways has recently been recognized 33,34. Autophagy is the process by which cytoplasmic components are sequestered into double membrane vacuoles which then fuse with lysosomes, and this process is important in microbial defence processes such as capture of intracellular bacteria during phagocytosis, antigen presentation, and inflammasome activation 35. ATG16L1 encodes an autophagy protein which is part of a complex responsible for proper subcellular localization of the autophagy machinery 36,37. The ATG16L1 polymorphism is associated with an increased risk of Crohn's disease 38 and similar to NOD2, shows an association with terminal ileal disease 38,39. Interestingly, the affected domain of ATG16L1 is non-conserved and is not required for all of its functions 40.
In human dendritic cells, autophagy is induced through NOD2 stimulation, and dendritic cells isolated from Crohn's disease patients with ATG16L1 and/or NOD2 polymorphisms have defective autophagy induction in addition to altered antigen presentation and bacterial handling 33 (Figure 1). Interestingly, promoter polymorphism in the autophagy gene IRGM has also been associated with increased risk of developing Crohn's disease 41, although characterization of the specific effects on dendritic cells is still preliminary. In aggregate, these data suggest that autophagy may be an attractive drug target area for modifying dendritic cell function in the treatment of Crohn's disease. Preliminary evidence in support of this concept has been generated using rapamycin, an antibiotic which triggers autophagy by forming a complex with FKB12, which then inhibits mTOR and is commonly used to up-regulate autophagy in cell culture 42. Rapamycin has been used successfully to treat a patient with severe refractory Crohn's disease and has also shown protection in a murine colitis model, suggesting that this therapeutic approach may be promising 43,44.
Genome-wide association studies (GWAS) have shown strong associations of polymorphisms in the IL23R and IL12B gene loci with Crohn's disease and ulcerative colitis 45,46. IL12B encodes the IL-12p40 subunit, which is a component of both the IL-12 and the IL-23 cytokines, while IL23R encodes one of two subunits of the IL-23R 47. IL-23 is induced in dendritic cells (DCs) by PPR stimulation and can promote a wide range of pathological responses in the intestine, mediated either through T cells or through excessive innate immune cell activation 48,49 (Figure 1). DC IL-23 production, which is augmented under conditions of endoplasmic reticulum stress and activation of the unfolded protein response (UPR), is an important component of anti-microbial defence linking innate and adaptive immune responses 50. However, excessive or inappropriate DC IL-23 production favours pro-inflammatory T-cell responses including enhanced proliferation of effector T cells, reduced differentiation of forkhead box P3 (FOXP3)-positive regulatory T cells, and emergence of the IL-17- and IFN-γ-producing cells associated with chronic intestinal inflammation 49. Conversely, IL-23R polymorphisms, which are associated with protection from IBD risk, have been functionally characterized and result in reduced T-cell activation 51–53. Studies in mouse colitis models have shown that IL-23 plays an important role in chronic intestinal inflammation 54. In addition, the anti-IL-12p40 monoclonal antibody ustekinumab has demonstrated some clinical efficacy in a subset of Crohn's disease patients who were resistant to TNF antagonists 55.
Some dendritic cell processes linked to IBD genetic susceptibility have been identified as potential therapeutic targets. For example, blocking interactions between dendritic cells and T cells has been proposed as a means of decreasing activity of the IL-23/IL-17 pro-inflammatory pathway important in IBD pathogenesis 56. Activated dendritic cells express the TNF family cytokine TL1A (TNFSF15), which interacts with the DR3 receptor (TNFRSF25) on lymphocytes 57,58 (Figure 1). Polymorphisms in the TNFSF15 gene have been shown to contribute to the risk of both ulcerative colitis and Crohn's disease 59,60. Increased expression of TL1A has been shown in Crohn's disease tissue 61, and antibody to TL1A prevents colitis development in mouse models 62. In addition, mice overexpressing TL1A develop spontaneous intestinal inflammation 63,64. These results suggest that TL1A may be a viable dendritic cell target in IBD.
A role for IL-10 in IBD pathogenesis was first identified in mice deficient in either IL-10 or the IL-10 receptor 2 (IL-10R2) subunit, as these mice develop spontaneous colitis 65,66. This has more recently been extended into humans, with the association of very early onset IBD with IL-10 or IL-10 receptor (IL-10R) deficiencies 67–69. Patients with IL-10/IL-10R loss-of-function mutations present, often in the first 3 months of life, with severe and progressive colitis complicated by perianal disease 70. While IL-10 and IL-10R deficiencies are fortunately rare, GWAS findings suggest that IL-10 may play a broader role in IBD pathogenesis. IL-10 SNPs have been associated with both ulcerative colitis and Crohn's disease in paediatric and adult populations 71–73.
IL-10 is secreted by a variety of immune cell types including dendritic cells 74 and plays an important anti-inflammatory role through interactions with regulatory T cells, leading to inhibition of effector T-cell response 75 (Figure 1). This effect of IL-10 on T cells is mediated primarily by antigen-presenting cells such as dendritic cells and macrophages 76. This suggests that dendritic cells may be important in the pathogenesis of IL-10 SNP-related IBD. IL-10 has been explored as a potential therapeutic in inflammatory bowel disease with limited success 77 and is currently of most interest in association with probiotic therapeutic strategies 78.
