

Abstract
The manipulation of chromatin structure regulates gene expression and the flow of genetic information. Histone modifications and ATP-dependent chromatin remodeling together with DNA methylation are dynamic processes that modify chromatin architecture and profoundly modulate gene expression. Their coordinated control is key to ensuring proper cell commitment and organ development, as well as adaption to environmental cues. Recent studies indicate that abnormal epigenetic status of the genome, in concert with alteration of transcriptional networks, contribute to the development of adult cardiomyopathy such as pathological cardiac hypertrophy. Here we consider the emerging role of different classes of chromatin regulators and how their dysregulation in the adult heart alters specific gene programs with subsequent development of major cardiomyopathies. Understanding the functional significance of the different epigenetic marks as points of genetic control may represent a promising future therapeutic tool.
Keywords: Histone modification, chromatin remodeling, epigenetics, gene regulation, cardiac hypertrophy, adult cardiomyopathy
Introduction
In eukaryotes, the manipulation of chromatin structure represents a means of regulating access to genetic information. One way to achieve this is via packaging of nuclear DNA. This process involves folding of approximately 2 metres of linear DNA around histone octamers comprising histone H2A, H2B, H3 and H4, which forms the basic unit of chromatin named the nucleosome 1. DNA packaging is achieved, in part, by the strength of DNA interaction with histone proteins that maintain chromatin in a condensed (silenced) or relaxed (active) state. Chromatin undergoes a number of structural re-arrangements that regulate gene activity and manifest as specific cellular phenotypes 2,3. The first level of control is the DNA sequence subjected to modification by methylation. The next layer is at the level of the nucleosome whose positioning together with the expression of histone variants (e.g. H2A.Z, H2A.X, H3, etc…) and histone post-translational modifications alter chromatin architecture. Chromatin is further organized through nuclear compartmentalization; specific nuclear domains are associated with genes of variable densities that correlate with active or silenced chromatin states. Changes in chromatin structure are the basis of many regulatory processes such as transcription, replication and DNA repair.
Enzymatic post-translational histone modification and ATP-dependent chromatin remodeling enzymes alter chromatin structure 4–6. Histone chaperones, inclusion of histone variants, DNA methylation (in some species) and recruitment of chromatin binding proteins further influence chromatin architecture 7. It has become clear that chromatin is a highly dynamic structure and that histone modifications are not as “static” as once thought. In fact, most histone modifications are reversible. Enzymes removing the histone “marks” act in pathways that oppose those involved in placing the marks. The existence of a plethora of factors that modify chromatin illustrates the extent to which chromatin conformation is integrated into gene regulatory pathways. These non-genetic changes that alter gene expression without affecting the DNA sequence are the basis of epigenetics.
Epigenetic mechanisms have different outcomes in different organs. Cell-type specificity is achieved by the selective recruitment of enzymes involved in chromatin remodeling, histone modification and DNA methylation that dictate gene expression patterns temporally and spatially. One organ that illustrates how changes in the epigenome results in disease associated phenotypes is the heart. Inadequate orchestration of epigenetic mechanisms in the developing heart results in cardiac malformations 8–11 12,13 14 15 16 17. Recent reports also indicate that chromatin remodeling and modifications play vital roles in adult cardiomyopathy. Here, we summarize this emerging area of research related to abnormal epigenetic interactions in adult cardiomyopathy. We particularly focus on epigenetic changes due to histone modifications and their role in cardiac hypertrophy and heart failure.
Pathological Cardiac Hypertrophy
Pathological hypertrophy is a maladaptive growth of the heart in response to stress resulting from several common disorders such as long-standing hypertension, atherosclerosis, myocardial infarction or mutations in genes encoding proteins of the sarcomere. The final outcome is reduced cardiac output with increased risk of heart failure and sudden death 18–20. The hypertrophic response is characterized by ventricular remodeling associated with increased fibrosis and cardiomyocyte size resulting in enlargement of the heart and depressed contractile function. At the molecular level, cardiac hypertrophy involves activation of a wide range of signaling pathways that culminate in the cell nucleus 21,22. There, changes in the activity of transcription factors re-activate fetal cardiac genes while their adult isoforms are down-regulated. Studies now indicate that changes in chromatin structure parallel the “classical” transcriptional reprogramming occurring during pathological cardiac hypertrophy. Below, we discuss abnormalities of the epigenome implicated in pathological cardiac growth, especially chromatin remodelers and histone modifications.
Histone Modifications
Studies from the past two decades have established that local changes in chromatin structure regulate the expression of many eukaryotic genes 23. Chromatin modifications have become integrated into normal gene regulatory pathways. Histones can be modified by post-translational modifications such as acetylation, methylation, phosphorylation and ADP-ribosylation (Figure 1) 4–6. While it is still difficult to decode the specific post-translational modifications at the level of single histones and single nucleosomes, mounting evidence suggests that histone modifications “communicate” and influence one another 23. Epigenomic studies indicate that local changes in chromatin architecture alter specific transcriptional programs and contribute to the development of cardiac pathologies in the adult (Figure 2, Table1).
Figure 1.
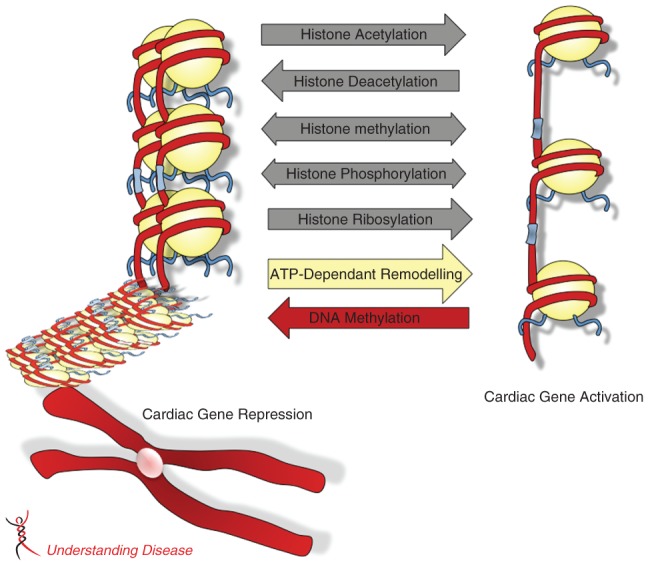
Schematic representation of epigenetic mechanisms altering chromatin structure and cardiac gene expression. DNA is packaged in chromatin composed of nucleosomes each containing an octamer of histone H3, H4, H2A and H2B. The flexible N-terminal histone “tails” are subjected to post-translational modifications such as acetylation, phosphorylation, methylation and ribosylation. These covalent modifications alter DNA-histone interactions which change chromatin conformation from an “inactive”/repressed state to an “active”/open state, allowing for transcription of cardiac genes. Chromatin conformation can be dynamically regulated by ATP-dependent chromatin remodeling complexes. Changes in DNA methylation can occur in response to stress and contribute to changes in cardiac gene expression.
Figure 2.
