

Abstract
Background
The 5' end of the Rous sarcoma virus (RSV) RNA around the primer-binding site forms a series of RNA secondary stem/loop structures (U5-IR stem, TψC interaction region, U5-leader stem) that are required for efficient initiation of reverse transcription. The U5-IR stem and loop also encode the U5 integrase (IN) recognition sequence at the level of DNA such that this region has overlapping biological functions in reverse transcription and integration.
Results
We have investigated the ability of RSV to tolerate mutations in and around the U5 IR stem and loop. Through the use of viral libraries with blocks of random sequence, we have screened for functional mutants in vivo, growing the virus libraries in turkey embryo fibroblasts. The library representing the U5-IR stem rapidly selects for clones that maintain the structure of the stem, and is subsequently overtaken by wild type sequence. In contrast, in the library representing the U5-IR loop, wild type sequence is found after five rounds of infection but it does not dominate the virus pool, indicating that the mutant sequences identified are able to replicate at or near wild type levels.
Conclusion
These results indicate that the region of the RNA genome in U5 adjacent to the PBS tolerates much sequence variation even though it is required for multiple biological functions in replication. The in vivo selection method utilized in this study was capable of detecting complex patterns of selection as well as identifying biologically relevant viral mutants.
Background
Rous sarcoma virus (RSV) is one of the most studied and best characterized members of the retrovirus family. As with all retroviruses, the RNA genome of RSV must be reverse transcribed into a double stranded DNA copy after entry into the host cell. This reverse transcription step is followed by the insertion of the viral DNA into the host cell chromosome. Both of these steps are mediated by viral enzymes, and are dependent on overlapping regions at the 5' end of the viral genome [1-3]. Reverse transcription is primed by a host cell tRNATrp, which anneals to an 18 nucleotide complimentary region known as the primer binding site (PBS) located at RNA positions 102–119 [4]. The surrounding region forms a complex RNA secondary structure, which includes the U5-leader stem, the U5-IR stem and loop, the tRNA-PBS interaction site, and a second primer interaction site between the viral RNA and the TΨC loop of the tRNA [4-6] (see Figure 1). A variation of this structure is present in all retroviruses [7-9].
Figure 1.
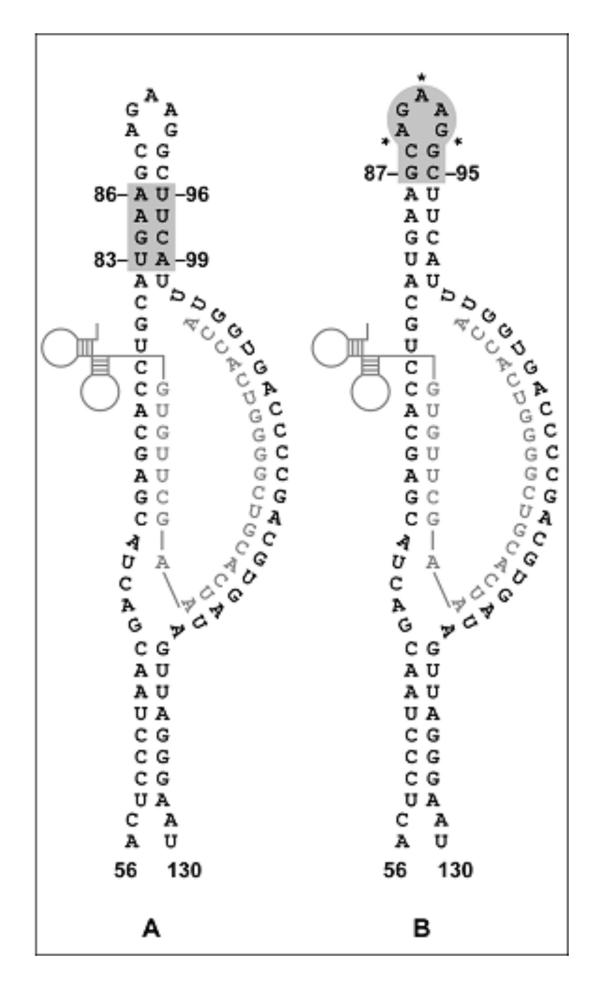
The secondary structure surrounding the primer-binding site of RSV RNA. The nucleotide sequence of wild type RSV, nucleotides 56 to 130 (bold) are shown annealed to the tRNATrp (gray). The shaded RSV RNA sequence indicates nucleotides that were randomized in the U5-IR stem (83–86 and 96–99) (Panel A) and U5-IR loop (87–95) (Panel B) libraries. The asterisks denote positions 89, 91, and 93 found in the U5-IR stem loop.
Previous studies have shown that placing mutations into the U5-IR stem that disrupt the RNA structure cause a partial defect in initiation of reverse transcription. Compensatory mutations that restore the RNA structure rescue these viruses [10]. Placing extensions into the stem also reduces the amount of RNA incorporated into virions, suggesting that this region may have additional role in RNA packaging [10]. In addition to these structural requirements at the RNA level, the same region, after reverse transcription, encompasses the integrase recognition sequence at the end of the U5 viral DNA. Base pair substitutions in the terminal 20 base pairs of the viral DNA can have dramatic effects on integration efficiency [3,11-13].
Results
Construction and analysis of randomized libraries
PCR based mutagenesis was used to introduce random nucleotide sequence into short stretches of the RSV genome in the RCAS [14] vector, within the U5-IR stem and U5-IR loop RNA structures (Figure 1). The U5-IR stem library affected positions 83–86 and 96–99 of the viral genome, with a predicted library size of 48, or about 262,000 individual clones. The U5-IR loop library included positions 87–95, with an estimated library size of just over 1,000,000 clones. At each step of the library construction, transfection, and in vivo culture, at least 1 × 106 clones were sampled [15]so that our libraries could include a majority of the possible sequence combinations. Adequate sampling was ensured during library construction by using an excess of both vector and insert (at least 1 × 108 molecules of each) in each of the cloning reactions and by verifying the number of vector plus insert plasmids recovered by quantifying the number of bacterial colonies transformed during the cloning process. Adequate sampling during transfection of the turkey embryo fibroblasts (TEFs) was evaluated by determining the transfection efficiency of a control vector carrying a beta galactoside gene driven by the promoter in the RSV long terminal repeat (LTR) during each experiment. Calculation of the multiplicity of infection (MOI) using a wild type virus control in parallel with each randomized library virus infection ensured adequate sampling of each library during infection.