Recently, an association has been identified between polymorphism in the dectin-1 gene (CLEC7A) and severe ulcerative colitis 79. The risk haplotype was strongly associated with the development of medically refractory ulcerative colitis and overrepresented in patients requiring colectomy. Dectin-1 is a C-type lectin receptor expressed by dendritic cells and macrophages which recognizes β-1,3-glucans found in fungal cell walls 80. Dectin-1 deficiency had previously been associated with increased susceptibility to fungal disease in humans and mice 81,82. The mechanisms by which dectin-1 influences fungal control and IBD pathogenesis are still being delineated, but evidence is emerging that it may be necessary to enable dendritic cell antigen presentation to T cells 83. Therapeutic strategies for enhancing the control of microflora in IBD, except for antibiotics and probiotics, are largely unexplored.
Microbiota in IBD
Intestinal microbes represent the largest microbial community in the body, where up to 100 trillion (1014) microbes may exist in a commensal relationship with the human host 84–86. This relationship is facilitated by the intestinal mucus layer which creates a physical boundary between the host and microbe, by specific characteristics of the microbial community which reduce their immune cell-activating properties, and by direct influence on the immune cells. The intestinal microflora develops in early life and then the composition remains largely stable under healthy conditions 87–90. Despite the large number of individual organisms, the number of bacterial species is estimated to be about 1000 per individual and represents only a small fraction of the existing phyla, which supports the idea that the commensal relationship is the product of a tight co-evolutionary history between the host and microbiota 86.
Shifts in the bacterial makeup on human intestinal microflora have been documented in IBD using 16S rRNA sequencing. These studies show that a subset of Crohn's disease and ulcerative colitis patients have depletion of commensal bacteria, especially members of the phyla Firmicutes and Bacteroidetes 91. Overall bacterial burden is typically lower in patients with Crohn's disease than in those with ulcerative colitis but both are lower than healthy control individuals 92. Even within the same patient, bacterial diversity is reduced in inflamed regions when compared with non-inflamed regions. These data suggest that not only is the host inflammatory response likely contributing to the loss of diversity characteristic of IBD patients but it is also providing a selective advantage to the subset of microbiota which have an increased presence in the disease state 93.
A role for microflora in IBD pathogenesis is supported by studies showing that exposure to the faecal stream is required for endoscopic and histological disease manifestations 94–96 and that faecal transplants from healthy individuals can also have therapeutic benefit 97,98. In addition, antibiotic therapy has shown some efficacy in clinical trials, with predominantly symptomatic improvement but also some endoscopic improvement and induction of remission, depending on the antibiotic and on the trial design 99–101. Experimental colitis models also support the contention that microflora is a key component, as most mouse models of IBD require the presence of intestinal microbiota for colitis to develop 102,103.
Enteric flora plays an important role in intestinal immune cell development including dendritic cells. Dendritic cells discriminate between pathogenic and commensal bacteria by utilizing PRRs which recognize specific pathogen-associated molecular patterns. These include the toll-like receptors (TLRs) and C-type lectins such as mannose receptor and dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin (DC-SIGN) 104. The microbial environment encountered by an immature dendritic cell can determine the ability of the mature dendritic cell to drive T-cell differentiation towards T helper 1 (TH1), T helper 2 (TH2) or regulatory polarization 105–110. This makes the dendritic cell a key player in determining whether a tolerant or protective immune response is mounted at the mucosal surface in response to specific microflora 111.
Tolerogenic properties of dendritic cells are potentially attractive in a therapeutic setting. This is one of the mechanistic tenants underlying the interest in probiotic therapy. For example, probiotics might help to restore normal microbial populations in the intestine 112; however, to date, these efforts have not generated consistently promising results in IBD, particularly in Crohn's disease 113–115. Therefore, many investigators have turned to investigating the immunomodulatory effects of specific bacteria or bacterial components in murine models to identify promising candidates. There is emerging evidence that alterations of cell surface components of lactobacilli can alter the immunoregulatory responses of dendritic cells, and this might provide a better defined therapeutic pathway in inflammatory diseases of the gastrointestinal tract 116. The feasibility of utilizing genetically modified bacteria as a therapeutic agent has been established 117.
Mucosal dendritic cells and IBD
The innate immune system exists to provide a rapid initial response to pathogens but it must also identify and minimize immune response to commensal microflora. Initiation of the gut mucosal immune response takes place following antigen uptake by dendritic cells and presentation to adaptive immune effector cells. However, in the absence of pathogen recognition, mucosal dendritic cells primarily function to regulate immune responsiveness 118. While there is general consensus that dendritic cells from a healthy intestinal tract are hyporesponsive to commensal bacterial components, the mechanisms by which this is achieved are not fully understood. In fact, the scientific literature surrounding this phenomenon is becoming increasingly more complex. One example of this is the evolving understanding of the role of CD103+ dendritic cells in mucosal homeostasis.
Dendritic cell subsets are typically defined based on expression of surface markers, especially CD11b (integrin αM) and CD103 (αE integrin). The majority of this work has been done in mice and dendritic cell subsets are not identical between mice and humans. CD103+ dendritic cells comprise a substantial subset of murine mucosal dendritic cells and develop from classical precursors 119,120. These have been considered ‘tolerogenic’ DCs in mice 121, which are believed to be important in initiating T-cell responses including induction of FOXP3+ regulatory T cells (Tregs), and in the establishment of oral tolerance 122.