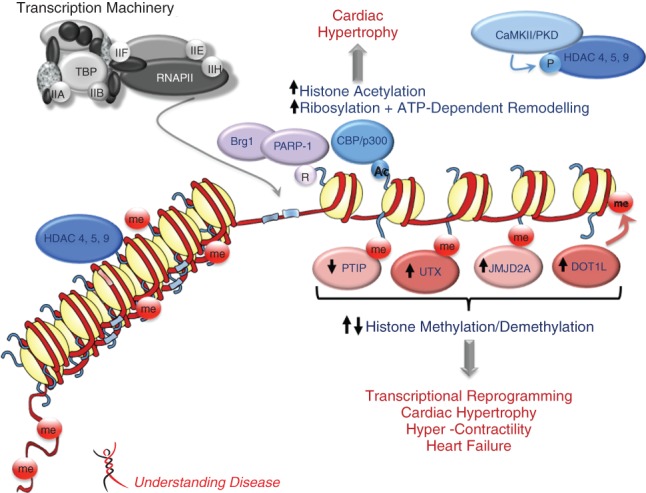
Epigenetic changes in cardiomyopathy. Epigenetic mechanisms such as DNA methylation, histone modifications and ATP-dependent chromatin remodeling alter chromatin structure and modulate gene expression. Histone acetylation by p300/CBP histone acetyltransferases (HATs) changes nucleosome conformation and increases accessibility of transcription factors, resulting in relief of transcriptional repression. Histone deacetylases (HDACs) have the opposite effect and are usually involved in gene repression. Both HATs and HDACs play a role in cardiac hypertrophy. Histone methylation is associated with activation or repression of transcription depending on the residue and the degree of methylation. The functional outcomes of histone methylation and the action of histone demethylases on cardiac gene transcription are shown. Chromatin remodelers such as Brg1 use the energy of ATP to change chromatin structure. These different epigenetic mechanisms are reversible and act in concert to regulate cardiac transcription. Dysregulation of these processes can result in cardiac abnormalities. HDAC4/5/9: histone deacetylases 4,5,9; CBP/p300: histone acetyltransferases; PTIP: PAX interacting protein 1; UTX: histone H3K27 demethylase; JMJD2A: jumonji C domain containing histone demethylase; DOT1L: histone H3 methyltransferase; Brg1: brahma-related gene 1; PARP-1: poly(ADP-ribose) polymerase 1; RNAPII: RNA polymerase II; CaMKII: calcium/calmodulin-dependent protein kinase II; PKD: protein kinase D.
Table 1.
Summary of Epigenetic Modifications Involved in Adult Cardiomyopathy
| Epigenetic Mark | Complexes | Targets | Modifications | Effect | References |
|---|---|---|---|---|---|
| Acetylation | CREB-binding protein(CBP) /p300 | H3 K4, K19 H4 K5, K8, K12, K16 | 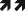 |
Cardiac hypertrophy regulation | 31,32,34 |
| Deacetylation | Class II HDACs (HDAC-4, -5, -9) | Histone tails |  |
Inhibit the activity of myocyte enhancer factor 2 (MEF2) Negative regulation of cardiac hypertrophy |
8,36 |
| Methylation | PTIP | H3 K4me3 |  |
Fetal cardiac gene activation | 43 |
| PTIP | H3K4me2 |  |
Angiogenesis and heart failure | 45 | |
| ------------- | H3 K9me3 |  |
Angiogenesis and heart failure | 45 | |
| ------------- | H3 K27me3 |  |
Suppresses angiogenesis | 9 | |
| DOT1L | H3 K79me |  |
Dilated cardiomyopathy | 46 | |
| Demethylation | JMJD2A | H3 K9me3 |  |
Cardiac hypertrophy stimulation | 51 |
| ------------- | H3 K4me3 |  |
Alters cardiac gene expression | 45 | |
| JMJD2A | H3 K36me3 |  |
Cardiac hypertrophy stimulation | 51 | |
| UTX | H3 K27me3 |  |
Heart malformation and embryonic lethality | 49,50 | |
| Phosphorylation | Aurora, Rsk2, Msk1, IKKα, PIM1, Akt, | H3 S10/S28 |  |
Mitotic activity, cellular proliferation, cardiac hypertrophy regulation | 75 |
| CaMKII | HDACs/ H3S10 |  |
Cardiac hypertrophy stimulation | 28,80 | |
| JAK2, AMPK | H4Y41 and H2B |  |
Transcriptional activation | 76,77 | |
| PKD | HDACs |  |
Cardiac hypertrophy stimulation | 81 | |
| Ribosylation | PARP-1 | Histones and PARP-1 |  |
Cardiac hypertrophy and heart failure | 88 |
| HDACs and Brg1 |  |
Form a complex with Brg1 and HDACs and increase expression of fetal β-MHC | 89 | ||
| DNA methylation | ------------- | CPG islands |  |
Modulate gene expression of angiogenic factors Heart Failure | 57,96 |
| ATP-Dependent Remodelers | SWI/SNF (Brg1 and Baf60c Subunits) | PARP-1/HDACs |  |
Fetal cardiac gene activation | 89 |
| Modulate extracellular matrix | 103 |
Histone acetylation
Histone acetylation and deacetylation are central mechanisms for the control of gene expression 24–26. With the discovery of coactivator complexes containing histone acetyltransferase (HAT) activity and co-repressor complexes containing histone deacetylase (HDAC) activity, acetylation evolved as a paradigm of gene regulation by histone modifications 27. The attachment of an acetyl group to lysine residues neutralizes the basic charge of the residue. Hence, histone acetylation by HATs disrupts intra- and internucleosomal interactions, which in turn “relaxes” chromatin structure and activates transcription. In contrast, deacetylation of histones by HDACs removes acetyl groups and consequently increases histone-DNA contacts, resulting in condensation of chromatin and gene repression. More than a decade ago, it was established that specific HATs and HDACS control cardiac growth in response to stress. Major findings showing that histone acetylation\deacetylation is critical to hypertrophic signaling pathways in heart muscle are summarized below (see also 8,28).
Histone Acetyltransferases
Acetylation of histones by p300 or the closely related CREB-binding protein (CBP) acetyltransfereases is required for proper heart development. Deletion of p300 or CBP is lethal in the embryo indicating that both co-activators are essential for mammalian heart development 29. The HAT domain is important in mediating p300/CBP function, as its' abolishment leads to abnormalities of the cardiovascular system 30. p300 also plays a positive role in cardiac hypertrophy and this function requires HAT activity 31,32. p300 interacts with histones and also specific transcription factors such as GATA4. Specific over-expression of p300 in mouse heart increases GATA4 acetylation, which results in eccentric cardiac dilation with depressed systolic function 31,33. p300 is selectively recruited at the ANF and BNP promoters in the left ventricle after pressure overload. This event is associated with increased H3K9/K14 and H4K5/K8/K12/K16 acetylation. The distinct histone acetylation pattern in the left and right ventricles indicates that ventricular cavities are epigenetically different 34.
Histone Deacetylases
HDACs are a class of chromatin remodeling enzymes involved in a broad range of biological functions. There are four major HDACs classified according to their sequence homology, enzymatic activity and tissue distribution. HDACs remove acetyl groups in the histone “tails”, thus increasing histone-DNA contacts and causing chromatin condensation and gene repression 24,25,35. Several reviews have recently summarized the function of HDACs in the cardiovascular system and the use of HDAC inhibitors in clinical setting to treat a wide variety of diseases 8,36.