As a control, the starting library pools were sequenced to look for initial bias in nucleotide ratios and were found not to contain any. Additionally, 37 individual clones from the U5-IR loop library were sequenced prior to transfection. No statistically significant deviations from a random pool were detected. Nor was there any tendency among these clones to preserve the base pairs between positions 87–95 and 88–94. This gives us confidence that the randomized regions in our starting libraries were evenly represented with no significant sequence bias, and that we were able to screen a majority of the potential sequences from these libraries.
Selection of sequences from the U5-IR stem library
Four paired bases on each side of the U5-IR stem were randomized, including positions 83–86 and 96–99 of the viral RNA (Figure 1A). After passage in TEFs for 3 rounds of infection, the pooled sequence data showed that wild type virus dominated (Figure 2A). To examine those mutant sequences that were best able to replicate in vivo, individual clones from the second round of infection were isolated and sequenced (Table 1). Of the 50 clones examined, 31 were wild type (62%). Notably, of the 19 recovered mutants, six were able to form the wild type U5-IR stem structure (31.5%). In a random pool, we would not expect this high percentage of clones to have the potential to form such a structure. A multinomial distribution analysis was used to determine if the presence of these mutant sequences was a statistically significant event, resulting in a p value of 4.2 × 10-9. The free energy for each of these stem structures was determined using mFOLD 3.0 software (Michael Zuker, Institute for Biomedical Computing, Washington University in St. Louis), and are presented with the structures in Figure 3. In addition to the enrichment for mutants maintaining the wild type stem, three clones have the potential to form an alternative stem structure with a four-nucleotide loop and a ΔG similar to the wild type stem. It should be noted that while the sequences of IRS-11 and IRS-19 are identical, we find a mutation in the flanking leader sequence in one of these clones suggesting that these are independent isolates. Six the remaining clones had the potential to form either a weak stem structure or a stem with a single mismatched position.
Figure 2.
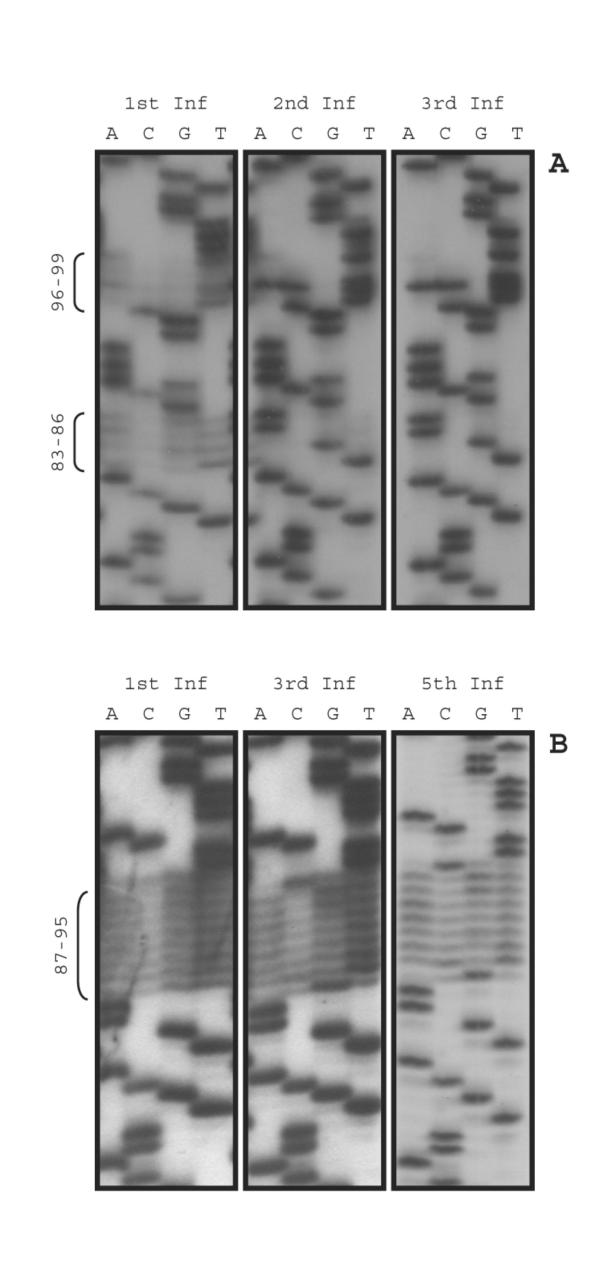
Sequences recovered from the pool of viruses derived from libraries with randomized bases in the U5-IR stem and loop. Panel A: Sequences of the U5-IR stem were randomized as described in Figure 1A. 1st Inf. refers to the sequence pool recovered after the first round of infection with virus derived from the transfected cells. 2nd and 3rd Inf. refers to the sequence pool recovered after the second and third rounds of infections, respectively. Viral DNA was recovered from cells and sequencing was carried out as described in Methods. Panel B: The sequences of the U5-IR loop and stem were randomized as described in Figure 1B. All other notations were as described in legend to panel A, except that 5 rounds of infection were carried out. Rounds 1, 3, and 5 are only shown.
Table 1.
Sequences of individual clones from the randomized U5-IR stem librarya.
| Clone ID | 83 | 84 | 85 | 86 | 87–95b | 96 | 97 | 98 | 99 |
| Wild Type | U | G | A | A | GCAGAAGGC | U | U | C | A |
| IRS-1 | U | G | A | G | U | U | C | A | |
| IRS-2 | U | A | A | G | U | U | U | A | |
| IRS-3 | C | A | A | U | G | C | A | U | |
| IRS-4 | G | A | G | U | C | A | U | U | |
| IRS-5 | U | G | C | C | A | G | G | A | |
| IRS-6 | U | G | G | G | C | G | C | A | |
| IRS-7 | U | G | G | A | U | U | C | A | |
| IRS-8 | G | G | A | U | U | G | C | A | |
| IRS-9 | U | A | U | G | C | A | U | A | |
| IRS-10 | U | A | A | U | A | C | A | U | |
| IRS-11 | C | G | A | U | U | G | C | A | |
| IRS-12 | U | G | A | U | A | U | C | A | |
| IRS-13 | A | C | C | G | G | A | C | U | |
| IRS-14 | U | U | G | G | A | C | C | A | |
| IRS-15 | A | G | U | G | U | A | U | U | |
| IRS-16 | G | A | U | A | U | U | C | A | |
| IRS-17 | U | G | U | G | G | G | C | A | |
| IRS-18 | A | A | A | C | A | U | G | A | |
| IRS-19 | C | G | A | U | U | G | C | A |
aIndividual clones were recovered after two rounds of infection and sequenced as described under "Methods". bRNA nucleotide positions 87–95 were not randomized in this library and were included to indicate the nucleotides between the two randomized regions.