While CD103+ dendritic cells have been credited with a major role in the maintenance of mucosal hyporesponsiveness, in part due to their ability to produce retinoic acid necessary for the development of FoxP3+ Tregs 123, there remains some controversy on this point. Recently, Batf3 KO mice that lack a CD103+ DC subset have been reported to have normal Treg populations in the lamina propria 124, suggesting that other dendritic subsets have the potential to support Treg development. Also, recent work has shown that CD103+ dendritic cells are capable of directly sampling, transporting, and presenting luminal antigens, and therefore are not restricted to a purely immunoregulatory role in the mucosa 125,126. Moreover, CD103+ mucosal dendritic cell populations are heterogeneous and can be further subdivided into two major populations of CD11b+ and CD11b− subsets, which vary in terms of transcription factors required for their development as well as in geographical distribution within the intestinal mucosa 127,128.
Substantially less is known about the origin and function of human intestinal dendritic cells. CD103+ dendritic cells have been identified in the human colon 129 but, unlike in mice, these cells do not constitute the dominant mucosal subset. This suggests that other distinct dendritic cell subsets may contribute to the tolerogenic intestinal mucosal environment. Therefore, although the data suggest that a subset of CD103+ dendritic cells may be very important for maintaining mucosal immune hyporesponsiveness in normal individuals, the scientific understanding of these aspects of dendritic cell biology is still evolving.
As noted previously, dendritic cells are implicated in IBD pathogenesis by both genetics and their central role in the control of microbial interactions. Activated dendritic cells accumulate at sites of intestinal inflammation in human IBD and in murine models of intestinal inflammation 130–132. These cells express increased levels of a variety of activation markers, enhanced TLR responsiveness, and are phenotypically distinct from the hyporesponsive dendritic cells which help to mediate mucosal homeostasis 119,120,133–135. These activated dendritic cells likely contribute to intestinal pathology and may prove to be valuable therapeutic targets in IBD. As our understanding of dendritic cell biology continues to grow and with increasing definition of mechanistic pathways, we expect to see the emergence of new dendritic cell-related drug targets.
Author contribution statement
Both authors contributed to the conceptualization and writing of this review paper.
References
- 1.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361::2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levine A, Griffiths A, Markowitz J, et al. Pediatric modification of the Montreal classification for inflammatory bowel disease: the Paris classification. Inflamm Bowel Dis. 2011;17::1314–1321. doi: 10.1002/ibd.21493. [DOI] [PubMed] [Google Scholar]
- 3.Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28::573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448::427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 5.Danese S, Fiocchi C. Ulcerative colitis. N Engl J Med. 2011;365::1713–1725. doi: 10.1056/NEJMra1102942. [DOI] [PubMed] [Google Scholar]
- 6.Farmer RG, Hawk WA, Turnbull RB. Clinical patterns in Crohn's disease: a statistical study of 615 cases. Gastroenterology. 1975;68::627–635. [PubMed] [Google Scholar]
- 7.Gramlich T, Petras RE. Pathology of inflammatory bowel disease. Semin Pediatr Surg. 2007;16::154–163. doi: 10.1053/j.sempedsurg.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Keighley MRB, Eastwood D, Ambrose NS. Incidence and microbiology of abdominal and pelvic abscess in Crohn's disease. Gastroenterology. 1982;83::1271–1275. [PubMed] [Google Scholar]
- 9.Maeda K, Okada M, Yao T, et al. Intestinal and extraintestinal complications of Crohn's disease: predictors and cumulative probability of complications. J Gastroenterol. 1994;29::577–582. doi: 10.1007/BF02365438. [DOI] [PubMed] [Google Scholar]
- 10.Jenkins D, Balsitis M, Gallivan S, et al. Guidelines for the initial biopsy diagnosis of suspected chronic idiopathic inflammatory bowel disease. The British Society of Gastroenterology Initiative. J Clin Pathol. 1997;50::93–105. doi: 10.1136/jcp.50.2.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scholmerich J, Warren B. Differential diagnosis and other forms of inflammatory bowel disease. In: Satsangi J, Sutherland LR, editors. Inflammatory Bowel Diseases. London: Churchill Livingstone; 2003. pp. 199–215. [Google Scholar]
- 12.Guindi M, Riddell RH. Indeterminante colitis. J Clin Pathol. 2004;57::1233–1244. doi: 10.1136/jcp.2003.015214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tremaine WJ. Diagnosis and treatment of indeterminate colitis. Gastroenterol Hepatol. 2011;7::826–828. [PMC free article] [PubMed] [Google Scholar]
- 14.Loftus CG, Loftus EVJ, Harmsen SW, et al. Update on the incidence and prevalence of Crohn's disease and ulcerative colitis in Olmsted County, Minnesota, 1940–2000. Inflamm Bowel Dis. 2007;13::254–261. doi: 10.1002/ibd.20029. [DOI] [PubMed] [Google Scholar]