Histone methylation
Histone methylation was described for the first time in 1964 37. Thirty-five years later, definite evidence linking histone methylation and transcription was provided when the histone H3 arginine-specific methyltransferase CARM1 was found to interact with the steroid-hormone-receptor coactivator GRIP-1 in transcriptional initiation 38. Histones can be methylated on arginine but also on lysine residues. The majority of studies on cardiac hypertrophy and heart failure have focused on histone lysine methylation. Sites predominantly methylated in histones H3 include lysines 4, 9 and 27, and 36. Lysine 20 is methylated in H4 39. These lysines can be mono-, di- or tri- methylated in histones, adding another level of complexity to the posttranslational status of the histone tails. While histone acetylation is universally linked to an “open” chromatin state and to gene activation, methylation of histones has a more diverse outcome; it can be associated with active, “poised” and repressive states of chromatin. This has resulted in considerable attention in the scientific community with an explosion of studies investigating the biological function of histone methylation. The specific epigenetic changes in embryonic stem cells (ESCs) were recently reviewed as well as the role of histone methylation in cardiac development 2,8,40,41. Below, we focus on the less well-studied role of histone lysine methylation in cardiac hypertrophy and heart failure.
H3K4 and K9 methylation
Recent studies indicate that H3K4me3 is important for the normal function of adult cardiomyocytes. Reduction of H3K4me3 level in the adult heart after cardiac-specific deletion of the PAX interacting protein 1 (PTIP), a cofactor for H3K4 methylation, alters cardiac gene expression. Among the genes affected by PTIP ablation is the Kv channel-interacting protein 2 (Kcnip2). PTIP deletion reduces cardiac repolarization (Ito) gradient and sodium current (INa). This also prolongs the duration of action potentials, which increases L-type calcium current (Ica,L) and increases intracellular calcium, resulting in increased cardiac contractility 42. Thus, appropriate H3K4me3 level is critical to maintain cellular homeostasis in differentiated cardiac cells. Few studies have tackled the involvement of histone methylation on hypertrophic phenotype induction. H3K4 trimetylation (H3K4me3) is a “mark” typically associated with gene activation. During cardiac hypertrophy, H3K4me3 increases at the promoter and transcribed regions of the β-MHC (myosin heavy chain) gene while H3K4me3 decreases at the α-MHC promoter 43. This indicates that the “classic” transcriptional reprogramming occurring during cardiac hypertrophy not only involves re-activation of fetal cardiac genes but also changes in methylation patterns at the very same gene loci re-activated under pathological stress. H3K4me3 is also elevated at the angiotensin-converting enzyme 1 promoter in the heart of spontaneous hypertensive rats. This correlates with H3 acetylation status and reduced H3K9me2 44. Different H3K4me3 and H3K9me3 levels are observed in rodent failing hearts and in patients with end-stage heart failure 45,46.
H3K27 methylation
H3K27me3 is a “mark” associated with gene repression 47,48. This modification has been particularly well studied in self-renewing ESCs where it down-regulates the expression of several developmental genes. During angiogenesis triggered by hypoxic conditions, H3K27me3 decreases globally and locally at the endothelial nitric oxide synthase (eNOS) promoter. This increases the ratio of active H3K4me3 to inactive H3K27me3 marks and enhances the expression of histone demethylase JMJD3 which in turn suppresses angiogenesis 9. The JumonjiC-domain-containing protein UTX is a H3K27 demethylase that regulates Hox genes 49 and interacts with a panel of cardiac-specific transcription factors (i.e. SRF, Tbx5, NKX2.5 and GATA4). UTX facilitates recruitment of Brg-1, a component of the ATP-dependent chromatin remodeling Swi/Snf complex, to cardiac specific enhancers. UTX-deficient mice exhibit severe heart malformation showing that removal of H3K27me3 by UTX is critical for proper heart development 50.
H3K36 and H3K9 demethylation
Demethylation of H3K9me3 and H3K36me3 is catalyzed by trimethyl lysine demethylase JMJD2A, another member of JumonjiC-domain containing family of demethylases. The impact of these modifications in adult cardiomyopathy was studied indirectly by investigating the effect of modulating JMJD2 expression. The generation of mice lacking or over-expressing JMJD2 in cardiac muscle revealed a role of JMJD2 in pathological cardiac hypertrophy 51. JMJD2A deficient mice display increased level of H3K9me or H3K36 methylation and have a normal phenotype under basal conditions. However, JMJD2A-null mice are resistant to cardiac stress. Conversely, JMJD2 transgenic mice have an exacerbated hypertrophic response after pressure overload hypertrophy 51. JMJD2 enhances cardiac hypertrophy by binding and activating the target gene, four-and-a-half LIM domains 1 (FHL1), and by increasing the binding of myocardin and SRF to the FHL1 promoter. This effect is associated with reduced H3K9me, emphasizing the importance of H3 tri-methylation as a major epigenetic mark critical for proper cardiac transcription and remodeling after pathological insult.
H3K79 methylation
While the majority of methylated sites are located in the histone H3 tail, additional residues such as H3K79 are located in the histone globular domain. Methylation of H3K79 is catalyzed by the disruptor of telomeric silencing protein DOT1L. Cardiac-specific deletion of Dot1L in the mouse increases lethality at the postnatal and adult stages and causes dilation of the cardiac chambers. Cardiac remodeling in Dot1L deficient mice is associated with re-activation of fetal cardiac genes, increased fibrosis and enhanced apoptosis. Dot1L knockout mice also have increased volume of the cardiac chambers and reduced contractility. These alterations are reminiscent of patients with dilated cardiomyopathy (DCM) 46. Mechanistically, Dot1L deletion selectively decreases transcription of the dystrophin gene and reduces H3K79me2/3 at the dystrophin promoter. Since Dot1L is down-regulated in patients with idiopathic DCM, impaired H3K79 methylation may also contribute to reduced cardiac contractility and DCM in humans.
Genome-wide histone modifications
The rapid expansion of genome-wide studies with the development of new technologies has allowed interrogation of histone modifications across the genome. Common methods used to address the role of histone modifications genome-wide combine chromatin immunoprecipitation (ChIP) with microarray analysis (ChIP-chip). More recently, ChIP followed by massive sequencing (ChIP-seq) has become the method of choice to understand how specific histone marks affect gene expression on a large-scale. One important finding from these studies is that different genomic regions exhibit distinct patterns of histone modifications and are associated with different gene activity. Acetylation, a universal mark for transcription activation, is clearly detected in the promoter region of active genes. Methylation of histones can be associated with gene activation or repression, depending on the residue targeted and the degree of methylation. Thus, an important finding that came from large-scale epigenomic studies is that “active” and “repressive” marks can co-exist within inactive promoters 47,48,52,53 and in self-renewing ESCs 54,55. This has led to the hypothesis that bivalent modifications maintain genes in a repressive but poised state, ready for future activation.
Until now, two genome-wide studies have evaluated histone methylation in the normal and failing heart. H3K4me3 and to a lesser extent H3K9me3, exhibits differential methylation patterns in the vicinity of genes regulating calcium signaling and cardiac contractility during the development of heart failure 56. Changes in the epigenome are also evident in patients with end-stage heart failure where H3K36me3 is enriched in actively transcribed regions of the genome 57. Thus, distinct epigenetic changes occur in human heart failure.
Histone phosphorylation
Protein kinases transmit extracellular signals from the cell surface to the nucleus. Kinases not only phosphorylate cellular proteins, transcription factors and components of the transcription machinery, but also signal to chromatin to regulate major cellular processes such as transcription, mitosis, DNA damage and apoptosis (reviewed in 58). While the majority of studies have focused on the role of histone acetylation and methylation in the control of cardiac gene transcription, histone phosphorylation remains less studied and understood. Histones are phosphorylated upon stress signals and many kinases are critical mediators of cardiac hypertrophy. However whether they act as true physiological histone kinases to modulate transcription remains controversial.