Figure 3.
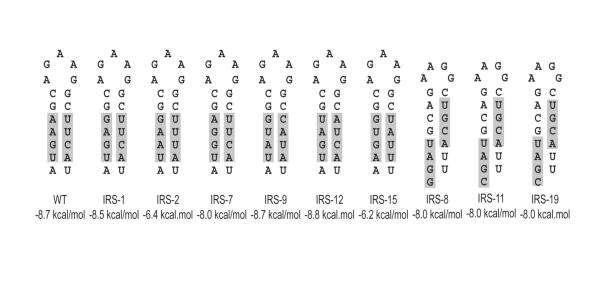
Base pairing in the U5-IR stem from individual clones selected from the randomized library after the second round of infection. The potential base pairing structure for the U5-IR stem is shown for individual mutant clones presented in Table 1. The bases at those positions randomized in the initial virus library are shaded. The relative stability of the stem structures is calculated below each.
In vitro analysis of U5-IR stem mutants
Some of the recovered mutants from the stem library were tested in vitro for an impact on initiation of reverse transcription or on processing by RSV integrase. To examine the reverse transcription step, a cDNA101 initiation and elongation assay was used. In this assay, we use PCR based mutagenesis to produce T7 templates including the mutations of interest, and then use these DNAs to produce the mutant RNA templates. A synthetic RNA primer is added, along with purified reverse transcriptase, and products detected by the incorporation of radio labeled deoxyribonucleotides after separation on a polyacrylamide gel. Six mutants were tested, one which maintains the stem structure similar to that of wild type (IRS-15), one which has the potential to form the alternative stem structure seen in Figure 3 (IRS-11), and four other mutants which have some potential to form weak alternative stem structures (IRS-3, IRS-4, IRS-16, and IRS-18). All of the mutants were able to serve as templates for initiation and elongation catalyzed by reverse transcriptase, with efficiencies approximately equal to that of a wild type RNA (Figure 4A).
Figure 4.
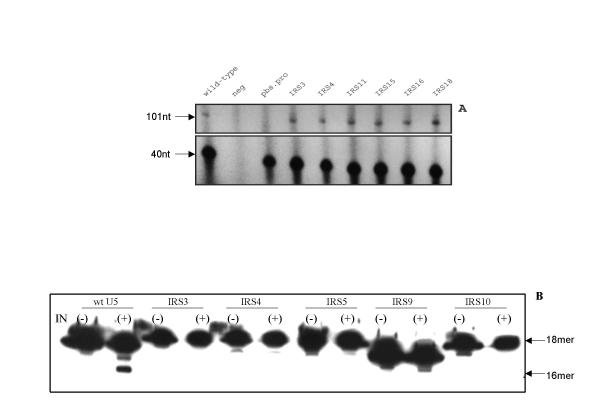
In vitro analysis of mutants from the U5-IR stem library. Panel A: Initiation of reverse transcription was reconstituted with viral RNA templates as indicated, an 18-nucleotide RNA primer, and reverse transcriptase as described in Methods. PAGE was used to separate the products of the reaction. The arrow denotes the migration position of strong stop cDNA, which is 101 deoxyribonucleotides in length. The 40 nt DNA product was initiated using a separate 20 mer DNA primer as a control for the amount of RNA added to the reaction [1]Neg, refers to no RNA added. Pbs.pro, denotes the substitution of an 18-nucleotide primer that anneals to a PBS complementary to tRNAPro rather than to the wild type tRNATrp. Panel B: The 3' end processing reactions were reconstituted with 18 base pair oligodeoxyribonucleotide duplexes and purified RSV integrase as described in Methods. The faster migrating band represents the cleavage product with 2 bases removed from the 3' end of the CA containing strand. The minus sign denotes IN not added. In both panels, the sample numbers refer to clones listed in Table 1.
Screening mutants for processing by integrase involved preparing oligodeoxyribonucleotide duplexes corresponding to the recovered mutant sequences, end labeling the plus strand oligonucleotide, and incubating with purified RSV integrase (IN). Products are analyzed by polyacrylamide gel elctrophoresis (PAGE), and are evidenced by a decrease in size of the substrate after IN removes two deoxyribonucleotides from the 3' end. RNA positions 98–99 correspond to positions 3–4 of the IN recognition sequence, which is the 'CA' dinucleotide conserved in all known retroviruses and retrotransposons. Mutations at these positions are known to cause a dramatic decrease in integrase processing. Included in this experiment were five mutants that were lacking the original CA dinucleotide at positions 98–99 (IRS3, IRS4, IRS5, IRS9, and IRS10). None of these mutant substrates were processed by RSV integrase at a detectable level compared to wild type (Figure 4B).
Selection of sequences from the U5-IR loop library
The nine bases randomized in the U5-IR loop library (RNA positions 87–95) include the five nucleotides of the single stranded loop as well as the top two base pairs of the U5-IR stem. Even after five rounds of infection in TEFs, there was significant degeneracy in the library (Figure 2B). The wild type base dominated only positions 87 and 95. To examine the mutant sequences selected, individual clones from the fifth round of infection were isolated and sequenced. Of the 37 clones examined, there were five wild type sequences recovered, indicating that the failure of this library to revert to wild type was not due to a lack of wild type sequence, but due to strong competition by the mutant viruses present in the pool with wild type. Of the 37 sequenced clones, 27 (73%) maintained both the 87–95 and 88–94 base pairs, and only one clone failed to maintain either base pair. A multinomial distribution analysis on the frequency with which these base pairs were selected yielded a p value of 1.4 × 10-17. This data was also compared to individual clones sequenced from the U5-IR loop library prior to selection. In the starting pool only 19% of the clones had the potential to form both base pairs, and 32% of the starting clones did not have any base pairing potential. Of the 32 fifth round clones that maintain the 87–95 base pair, 24 clones (75%) do so with the wild type G-C base pair. In contrast, the 88–94 base pair is maintained in 31 clones, but only 9 of those (29%) do so with the wild type C-G base pair (including the five wild type clones). There is not a wild type C at position 88 in any of the other 22 clones. In fact, of the 27 clones, which maintain both base pairs, 15 selected a U at position 88 (55%). Interestingly, there are six clones which fail to maintain the 88–94 base pair, and of these, five selected for a C at position 88 (83%), so it seems that there may be a difference in the selection of the wild type base depending on whether a base pair is present.