- 15.Herrinton LJ, Liu L, Lafata JE, et al. Estimation of the period prevalence of inflammatory bowel disease among nine health plans using computerized diagnoses and outpatient pharmacy dispensings. Inflamm Bowel Dis. 2007;13::451–461. doi: 10.1002/ibd.20021. [DOI] [PubMed] [Google Scholar]
- 16.Kappelman MD, Rifas-Shiman SL, Kleinman K, et al. The prevalence and geographic distribution of Crohn's disease and ulcerative colitis in the United States. Clin Gastroenterol Hepatol. 2007;5::1424–1429. doi: 10.1016/j.cgh.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 17.Lambrecht BN, Hammad H. The role of dendritic and epithelial cells as master regulators of allergic airway inflammation. Lancet. 2010;376::835–843. doi: 10.1016/S0140-6736(10)61226-3. [DOI] [PubMed] [Google Scholar]
- 18.Hugot J-P, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411::599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 19.Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411::603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 20.Jostins L, Ripke S, Weersma RK, et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491::119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhernakova A, van Diemen CC, Wijmenga C. Detecting shared pathogenesis from the shared genetics of immune-related diseases. Nature Rev Genet. 2009;10::43–55. doi: 10.1038/nrg2489. [DOI] [PubMed] [Google Scholar]
- 22.Budarf ML, Labbé C, David G, et al. GWA studies: rewriting the story of IBD. Trends Genet. 2009;25::137–146. doi: 10.1016/j.tig.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 23.Inohara N, Chamaillard M, McDonald C, et al. NOD-LRR proteins: role in host–microbial interactions and inflammatory disease. Annu Rev Biochem. 2005;74::355–383. doi: 10.1146/annurev.biochem.74.082803.133347. [DOI] [PubMed] [Google Scholar]
- 24.Strober W, Watanabe T. NOD2, an intracellular innate immune sensor involved in host defense and Crohn's disease. Mucosal Immunol. 2011;4::484–495. doi: 10.1038/mi.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho JH. The genetics and immunopathogenesis of inflammatory bowel disease. Nature Rev Immunol. 2008;8::458–466. doi: 10.1038/nri2340. [DOI] [PubMed] [Google Scholar]
- 26.Lesage S, Zouali H, Cézard J-P, et al. CARD15/NOD2 mutational analysis and genotype–phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet. 2002;70::845–857. doi: 10.1086/339432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abraham C, Cho JH. Functional consequences of NOD2 (CARD15) mutations. Inflamm Bowel Dis. 2006;12::641–650. doi: 10.1097/01.MIB.0000225332.83861.5f. [DOI] [PubMed] [Google Scholar]
- 28.Girardin SE, Travassos LH, Hervé M, et al. Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J Biol Chem. 2003;278::41702–41708. doi: 10.1074/jbc.M307198200. [DOI] [PubMed] [Google Scholar]
- 29.Inohara N, Ogura Y, Fontalba A, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn's disease. J Biol Chem. 2003;278::5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 30.Watanabe T, Asano N, Murray PJ, et al. Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J Clin Invest. 2008;118::545–559. doi: 10.1172/JCI33145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hedl M, Li J, Cho JH, et al. Chronic stimulation of Nod2 mediates tolerance to bacterial products. Proc Natl Acad Sci U S A. 2007;104::19440–19445. doi: 10.1073/pnas.0706097104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strober W, Murray PJ, Kitani A, Watanabe T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol. 2006;6:9–20. doi: 10.1038/nri1747. [DOI] [PubMed] [Google Scholar]
- 33.Cooney R, Baker J, Brain O, et al. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nature Med. 2010;16::90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 34.Travassos LH, Carneiro LAM, Ramjeet M, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nature Immunol. 2010;11::55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 35.Deretic V. Autophagy: an emerging immunological paradigm. J Immunol. 2012;189::15–20. doi: 10.4049/jimmunol.1102108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuma A, Mizushima N, Ishihara N, et al. Formation of the approximately 350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J Biol Chem. 2002;277::18619–18625. doi: 10.1074/jbc.M111889200. [DOI] [PubMed] [Google Scholar]
- 37.Fujita N, Itoh T, Omori H, et al. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008;19::2092–2100. doi: 10.1091/mbc.E07-12-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nature Genet. 2007;39::596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nature Genet. 2006;39::207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 40.Kuballa P, Huett A, Rioux JD, et al. Impaired autophagy of an intracellular pathogen induced by a Crohn's disease associated ATG16L1 variant. PLoS One. 2008;3::e3391. doi: 10.1371/journal.pone.0003391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCarroll SA, Huett A, Kuballa P, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nature Genet. 2008;40::1107–1112. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sabers CJ, Martin MM, Brunn GJ, et al. Isolation of a protein target of the FKBP12–rapamycin complex in mammalian cells. J Biol Chem. 1995;270::815–822. doi: 10.1074/jbc.270.2.815. [DOI] [PubMed] [Google Scholar]