H3 S10 and S28 phosphorylation
Phosphorylation of H3 serine-10 (S10) is a mark originally associated with mitosis and chromosome condensation 58–61. Subsequently, H3S10 was linked to activation of transcription by the observation that H3S10 phosphorylation increases rapidly in response to growth factors 62. The condensation of chromatin during mitosis and its relaxation during transcriptional activation initially appeared as a paradox. This contradiction was reconciled by the discovery that H3S10 is phosphorylated in every H3 during mitosis whereas H3S10 phosphorylation after stress occurs transiently and does not involve every H3 58,62. Serine 28 in H3 is another site phosphorylated upon cellular stress 58,60. The mechanism by which histone phosphorylation contributes to transcriptional activation involves the generation of negatively charged phosphate groups that neutralize basic charges on the histone tails. As a result, the affinity of histones for DNA is reduced in a similar way to that proposed for acetylation. Increased accessibility to nuclear factors and coupling of histone phosphorylation with acetylation is another suggested mechanism linking histone phosphorylation and transcription activation 62,63. Histone acetylation may also create epitopes recognized by phospho-specific interaction partners such as 14-3-3 proteins 64.
In cardiac muscle, phosphorylation events occur in actively proliferating cardiomyocytes during fetal life and in the diseased adult heart 65. This mitotic activity is indicative of regenerative capacity of the adult myocardium mostly attributed to a small pool of cardiac progenitor cells that replace damaged tissue after pathological insult 66,67. H3 phosphorylation is also high in cardiomyocytes during the first week of post-natal life, which indicates a unique regenerative capacity of the neonatal heart 68. H3S10 phosphorylation is detected in the heart of cardiomyopathic animals in both the hypertrophic and failing phase of the disease 69.
Histone Kinases
Aurora kinase and its counterpart protein phosphatase 1 maintain the balance of H3S10 during the condensation/decondensation cycle of chromosomes 70. NIMA is another kinase that phosporylates H3S10 during mitosis 71. Central kinases targeting H3S10 during stress and growth factor stimulation include Rsk2, Msk1, IKKα, PIM1 and Akt 62,63,72–74 (for review 75). JAK2 and adenosine monophosphate-activated protein kinase (AMPK) directly activate transcription by phosphorylating H4Y41 and H2B respectively 76,77. Although recruitment of these enzymes to specific chromatin regions clearly contributes to transcriptional activation and many central kinases are critical regulators of cardiac hypertrophy, direct signaling to histones has not been established in the heart 21,78,79. Recently, a specific nuclear isoform of calcium-dependent protein kinase II (CaMKIIδB) was shown to bind and phosphorylate histone H3 at serine-10. Increased H3 phosphorylation is detected in ventricular myocytes during cardiac hypertrophy and at hypertrophic gene loci. This phosphorylation event increases chromatin accessibility and is required for chromatin-mediated transcription of the Mef2 transcription factor 80.
Kinases targeting chromatin remodelers
One mechanism by which kinases modify chromatin architecture is by interfering with the activity of HDACs. Protein kinase D (PKD), protein kinase C (PKC) and CaMKII regulate cardiac hypertrophy by signaling to class II HDACs. This promotes HDACs nuclear export and prevents them from acting as transcriptional repressors 81–84. Casein kinase-2α1 (CK2α1)-dependent phosphorylation of class I HDAC2 is also essential for the development of cardiac hypertrophy 85. Thus, kinases regulate pathological cardiac hypertrophy by many mechanisms including interfering with the activity of chromatin remodeling enzymes.
Histone Ribosylation
Histone ribosylation is mediated by Poly(ADP-ribose) polymerase-1 (PARP-1), which transfers ADP-ribose groups from nicotinamide adenine dinucleotide (NAD+) onto acceptor substrates 54. Substrates of PARP-1 include PARP-1 itself and the tail of histones H1, H2A, H2B, H3 and H4 86. Histone ribosylation induces relaxation of chromatin, which facilitates the recruitment of repair enzymes to the site of damaged DNA. Besides a role in DNA repair, PARP regulates cell death, cell cycle progression and genome stability 54. In cardiac muscle, PARP-1 promotes cardiac hypertrophy and increased PARP-1 activity leads to heart failure in both mice and humans 87. In contrast, Parp-1 null mice are resistant to angiotensin II-induced cardiac hypertrophy 87,88. The mechanism by which PARP-1 induces pathological hypertrophy is by interacting with HDACs and with the chromatin remodeller Brg-1 and by inducing α-MHC to β-MHC switch 89. In addition to its effect on fetal cardiac genes, PARP controls cardiac hypertrophy by modulating p38 MAP kinase, ERK1/2, PI3 kinase-Akt-Gsk3β and JNK signaling pathways 90,91. The protective effects of PARP inhibitors suggest the potential use of PARP blockade for the treatment of cardiomyopathies.
Dna Methylation
DNA methylation is an important epigenetic mechanism, which, like histone modifications, alters chromatin structure and affects gene expression. In differentiated mammalian cells, DNA methylation specifically targets cytosines mainly located in CpG dinucleotides. It involves the covalent addition of a methyl group at the 5th position of cystosine and is catalyzed by DNA methyltransferases 92. DNA methylation was first regarded as a stable heritable mark associated with silent chromatin. However, DNA methylation is now known to be a dynamic process that increases with age and can be reversed by demethylation 93.
The involvement of epigenetic modifications due to disturbed DNA methylation patterns in cardiac disorders remains elusive 94. Interestingly, differential DNA methylation correlates with expression of angiogenic factor genes, mainly PECAM1, ARHGAP24 and AMOTL2 95. Moreover, DNA methylation patterns differ in promoters and gene bodies from normal individuals and patients with end-stage heart failure. Reduction of global DNA methylation patterns correlates with the expression of genes that are up-regulated in heart failure 57,96. Nevertheless, the causal role of altered DNA methylation on the progression of adult cardiomyopathy remains unclear.
Atp-Dependent Chromatin Remodeling Complexes
ATP-dependent chromatin remodeling complexes use energy derived from ATP hydrolysis to reconfigure chromatin structure. These enzymes share sequence homology with the RecA domains of Superfamily II helicase related proteins 97. The first ATP-dependent chromatin remodeling protein identified is SWI/SNF (switching defective/sucrose nonfermenting) 97–99. ATP-dependent proteins can be further classified into 24 subfamilies based on their biochemical properties and the overall sequence similarity of their ATPase subunits (reviewed in 77,100). While most of the subfamilies are conserved across a broad evolutionary scale, each has a distinct function. Among the subfamilies, only four are reported to be involved in heart development. These are SWI/SNF, ISWI (imitation switch), CHD (chromodomain, helicase, DNA binding) and INO80 (inositol requiring 80) complexes 100. The ATP-dependent chromatin remodelers that play a role in adult cardiomyopathy are limited to members of the SWI/SNF complex.
SWI/SNF complex
The mammalian homologue of yeast SWI/SNF complex is the brahma-associated factor (BAF) complex. It is a large protein complex composed of 12 subunits, containing a catalytic ATPase subunit encoded by Brm (brahma) or Brg1 (brahma-related gene 1). Assembly of various subunits dictates certain cellular phenotypes and drives tissue specificity 101.