Positions 89–93 of the U5-IR loop library make up the single stranded loop region. There was a large amount of degeneracy in this region with no single base dominating any of these positions. Further analysis, however, revealed a statistically significant pattern of selection linked to positions 89, 91, and 93. For each of these positions, there were two slightly enriched bases selected. The A/C/G/T ratios of these positions are as follows: position 89, 13/5/5/14; position 91, 14/11/5/7; and position 93, 12/5/14/6 (The enriched bases are in bold, and the wild type base is underlined). In Figure 5, the recovered clones are grouped based on the selection at each of these three positions independently. A chi square statistical analysis was applied to these data sets, and we found these relationships to be significant, with p-values ranging from 0.014 to 0.0007. We found that the base selected at each of these positions dramatically influences base pair distribution at the other two. The general pattern seen was that if a wild type base was present at one position, the other two positions also tended towards wild type. For example, in 14 of 37 clones there was a wild-type A at position 91; 10 of those 14 clones also contained both a wild type A at position 89 and a wild type G at position 93 (Table 2 and Figure 5). In contrast, when a non-wild type base was selected at one of these positions, wild type bases were specifically excluded from the other two positions. For example, in 11 of 37 clones a mutant C was selected at position 91; none of these clones had a wild type A at position 89, and in only one case was there a wild type G at position 93 (Table 2 and Figure 5). Strikingly, the base selected at 89, 91, or 93 had no effect on which base was selected at positions 90 or 92 (p-values for these analyses were between 0.19 and 0.45). Positions 90 and 92 also had no influence on each other (data not shown).
Figure 5.
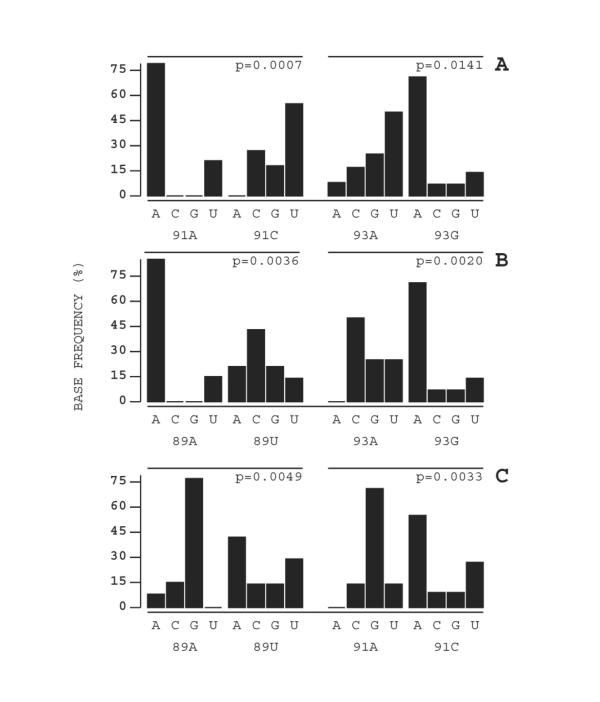
Certain nucleotides in the U5-IR loop influence selection of nucleotides at nearby positions. The sequences listed in Table 2 were analyzed to show patterns of selection. Panel A: Effects on selection at position 89. Panel B: Effects on selection at position 91. Panel C: Effects on selection at position 93. These data sets were analyzed using a 2-way chi square analysis, and the obtained p-value is indicated for each panel. A p value of <0.05 is considered significant. The number scale along the vertical axis represents the percentage of clones carrying a particular nucleotide at that position.
Table 2.
Sequences of individual clones from the randomized U5-IR loop librarya.
| Clone ID | 87 | 88 | 89 | 90 | 91 | 92 | 93 | 94 | 95 |
| Wild Type | G | C | A | G | A | A | G | G | C |
| IRL-1b | G | C | A | G | A | A | G | G | C |
| IRL-2b | G | C | A | G | A | A | G | G | C |
| IRL-3b | G | C | A | G | A | A | G | G | C |
| IRL-4b | G | C | A | G | A | A | G | G | C |
| IRL-5b | G | C | A | G | A | A | G | G | C |
| IRL-6 | G | C | C | C | G | A | U | G | C |
| IRL-7 | G | C | G | G | C | U | U | G | C |
| IRL-8 | G | U | A | U | A | C | G | G | C |
| IRL-9 | G | U | C | U | C | A | G | G | C |
| IRL-10 | G | U | U | A | C | G | A | G | C |
| IRL-11 | G | U | C | A | C | A | A | G | C |
| IRL-12 | G | U | U | G | A | G | T | G | C |
| IRL-13 | G | U | U | A | U | G | A | G | C |
| IRL-14 | G | U | G | A | U | G | A | G | C |
| IRL-15 | G | U | A | A | A | U | G | A | C |
| IRL-16 | G | U | U | A | A | A | U | A | C |
| IRL-17 | G | U | A | G | U | U | A | A | C |
| IRL-18 | G | U | A | U | A | G | G | A | C |
| IRL-19 | G | U | U | U | G | A | A | A | C |
| IRL-20 | G | A | U | G | C | G | A | U | C |
| IRL-21 | G | A | A | G | A | A | G | U | C |
| IRL-22 | G | A | U | U | A | A | C | U | C |
| IRL-23 | A | G | U | G | C | A | U | C | U |
| IRL-24 | C | G | G | G | C | G | A | U | G |
| IRL-25 | C | U | C | G | C | G | A | A | G |
| IRL-26 | U | U | A | U | U | A | C | A | A |
| IRL-27 | U | U | G | C | G | C | A | G | A |
| IRL-28 | U | C | U | G | C | A | U | G | C |
| IRL-29 | U | C | A | G | A | A | C | G | C |
| IRL-30 | G | C | U | U | G | U | G | A | C |
| IRL-31 | C | G | C | C | U | A | C | A | C |
| IRL-32 | G | C | G | A | U | G | G | U | C |
| IRL-33 | C | A | U | A | C | C | C | U | C |
| IRL-34 | C | C | U | A | U | G | G | U | G |
| IRL-35 | C | C | U | G | C | G | A | C | G |
| IRL-36 | C | C | A | U | A | G | G | A | G |
| IRL-37 | C | G | U | C | G | U | A | U | U |
aIndividual clones were recovered after five rounds of infection and sequenced as described under "Methods". bwild type sequence found in the clone.