- 43.Massey DCO, Bredin F, Parkes M. Use of sirolimus (rapamycin) to treat refractory Crohn's disease. Gut. 2008;57::1294–1296. doi: 10.1136/gut.2008.157297. [DOI] [PubMed] [Google Scholar]
- 44.Yin H, Li X, Zhang B, et al. Sirolimus ameliorates inflammatory responses by switching the regulatory T/T helper type 17 profile in murine colitis. Immunology. 2013;139::494–502. doi: 10.1111/imm.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duerr RH, Taylor KD, Brant SR, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314::1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang K, Zhang H, Kugathasan S, et al. Diverse genome-wide association studies associate the IL12/IL23 pathway with Crohn disease. Am J Hum Genet. 2009;84::399–405. doi: 10.1016/j.ajhg.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parham C, Chirica M, Timans J, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rβ1 and a novel cytokine receptor subunit, IL-23R. J Immunol. 2002;168::5699–5708. doi: 10.4049/jimmunol.168.11.5699. [DOI] [PubMed] [Google Scholar]
- 48.Maloy KJ, Kullberg MC. IL-23 and Th17 cytokines in intestinal homeostasis. Mucosal Immunol. 2008;1::339–349. doi: 10.1038/mi.2008.28. [DOI] [PubMed] [Google Scholar]
- 49.Ahern PP, Schiering C, Buonocore S, et al. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity. 2010;33::279–288. doi: 10.1016/j.immuni.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goodall JC, Wu C, Zhang Y, et al. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proc Natl Acad Sci U S A. 2010;107::17698–17703. doi: 10.1073/pnas.1011736107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Di Meglio P, Di Cesare A, Laggner U, et al. The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced Th17 effector response in humans. PLoS One. 2011;6::e17160. doi: 10.1371/journal.pone.0017160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sarin R, Wu X, Abraham C. Inflammatory disease protective R381Q IL23 receptor polymorphism results in decreased primary CD4+ and CD8+ human T-cell functional responses. Proc Natl Acad Sci U S A. 2011;108::9560–9565. doi: 10.1073/pnas.1017854108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pidasheva S, Trifari S, Phillips A, et al. Functional studies on the IBD susceptibility gene IL23R implicate reduced receptor function in the protective genetic variant R381Q. PLoS One. 2011;6::e25038. doi: 10.1371/journal.pone.0025038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maloy KJ, Kullberg MC. IL-23 and Th17 cytokines in intestinal homeostasis. Mucosal Immunol. 2008;1::339–349. doi: 10.1038/mi.2008.28. [DOI] [PubMed] [Google Scholar]
- 55.Sandborn WJ, Gasink C, Gao L-L, et al. Ustekinumab induction and maintenance therapy in refractory Crohn's disease. N Engl J Med. 2012;367::1519–1528. doi: 10.1056/NEJMoa1203572. [DOI] [PubMed] [Google Scholar]
- 56.Fitzpatrick LR. Novel pharmacological approaches for inflammatory bowel disease: targeting key intracellular pathways and the IL-23/IL-17 axis. Int J Inflamm. 2012;2012::389404. doi: 10.1155/2012/389404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meylan F, Richard AC, Siegel RM. TL1A and DR3, a TNF family ligand-receptor pair that promotes lymphocyte costimulation, mucosal hyperplasia, and autoimmune inflammation. Immunol Rev. 2011;244::188–196. doi: 10.1111/j.1600-065X.2011.01068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shih DQ, Kwan LY, Chavez V, et al. Microbial induction of inflammatory bowel disease associated gene TL1A (TNFSF15) in antigen presenting cells. Eur J Immunol. 2009;39::3239–3250. doi: 10.1002/eji.200839087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yamazaki K, McGovern D, Ragoussis J, et al. Single nucleotide polymorphisms in TNFSF15 confer susceptibility to Crohn's disease. Hum Mol Genet. 2005;14::3499–3506. doi: 10.1093/hmg/ddi379. [DOI] [PubMed] [Google Scholar]
- 60.Kakuta Y, Kinouchi Y, Negoro K, et al. Association study of TNFSF15 polymorphisms in Japanese patients with inflammatory bowel disease. Gut. 2006;55::1527–1528. doi: 10.1136/gut.2006.100297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shih DQ, Targan SR. Insights into IBD pathogenesis. Curr Gastroenterol Rep. 2009;11::473–480. doi: 10.1007/s11894-009-0072-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takedatsu H, Michelsen KS, Wei B, et al. TL1A (TNFSF15) regulates the development of chronic colitis by modulating both T-helper 1 and T-helper 17 activation. Gastroenterology. 2008;135::552–567. doi: 10.1053/j.gastro.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meylan F, Song YJ, Fuss I, et al. The TNF-family cytokine TL1A drives IL-13-dependent small intestinal inflammation. Mucosal Immunol. 2011;4::172–185. doi: 10.1038/mi.