Brg1
Brg1 plays a critical role in cardiac development and also in cardiac hypertrophy 89. Brg1 is highly expressed in embryonic hearts where it represses α-MHC expression by interacting with HDACs and PARP, allowing β-MHC expression. In the adult heart, Brg1 expression is turned off. Once induced by myocardial stress, Brg1 forms a large protein complex with other chromatin regulators (i.e. HDACs, PARP). This induces pathological hypertrophy by switching MHC to its fetal β-MHC isoform 89. Preventing Brg1 up-regulation inhibits cardiac hypertrophy and reverses the switch of MHC. Brg1 expression is also elevated in patients with hypertrophic cardiomyopathy 89. Since Brg1 is part of a large protein complex and interacts with several chromatin regulators, cooperation of these factors at specific gene loci is a likely epigenetic mechanism that controls pathological cardiac hypertrophy.
Baf60c
Another subunit of the BAF complex, Baf60c, is required for proper heart morphogenesis. Baf60C mediates the interaction of several cardiac-specific transcription factors (i.e.Tbx5, Gata4 and Nkx2.5) with Brg1 to activate a set of cardiac-specific genes 102. Since many members of the SWI/SNF complex are well characterized chromatin modifying factors regulating cardiac hypertrophy, this suggests that chromatin may be the ultimate effector that controls the regulation of MHC genes during pathological hypertrophy.
In addition to their direct effect on chromatin components, chromatin regulators indirectly affect cardiac development and disease through modulation of the extracellular matrix. For instance, Brg1 suppresses the expression of Adamts1, a secreted metalloproteinase. This repression permits the development of cardiac jelly that triggers myocardial trabeculation 103. Improper trabeculation results in cardiomyopathy and heart failure 104. Thus, an exciting topic for future investigation is to understand Brg1 interaction with other chromatin regulators, and whether such large protein complexes modulate extracellular matrix and contributes to specific cardiomyopathies.
Future Directions And Challenges
Chromatin regulation provides a mechanism to control cardiac gene expression (Table1). While many studies have documented correlations between the different epigenetic marks and end-stage heart failure 57, there is now mounting evidence that changes in the epigenome contribute to the development of cardiomyopathies in the adult. Modulation of JMJD2 demethylase in the heart modifies H3 methylation status and alters the hypertrophic response after pressure overload 51. Dot1L deficiency in mouse heart results in adult lethality from dilated cardiomyopathy and heart failure 46. Chromatin regulators form large protein complexes and interact with a variety of cardiac transcription factors to suppress fetal cardiac genes in the postnatal heart. For instance, Brg1/Baf interacts with HDAC and PARP protein to regulate MHC in the developing and hypertrophic heart 89. Perturbation of these mega-complexes secondary to increased Brg-1 or PARP expression results in pathological cardiac hypertrophy. Collectively, these observations indicate that changes in the epigenome contribute to the development of cardiomyopathies. However, how chromatin regulators together with post-translational modifications of histones and recruitment of transcription factors talk to one another to alter gene transcription is just beginning to emerge. According to the “histone code hypothesis” connections exist between the different covalent histone modifications. Early studies defined the histone code in a relatively simple manner where chromatin marks were associated with activation or repression of transcription. More recent studies now indicate that transcription occurs in response to many marks, some of which are conflicting. Rather than a simple “code” where the histone marks are turned “on” or “off”, chromatin is viewed by some as a “language” in which various histone modifications coexist and are highly dynamic 3. The histone code embraces the idea that some modifications act as docking sites that enable other modifications or remodeling factors to bind chromatin, thus introducing gene specificity and regulatory control. For instance, pathological cardiac hypertrophy requires the nuclear export of HDACs subsequent to phosphorylation by CaMKII. Cooperation between ATP-dependent chromatin remodeling and histone modifications represents another mechanism of transcriptional control. In addition to their direct influence on chromatin components, chromatin regulators indirectly affect cardiac development and diseases through modulation of the extracellular matrix environment. This is illustrated by the observation that Brg1 suppresses Adamts1, and this repression permits the development of cardiac jelly that triggers myocardial trabeculation 103. However, it is not yet known if the role of chromatin regulation in modulating extracellular matrix in the adult heart plays a role in the responses of cardiac tissue to pathological stress.
An obvious challenge for the future is to understand the complexity of these interactions in vivo. The introduction of specific mutations in histone proteins to dissect their physiological role is possible in simple organisms but still represents major difficulties in model organisms due to the large copy number of histone genes. The use of in vitro transcription studies with reconstituted chromatin templates together with the development of animal models lacking or over-expressing chromatin regulators in a tissue-specific manner should help understanding the true biological function of the various histone “marks”. Also, the variability in penetrance and expressivity of human phenotypes indicate a major influence of modifier genes and environmental factors. There is evidence of inter-individual epigenetic variations among twins with identical genotypes and proof that environmental factors can alter the epigenome to contribute to disease 105–108. That epigenetic variations and changes in the environment contribute to cardiac diseases is now becoming evident. Understanding the molecular basis for such variability will necessitate integration of different networks. This represents another challenge for the future but also an exciting topic of investigation.
Assessing global and local epigenetic changes in the normal and diseased heart should provide a deeper understanding of epigenetic contributions to cardiovascular diseases. The identification of major players controlling chromatin architecture and cardiac regulatory networks represents another step towards understanding epigenetic mechanisms in the heart. Tracking epigenetic marks may help in monitoring disease progression. Targeting specific chromatin remodeling factors and histone modifiers may represent a promising strategy for the prevention or at least partial reversion of some cardiomyopathic processes.
Acknowledgments
We apologize to the many authors that contributed to the epigenetics field and could not be cited for space constraint. CP thanks all the members of her laboratory for their dedication and hard work. CP is supported by two grants from King Abdulaziz City for Science & Technology (10-BIO 1350–20 and 10-BIO 1347–20).
Glossary
- AKT
protein kinase B
- AMPK
adenosine monophosphate-activated protein kinase
- AMOTL2
angiomotin like 2
- ARHGAP24
Rho GTPase activating protein 24
- BAF
brahma-associated factor
- BRG1
brahma-related gene 1
- CaMKII
calcium/calmodulin-dependent protein kinase II
- CBP
CREB-binding protein
- CK2α1
casein kinase 2α1
- DCM
dilated cardiomyopathy
- DOT1L
Dot1like histone H3 methyltransferase
- eNOS
endothelial nitric oxide synthase
- ERK
extracellular-signal-regulated kinase
- ESCs
embryonic stem cells
- FHL1
four-and-a-half LIM domains 1
- GSK
glycogen synthase kinase 3
- HATs
histone acetyl transferases
- HDACs
histone deacetylases
- IKKα
inhibitor of nuclear factor kappa-B kinase alpha
- INO80
inositol requiring 80
- ISWI
imitation switch
- JAK2
janus kinase 2
- JMJD2
jumonji domain containing 2
- Kcnip
Kv channel-interacting protein 2
- MHC
myosin heavy chain
- MSK1
mitogen- and stress-activated protein kinase 1
- NIMA
never in mitosis gene A
- PARP-1
Poly(ADP-ribose) polymerase 1
- PECAM1
platelet/endothelial cell adhesion molecule 1
- PI3K
phosphatidylinositol 3 kinase
- PKC
protein kinase C
- PKD
protein kinase D
- PIM1
pim1 kinase
- PTIP
Pax interacting protein 1
- RSK2
ribosomal S6 kinase
- SWI/SNF
switch/sucrose nonfermentable
- TFF1
trefoil factor 1
- UTX
histone H3K27 demethylase
Author Contributions
SMA and CP wrote the manuscript.