In vitro analysis of U5-IR loop mutants
Some of the recovered mutants from the loop library were tested in vitro for any impact on synthesis of cDNA101 catalyzed by reverse transcriptase and processing of duplex oligos by RSV integrase, as described for the stem library. Two mutants were tested for synthesis of cDNA101, IRL10 and IRL18. IRL10 includes the 89U-91C-93A mutations, whereas IRL18 is wild type at all three positions (Table 2). Both of these substrates performed as well as, if not better, than a wild type substrate in the reverse transcription assay (Figure 6A).
Figure 6.
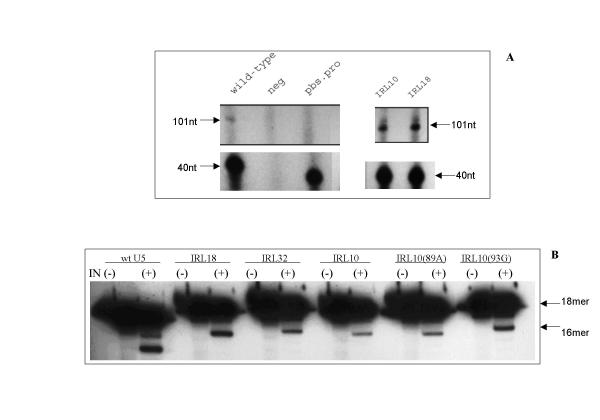
In vitro analysis of mutants from the U5-IR loop library. Panel A: Initiation of reverse transcription. Panels B: End processing by RSV integrase. Experiments were carried out as described in the legend to Figure 4. In all panels, the sample numbers refer to clones listed in Table 2. Two additional substrates derived from clone IRL-10 were examined, which substitute the wild type base pair at position 89 and 93, respectively.
Three mutants were tested in the integrase-processing assay; those were IRL10, IRL18, and IRL32. All of them were processed but less than the wild type level (Figure 6B). To see if the pattern exhibited at positions 89–91–93 was due to selection by integrase, derivatives of clone IRL10 were made which reintroduced a wild type base either at position 89 or 93 (Figure 6B). The substitution at position 89 and 93 increased detectable activity on the substrates by different amounts, respectively, but still not to the wild type level.
Discussion
In this study we examined the region containing RNA nucleotides 83–99, which includes the entire U5-IR stem and loop. This region of the RNA corresponds to positions 3–19 on the U5 DNA end. It provides an RNA structure involved in the proper reverse transcription and packaging of the viral genome, as well as the sequences required for recognition by the viral integrase [3] To look at the ability of RSV to tolerate mutations in this region, we employed an in vivo rapid evolution procedure, whereby random nucleotides are inserted into short stretches of the genome, and viral replication selects those sequences that are functional. This type of randomization of select regions of an RNA genome have been previously reported [15-18]. Using this method, we found that the U5-IR stem library is rapidly enriched for mutant viruses that maintain the proper RNA structure of the region. By the third round of infection in vivo, these mutant sequences are out-competed by wild type. In contrast, the U5-IR loop region was able to tolerate significant sequence variation. After five rounds of infection, wild type accounted for approximately 13% of the recovered sequences. Though present, it failed to dominate the library, suggesting that the selected mutants were able to replicate at or above wild type levels. We were also able to detect more complex patterns of selection within the loop, including a relationship between selection at three positions of the single stranded region, and differential selection of position 88 depending on whether the 88–94 base pair is intact. Our results demonstrate that RSV is able to tolerate a surprising amount of sequence variation in a region containing multiple overlapping biological functions.
One concern in designing libraries for analysis by an in vivo rapid evolution procedure was the number of nucleotides that should be randomized. Previous studies have utilized large libraries, up to 28 bases in size [17,18]. Had we randomized all 17 base pairs of the U5-IR stem and loop simultaneously, it would have resulted in a library of 1.7 × 1010 unique sequences, making it possible to screen only a small percentage of the possible clones, and unlikely for wild type to be represented in the library at all. The library was therefore split into two separate regions, of 8 base pairs (stem) and 9 base pairs (loop), resulting in library sizes of 2.62 × 105 and 1.05 × 106, respectively. These libraries are manageable enough that the majority of sequences would be represented in each experiment. The representative nature of these libraries is evidenced by the fact that wild type sequences were obtained in each case, and that in the stem library two independent clones were selected with the same sequence.
A second concern was whether sequences would be selected from the randomized region of the RNA during replication rather than by recombination between the two LTRs. The use of TEFs, which do not contain endogenous RSV sequences, precludes recombination between exogenous and endogenous sequences. Initial experiments with the randomized libraries in an unmodified RCAS vector did show immediate reversion to wild type sequence after only one round of selection (data not shown). However, once the RCAS vector was modified and a redundant PBS downstream of the 3' LTR of RCAS was removed, the opportunity for recombination between the two LTRs was significantly reduced and the randomized targets could be subjected to selection for replication competency. Additional evidence suggesting that selection rather than recombination explains our experimental findings is the large sequence heterogeneity found in the clones sequenced and the different rates to which wild type sequence appeared to replace the randomized regions. Moreover, when the adjacent TψC interacting region of the RNA was randomized, selection of sequence complementary to the tRNATrp occurred only when the PBS was complementary to the wild type RNA. When the PBS was changed to be complementary to tRNAPro, sequences selected from a randomized TψC interacting region of the RNA were complementary to tRNAPro, which could arise only by selection and not recombination between the LTRs [6] Finally, there was a small sequence difference between the U5 and the U3 LTR sequences. When the wild type sequence was recovered, we did not find the sequence marker from U3 in the targeted U5 LTR sequence, arguing that there was selection and not recombination. Alternatively, the wild type sequence could have arisen from the random library in part by error prone reverse transcription. However, we believe that this is less likely since it would require multiple misincorporations of deoxyribonucleotides across the randomized region of the genome and we have restricted the number of rounds of replication in these experiments to 5.