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taraban VY, Slebioda TJ, Willoughby JE, et al. Sustained TL1A expression modulates effector and regulatory T-cell responses and drives intestinal goblet cell hyperplasia. Mucosal Immunol. 2011;4::186–196. doi: 10.1038/mi.2010.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Spencer SD, DiMarco F, Hooley J, et al. The orphan receptor CRF2-4 is an essential subunit of the interleukin 10 receptor. J Exp Med. 1998;187::571–578. doi: 10.1084/jem.187.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kühn R, Löhler J, Rennick D, et al. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75::263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 67.Kotlarz D, Beier R, Murugan D, et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology. 2012;143::347–355. doi: 10.1053/j.gastro.2012.04.045. [DOI] [PubMed] [Google Scholar]
- 68.Glocker E-O, Kotlarz D, Boztug K, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361::2033–2045. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Glocker E-O, Frede N, Perro M, et al. Infant colitis – it's in the genes. Lancet. 2010;376::1272. doi: 10.1016/S0140-6736(10)61008-2. [DOI] [PubMed] [Google Scholar]
- 70.Engelhardt KR, Shah N, Faizura-Yeop I, et al. Clinical outcome in IL-10- and IL-10 receptor-deficient patients with or without hematopoietic stem cell transplantation. J Allergy Clin Immunol. 2013;131::825–830. doi: 10.1016/j.jaci.2012.09.025. [DOI] [PubMed] [Google Scholar]
- 71.Franke A, Balschun T, Karlsen TH, et al. Sequence variants in IL-10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nature Genet. 2008;40::1319–1323. doi: 10.1038/ng.221. [DOI] [PubMed] [Google Scholar]
- 72.Amre DK, Mack DR, Morgan K, et al. Interleukin 10 (IL-10) gene variants and susceptibiltiy for paediatric onset Crohn's disease. Aliment Pharmacol Ther. 2009;29::1025–1031. doi: 10.1111/j.1365-2036.2009.03953.x. [DOI] [PubMed] [Google Scholar]
- 73.Noguchi E, Homma Y, Kang X, et al. A Crohn's disease-associated NOD2 mutation suppresses transcription of human IL10 by inhibiting activity of the nuclear ribonucleoprotein hnRNP-A1. Nature Immunol. 2009;10::471–479. doi: 10.1038/ni.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moore KW, de Waal MalefytR, Coffman RL, et al. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19::683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 75.Chaudhry A, Samstein RM, Treuting P, et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity. 2011;34::566–578. doi: 10.1016/j.immuni.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grütz G. New insights into the molecular mechanism of interleukin-10-mediated immunosuppression. J Leukoc Biol. 2005;77::3–15. doi: 10.1189/jlb.0904484. [DOI] [PubMed] [Google Scholar]
- 77.Braat H, Peppelenbosch MP, Hommes DW. Interleukin-10-based therapy for inflammatory bowel disease. Expert Opin Biol Ther. 2003;3::725–731. doi: 10.1517/14712598.3.5.725. [DOI] [PubMed] [Google Scholar]
- 78.Martín R, Miquel S, Ulmer J, et al. Role of commensal and probiotic bacteria in human health: a focus on inflammatory bowel disease. Microb Cell Fact. 2013;12::71. doi: 10.1186/1475-2859-12-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Iliev ID, Funari VA, Taylor KD, et al. Interactions between commensal fungi and C-type lectin receptor dectin-1 influence colitis. Science. 2012;336::1314–1317. doi: 10.1126/science.1221789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brown GD, Gordon S. Immune recognition: a new receptor for β-glucans. Nature. 2001;413::36–37. doi: 10.1038/35092620. [DOI] [PubMed] [Google Scholar]
- 81.Ferwerda B, Ferwerda G, Plantinga TS, et al. Human dectin-1 deficiency and mucocutaneous fungal infections. N Engl J Med. 2009;361::1760–1767. doi: 10.1056/NEJMoa0901053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Taylor PR, Tsoni SV, Willment JA, et al. Dectin-1 is required for β-glucan recognition and control of fungal infection. Nature Immunol. 2006;8::31–38. doi: 10.1038/ni1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ma J, Becker C, Lowell CA, et al. Dectin-1 triggered recruitment of light chain 3 protein to phagosomes facilitates major histocompatibility complex class II presentation of fungal-derived antigens. J Biol Chem. 2012;287::34149–34156. doi: 10.1074/jbc.M112.382812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science. 2005;308::1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124::837–848. doi: 10.1016/j.cell.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 86.Lepage P, Leclerc MC, Joossens M, et al. A metagenomic insight into our gut's microbiome. Gut. 2013;62::146–158. doi: 10.1136/gutjnl-2011-301805. [DOI] [PubMed] [Google Scholar]