References
- 1.Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- 2.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 3.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 4.Vignali M, Hassan AH, Neely KE, et al. ATP-dependent chromatin-remodeling complexes. Mol Cell Biol. 2000;20:1899–1910. doi: 10.1128/mcb.20.6.1899-1910.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson CN, Adkins NL, Georgel P. Chromatin remodeling complexes: ATP-dependent machines in action. Biochem Cell Biol. 2005;83:405–417. doi: 10.1139/o05-115. [DOI] [PubMed] [Google Scholar]
- 6.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 7.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–398. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 8.Chang CP, Bruneau BG. Epigenetics and cardiovascular development. Annu Rev Physiol. 2012;74:41–68. doi: 10.1146/annurev-physiol-020911-153242. [DOI] [PubMed] [Google Scholar]
- 9.Ohtani K, Vlachojannis GJ, Koyanagi M, et al. Epigenetic regulation of endothelial lineage committed genes in pro-angiogenic hematopoietic and endothelial progenitor cells. Circ Res. 2011;109:1219–1229. doi: 10.1161/CIRCRESAHA.111.247304. [DOI] [PubMed] [Google Scholar]
- 10.Shirodkar AV, Marsden PA. Epigenetics in cardiovascular disease. Curr Opin Cardiol. 2011;26:209–215. doi: 10.1097/HCO.0b013e328345986e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baccarelli A, Ghosh S. Environmental exposures, epigenetics and cardiovascular disease. Curr Opin Clin Nutr Metab Care. 2012;15:323–329. doi: 10.1097/MCO.0b013e328354bf5c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J, Yang X. The function of miRNA in cardiac hypertrophy. Cell Mol Life Sci. 2012;69:3561–3570. doi: 10.1007/s00018-012-1126-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Da Costa Martins PA, De Windt LJ. MicroRNAs in control of cardiac hypertrophy. Cardiovasc Res. 2012;93:563–572. doi: 10.1093/cvr/cvs013. [DOI] [PubMed] [Google Scholar]
- 14.Zhu H, Fan GC. Role of microRNAs in the reperfused myocardium towards post-infarct remodelling. Cardiovasc Res. 2012;94:284–292. doi: 10.1093/cvr/cvr291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dorn GW., 2nd MicroRNAs in cardiac disease. Transl Res. 2011;157:226–235. doi: 10.1016/j.trsl.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Small EM, Frost RJ, Olson EN. MicroRNAs add a new dimension to cardiovascular disease. Circulation. 2010;121:1022–1032. doi: 10.1161/CIRCULATIONAHA.109.889048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Latronico MV, Condorelli G. MicroRNAs and cardiac pathology. Nat Rev Cardiol. 2009;6:419–429. doi: 10.1038/nrcardio.2009.56. [DOI] [PubMed] [Google Scholar]
- 18.Irons CE, Sei CA, Hidaka H, et al. Protein kinase C and calmodulin kinase are required for endothelin- stimulated atrial natriuretic factor secretion from primary atrial myocytes. J Biol Chem. 1992;267:5211–5216. [PubMed] [Google Scholar]
- 19.Zhu W, Zou Y, Shiojima I, et al. Ca2+/calmodulin-dependent kinase II and calcineurin play critical roles in endothelin-1-induced cardiomyocyte hypertrophy. J Biol Chem. 2000;275:15239–15245. doi: 10.1074/jbc.275.20.15239. [DOI] [PubMed] [Google Scholar]
- 20.Spirito P, Autore C. Management of hypertrophic cardiomyopathy. BMJ. 2006;332:1251–1255. doi: 10.1136/bmj.332.7552.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 22.Clerk A, Cullingford TE, Fuller SJ, et al. Signaling pathways mediating cardiac myocyte gene expression in physiological and stress responses. J Cell Physiol. 2007;212:311–322. doi: 10.1002/jcp.21094. [DOI] [PubMed] [Google Scholar]
- 23.Berger SL. Histone modifications in transcriptional regulation. Curr Opin Genet Dev. 2002;12:142–148. doi: 10.1016/s0959-437x(02)00279-4. [DOI] [PubMed] [Google Scholar]
- 24.Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 25.Vogelauer M, Wu J, Suka N, et al. Global histone acetylation and deacetylation in yeast. Nature. 2000;408:495–498. doi: 10.1038/35044127. [DOI] [PubMed] [Google Scholar]
- 26.Turner BM. Histone acetylation and an epigenetic code. Bioessays. 2000;22:836–845. doi: 10.1002/1521-1878(200009)22:9<836::AID-BIES9>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 27.Eberharter A, Becker PB. Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep. 2002;3:224–229. doi: 10.1093/embo-reports/kvf053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Backs J, Olson EN. Control of Cardiac Growth by Histone Acetylation/Deacetylation. Circulation Research. 2006;98:15–24. doi: 10.1161/01.RES.0000197782.21444.8f. [DOI] [PubMed] [Google Scholar]
- 29.Yao TP, Oh SP, Fuchs M, et al. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell. 1998;93:361–372. doi: 10.1016/s0092-8674(00)81165-4. [DOI] [PubMed] [Google Scholar]
- 30.Shikama N, Lutz W, Kretzschmar R, et al. Essential function of p300 acetyltransferase activity in heart, lung and small intestine formation. The EMBO Journal. 2003;22:5175–5185. doi: 10.1093/emboj/cdg502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yanazume T, Hasegawa K, Morimoto T, et al. Cardiac p300 is involved in myocyte growth with decompensated heart failure. Mol Cell Biol. 2003;23:3593–3606. doi: 10.1128/MCB.23.10.3593-3606.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gusterson RJ, Jazrawi E, Adcock IM, et al. The transcriptional co-activators CREB-binding protein (CBP) and p300 play a critical role in cardiac hypertrophy that is dependent on their histone acetyltransferase activity. J Biol Chem. 2003;278:6838–6847. doi: 10.1074/jbc.M211762200. [DOI] [PubMed] [Google Scholar]
- 33.Miyamoto S, Kawamura T, Morimoto T, et al. Histone acetyltransferase activity of p300 is required for the promotion of left ventricular remodeling after myocardial infarction in adult mice in vivo. Circulation. 2006;113:679–690. doi: 10.1161/CIRCULATIONAHA.105.585182. [DOI] [PubMed] [Google Scholar]
- 34.Mathiyalagan P, Chang L, Du XJ. Cardiac ventricular chambers are epigenetically distinguishable. Cell Cycle. 2010;9:612–617. doi: 10.4161/cc.9.3.10612. [DOI] [PubMed] [Google Scholar]
- 35.Heo K, Kim H, Choi SH, et al. FACT-mediated exchange of histone variant H2AX regulated by phosphorylation of H2AX and ADP-ribosylation of Spt16. Mol Cell. 2008;30:86–97. doi: 10.1016/j.molcel.2008.02.029. [DOI] [PubMed] [Google Scholar]
- 36.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murray K. The Occurrence Of Epsilon-N-Methyl Lysine In Histones. Biochemistry. 1964;3:10–15. doi: 10.1021/bi00889a003. [DOI] [PubMed] [Google Scholar]
- 38.Chen D, Ma H, Hong H, et al. Regulation of transcription by a protein methyltransferase. Science. 1999;284:2174–2177. doi: 10.1126/science.284.5423.2174. [DOI] [PubMed] [Google Scholar]