The ability of RSV to tolerate mutations within a defined region of the genome is evidenced primarily by the speed at which the library returns to wild type. Previous work by this lab randomized part of the primer binding site of RSV, and found that after only a single round of infection the library had selected exclusively for wild type sequence, showing that such immediate selection is possible if the biological pressure is strong enough [6] Therefore, the fact that it took three rounds of infection for the stem library to return to wild type demonstrates that RSV can replicate with mutations in this region. The mutant sequences, however, are assumed to be less fit than wild type since they disappeared by the third round of infection. While it is entirely possible that some mutant sequences persisted, they would represent such a small fraction of the library at this stage that it would be impractical to attempt to clone them. In contrast, the loop library consisted of mainly mutant sequences through five rounds of infection, and these mutant viruses continued to thrive despite the presence of wild type clones in the fifth round library. This indicates that the mutants identified in the loop library are not replication defective compared to wild type.
It is significant that within the U5-IR stem library there was selection of the structure prior to reversion to wild type. Six out of 19 mutant clones from the second round maintained the ability to form a stem structure similar to that of wild type. The statistical analysis proves that this was the result of selection by the virus, demonstrating that even in the absence of the wild type sequence the ability to preserve the structure of the stem imparts a survival advantage to the virus in vivo. We believe the alternative stem structure presented in Figure 3 is significant as well, since three clones from the library have the potential to form such a structure.
In vitro, six clones were tested as templates for initiation and elongation of reverse transcription. Even those clones that failed to maintain the U5-IR stem were able to serve as templates in this assay. Previous work identified mutations in this region that caused only partial defects in initiation in vitro [10]. The fact that all of the clones were functional is consistent with the biological selection. Additionally, five clones were tested for their ability to serve as substrates for 3' processing by integrase. All of these clones were defective compared to wild type. This was not unexpected, as many of the clones from this library lacked the 'CA' dinucleotide at positions 98–99. These positions are conserved in all retroviruses, and mutations at these sites are known to cause a defect in processing by IN. It has been reported that, in vitro, mutations within the IN recognition sequence can cause integrase to use an internal site for cleavage, deleting a portion of the viral DNA end in the process [11]. In vivo, these sequences would be outside of the transcribed RNA genome, so they should not impact subsequent steps of the viral life cycle. It is likely that these mutations persisted in vivo for a few rounds of replication in our experiments by using an internal site in this manner.
The U5-IR loop library has revealed an extremely complex pattern of selection, which highlights the sensitivity of this in vivo approach. Within a library of only nine targeted nucleotides, we detected at least four distinct levels of selection. Positions 87 and 95 selected primarily for wild type sequence while maintaining base pairing. Positions 88 and 94 maintained base pairing, but did not preserve the wild type bases. Within the single stranded loop region, positions 90 and 92 were largely random, and not linked to any other positions in the library. In contrast, positions 89, 91, and 93 displayed an interdependent pattern of selection, such that the base present at one position influenced base pair selection at the other two. Both statistical analysis, and an examination of individual clones from the starting library prove that all of these features of the IR-loop clones from the fifth round of infection were the result of selection by RSV. It also appears as if another level of selection is present for position 88; in 31 of 37 clones that maintain base pairing for 88–94, only 9 maintain the wild type 88C, whereas 15 clones selected for 88U. In contrast, 5 of the 6 clones which failed to maintain base pairing selected for the wild type 88C. It is possible that a C at position 88 imparts a selective advantage of its own, but is not optimal for base pairing. In vitro analysis of select clones from the fifth round pool show that these mutant sequences are efficient substrates for both initiation of reverse transcription and integrase 3' processing. This is consistent with the fact that these mutant viruses competed well with wild type.
Conclusions
It was surprising that so much variation was tolerated in a region of the RNA genome with multiple overlapping biological functions. The in vivo selection method utilized in this research has demonstrated the ability to detect highly complex patterns of selection, and to identify biologically relevant viral mutants. Key to this is keeping the library size small and sampling a large enough pool so that the majority of sequences are represented. In this study we were able to identify replication competent viruses, in the absence of any selective pressure against wild type reversion. In the future, combining such an approach with drug selection should make it possible to identify mutations that confer drug resistance before they appear in patients.
Methods
Reagents and cells
All enzymes except reverse transcriptase were purchased from New England Biolabs (Beverly, MA). AMV reverse transcriptase was purchased from Molecular Genetic Research (Tampa, FL). Deoxyribonucleotides were purchased from Roche Applied Science (Indianapolis, IN). Oligodeoxyribonucleotides were obtained from Genosys Biotechnologies, Inc. (Woodslands, TX) and from Integrated DNA Technologies Inc (Coralville, Iowa). Oligoribonucleotides and mutagenic oligodeoxyribonucleotides, which were prepared with equimolar mixes of all four nucleotides at each randomized position, were from Integrated DNA Technologies, Inc. E. coli DH10B bacteria and lipofectamine transfection reagent were purchased from Invitrogen (Gathersburg, MD). Primary TEFs, which do not contain endogenous retroviruses capable of recombining with RSV, were a generous gift from Rebecca Craven (Pennsylvania State Medical Center, Hershey PA). The plasmids pDC101S and RCAS, and their derivatives pDC101S.linker and RCAS.linkerΔ3'PBS have been described previously [6,10,14].
Construction of randomized libraries
Wild type pDC101S was amplified with mutagenic primers to create two randomized PCR products rIR-stem and rIR-loop. After amplification, the PCR products were purified by agarose gel electrophoresis using a QIAquick agarose gel extraction kit from Qiagen (Valencia, CA). The purified DNAs were then digested with SacI and SalI, and treated with shrimp alkaline phosphatase (SAP) to remove the 5' phosphate groups. The PDC101S.linker was digested with SalI and SacI, and the linker was removed using Microcon 50 spin columns from Amicon Bioseparations (Billerica, MA). The mutant inserts were ligated to the digested vector, using T4 DNA ligase with an insert to vector ratio of 4:1. After ligation, the reactions were heated to 65°C for 20 min to inactivate the T4 ligase and were then subjected to KpnI digestion. These products were introduced into E. coli DH10B by electroporation. Each plasmid was prepared from bacterial cultures and sequenced to confirm the location of the randomization. RCAS.linkerΔ3'PBS and the randomized pDC101S plasmids were digested with BsmI and SacI. The plasmid and inserts were prepared as described for the first cloning step. After ligation, the plasmid DNA was digested with SpeI to cut any residual RCAS.linker.Δ3'PBS. This product was then electroporated into E. coli DH10B. Plasmid DNA was prepared using the Qiagen EndoFree Plasmid Maxi kit.