- 87.Koenig JE, Spor A, Scalfone N, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4578–4585. doi: 10.1073/pnas.1000081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Matamoros S, Gras-Leguen C, Le Vacon F, et al. Development of intestinal microbiota in infants and its impact on health. Trends Microbiol. 2013;21::167–173. doi: 10.1016/j.tim.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 89.De La Cochetière MF, Durand T, Lepage P, et al. Resilience of the dominant human fecal microbiota upon short-course antibiotic challenge. J Clin Microbiol. 2005;43::5588–5592. doi: 10.1128/JCM.43.11.5588-5592.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4554–4561. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Frank DN, St Amand AL, Feldman RA, et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104::13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Walker AW, Sanderson JD, Churcher C, et al. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011;11::7. doi: 10.1186/1471-2180-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nagalingam NA, Lynch SV. Role of the microbiota in inflammatory bowel diseases. Inflamm Bowel Dis. 2012;18::968–984. doi: 10.1002/ibd.21866. [DOI] [PubMed] [Google Scholar]
- 94.Harper PH, Lee EC, Kettlewell MG, et al. Role of the faecal stream in the maintenance of Crohn's colitis. Gut. 1985;26::279–284. doi: 10.1136/gut.26.3.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rutgeerts P, Goboes K, Peeters M, et al. Effect of faecal stream diversion on recurrence of Crohn's disease in the neoterminal ileum. Lancet. 1991;338::771–774. doi: 10.1016/0140-6736(91)90663-a. [DOI] [PubMed] [Google Scholar]
- 96.Winslet MC, Allan A, Poxon V, et al. Faecal diversion for Crohn's colitis: a model to study the role of the faecal stream in the inflammatory process. Gut. 1994;35::236–242. doi: 10.1136/gut.35.2.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bennet JD, Brinkman M. Treatment of ulcerative colitis by implantation of normal colonic flora. Lancet. 1989;1::164. doi: 10.1016/s0140-6736(89)91183-5. [DOI] [PubMed] [Google Scholar]
- 98.Borody TJ, Warren EF, Leis S, et al. Treatment of ulcerative colitis using fecal bacteriotherapy. J Clin Gastroenterol. 2003;37::42–47. doi: 10.1097/00004836-200307000-00012. [DOI] [PubMed] [Google Scholar]
- 99.Rietdijk ST, D'Haens GR. Recent developments in the treatment of inflammatory bowel disease. J Digest Dis. 2013;14::282–287. doi: 10.1111/1751-2980.12048. [DOI] [PubMed] [Google Scholar]
- 100.Prantera C, Lochs H, Grimaldi M, et al. Rifaximin-extended intestinal release induces remission in patients with moderately active Crohn's disease. Gastroenterology. 2012;142::473–481. doi: 10.1053/j.gastro.2011.11.032. [DOI] [PubMed] [Google Scholar]
- 101.Gionchetti P, Rizzello F, Ferrieri A, et al. Rifaximin in patients with moderate or severe ulcerative colitis refractory to steroid-treatment: a double-blind, placebo-controlled trial. Dig Dis Sci. 1999;44::1220–1221. doi: 10.1023/a:1026648812439. [DOI] [PubMed] [Google Scholar]
- 102.Saleh M, Elson CO. Experimental inflammatory bowel disease: insights into the host–microbiota dialog. Immunity. 2011;34::293–302. doi: 10.1016/j.immuni.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nell S, Suerbaum S, Josenhans C. The impact of the microbiota on the pathogenesis of IBD: lessons from mouse infection models. Nature Rev Microbiol. 2010;8::564–577. doi: 10.1038/nrmicro2403. [DOI] [PubMed] [Google Scholar]
- 104.Gordon S. Pattern recognition receptors: doubling up for the innate immune response. Cell. 2002;111::927–930. doi: 10.1016/s0092-8674(02)01201-1. [DOI] [PubMed] [Google Scholar]
- 105.Kalinski P, Hilkens CMU, Wierenga EA, et al. T-cell priming by type-1and type-2 polarized dendritic cells: the concept of a third signal. Immunol Today. 1999;20::561–567. doi: 10.1016/s0167-5699(99)01547-9. [DOI] [PubMed] [Google Scholar]
- 106.de Jong EC, Vieira PL, Kalinski P, et al. Microbial compounds selectively induce Th1 cell-promoting or Th2 cell-promoting dendritic cells in vitro with diverse Th cell-polarizing signals. J Immunol. 2002;168::1704–1709. doi: 10.4049/jimmunol.168.4.1704. [DOI] [PubMed] [Google Scholar]
- 107.Dolganiuc A, Kodys K, Kopasz A, et al. Hepatitis C virus core and nonstructural protein 3 proteins induce pro- and anti-inflammatory cytokines and inhibit dendritic cell differentiation. J Immunol. 2003;170::5615–5624. doi: 10.4049/jimmunol.170.11.5615. [DOI] [PubMed] [Google Scholar]
- 108.Smits HH, Engering A, van der Kleij D, et al. Selective probiotic bacteria induce IL-10-producing regulatory T cells in vitro by modulating dendritic cell function through dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin. J Allergy Clin Immunol. 2005;115::1260–1267. doi: 10.1016/j.jaci.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 109.Uematsu S, Fujimoto K, Jang MH, et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nature Immunol. 2008;9::769–776. doi: 10.1038/ni.1622. [DOI] [PubMed] [Google Scholar]
- 110.Schlitzer A, McGovern N, Teo P, et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity. 2013;38::970–983. doi: 10.1016/j.immuni.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21::685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 112.Reid G, Younes JA, Van der Mei HC, et al. Microbiota restoration: natural and supplemented recovery of human microbial communities. Nature Rev Microbiol. 2011;9::27–38. doi: 10.1038/nrmicro2473. [DOI] [PubMed] [Google Scholar]