- 39.Strahl BD, Ohba R, Cook RG, et al. Methylation of histone H3 at lysine 4 is highly conserved and correlates with transcriptionally active nuclei in Tetrahymena. Proc Natl Acad Sci U S A. 1999;96:14967–14972. doi: 10.1073/pnas.96.26.14967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Weerd JH, Koshiba-Takeuchi K, Kwon C, et al. Epigenetic factors and cardiac development. Cardiovasc Res. 2011;91:203–211. doi: 10.1093/cvr/cvr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vallaster M, Vallaster CD, Wu SM. Epigenetic mechanisms in cardiac development and disease. Acta Biochim Biophys Sin (Shanghai) 2012;44:92–102. doi: 10.1093/abbs/gmr090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stein AB, Jones TA, Herron TJ, et al. Loss of H3K4 methylation destabilizes gene expression patterns and physiological functions in adult murine cardiomyocytes. J Clin Invest. 2011;121:2641–2650. doi: 10.1172/JCI44641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pandya K, Kohro T, Mimura I, et al. Distribution of histone3 lysine 4 trimethylation at T3-responsive loci in the heart during reversible changes in gene expression. Gene Expr. 2012;15:183–198. doi: 10.3727/105221612x13372578119698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Easley CA, Faison MO, Kirsch TL, et al. Laminin activates CaMK-II to stabilize nascent embryonic axons. Brain Res. 2006;1092:59–68. doi: 10.1016/j.brainres.2006.03.099. [DOI] [PubMed] [Google Scholar]
- 45.Lorenzen JM, Martino F, Thum T. Epigenetic modifications in cardiovascular disease. Basic Res Cardiol. 2012;107:245–245. doi: 10.1007/s00395-012-0245-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen AT, Xiao B, Neppl RL, et al. DOT1L regulates dystrophin expression and is critical for cardiac function. Genes Dev. 2011;25:263–274. doi: 10.1101/gad.2018511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boyer LA, Plath K, Zeitlinger J, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. doi: 10.1038/nature04733. [DOI] [PubMed] [Google Scholar]
- 48.Roh TY, Cuddapah S, Cui K, et al. The genomic landscape of histone modifications in human T cells. Proc Natl Acad Sci U S A. 2006;103:15782–15787. doi: 10.1073/pnas.0607617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agger K, Cloos PA, Christensen J, et al. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449:731–734. doi: 10.1038/nature06145. [DOI] [PubMed] [Google Scholar]
- 50.Lee S, Lee JW, Lee SK. UTX, a histone H3-lysine 27 demethylase, acts as a critical switch to activate the cardiac developmental program. Dev Cell. 2012;22:25–37. doi: 10.1016/j.devcel.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang QJ, Chen HZ, Wang L, et al. The histone trimethyllysine demethylase JMJD2A promotes cardiac hypertrophy in response to hypertrophic stimuli in mice. J Clin Invest. 2011;121:2447–2456. doi: 10.1172/JCI46277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bernstein BE, Kamal M, Lindblad-Toh K, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 53.Kim TH, Barrera LO, Zheng M, et al. A high-resolution map of active promoters in the human genome. Nature. 2005;436:876–880. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 55.Azuara V, Perry P, Sauer S, et al. Chromatin signatures of pluripotent cell lines. Nature cell biology. 2006;8:532–538. doi: 10.1038/ncb1403. [DOI] [PubMed] [Google Scholar]
- 56.Kaneda R, Takada S, Yamashita Y, et al. Genome-wide histone methylation profile for heart failure. Genes Cells. 2009;14:69–77. doi: 10.1111/j.1365-2443.2008.01252.x. [DOI] [PubMed] [Google Scholar]
- 57.Movassagh M, Choy MK, Knowles DA, et al. Distinct epigenomic features in end-stage failing human hearts. Circulation. 2011;124:2411–2422. doi: 10.1161/CIRCULATIONAHA.111.040071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cheung P, Allis CD, Sassone-Corsi P. Signaling to chromatin through histone modifications. Cell. 2000;103:263–271. doi: 10.1016/s0092-8674(00)00118-5. [DOI] [PubMed] [Google Scholar]
- 59.Al-Nasser IA. In vivo prevention of adriamycin cardiotoxicity by cyclosporin A or FK506. Toxicology. 1998;131:175–181. doi: 10.1016/s0300-483x(98)00128-0. [DOI] [PubMed] [Google Scholar]
- 60.Bode AM, Dong Z. Inducible covalent posttranslational modification of histone H3. Sci STKE. 2005;2005:re4. doi: 10.1126/stke.2812005re4. [DOI] [PubMed] [Google Scholar]
- 61.Goto H, Tomono Y, Ajiro K, et al. Identification of a novel phosphorylation site on histone H3 coupled with mitotic chromosome condensation. J Biol Chem. 1999;274:25543–25549. doi: 10.1074/jbc.274.36.25543. [DOI] [PubMed] [Google Scholar]
- 62.Thomson S, Clayton AL, Hazzalin CA, et al. The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. EMBO J. 1999;18:4779–4793. doi: 10.1093/emboj/18.17.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yamamoto Y, Verma UN, Prajapati S, et al. Histone H3 phosphorylation by IKK-alpha is critical for cytokine-induced gene expression. Nature. 2003;423:655–659. doi: 10.1038/nature01576. [DOI] [PubMed] [Google Scholar]
- 64.Macdonald N, Welburn JP, Noble ME, et al. Molecular basis for the recognition of phosphorylated and phosphoacetylated histone h3 by 14-3-3. Mol Cell. 2005;20:199–211. doi: 10.1016/j.molcel.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 65.Anversa P, Kajstura J. Ventricular myocytes are not terminally differentiated in the adult mammalian heart. Circ Res. 1998;83:1–14. doi: 10.1161/01.res.83.1.1. [DOI] [PubMed] [Google Scholar]
- 66.Kajstura J, Urbanek K, Perl S, et al. Cardiomyogenesis in the adult human heart. Circ Res. 2010;107:305–315. doi: 10.1161/CIRCRESAHA.110.223024. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 67.Cottage CT, Bailey B, Fischer KM, et al. Cardiac progenitor cell cycling stimulated by pim-1 kinase. Circ Res. 2010;106:891–901. doi: 10.1161/CIRCRESAHA.109.208629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Porrello ER, Mahmoud AI, Simpson E, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–1080. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liew CC, Sole MJ. Studies of nuclear proteins in the heart of the cardiomyopathic Syrian hamster--phosphorylation of histones. J Mol Cell Cardiol. 1978;10:847–855. doi: 10.1016/0022-2828(78)90393-0. [DOI] [PubMed] [Google Scholar]
- 70.Hsu JY, Sun ZW, Li X, et al. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell. 2000;102:279–291. doi: 10.1016/s0092-8674(00)00034-9. [DOI] [PubMed] [Google Scholar]
- 71.De Souza CP, Osmani AH, Wu LP, et al. Mitotic histone H3 phosphorylation by the NIMA kinase in Aspergillus nidulans. Cell. 2000;102:293–302. doi: 10.1016/s0092-8674(00)00035-0. [DOI] [PubMed] [Google Scholar]
- 72.Sassone-Corsi P, Mizzen CA, Cheung P, et al. Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science. 1999;285:886–891. doi: 10.1126/science.285.5429.886. [DOI] [PubMed] [Google Scholar]
- 73.Anest V, Hanson JL, Cogswell PC, et al. A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature. 2003;423:659–663. doi: 10.1038/nature01648. [DOI] [PubMed] [Google Scholar]
- 74.Zippo A, De Robertis A, Serafini R, et al. PIM1-dependent phosphorylation of histone H3 at serine 10 is required for MYC-dependent transcriptional activation and oncogenic transformation. Nature cell biology. 2007;9:932–944. doi: 10.1038/ncb1618. [DOI] [PubMed] [Google Scholar]
- 75.Baek SH. When signaling kinases meet histones and histone modifiers in the nucleus. Mol Cell. 2011;42:274–284. doi: 10.1016/j.molcel.2011.03.022. [DOI] [PubMed] [Google Scholar]