Transfection and infection of cells
TEFs were transfected with mutant RCAS.linker.Δ3'PBS plasmids using the Lipofectamine PLUS reagent, as described by Invitrogen. Cells (2 × 106) in a 100 mm dish were transfected with 8 μg of DNA. Transfection efficiency was estimated using parallel experiments with a control vector encoding the beta-galactosidase gene driven by the RSV LTR. One-day post-lipofection, the media on the cells was changed in preparation for a 48 h virus collection period. Three days post-lipofection, mutant virus was harvested from the media by centrifugation through 20% sucrose gradients in the presence of STE (0.1 M NaCl, 10 mM Tris-HCl, pH 8.0, 0.1 mM EDTA) at 4°C for 90 min at 26,000 rpm in a SW27 rotor from Beckman Instruments (Palo Alto, CA). Virus pellets were suspended in STE and aliquots were assayed for reverse transcriptase (RT) activity as described previously [6]. Mock-transfected control cells were treated in an identical fashion. Equal quantities of virus (as measured by RT activity) in 1 ml of serum-free dMEM were used to infect polybrene-treated (5 μg/ml) TEFs for 1 h (2 × 106 cells in 100 mm dishes) at an MOI of 0.2. At three days post-infection, virus was harvested from the media, as was done following lipofection. Serial passage into uninfected TEFs was performed after each harvest, and continued through five rounds of infection, or until the library reverted to wild type.
Analysis of selected viral sequences
Infected TEF cells were trypsinized, washed with phosphate-buffered saline (PBS), suspended in 200 μl PBS and frozen at -20°C. Cellular DNA was purified from these cell samples using the QIAamp Tissue kit from Qiagen. The region of viral DNA surrounding the randomized region was PCR amplified from the cellular DNA. The PCR products, which represent the pool of sequences still present within the library, were recovered from a 1% agarose gel, purified using the QIAquick agarose gel extraction kit (Qiagen), and sequenced using the Thermo Sequenase radio labeled terminator cycle sequencing kit from USB Corporation (Cleveland, OH). Equimolar amounts of each PCR product were used in each sequencing reaction so that direct comparisons could be made between samples. Additionally, the purified PCR products were digested with SacI and ligated into pUC19 linearized with SacI. Ligation products were electroporated into E. coli DH10B, and the transformed bacteria were plated onto ampicillin-selection media. Individual colonies were picked, suspended in 10 μl of distilled water, heated to 95°C for 5 min, cooled on ice for 5 min, and debris removed by centrifugation for 3 min. DNA from each of these colony preparations was individually sequenced.
Making RNA templates for RT initiation
RNA templates carrying mutations in the U5-IR stem and loop were constructed to examine reverse transcription in vitro. A DNA template was assembled from two PCR products. The first product extends from position 1–119 of the viral sequence, and includes a T7 promoter sequence at the 5' end. The reverse primer in this PCR product introduces any desired mutations (ASV 119-70 5'-ATCACGTCGGGGTCACCAAATGAAGCCTTCTGCTTCATGCAGGTGCTCGT-3' where the underlined sequence indicates the location of the U5-IR stem and loop). The second product extends from position 100–1306, overlapping the first product in the PBS sequence. These two products were gel-purified and combined in an overlap extension PCR step to produce a full 1306 base pair T7 template. RNAs were prepared using the MEGAscript T7 kit from Ambion (Austin, TX), and purified using the Qiagen RNeasy kit according to manufacturer's instructions.
Initiation of reverse transcription
Reverse transcription initiation and elongation assays were carried out using a 1306 nucleotide RNA template and a 18mer nucleotide synthetic RNA primer. The reactions were set up in two steps. In the first step, 1.5 μg of template RNA was incubated with 1 mM each of dATP, dGTP, and dTTP, 10 μM dCTP, 0.003 pmol of α-32P dCTP, and 50 pmols of RNA primer in a volume of 16.5 μl for 10 min at 75°C. Also included was 1 pmol of an internal DNA primer, ASV 40-20, which yields a 40-nucleotide run-off product to control for template amount [1]. After this initial incubation, 7.5 U of AMV reverse transcriptase (Molecular Genetic Resources), 20 U RNase Inhibitor, and buffer was added, bringing the reactions to a final volume of 20 μl containing 50 mM Tris-HCl [pH8.3], 40 mM KCl, 8 mM MgCl2, and 1 mM DTT. The reactions were then incubated at 25°C for 15 min, followed by a second 50 min incubation at 42°C. Products were analyzed by polyacrylamide gel electrophoresis (PAGE) on a 5% gel and visualized using KODAK MR film.
Integrase end processing assays
The 3' end processing activity of RSV integrase (IN) was assayed using 18 base pair synthetic substrate mimicking the viral DNA end. An 100 pmol of the plus strand oligodeoxyribonucleotide (containing the conserved 'CA' dinucleotide) was 5' end labeled using 30 U T4 polynuclotide kinase (Amersham BioSciences, Arlington Heights, IL)and 2 μl γ-33P-ATP (2,500 Ci/mmole)(Amersham BioSciences). The labeled product was purified by PAGE by migration though a 20% gel. This was then annealed to an excess of unlabeled complimentary strand for the processing assays. Reaction conditions were as follows (prepared on ice). All components were added individually, the final reaction mixture contained 20 mM MOPs PH7.2, 3 mM DTT, 100 μg/ml BSA, 1 μg RSV integrase, 0.5 pmol of labeled duplex substrate (specific activity of 105cpm/pmol). The mixture was preincubated on ice at 4°C overnight. Before processing, MgCl2 was added to final concentration 10 mM, then the reaction mixture incubated at 37°C for 90 min. Samples were mixed with stop buffer (95% formamide, 20 mM EDTA, 0.1% xylene cyanol, 0.1%bromphenol blue), heated 95°C for 5 min, placed on ice, and then separated through a 20% polyacrylamide gel. Labeled reaction products were visualized using KODAK MR film.