- 113.Naidoo K, Gordon M, Fagbemi AO, et al. Probiotics for maintenance of remission in ulcerative colitis. Cochrane Database Syst Rev. 2011;12::CD007443. doi: 10.1002/14651858.CD007443.pub2. [DOI] [PubMed] [Google Scholar]
- 114.Butterworth AD, Thomas AG, Akonbeng AK. Probiotics for remission in Crohn's disease. Cochrane Database Syst Rev. 2008:3. doi: 10.1002/14651858.CD006634.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.De Greef E, Vandenplas Y, Hauser B, et al. Probiotics and IBD. Acta Gastroenterol Belg. 2013;76::15–19. [PubMed] [Google Scholar]
- 116.Klaenhammer TR, Kleerebezem M, Kopp MV, et al. The impact of probiotics and prebiotics on the immune system. Nature Rev Immunol. 2012;12::728–734. doi: 10.1038/nri3312. [DOI] [PubMed] [Google Scholar]
- 117.Braat H, Rottiers P, Hommes DW, et al. A phase I trial with transgenic bacteria expressing interleukin-10 in Crohn's disease. Clin Gastroenterol Hepatol. 2006;4::754–759. doi: 10.1016/j.cgh.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 118.Iwasaki A. Mucosal dendritic cells. Annu Rev Immunol. 2007;25::381–418. doi: 10.1146/annurev.immunol.25.022106.141634. [DOI] [PubMed] [Google Scholar]
- 119.Bogunovic M, Ginhoux F, Helft J, et al. Origin of the lamina propria dendritic cell network. Immunity. 2009;31::513–525. doi: 10.1016/j.immuni.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Varol C, Vallon-Eberhard A, Elinav E, et al. Intestinal lamina propria dendritic cell subsets have different origin and functions. Immunity. 2009;31::502–512. doi: 10.1016/j.immuni.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 121.Johansson-Lindbom B, Svensson M, Pabst O, et al. Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J Exp Med. 2005;202::1063–1073. doi: 10.1084/jem.20051100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Scott CL, Aumeunier AM, Mowat AM. Intestinal CD103+ dendritic cells: master regulators of tolerance? Trends Immunol. 2011;32::412–419. doi: 10.1016/j.it.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 123.Agace WW, Persson EK. How vitamin A metabolizing dendritic cells are generated in the gut mucosa. Trends Immunol. 2012;33::42–48. doi: 10.1016/j.it.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 124.Edelson BT, Wumesh KC, Juang R, et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8α+ conventional dendritic cells. J Exp Med. 2010;207::823–836. doi: 10.1084/jem.20091627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Farache J, Koren I, Milo I, et al. Luminal bacteria recruit CD103+ dendritic cells into the intestinal epithelium to sample bacterial antigens for presentation. Immunity. 2013;38::581–595. doi: 10.1016/j.immuni.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kinnebrew MA, Charlie G, Buffie CG, et al. Intestinal CD103+ CD11b+ lamina propria dendritic cells instruct intestinal epithelial cells to express antimicrobial proteins in response to Toll-like receptor 5 activation. Immunity. 2012;36::276–287. doi: 10.1016/j.immuni.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Plantinga M, Guilliams M, Vanheerswynghels M, et al. Conventional and monocyte-derived CD11b+ dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity. 2013;38::322–335. doi: 10.1016/j.immuni.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 128.Ohtani M, Takayuki Hoshii T, Fujii H, et al. Cutting edge: mTORC1 in intestinal CD11c+CD11b+ dendritic cells regulates intestinal homeostasis by promoting IL-10 production. J Immunol. 188::4736–4740. doi: 10.4049/jimmunol.1200069. [DOI] [PubMed] [Google Scholar]
- 129.Mann ER, Landy JD, Bernardo D, et al. Intestinal dendritic cells: their role in intestinal inflammation, manipulation by gut microbiota and differences between mice and men. Immunol Lett. 2012;150:30–40. doi: 10.1016/j.imlet.2013.01.007. 1938. [DOI] [PubMed] [Google Scholar]
- 130.Bell SJ, Rigby R, English N, et al. Migration and maturation of human colonic dendritic cells. J Immunol. 2001;166::4958–4967. doi: 10.4049/jimmunol.166.8.4958. [DOI] [PubMed] [Google Scholar]
- 131.Silva MA. Intestinal dendritic cells and epithelial barrier dysfunction in Crohn's disease. Inflamm Bowel Dis. 2009;15::436–453. doi: 10.1002/ibd.20660. [DOI] [PubMed] [Google Scholar]
- 132.Varol C, Zigmond E, Jung S. Securing the immune tightrope: mononuclear phagocytes in the intestinal lamina propria. Nature Rev Immunol. 2010;10::415–426. doi: 10.1038/nri2778. [DOI] [PubMed] [Google Scholar]
- 133.te Velde AA, van Kooyk Y, Braat H, et al. Increased expression of DC-SIGN+IL-12+IL-18+ and CD83+IL-12–IL-18– dendritic cell populations in the colonic mucosa of patients with Crohn's disease. Eur J Immunol. 2003;33::143–151. doi: 10.1002/immu.200390017. [DOI] [PubMed] [Google Scholar]
- 134.Hart AL, Al-Hassia HO, Rigby RJ, et al. Characteristics of intestinal dendritic cells in inflammatory bowel diseases. Gastroenterology. 2005;129::50–65. doi: 10.1053/j.gastro.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 135.Takenaka S, Safroneeva E, Xing Z, et al. Dendritic cells derived from murine colonic mucosa have unique functional and phenotypic characteristics. J Immunol. 2007;178::7984–7993. doi: 10.4049/jimmunol.178.12.7984. [DOI] [PubMed] [Google Scholar]