- 76.Dawson MA, Bannister AJ, Gottgens B, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009;461:819–822. doi: 10.1038/nature08448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bungard D, Fuerth BJ, Zeng PY, et al. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science. 2010;329:1201–1205. doi: 10.1126/science.1191241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Booz GW, Day JN, Baker KM. Interplay between the cardiac renin angiotensin system and JAK-STAT signaling: role in cardiac hypertrophy, ischemia/reperfusion dysfunction, and heart failure. J Mol Cell Cardiol. 2002;34:1443–1453. doi: 10.1006/jmcc.2002.2076. [DOI] [PubMed] [Google Scholar]
- 79.Karmazyn M, Purdham DM, Rajapurohitam V, et al. Leptin as a cardiac hypertrophic factor: a potential target for therapeutics. Trends Cardiovasc Med. 2007;17:206–211. doi: 10.1016/j.tcm.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 80.Mahmoud SA KM, Little GH, Bai Y, An W, Bers D, Kedes L, Poizat C. Nuclear CaMKII Enhances Histone H3 Phosphorylation and Remodels Chromatin During Cardiac Hypertrophy. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt500. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 81.Vega RB, Harrison BC, Meadows E, et al. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol. 2004;24:8374–8385. doi: 10.1128/MCB.24.19.8374-8385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Backs J, Song K, Bezprozvannaya S, et al. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. The Journal of Clinical Investigation. 2006:1–12. doi: 10.1172/JCI27438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Little GH, Bai Y, Williams T, et al. Nuclear calcium/calmodulin-dependent protein kinase IIdelta preferentially transmits signals to histone deacetylase 4 in cardiac cells. J Biol Chem. 2007;282:7219–7231. doi: 10.1074/jbc.M604281200. [DOI] [PubMed] [Google Scholar]
- 84.Backs J, Backs T, Neef S, et al. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci U S A. 2009;106:2342–2347. doi: 10.1073/pnas.0813013106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Eom GH, Cho YK, Ko JH, et al. Casein kinase-2alpha1 induces hypertrophic response by phosphorylation of histone deacetylase 2 S394 and its activation in the heart. Circulation. 2011;123:2392–2403. doi: 10.1161/CIRCULATIONAHA.110.003665. [DOI] [PubMed] [Google Scholar]
- 86.Ogata N, Ueda K, Hayaishi O. ADP-ribosylation of histone H2B. Identification of glutamic acid residue 2 as the modification site. J Biol Chem. 1980;255:7610–7615. [PubMed] [Google Scholar]
- 87.Pillai JB, Gupta M, Rajamohan SB, et al. Poly(ADP-ribose) polymerase-1-deficient mice are protected from angiotensin II-induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2006;291:H1545–1553. doi: 10.1152/ajpheart.01124.2005. [DOI] [PubMed] [Google Scholar]
- 88.Bartha E, Solti I, Kereskai L, et al. PARP inhibition delays transition of hypertensive cardiopathy to heart failure in spontaneously hypertensive rats. Cardiovasc Res. 2009;83:501–510. doi: 10.1093/cvr/cvp144. [DOI] [PubMed] [Google Scholar]
- 89.Hang CT, Yang J, Han P, et al. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature. 2010;466:62–67. doi: 10.1038/nature09130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Palfi A, Toth A, Kulcsar G, et al. The role of Akt and mitogen-activated protein kinase systems in the protective effect of poly(ADP-ribose) polymerase inhibition in Langendorff perfused and in isoproterenol-damaged rat hearts. J Pharmacol Exp Ther. 2005;315:273–282. doi: 10.1124/jpet.105.088336. [DOI] [PubMed] [Google Scholar]
- 91.Palfi A, Toth A, Hanto K, et al. PARP inhibition prevents postinfarction myocardial remodeling and heart failure via the protein kinase C/glycogen synthase kinase-3beta pathway. J Mol Cell Cardiol. 2006;41:149–159. doi: 10.1016/j.yjmcc.2006.03.427. [DOI] [PubMed] [Google Scholar]
- 92.Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 93.Kangaspeska S, Stride B, Metivier R, et al. Transient cyclical methylation of promoter DNA. Nature. 2008;452:112–115. doi: 10.1038/nature06640. [DOI] [PubMed] [Google Scholar]
- 94.Handy DE, Castro R, Loscalzo J. Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation. 2011;123:2145–2156. doi: 10.1161/CIRCULATIONAHA.110.956839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Movassagh M, Choy MK, Goddard M, et al. Differential DNA methylation correlates with differential expression of angiogenic factors in human heart failure. PLoS One. 2010;5:e8564. doi: 10.1371/journal.pone.0008564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee SH, Wolf PL, Escudero R, et al. Early expression of angiogenesis factors in acute myocardial ischemia and infarction. N Engl J Med. 2000;342:626–633. doi: 10.1056/NEJM200003023420904. [DOI] [PubMed] [Google Scholar]
- 97.Eisen JA, Sweder KS, Hanawalt PC. Evolution of the SNF2 family of proteins: subfamilies with distinct sequences and functions. Nucleic Acids Res. 1995;23:2715–2723. doi: 10.1093/nar/23.14.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Winston F, Carlson M. Yeast SNF/SWI transcriptional activators and the SPT/SIN chromatin connection. Trends Genet. 1992;8:387–391. doi: 10.1016/0168-9525(92)90300-s. [DOI] [PubMed] [Google Scholar]
- 99.Cote J, Quinn J, Workman JL, et al. Stimulation of GAL4 derivative binding to nucleosomal DNA by the yeast SWI/SNF complex. Science. 1994;265:53–60. doi: 10.1126/science.8016655. [DOI] [PubMed] [Google Scholar]
- 100.Flaus A, Martin DMA, Barton GJ, et al. Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res. 2006;34:2887–2905. doi: 10.1093/nar/gkl295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lessard J, Wu JI, Ranish JA, et al. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron. 2007;55:201–215. doi: 10.1016/j.neuron.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lickert H, Takeuchi JK, Von Both I, et al. Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature. 2004;432:107–112. doi: 10.1038/nature03071. [DOI] [PubMed] [Google Scholar]
- 103.Stankunas K, Hang CT, Tsun ZY, et al. Endocardial Brg1 represses ADAMTS1 to maintain the microenvironment for myocardial morphogenesis. Dev Cell. 2008;14:298–311. doi: 10.1016/j.devcel.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jenni R, Rojas J, Oechslin E. Isolated noncompaction of the myocardium. N Engl J Med. 1999;340:966–967. doi: 10.1056/NEJM199903253401215. [DOI] [PubMed] [Google Scholar]
- 105.Fraga MF, Ballestar E, Paz MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bradshaw LA, Irimia A, Sims JA, et al. Biomagnetic characterization of spatiotemporal parameters of the gastric slow wave. Neurogastroenterol Motil. 2006;18:619–631. doi: 10.1111/j.1365-2982.2006.00794.x. [DOI] [PubMed] [Google Scholar]
- 107.Javierre BM, Fernandez AF, Richter J, et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome research. 2010;20:170–179. doi: 10.1101/gr.100289.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Turan N, Katari S, Coutifaris C, et al. Explaining inter-individual variability in phenotype: is epigenetics up to the challenge? Epigenetics. 2010;5:16–19. doi: 10.4161/epi.5.1.10557. [DOI] [PMC free article] [PubMed] [Google Scholar]