Statistical analysis
Multinomial distribution was used to evaluate the probability of recovering multiple consecutive base pairs from a random library. The p-value P(r1, r2,..., rn) = N!/[(r1!)(r2!)...(rn!)] × (p1r1)(p2r2)(pnrn), where "r" represents the reported frequencies, "p" represents the probability of each frequency, and N equals the sample size. Chi square analysis was used to compare the base pair distribution of positions 89, 91, and 93 of the U5-IR loop library. The chi square value is determined by the formula χ2 = Σ[(O-E)2/E], where "O" is the observed frequency, and "E" is the expected frequency based on the assumption that both samples are identical.
Authors' contributions
MJ performed the selection of libraries, sequence analysis of mutants, cDNA100 synthesis assays. SM constructed the random libraries and established the methods for selection. AC performed in vitro IN assays. ES participated in the design of the study. JL is the senior and corresponding author.
Acknowledgments
Acknowledgements
This work was supported in part by United States Public Health Grants CA38046 and CA52047 (to J.L.) and CA43600 (to E.S.). S.M. is a Medical Scientist Trainee supported by grant GM07250 from the National Institutes of Health. M.J. is supported by Carcinogenesis Training Program grant CA09560 from the National Institutes of Health. We would like to thank Rebecca Craven for the generous gift of primary Turkey Embryo Fibroblasts.
Contributor Information
Michael Johnson, Email: michaeljohnson@northwestern.edu.
Shannon Morris, Email: srmorris11@yahoo.com.
Aiping Chen, Email: a-chen2@northwestern.edu.
Ed Stavnezer, Email: exs44@po.cwru.edu.
Jonathan Leis, Email: j-leis@northwestern.edu.
References
- Aiyar A, Ge Z, Leis J. A specific orientation of RNA secondary structures is required for initiation of reverse transcription. J Virol. 1994;68: 611–618. doi: 10.1128/jvi.68.2.611-618.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobrinik D, Soskey L, Lei J. A retroviral RNA secondary structure required for efficient initiation of reverse transcription. J Virol. 1988;62: 3622–3630. doi: 10.1128/jvi.62.10.3622-3630.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobrinik D, Aiyar A, Ge Z, Katzman M, Huang H, Leis J. Overlapping retrovirus U5 sequence elements are required for efficient integration and initiation of reverse transcription. J Virol. 1991;65: 3864–3872. doi: 10.1128/jvi.65.7.3864-3872.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris S, Leis J. Changes in Rous sarcoma virus RNA secondary structure near the primer binding site upon tRNATrp primer annealing. J Virol. 1999;73: 6307–6318. doi: 10.1128/jvi.73.8.6307-6318.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiyar A, Cobrinik D, Ge Z, Kung HJ, Leis J. Interaction between Retroviral U5 RNA and the TC Loop of the tRNATrp Primer Is Required for Efficient Initiation of Reverse Transcription. J Virol. 1992;66: 2464–2472. doi: 10.1128/jvi.66.4.2464-2472.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris S, Johnson M, Stavnezer E, Leis J. Replication of avian sarcoma virus in vivo requires an interaction between the viral RNA and the TpsiC loop of the tRNA(Trp) primer. J Virol. 2002;76: 7571–7577. doi: 10.1128/JVI.76.15.7571-7577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerens N, Klaver B, Berkhout B. A structured RNA motif is involved in correct placement of the tRNA(3)(Lys) primer onto the human immunodeficiency virus genome. J Virol. 2000;74: 2227–2238. doi: 10.1128/JVI.74.5.2227-2238.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerens N, Groot F, Berkhout B. Initiation of HIV-1 reverse transcription is regulated by a primer activation signal. J Biol Chem. 2001;276:31247–31256. doi: 10.1074/jbc.M102441200. [DOI] [PubMed] [Google Scholar]
- Leis J, Aiyar A, Cobrinik D. Reverse Transcriptase. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.; 1993. [Google Scholar]
- Miller J, Ge Z, Morris S, Das K, Leis J. Multiple biological roles associated with the Rous sarcoma virus 5' untranslated RNA U5-IR stem and loop. J Virol. 1997;71: 7648–7656. doi: 10.1128/jvi.71.10.7648-7656.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindmarsh P, Johnson M, Reeves R, Leis J. Base-pair substitutions in avian sarcoma virus U5 and U3 long terminal repeat sequences alter the process of DNA integration in vitro. J Virol. 2001; 75:1132–1141. doi: 10.1128/JVI.75.3.1132-1141.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzman M, Katz R, Skalka AM, Leis J. The avian retroviral integration protein cleaves the terminal sequences of linear viral DNA at the in vivo sites of integration. J Virol. 1989;63:5319–5327. doi: 10.1128/jvi.63.12.5319-5327.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AL, Katzman M. Subterminal viral DNA nucleotides as specific recognition signals for human immunodeficiency virus type 1 and visna virus integrases under magnesium-dependent conditions. J Gen Virol. 2000;81:839–849. doi: 10.1099/0022-1317-81-3-839. [DOI] [PubMed] [Google Scholar]
- Hughes SH, Greenhouse J, Petropoulos J, Sutrave P. Adaptor plasmids simplify the insertion of foreign DNA into helper-independent retroviral vectors. J Virol. 1987;61: 3004–3012. doi: 10.1128/jvi.61.10.3004-3012.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doria-Rose Na Voge VM. in vivo selection of Rous sarcoma virus mutants with randomized sequences in the packaging signal. JVirol. 1998;72:8073–8082. doi: 10.1128/jvi.72.10.8073-8082.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkhout B Klaver B. in vivo selection of randomly mutated retroviral genomes. nucleic acids Res. 1993;21:5020–5024. doi: 10.1093/nar/21.22.5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan H, Carpenter C, Simon A. Analysis of cis-acting sequences involved in plus-strand synthesis of a turnip crinkle virus-associated satellite RNA identifies a new carmovirus replication element. Virology. 2000;268: 345–354. doi: 10.1006/viro.1999.0153. [DOI] [PubMed] [Google Scholar]
- Zhang G. Simon A. A multifunctional turnip crinkle virus replication enhancer revealed by in vivo functional SELEX. J Mol Biol. 2003;326: 35–48. doi: 10.1016/S0022-2836(02)01366-9. [DOI] [PubMed] [Google Scholar]